Synthesis and Ab Initio/DFT Studies on 2-(4-methoxyphenyl)benzo[d]thiazole

1
Department of Chemistry, Faculty of Arts and Science, Mersin University, 33343-Mersin, Turkey
2
Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Mersin University, 33169-Mersin, Turkey
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2007, 8(8), 760-776; https://0-doi-org.brum.beds.ac.uk/10.3390/i8080760
Submission received: 29 June 2007
/
Revised: 18 July 2007
/
Accepted: 18 July 2007
/
Published: 3 August 2007
(This article belongs to the Section Physical Chemistry, Theoretical and Computational Chemistry)
Abstract
:2-aminothiophenol and 4-methoxybenzaldehyde were cyclized under microwave irridation and solvent free conditions to synthesize 2-(4-methoxyphenyl)benzo[d]thiazole. The molecular structure and vibrational frequencies of the title compound in the ground state have been investigated with ab initio (HF) and density functional theory methods (BLYP, B3LYP, B3PW91 and mPW1PW91) implementing the standard 6–311G(d,p) basis set. Comparison of the observed fundamental vibrational frequencies of title compound and calculated results by HF and DFT methods indicate that B3LYP is superior to the scaled HF approach for molecular problems.
1. Introduction
The synthesis of benzothiazoles and their derivatives has been of considerable interest to organic and medicinal chemists for many years as indicated by large number of drugs including this group [1–4]. This heterocyclic nucleus is a very important group because of its potent antitumor activity [5–8] and other important pharmaceutical utilities, such as treatment of inflammatory diseases, epilepsy, analgesia, viral infections, cancer, and tuberculosis [9–15]. Particularly, there is significant interest in the synthesis of 2-substituted benzothiazoles in recent years [16,17]. The benzothiazoyl-moiety is a structure element of compounds with potent and selective antitumor activity. For instance, 2-(4-aminophenyl)benzothiazoles exhibit nanomolar inhibitory activity against a range of human breast, ovarian, colon, and renal cell lines in vitro [18,19]. Structure-activity relationships derived using these cell types has revealed that activity follows the heterocyclic sequence benzothiazole>benzoxazole>benzimidazole and the 2-(4-amino-3-methylphenyl)benzothiazole derivative is found as the most potent compound in this series with its activity extending to ovarian, lung and renal cell lines. Our earlier work led to the development of new antitumor active compounds with a benzothiazole skeleton.
IR spectroscopy is usually considered as an important experimental method for chemists. The experimental and theoretical vibrational spectrum assignments of free benzothiazole have not been published so far. In the present work, we have synthesized and calculated the geometric parameters and vibrational frequencies of the title compound in the ground state to distinguish the fundamental from the experimental vibrational frequencies using the Hartree-Fock (HF) [20], density functional by employing Becke’s three-parameter hybrid method [21] with the Lee, Yang, and Parr correlation functional methods [22] (B3LYP), Becke’s exchange functional in combination with the Lee, Yang and Parr correlation functional methods (BLYP) [22,23], the Barone and Adamo's Becke-style one-parameter functional using the modified Perdew–Wang exchange and Perdew–Wang 91 correlation method, (mPW1PW91) [24,25], Becke's three parameter exchange functional combined with gradient corrected correlation functional of Perdew and Wang's 1991 (B3PW91) [23,26], and 6–311G(d,p) basis set. Furthermore, we interpreted the calculated spectra in terms of potential energy distributions (PEDs) and made the assignment of all experimental bands based on PED analysis results.
2. Results and Discussion
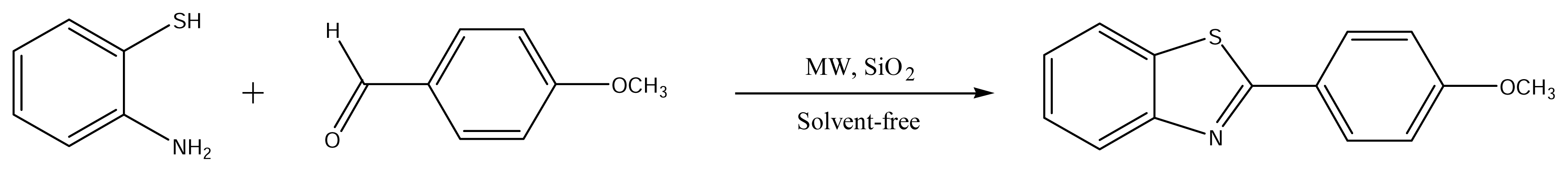
The simplest route to substitute 2-phenylbenzothiazoles involves various reactions between o-aminothiophenols and substituted benzoic acid derivatives (e.g., in polyphosphoric acid) [27]. In recent years, the use of microwave irridation to simplify and improve classical organic reactions has become a very popular method [28] because it often leads to high yields, clean reactions, and shorter reaction times. We have carried out the reaction of aromatic aldehydes with o-aminothiophenol in the presence of silica gel under microwave irradiation and solvent free conditions. It has been found that the 2-(4-methoxyphenyl)benzo[d]thiazole was obtained in good yield. The optimum conditions employed are that a molar ratio of aldehyde and o-aminothiophenol is 1:2 and irridation time and power levels of microwave set-up are 6 min and 300 W.
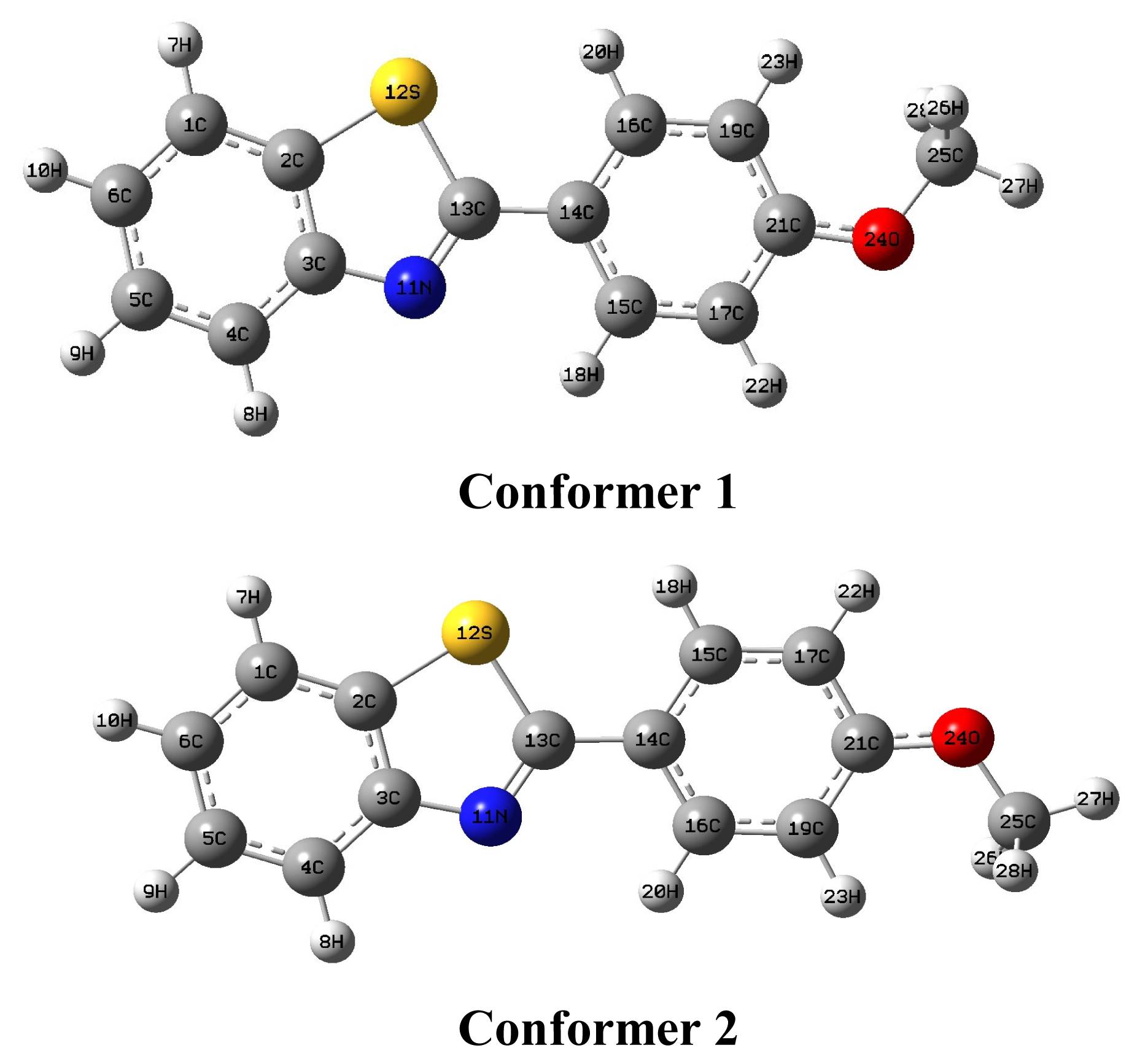
2.1. Conformational stability
To establish the most stable conformation as the initial point for further calculations, the molecule was submitted to a rigorous conformation analysis around the free rotation bonds. This study was performed with the software Spartan 06 [29]. The structure of the title compound shown in Figure 1 has two conformations. Energetics, gathered in Table 1, show that the Conformer 1 is the most stable. Therefore, further in this paper, we focus on this particular form of the title compound.
2.2. Molecular geometry
In this work, we performed full geometry optimization of the title compound. The optimized structure parameters of the title compound calculated by ab initio and DFT methods listed in Table 2 are in accordance with atom numbering scheme given in Figure 1. To the best of our knowledge, crystal data of the 2-(4-methoxyphenyl)benzo[d]thiazole are not available in the literature. Therefore, the optimized structure can only be compared with crystal structure of other similar systems [30–32]. We have noticed that the DFT calculations are consistent with the results of X-ray data. From Table 2, it was found that the bond angles calculated by DFT methods are consistent with those by HF method. However the bond lengths calculated by HF method are little shorter than those obtained by DFT method.
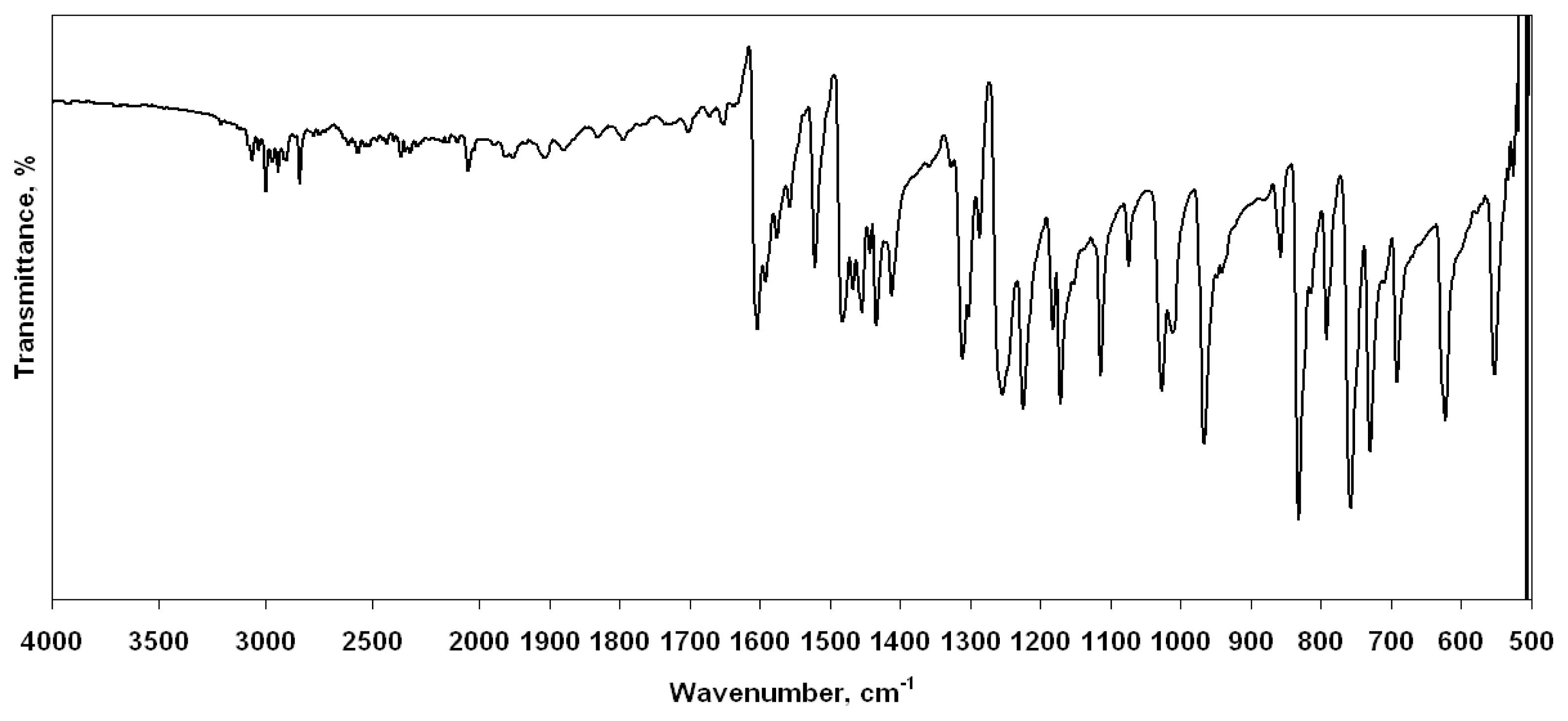
2.3. Vibrational assignments
The vibrational spectra of benzo[d]thiazole derivatives have not been described in detail in any literature. Therefore, we focused on a benzo[d]thiazole derivative, 2-(4-methoxyphenyl) benzo[d]thiazole. The FT-IR spectrum of the title compound is shown in Fig. 2. We have calculated the theoretical vibrational spectra of 2-(4-methoxyphenyl)benzo[d]thiazole by using HF, B3LYP, BLYP, B3PW91 and mPW1PW91 methods with 6–311G(d,p) basis set. None of the predicted vibrational spectra has no imaginary frequency, implying the optimized geometry is located at the local lowest point on the potential energy surface. It is known that ab initio and DFT potentials systematically overestimate the vibrational wavenumbers. These discrepancies can be corrected either by computing anharmonic corrections explicitly or by introducing a scaled field [33] or directly scaling the calculated wavenumbers with the proper factor [34]. Considering systematic errors with scaling factor of 0.9051, 0.9679, 0.9934, 0.9631 and 0.9567, we calibrated the vibrational wavenumbers calculated by HF, B3LYP, BLYP, B3PW91 and mPW1PW91, respectively. After scaled with a scaling factor, the deviation from experiments is less than 10 cm−1 with a few exceptions. Theoretical and experimental results of the title compound are shown in Table 3. The vibrational bands’ assignments have been made by using both the animation option of GaussView 3.0 graphical interface for Gaussian programs [35] and VEDA 4 program [36]. All the calculated spectra are in a good agreement with the experimental data. All DFT methods are superior to HF method in terms of realistic reproduction of both band intensity distribution and general spectral features.
The characteristic νCH stretching vibrations of heteroaromatic structure are expected to appear in 3000–3100 cm−1 frequency ranges. The νCH stretching vibrations of the title compound were assigned to four bands observed at 3111, 3084, 3076, and 3062 cm−1 in the infrared spectrum. The B3LYP calculated wavenumbers of these bands very well reproduced the experimental ones. The difference between experimental and calculated νCH is about 3, 7, 4, and 4 cm−1[37].
For the assignments of CH3 group frequencies, eight fundamental vibrations can be associated to CH3 groups. Three stretching, one umbrella, one rocking and three deformation vibration mode designated the motion of the methyl group. The CH3 asymmetric and symmetric stretching frequencies are established at 3034, 2968, and 2910 cm−1 in infrared spectra. The three methyl hydrogen deformation modes are also well established in the spectra. We have observed the methyl deformation mode at 1483, 1467, and 1454 cm−1 in the infrared. The calculated band at 1433 and 1122 cm−1 are attributed to an umbrella and a CH3 rocking vibration, respectively [38].
The characteristic region of the benzothiazole derivatives on the spectrum is 1500–1650 cm−1 as benzimidazole derivatives. The vibrational frequencies and intensities for all substituted benzothiazole derivatives in this range differ from the position of the substituent [39]. Generally, two bands observed in 1500–1650 cm−1 range for benzothiazole derivatives. The observed vibrational frequencies (1591 and 1558 cm−1) are generally intense because of the conjugation between the benzene and thiazole rings. In addition, the vibrational frequencies in this range vary with the electronegativity of the substituent. The 4-methoxyphenyl group in position 2 is less apt to influence the vibrational frequencies in this range. Additionally, we obtained another vibrational frequency at 1604 and 1571 cm−1 which comes from 4-methoxyphenyl group.
The identification of νC-N vibrations is a difficult task since the mixing of vibrations is possible in this region. However, with the help of the animation option of GaussView 3.0 graphical interface for gaussian programs and VEDA 4 program, the νC-N vibrations are identified and assigned in this study. The IR bands appearing at 1521, 1483, and 1467 cm−1 are assigned to νC=N vibrations and 1267, 1253, and 1207 cm−1 are assigned to νC-N vibrations with the δCH for the title compound. All these results agree with Sundaraganesan et al. [40] and Krishnakumar et al. [41,42].
The O–CH3 mode is assigned in the region 1000–1100 cm−1 for anisole and its derivatives [38,40,43–45]. In this work, the O–CH3 stretching mode is assigned to medium IR band at 1010 cm−1. The theoretically computed value at 1005cm−1 exactly coincides with the experimental results. The C–O–CH3 angle bending mode is assigned near 300 cm−1 for anisole by and at 421 cm−1 for p-methoxy benzaldehyde by Campagnaro and Wood [46]. Owen and Hester [47], Sundaraganesan et al. [40] and Ramana Rao et al. [43–45,48] have proposed assignment for C–O–CH3 angle bending mode in the region 300–670 cm−1 for anisole and its derivatives. As this mode lies in the region of the ring planar C–C–C angle bending modes, a strong mixing amongst these two modes and other planar modes is expected. Accordingly, we have assigned the theoretically calculated value by B3LYP/6–311G(d,p) at 461 cm−1 as C–O–CH3 angle bending mode.
A better performance of B3LYP, BLYP, B3PW91 and mPW1PW91 versus HF in general can be quantitatively characterized by using the mean absolute deviation, root mean square values and coefficients of correlation (cc) between the calculated and observed vibration frequencies and given in Table 4. The root mean square (RMS) values were obtained in this study using the equations (12) and (13) from Ref. [22]. The cc values for all three DFT methods were bigger than 0.9998, whereas for HF it was 0.9997. These values are very close to those reported for the literature data [49–59].
These results indicate that the fundamental frequencies calculated (DFT) for the title compound show quite good agreement with experimental values. Furthermore, the B3LYP method calculations approximate the observed fundamental frequencies much better than results of the other investigated DFT methods. This is also proved by the low RMS deviation values of about 7.7 cm−1. The RMS value obtained with the B3LYP method is smaller than those obtained by Rauhut and Pulay [60] for a group of 20 molecules (RMS = 13 cm−1). The small difference between experimental and calculated vibrational modes is observed. This discrepancy can come from the formation of intermolecular hydrogen bonding. Also, we note that the experimental results obtained from solid phase and theoretical calculations belong to gaseous phase.
3. Experimental Section
3.1. Synthesis of 2-(4-methoxyphenyl)benzothiazol
All chemicals used for the preparation of the title compound were reagent grade quality. To a solution of 4-methoxybenzaldehyde (3 mmol) and o-aminothiophenol (6 mmol) in diethylether (10 mL) silica gel (3 g) was added (Scheme 1). The slurry was mixed thoroughly and the solvent was removed by rotary evaporation. The resultant solid was subjected to microwave irridation using microwave oven operating at 300W for 6 min. After cooling, the product was extracted with ethyl acetate. The extract was then filtered and the filtrate was evaporated under reduced pressure to yield the crude product. The product was purified by recrystallization in methanol. (Yield 94%), M.p. 393 K (392–394 K) [61]. 1H NMR (DMSO-d6) δ: 8.04 (J=2, d, 1H), 8.02 (J=2.4, 1H, s), 7.87 (J=8, 1H, d), 7.46 (1H, t), 7.35 (1H, t), 7.00 (J=2, d, 1H), 6.90 (J=2, 2H, d), 3.87 (3H, s, OMe). MS (70 eV) m/z (%): 242 (M+1, 80). Anal. required for C14H11NSO: C, 69.68; H, 4.59; N, 5.80; Found: C, 69.45; H, 4.51; N, 5.82% [62].
3.2. Instrumentation
Microwave reaction was carried out in 10 mL sealed glass tubes in focused mono-mode microwave oven (Discover by CEM). Melting points were determined using an Electrothermal-9300 Digital Melting Points Apparatus (Electrothermal Inc., Essex, UK). The 1H-NMR spectra were recorded on Bruker 400 MHz NMR spectrometer. Chemical shifts are reported in parts per million relative to internal standard tetramethylsilane. Microanalyses were performed by TUBITAK Analytical Laboratory (Ankara, Turkey). Mass spectra were recorded on a VG Autospec, using the FAB technique. The room temperature attenuated total reflection Fourier transform infrared (FT-IR ATR) spectrum of the 2-(4-methoxyphenyl)benzo[d]thiazole was recorded using Varian FTS1000 FT-IR spectrometer with Diamond/ZnSe prism (4000–525 cm−1; number of scans: 250; resolution: 1 cm−1) in the solid (Fig. 2).
3.3. Calculation details
The conformation analysis study was carried out by Spartan 06 program package [29]. All the other calculations were performed with the Gaussian 03W program package on a double Xeon/3.2 GHz processor with 8 GB Ram [63]. The molecular structure of the title compound, in the ground state are optimized by using the Hartree-Fock (HF) [20], density functional using Becke’s three-parameter hybrid method [21] with the Lee, Yang, and Parr correlation functional methods [22] (B3LYP), Becke’s exchange functional in combination with the Lee, Yang and Parr correlation functional methods (BLYP) [22,23], the Barone and Adamo's Becke-style one-parameter functional using the modified Perdew–Wang exchange and Perdew–Wang 91 correlation method, (mPW1PW91) [24,25], Becke's three parameter exchange functional combined with gradient corrected correlation functional of Perdew and Wang's 1991 (B3PW91) [23,26], and 6–311G(d,p) basis set. The vibrational frequencies were also calculated with these methods. The frequency values computed at these levels contain known systematic errors [64]. Therefore, we have used the scaling factor values of 0.9051, 0.9679, 0.9934, 0.9631 and 0.9567 for HF, B3LYP, BLYP, B3PW91 and mPW1PW91, respectively [34,49]. The assignment of the calculated wave numbers is aided by the animation option of GaussView 3.0 graphical interface for gaussian programs, which gives a visual presentation of the shape of the vibrational modes [35]. Furthermore, theoretical vibrational spectra of the title compound were interpreted by means of PEDs using VEDA 4 program [36].
Figure 1.
Two stable conformers of the title compound calculated at HF/6–311G(d,p) level.

Figure 2.
FT-IR spectrum of 2-(4-methoxyphenyl)benzo[d]thiazole recorded at room temperature.

Scheme 1.
Synthesis of the title compound.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Energies of the different conformations of the title compound calculated at the HF/6–311G(d,p) levela.
| Conformer | E (Hartree) | ΔE (kcal/mol) | Dipole moment (D) |
|---|---|---|---|
| 1 | −1063.40977 | 0.0000 | 2.2783 |
| 2 | −1063.40973 | 0.0249 | 1.3747 |
aTotal energies, E (with the zero-point vibrational energy correction).
| Parameters | Calculated | Experimental | ||||
|---|---|---|---|---|---|---|
| HF | DFT | |||||
| BLYP | B3LYP | B3PW91 | mPW1PW91 | |||
| Bond lengths (Å) | ||||||
| R(1,2) | 1.388 | 1.403 | 1.394 | 1.392 | 1.391 | 1.394(2)a |
| R(1,6) | 1.378 | 1.401 | 1.390 | 1.388 | 1.386 | 1.384(2)a |
| R(2,3) | 1.391 | 1.428 | 1.414 | 1.412 | 1.410 | 1.402(17)a |
| R(2,12) | 1.743 | 1.767 | 1.750 | 1.740 | 1.737 | 1.7367(13)a |
| R(3,4) | 1.393 | 1.411 | 1.401 | 1.399 | 1.397 | 1.3950(18)a |
| R(3,11) | 1.382 | 1.387 | 1.379 | 1.374 | 1.373 | 1.3854(16)a |
| R(4,5) | 1.377 | 1.397 | 1.387 | 1.384 | 1.382 | 1.381(2)a |
| R(5,6) | 1.397 | 1.412 | 1.403 | 1.401 | 1.400 | 1.390(2)a |
| R(11,13) | 1.270 | 1.308 | 1.295 | 1.295 | 1.293 | 1.3150(15)a |
| R(12,13) | 1.766 | 1.822 | 1.792 | 1.778 | 1.772 | 1.764(4)a |
| R(13,14) | 1.475 | 1.469 | 1.464 | 1.461 | 1.459 | 1.463(2)b |
| R(14,15) | 1.394 | 1.416 | 1.405 | 1.402 | 1.400 | 1.400(4)c |
| R(14,16) | 1.387 | 1.413 | 1.401 | 1.398 | 1.396 | 1.377(4)c |
| R(15,17) | 1.374 | 1.392 | 1.383 | 1.381 | 1.379 | 1.381(4)c |
| R(16,19) | 1.384 | 1.398 | 1.389 | 1.387 | 1.386 | 1.375(5)c |
| R(17,21) | 1.392 | 1.412 | 1.402 | 1.400 | 1.398 | 1.391(4)c |
| R(19,21) | 1.388 | 1.411 | 1.400 | 1.398 | 1.396 | 1.376(5)c |
| R(21,24) | 1.341 | 1.374 | 1.359 | 1.353 | 1.350 | 1.352(4)c |
| R(24,25) | 1.401 | 1.442 | 1.423 | 1.416 | 1.412 | 1.424(4)c |
| Bond angles (o) | ||||||
| A(2,1,6) | 118.08 | 118.16 | 118.12 | 118.05 | 118.03 | 117.95(14)a |
| A(1,2,3) | 121.65 | 121.50 | 121.54 | 121.61 | 121.62 | 121.44(12)a |
| A(1,2,12) | 129.51 | 129.12 | 129.28 | 129.27 | 129.29 | 129.55(10)a |
| A(3,2,12) | 108.84 | 109.39 | 109.18 | 109.12 | 109.09 | 109.01(9)a |
| A(2,3,4) | 119.73 | 119.26 | 119.37 | 119.33 | 119.35 | 119.31(1)a |
| A(2,3,11) | 115.26 | 115.51 | 115.35 | 115.33 | 115.34 | 115.97(11)a |
| A(4,3,11) | 125.00 | 125.23 | 125.28 | 125.34 | 125.31 | 124.70(11)a |
| A(3,4,5) | 118.86 | 119.20 | 119.12 | 119.10 | 119.08 | 119.31(13)a |
| A(4,5,6) | 120.90 | 120.87 | 120.88 | 120.92 | 120.92 | 120.74(14)a |
| A(1,6,5) | 120.78 | 121.01 | 120.97 | 121.00 | 121.00 | 121.22(14)a |
| A(3,11,13) | 112.11 | 112.43 | 112.21 | 111.89 | 111.78 | 110.72(10)a |
| A(2,12,13) | 88.78 | 88.38 | 88.66 | 88.85 | 88.88 | 89.50(6)a |
| A(11,13,12) | 115.01 | 114.30 | 114.59 | 114.80 | 114.92 | 114.78(9)a |
| A(11,13,14) | 123.50 | 124.51 | 124.16 | 124.07 | 123.95 | 122.78(19)b |
| A(12,13,14) | 121.49 | 121.19 | 121.26 | 121.13 | 121.13 | 121.84(15)b |
| A(13,14,15) | 122.42 | 122.64 | 122.53 | 122.51 | 122.46 | 121.80(17)b |
| A(13,14,16) | 119.38 | 119.35 | 119.30 | 119.24 | 119.21 | 119.73(18)b |
| A(15,14,16) | 118.20 | 118.01 | 118.17 | 118.25 | 118.33 | 118.48(17)b |
| A(14,15,17) | 121.07 | 121.13 | 121.05 | 121.02 | 120.98 | 119.71(18)b |
| A(14,16,19) | 121.36 | 121.37 | 121.30 | 121.26 | 121.22 | 121.56(19)b |
| A(15,17,21) | 120.19 | 120.23 | 120.23 | 120.21 | 120.19 | 120.0(3)c |
| A(16,19,21) | 119.74 | 119.81 | 119.81 | 119.80 | 119.78 | 120.0(3)c |
| A(17,21,19) | 119.45 | 119.45 | 119.45 | 119.46 | 119.50 | 118.9(3)c |
| A(17,21,24) | 115.93 | 115.54 | 115.79 | 115.86 | 115.88 | 115.8(3)c |
| A(19,21,24) | 124.62 | 125.00 | 124.75 | 124.68 | 124.62 | 125.1(3)c |
| A(21,24,25) | 120.11 | 118.40 | 118.70 | 118.37 | 118.30 | 118.3(3)c |
| No | Exp. | HF | DFT | Assignments, PED (%)g | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BLYP | B3LYP | B3PW91 | mPW1PW91 | ||||||||||||||
| Wave numbera | Rel. Inten. | Wave numbera | Rel. Inten. | Wave numbera | Rel. Inten. | Wave numbera | Rel. Inten. | Wave numbera | Rel. Inten. | ||||||||
| Unscal. | Scaledb | Unscal. | Scaledc | Unscal. | Scaledd | Unscal. | Scalede | Unscal. | Scaledf | ||||||||
| 1 | 3111 | 3380 | 3059 | 2 | 3131 | 3110 | 4 | 3211 | 3108 | 3 | 3219 | 3101 | 3 | 3238 | 3098 | 3 | νCH, sym, ring 1, (99) |
| 2 | 3084 | 3363 | 3044 | 1 | 3118 | 3097 | 2 | 3198 | 3095 | 1 | 3210 | 3092 | 4 | 3229 | 3090 | 4 | νCH, asym, ring 1, (93) |
| 3 | 3084 | 3362 | 3043 | 1 | 3117 | 3097 | 6 | 3198 | 3095 | 5 | 3210 | 3091 | 1 | 3229 | 3089 | 1 | νCH, sym, ring 2, (87) |
| 4 | 3084 | 3359 | 3040 | 3 | 3117 | 3096 | 5 | 3197 | 3094 | 2 | 3205 | 3087 | 1 | 3224 | 3084 | 1 | νCH, asym, ring 1, (89) |
| 5 | 3084 | 3351 | 3033 | 5 | 3110 | 3090 | 9 | 3190 | 3088 | 6 | 3203 | 3085 | 5 | 3223 | 3083 | 5 | νCH, asym, ring 2, (99) |
| 6 | 3076 | 3338 | 3021 | 2 | 3100 | 3079 | 4 | 3180 | 3078 | 2 | 3192 | 3075 | 2 | 3212 | 3073 | 2 | νCH, asym, ring 2, (100) |
| 7 | 3076 | 3337 | 3020 | 2 | 3093 | 3073 | 3 | 3172 | 3070 | 3 | 3180 | 3063 | 1 | 3200 | 3061 | 1 | νCH, asym, ring 1, (100) |
| 8 | 3062 | 3324 | 3008 | 0 | 3088 | 3068 | 0 | 3168 | 3066 | 0 | 3180 | 3063 | 2 | 3198 | 3060 | 3 | νCH, asym, ring 2, (95) |
| 9 | 3034 | 3289 | 2977 | 9 | 3061 | 3041 | 9 | 3137 | 3037 | 8 | 3152 | 3036 | 7 | 3171 | 3034 | 8 | νCH, asym, CH3, (92) |
| 10 | 2968 | 3230 | 2923 | 10 | 2988 | 2969 | 15 | 3067 | 2969 | 12 | 3082 | 2969 | 11 | 3100 | 2966 | 12 | νCH, asym, CH3, (100) |
| 11 | 2910 | 3167 | 2867 | 11 | 2932 | 2912 | 29 | 3007 | 2911 | 21 | 3015 | 2904 | 21 | 3032 | 2901 | 22 | νCH, sym, CH3, (92) |
| 12 | 1604 | 1797 | 1627 | 43 | 1585 | 1574 | 63 | 1651 | 1598 | 62 | 1666 | 1604 | 66 | 1681 | 1608 | 73 | νC=C, ring 1, (43) |
| 13 | 1591 | 1790 | 1620 | 4 | 1571 | 1560 | 2 | 1638 | 1586 | 2 | 1653 | 1592 | 2 | 1669 | 1597 | 2 | νC=C, ring 2, (59) + νCN (10) |
| 14 | 1571 | 1759 | 1592 | 11 | 1540 | 1530 | 2 | 1608 | 1556 | 3 | 1623 | 1563 | 4 | 1639 | 1568 | 4 | νC=C, ring 1, (42) + δCCC (12) |
| 15 | 1558 | 1749 | 1583 | 1 | 1533 | 1523 | 2 | 1597 | 1546 | 2 | 1611 | 1551 | 2 | 1626 | 1555 | 2 | νC=C, ring 2, (57) + νCN (13) |
| 16 | 1521 | 1728 | 1564 | 13 | 1495 | 1485 | 8 | 1560 | 1510 | 5 | 1575 | 1517 | 6 | 1591 | 1522 | 7 | νCN (38)+νCC (12)+ δCCC (23) |
| 17 | 1483 | 1676 | 1517 | 55 | 1471 | 1462 | 22 | 1533 | 1483 | 67 | 1540 | 1483 | 77 | 1552 | 1485 | 89 | νCN (31) + δCH3, deform. (26) |
| 18 | 1467 | 1630 | 1476 | 9 | 1463 | 1453 | 58 | 1505 | 1457 | 24 | 1501 | 1445 | 23 | 1509 | 1444 | 24 | νCN (12) + δCH3, deform. (58) |
| 19 | 1454 | 1622 | 1468 | 1 | 1453 | 1444 | 3 | 1494 | 1446 | 3 | 1493 | 1438 | 3 | 1504 | 1439 | 3 | δCH3, deform. (91) |
| 20 | 1442 | 1609 | 1457 | 4 | 1440 | 1430 | 3 | 1489 | 1441 | 3 | 1489 | 1434 | 3 | 1497 | 1432 | 3 | νCC, ring 2, (26) + δCH (47) |
| 21 | 1433 | 1608 | 1455 | 8 | 1435 | 1426 | 12 | 1480 | 1433 | 6 | 1475 | 1421 | 4 | 1484 | 1420 | 3 | δCH3, umbrella (76) |
| 22 | 1412 | 1600 | 1448 | 12 | 1413 | 1404 | 9 | 1465 | 1418 | 12 | 1470 | 1415 | 11 | 1481 | 1416 | 14 | δCH, ring 2, (45) |
| 23 | 1400 | 1560 | 1412 | 3 | 1404 | 1394 | 9 | 1450 | 1404 | 5 | 1456 | 1402 | 3 | 1465 | 1402 | 3 | νCC, ring 1, (40) + δCH (13) |
| 24 | 1309 | 1448 | 1310 | 6 | 1312 | 1304 | 3 | 1346 | 1303 | 2 | 1369 | 1318 | 5 | 1377 | 1317 | 5 | νCC, ring 2, (52) |
| 25 | 1303 | 1422 | 1287 | 100 | 1307 | 1299 | 27 | 1342 | 1299 | 39 | 1366 | 1316 | 28 | 1375 | 1315 | 35 | νCC, ring 1, (53) |
| 26 | 1286 | 1414 | 1280 | 1 | 1297 | 1288 | 1 | 1334 | 1291 | 1 | 1329 | 1280 | 15 | 1340 | 1282 | 25 | δCH, ring 1, (80) |
| 27 | 1267 | 1372 | 1242 | 11 | 1272 | 1264 | 3 | 1316 | 1273 | 9 | 1325 | 1276 | 10 | 1333 | 1275 | 11 | νCC,ring 2, (13)+δCH (15)+νCN(19) |
| 28 | 1253 | 1367 | 1237 | 8 | 1235 | 1227 | 21 | 1289 | 1247 | 100 | 1302 | 1254 | 100 | 1314 | 1257 | 100 | νCN (13) + δCH (18), ring 2 |
| 29 | 1224 | 1335 | 1208 | 1 | 1229 | 1221 | 100 | 1274 | 1234 | 3 | 1277 | 1230 | 2 | 1285 | 1230 | 2 | νCO (39) + δCH (20), ring 1 |
| 30 | 1207 | 1322 | 1196 | 11 | 1201 | 1193 | 15 | 1250 | 1210 | 24 | 1262 | 1216 | 18 | 1274 | 1219 | 20 | νCN (21) + νCC (29) |
| 31 | 1170 | 1287 | 1165 | 29 | 1168 | 1160 | 1 | 1206 | 1167 | 1 | 1205 | 1160 | 1 | 1213 | 1160 | 2 | δCH, (74) |
| 32 | 1157 | 1282 | 1161 | 1 | 1158 | 1151 | 83 | 1193 | 1155 | 51 | 1190 | 1146 | 38 | 1196 | 1145 | 40 | δCH (51), ring 1 |
| 33 | 1141 | 1270 | 1149 | 2 | 1155 | 1147 | 0 | 1183 | 1145 | 1 | 1183 | 1139 | 2 | 1188 | 1137 | 2 | δCH (73), ring 2 |
| 34 | 1122 | 1240 | 1122 | 2 | 1130 | 1123 | 0 | 1171 | 1134 | 0 | 1171 | 1128 | 0 | 1179 | 1128 | 0 | δCH3, rock. (99) |
| 35 | 1114 | 1209 | 1094 | 1 | 1110 | 1103 | 2 | 1144 | 1107 | 2 | 1146 | 1104 | 2 | 1153 | 1103 | 2 | νCC (28) + δCH (32), ring 2 |
| 36 | 1084 | 1200 | 1087 | 2 | 1108 | 1100 | 5 | 1139 | 1102 | 4 | 1136 | 1094 | 4 | 1141 | 1092 | 4 | δCH (29), ring 1 |
| 37 | 1037 | 1174 | 1062 | 7 | 1040 | 1033 | 3 | 1082 | 1048 | 2 | 1089 | 1049 | 2 | 1098 | 1050 | 3 | νSC (22) + δCCC (46) |
| 38 | 1026 | 1169 | 1058 | 5 | 1007 | 1000 | 3 | 1060 | 1026 | 20 | 1079 | 1039 | 18 | 1093 | 1046 | 18 | νCC (29) + δCH (52), ring 2 |
| 39 | 1010 | 1107 | 1002 | 1 | 997 | 990 | 13 | 1039 | 1005 | 2 | 1043 | 1005 | 2 | 1050 | 1005 | 2 | νCC (11) + νO-CH3 (45) |
| 40 | 985 | 1104 | 999 | 0 | 993 | 986 | 15 | 1026 | 993 | 1 | 1026 | 988 | 1 | 1033 | 988 | 1 | νO-CH3 (30) + δCCC (55) |
| 41 | 968 | 1104 | 999 | 0 | 946 | 940 | 0 | 989 | 957 | 0 | 990 | 953 | 0 | 998 | 955 | 0 | γCH, (85), ring 2 |
| 42 | 947 | 1103 | 998 | 1 | 943 | 936 | 0 | 985 | 953 | 0 | 983 | 947 | 0 | 992 | 949 | 0 | γCH, (89), ring 1 |
| 43 | 941 | 1072 | 970 | 0 | 927 | 921 | 25 | 970 | 939 | 20 | 976 | 940 | 18 | 985 | 942 | 19 | νCC (11) +δCNC (34) + δCCN (11) |
| 44 | 925 | 1069 | 967 | 0 | 916 | 910 | 0 | 957 | 926 | 0 | 956 | 921 | 0 | 964 | 922 | 0 | γCH, (86), ring 1 |
| 45 | 910 | 1058 | 958 | 10 | 906 | 900 | 1 | 950 | 919 | 0 | 952 | 916 | 0 | 960 | 918 | 1 | γCH, (81), ring 2 |
| 46 | 858 | 968 | 876 | 0 | 845 | 839 | 0 | 873 | 845 | 0 | 876 | 844 | 0 | 882 | 844 | 0 | νCN (12) + δCCC (32) |
| 47 | 831 | 942 | 853 | 14 | 830 | 825 | 1 | 866 | 838 | 1 | 866 | 834 | 1 | 873 | 835 | 1 | γCH, (87), ring 2 |
| 48 | 815 | 930 | 842 | 1 | 812 | 807 | 19 | 848 | 821 | 18 | 847 | 816 | 19 | 854 | 817 | 20 | γCH, (49), ring 1 |
| 49 | 791 | 918 | 831 | 0 | 787 | 782 | 0 | 822 | 795 | 0 | 821 | 791 | 0 | 828 | 793 | 0 | γCH, (98), ring 1 |
| 50 | 779 | 866 | 784 | 2 | 774 | 769 | 3 | 805 | 780 | 3 | 811 | 781 | 3 | 818 | 782 | 3 | νCN (34) +νCO (20) |
| 51 | 756 | 852 | 771 | 13 | 742 | 738 | 16 | 772 | 747 | 14 | 772 | 744 | 15 | 778 | 744 | 17 | γCH, (53), ring 2+ γC, (17) |
| 52 | 729 | 812 | 735 | 4 | 710 | 705 | 6 | 738 | 714 | 7 | 737 | 709 | 7 | 742 | 710 | 7 | γC (21) + γCH, (10) |
| 53 | 709 | 797 | 722 | 0 | 705 | 701 | 2 | 732 | 708 | 1 | 730 | 703 | 1 | 735 | 704 | 1 | γC, (46) |
| 54 | 692 | 771 | 698 | 0 | 690 | 686 | 1 | 717 | 694 | 0 | 721 | 694 | 0 | 726 | 695 | 0 | νCS (23) + δCCC (47) |
| 55 | 662 | 748 | 677 | 4 | 658 | 654 | 4 | 689 | 667 | 6 | 697 | 671 | 7 | 704 | 673 | 8 | δCCC (33) + δCCN (16) |
| 56 | 623 | 689 | 624 | 2 | 619 | 615 | 1 | 640 | 619 | 4 | 639 | 615 | 6 | 643 | 615 | 6 | δCCC (54) |
| 57 | 617 | 688 | 623 | 3 | 611 | 607 | 11 | 635 | 614 | 5 | 634 | 611 | 1 | 638 | 610 | 1 | δCNC (11) + δCCN (11) + δCCC (26) |
| 58 | 605 | 680 | 615 | 1 | 600 | 596 | 1 | 625 | 605 | 1 | 626 | 603 | 1 | 631 | 604 | 1 | γC (52) + τCN (25) |
| 59 | 551 | 613 | 555 | 2 | 546 | 543 | 12 | 564 | 546 | 8 | 562 | 542 | 7 | 565 | 541 | 8 | δCCC (15) + δCCO (15) + δCOC (18) |
| 60 | 540 | 604 | 546 | 4 | 540 | 536 | 2 | 561 | 543 | 2 | 560 | 539 | 2 | 564 | 539 | 2 | τCC (26) + γC (10) |
| 61 | - | 566 | 512 | 2 | 494 | 491 | 3 | 514 | 498 | 3 | 514 | 495 | 3 | 518 | 496 | 3 | τCN (10) + γC (35) |
| 62 | - | 550 | 497 | 0 | 490 | 486 | 1 | 509 | 493 | 0 | 512 | 493 | 0 | 516 | 494 | 0 | νCS (30) + δCCC (28) + δSCC (19) |
| 63 | - | 507 | 459 | 0 | 460 | 457 | 0 | 476 | 461 | 0 | 476 | 459 | 0 | 480 | 459 | 0 | δC-O-CH3 (25) +δCCC (10)+δCCO (15) |
| 64 | - | 482 | 436 | 1 | 422 | 419 | 2 | 438 | 424 | 1 | 436 | 420 | 2 | 439 | 420 | 2 | τCC (81), ring 2 |
| 65 | - | 459 | 416 | 0 | 406 | 403 | 0 | 421 | 407 | 0 | 422 | 406 | 1 | 425 | 407 | 1 | τCC (93), ring 1 |
| 66 | - | 451 | 408 | 1 | 399 | 396 | 0 | 418 | 405 | 0 | 419 | 403 | 0 | 422 | 403 | 0 | δSCC (38) + δCCN (12) |
| 67 | - | 398 | 360 | 0 | 349 | 347 | 0 | 363 | 352 | 0 | 364 | 350 | 0 | 367 | 351 | 0 | τCN (101) + τCC (26) |
| 68 | - | 349 | 316 | 1 | 314 | 312 | 1 | 326 | 315 | 1 | 326 | 314 | 1 | 329 | 314 | 1 | δCCN (19) + δCCC (21) + δCOC (16) |
| 69 | - | 300 | 272 | 0 | 262 | 260 | 0 | 274 | 265 | 0 | 275 | 264 | 0 | 277 | 265 | 0 | τCO (17) + τCC (17) + τCN (18) |
| 70 | - | 267 | 242 | 0 | 245 | 243 | 1 | 252 | 244 | 1 | 253 | 244 | 1 | 255 | 244 | 1 | τCC (18) + δCCC (22) + δCCO (11) |
| 71 | - | 252 | 228 | 0 | 219 | 217 | 0 | 230 | 222 | 0 | 232 | 223 | 0 | 234 | 224 | 0 | τCO (47) |
| 72 | - | 227 | 206 | 0 | 205 | 204 | 0 | 213 | 206 | 0 | 214 | 206 | 0 | 216 | 207 | 0 | δCNC (15) + δOCC (22) + δCOC (21) |
| 73 | - | 209 | 189 | 0 | 185 | 184 | 1 | 192 | 186 | 0 | 193 | 186 | 1 | 194 | 186 | 1 | τCC (57) + γC (30) |
| 74 | - | 147 | 133 | 1 | 136 | 135 | 1 | 140 | 136 | 1 | 142 | 137 | 1 | 143 | 137 | 1 | τCC (22) + τCO (31) |
| 75 | - | 90 | 81 | 0 | 92 | 91 | 0 | 93 | 90 | 0 | 94 | 91 | 0 | 95 | 91 | 0 | τCN (13) + τCO (51) |
| 76 | - | 87 | 79 | 0 | 81 | 80 | 0 | 83 | 81 | 0 | 83 | 79 | 0 | 83 | 79 | 0 | δCNC (42) + δCCC (38) |
| 77 | - | 47 | 42 | 0 | 43 | 43 | 0 | 45 | 43 | 0 | 45 | 43 | 0 | 45 | 43 | 0 | τCC (11) + τCN (35) +γC (26) |
| 78 | - | 17 | 16 | 0 | 30 | 30 | 0 | 29 | 28 | 0 | 30 | 29 | 0 | 31 | 30 | 0 | τCC (91) |
aHarmonic frequencies (in cm−1) and IR intensities (km mol−1).
bScaling factor = 0.9051.
cScaling factor = 0.9934.
dScaling factor = 0.9679.
eScaling factor = 0.9631.
fScaling factor = 0.9567.
gν, stretching; δ, bending; ipb, in-plane bending; γ, out-of-plane bending; τ, torsion; sym, symmetric; asym, asymmetric; Ring 1: C14-C15-C17-C21-C19-C16; Ring 1: C1-C2-C3-C4-C5-C6; PED less than 10% are not shown.
Table 4.
Mean absolute deviation, correlation coefficient, and root mean square between the calculated and observed fundamental vibrational frequencies for the title compound.
| HF | BLYP | B3LYP | B3PW91 | mPW1PW91 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean absolute deviation | 153.80 | 23.14 | 12.90 | 12.90 | 48.68 | 6.62 | 54.13 | 7.14 | 64.50 | 7.46 |
| RMSmol | 167.10 | 28.19 | 17.37 | 16.25 | 57.50 | 8.73 | 64.00 | 9.02 | 75.23 | 9.22 |
| RMSover | 146.56 | 24.72 | 15.23 | 14.25 | 50.43 | 7.65 | 56.13 | 7.91 | 65.98 | 8.09 |
| Scaling Factor | 1.0000 | 0.9051 | 1.0000 | 0.9934 | 1.0000 | 0.9679 | 1.0000 | 0.9631 | 1.0000 | 0.9567 |
| r | 0.9997 | 0.9999 | 0.9999 | 0.9999 | 0.9999 | |||||
Acknowledgements
This work was supported by the Mersin University Research Fund (Project no: BAP.ECZ.F.TB.(HA).2007-1 and BAP.ECZ.F.MB.(ÖA).2006-3).
References
- Horton, D.A.; Bourne, G.T.; Smythe, M.L. The combinatorial synthesis of bicyclic privileged structures or privileged substructures. Chem. Rev 2003, 103, 893–930. [Google Scholar]
- Chua, M.S.; Shi, D.F.; Wrigley, S.; Bradshaw, T.D.; Hutchinson, I.; Shaw, P.N.; Barrett, D.A.; Stanley, L.A.; Stevens, M.F.G. Antitumor benzothiazoles. 7. Synthesis of 2-(4-acylaminophenyl)benzothiazoles and investigations into the role of acetylation in the antitumor activities of the parent amines. J. Med. Chem 1999, 42, 381–392. [Google Scholar]
- Sharma, P.; Mandloi, A.; Pritmani, S. Synthesis of new 2-(substituted benzothiazolylcarbamoyl)benzimidazoles as potential CNS depressants. Indian J. Chem. B 1999, 38, 1289–1294. [Google Scholar]
- Kawakami, M.; Koya, K.; Ukai, T.; Tatsuta, N.; Ikegawa, A.; Ogawa, K.; Shishido, T.; Chen, L. B. Structure-activity of novel rhodacyanine dyes as antitumor agents. J. Med. Chem 1998, 41, 130–142. [Google Scholar]
- Bradshaw, T.D.; Westwell, A.D. The development of the antitumour benzothiazole prodrug, Phortress, as a clinical candidate. Curr. Med. Chem 2004, 11, 1009–1021. [Google Scholar]
- Kashiyama, E.; Hutchinson, I.; Chua, M.S.; Stinson, S.F.; Phillips, L.R.; Kaur, G.; Sausville, E. A.; Bradshaw, T.D.; Westwell, A.D.; Stevens, M.F.G. Antitumor benzothiazoles. 8. Synthesis, metabolic formation, and biological properties of the C- and N-oxidation products of antitumor 2-(4-aminophenyl)benzothiazoles. J. Med. Chem 1999, 42, 4172–4184. [Google Scholar]
- Hutchinson, I.; Jennings, S.A.; Vishnuvajjala, B.R.; Westwell, A.D.; Stevens, M.F.G. Antitumor benzothiazoles. 16. Synthesis and pharmaceutical properties of antitumor 2-(4-aminophenyl)benzothiazole amino acid prodrugs. J. Med. Chem 2002, 45, 744–747. [Google Scholar]
- Hutchinson, I.; Chua, M.S.; Browne, H.L.; Trapani, V.; Bradshaw, T.D.; Westwell, A.D.; Stevens, M.F.G. Antitumor benzothiazoles. 14. Synthesis and in vitro biological properties of fluorinated 2-(4-aminophenyl)benzothiazoles. J. Med. Chem 2001, 44, 1446–1455. [Google Scholar]
- Das, J.; Moquin, R.V.; Lin, J.; Liu, C.J.; Doweyko, A.M.; DeFex, H.F.; Fang, Q.; Pang, S.H.; Pitt, S.; Shen, D.R.; Schieven, G.L.; Barrish, J.C.; Wityak, J. Discovery of 2-amino-heteroaryl-benzothiazole-6-anilides as potent p56(lck) inhibitors. Bioorg. Med. Chem. Lett 2003, 13, 2587–2590. [Google Scholar]
- Hays, S.J.; Rice, M.J.; Ortwine, D.F.; Johnson, G.; Schwarz, R.D.; Boyd, D.K.; Copeland, L.F.; Vartanian, M.G.; Boxer, P.A. Substituted 2-Benzothiazolamine as Sodium Flux Inhibitors - Quantitative Structure-Activity-Relationships and Anticonvulsant Activity. J. Pharm. Sci 1994, 83, 1425–1432. [Google Scholar]
- Foscolos, G.; Tsatsas, G.; Champagnac, A.; Pommier, M. Synthesis and pharmacodynamic study of new derivatives of benzothiazole. Ann. Pharm. Fr 1977, 35, 295–307. [Google Scholar]
- Akbay, A.; Ören, I.; Arpaci, Ö.T.; Sener, E.A.; Yalçin, I. Synthesis and HIV-1 Reverse Transcriptase Inhibitor Activity of Some 2,5,6-Substituted Benzoxazole, Benzimidazole, Benzothiazole and Oxazolo(4,5-b)pyridine Derivatives. Arzneimittel-Forsch. 2003, 53(4), 266–271. [Google Scholar]
- Paget, C.J.; Kisner, K.; Stone, R.L.; DeLong, D.C. Heterocyclic substituted ureas. II. Immunosuppressive and antiviral activity of benzothiazolyl-and benzoxazolylureas. J. Med. Chem. 1969, 12(6), 1016–1018. [Google Scholar]
- Gong, B.Q.; Hong, F.; Kohm, C.; Bonham, L.; Klein, P. Synthesis and SAR of 2-arylbenzoxazoles, benzothiazoles and benzimidazoles as inhibitors of lysophosphatidic acid acyltransferase-beta. Bioorg. Med. Chem. Lett. 2004, 14(6), 1455–1459. [Google Scholar]
- Hutchinson, I.; Bradshaw, T.D.; Matthews, C.S.; Stevens, M.F.G.; Westwell, A.D. Antitumour benzothiazoles. Part 20: 3′-cyano and 3′-alkynyl-substituted 2-(4′-aminophenyl)benzothiazoles as new potent and selective analogues. Bioorg. Med. Chem. Lett. 2003, 13(3), 471–474. [Google Scholar]
- Hutchinson, I.; Stevens, M.F.G.; Westwell, A.D. The regiospecific synthesis of 5-and 7- monosubstituted and 5,6-disubstituted 2-arylbenzothiazoles. Tetrahedron Lett. 2000, 41(3), 425–428. [Google Scholar]
- Asit, K.C.; Santosh, R.; Gurmeet, K.; Lalima, S. An Efficient Conversion of Phenolic Esters to Benzothiazoles under Mild and Virtually Neutral Conditions. Synlett 2004, 1533–1537. [Google Scholar]
- Bradshaw, T. D.; Wrigley, S.; Shi, D. F.; Schultz, R. J.; Paull, K. D.; Stevens, M. F. G. 2-(4-aminophenyl)benzothiazoles: novel agents with selective profiles of in vitro anti-tumour activity. Brit. J. Cancer 1998, 77(5), 745–752. [Google Scholar]
- Shi, D.F.; Bradshaw, T.D.; Wrigley, S.; McCall, C.J.; Lelieveld, P.; Fichtner, I.; Stevens, M.F.G. Antitumor benzothiazoles .3. Synthesis of 2-(4-aminophenyl)benzothiazoles and evaluation of their activities against breast cancer cell lines in vitro and in vivo. J. Med. Chem. 1996, 39(17), 3375–3384. [Google Scholar]
- Moller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev 1934, 46, 618–622. [Google Scholar]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic-Behavior. Phys. Rev. A 1988, 38(6), 3098–3100. [Google Scholar]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron-Density. Phys. Rev. B 1988, 37(2), 785–789. [Google Scholar]
- Becke, A.D. Density-Functional Thermochemistry 3. The Role of Exact Exchange. J. Chem. Phys. 1993, 98(7), 5648–5652. [Google Scholar]
- Adamo, C.; Barone, V. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The mPW and mPW1PW models. J. Chem. Phys. 1998, 108(2), 664–675. [Google Scholar]
- Burke, K.; Perdew, J.P.; Wang, Y. Electronic Density Functional Theory: Recent Progress and New Directions; Dobson, JF, Vignale, G, Das, MP, Eds.; Plenum; New York, 1998. [Google Scholar]
- Predew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar]
- Hein, D.W.; Alheim, R.J.; Leavitt, J.J. The Used of Polyphoshoric Acid in the Synthesis of 2-Aryl- and 2-Alkyl-substituted Benzimidazoles, Benzoxazoles and Benzothiazoles. J. Am. Chem. Soc 1957, 79, 427–429. [Google Scholar]
- Microwaves in Organic Synthesis; Loupy, A (Ed.) Wiley-VCH: Weinheim, 2002.
- Shao, Y.; Molnar, L.F.; Jung, Y.; Kussmann, J.; Ochsenfeld, C.; Brown, S.T.; Gilbert, A.T.B.; Slipchenko, L.V.; Levchenko, S.V.; O’Neill, D.P.; DiStasio, R.A., Jr; Lochan, R.C.; Wang, T.; Beran, G.J.O.; Besley, N.A.; Herbert, J.M.; Lin, C.Y.; Van Voorhis, T.; Chien, S.H.; Sodt, A.; Steele, R.P.; Rassolov, V.A.; Maslen, P.E.; Korambath, P.P.; Adamson, R.D.; Austin, B.; Baker, J.; Byrd, E.F.C.; Dachsel, H.; Doerksen, R.J.; Dreuw, A.; Dunietz, B.D.; Dutoi, A.D.; Furlani, T.R.; Gwaltney, S.R.; Heyden, A.; Hirata, S.; Hsu, C-P.; Kedziora, G.; Khalliulin, R.Z.; Klunzinger, P.; Lee, A.M.; Lee, M.S.; Liang, W.Z.; Lotan, I.; Nair, N.; Peters, B.; Proynov, E.I.; Pieniazek, P.A.; Rhee, Y.M.; Ritchie, J.; Rosta, E.; Sherrill, C.D.; Simmonett, A.C.; Subotnik, J.E.; Woodcock, H.L., III; Zhang, W.; Bell, A.T.; Chakraborty, A.K.; Chipman, D.M.; Keil, F.J.; Warshel, A.; Hehre, W.J.; Schaefer, H.F.; Kong, J.; Krylov, A.I.; Gill, P.M.W.; Head-Gordon, M. Phys. Chem. Chem. Phys 2006, 8, 3172.Spartan 06; Wavefunction Inc: Irvine, CA, 92612 USA, 2006.
- Yildirim, S.O.; Akkurt, M.; Danisman, O.; Sekerci, M.; Fun, H.K. (1,3-benzothiazol-2-yl)[1-(2-chlorobenzyl)imidazolidin-2-ylidene]amine. Acta Crystallogr. E 2006, 62, O3812–O3813. [Google Scholar]
- Huang, Z.Q.; Du, Z.Q.; Du, G.S.; Yan, J. 3-(1,3-benzothiazol-2-yl)-2-naphthol. Acta Crystallogr. E 2006, 62, O2010–O2011. [Google Scholar]
- Li, J.; Liang, Z.; Wu, Q. (E, E)-N, N′-bis(4-methoxybenzylidene)-hexane-1,6-diamine. Acta Crystallogr. E 2007, 63, o1086–o1087. [Google Scholar]
- Pulay, P.; Fogarasi, G.; Pongor, G.; Boggs, J.E.; Vargha, A. Combination of Theoretical Abinitio and Experimental Information to Obtain Reliable Harmonic Force-Constants - Scaled Quantum-Mechanical (Sqm) Force-Fields for Glyoxal, Acrolein, Butadiene, Formaldehyde, and Ethylene. J. Am. Chem. Soc 1983, 105, 7037–7047. [Google Scholar]
- Scott, A.P.; Radom, L. Harmonic vibrational frequencies: An evaluation of Hartree-Fock, Moller-Plesset, quadratic configuration interaction, density functional theory, and semiempirical scale factors. J. Phys. Chem. -US 1996, 100, 16502–16513. [Google Scholar]
- GaussView, Version 3.07; Dennington, Roy, II; Keith, T.; Millam, J.; Eppinnett, K.; Hovell, W.L.; Gilliland, R. Semichem, Inc: Shawnee Mission, KS, 2003. [Google Scholar]
- Jamróz, M.H. Vibrational Energy Distribution Analysis VEDA 4; Warsaw, 2004. [Google Scholar]
- Krishnakumar, V.; Xavier, R.J. Normal coordinate analysis of 2-mercapto and 4,6-dihydroxy-2-mercapto pyrimidines. Indian J. Pure Ap. Phy 2003, 41, 597–601. [Google Scholar]
- Balfour, W.J. The Vibrational-Spectrum of Anisole. Spectrochim. Acta A 1983, 39, 795–800. [Google Scholar]
- Rabiger, D.J.; Joullie, M.M. The Ionization Constants, Ultraviolet and Infrared Spectra of Some Substituted Benzimidazoles. J. Org. Chem 1964, 29, 476–482. [Google Scholar]
- Sundaraganesan, N.; Kumar, K.S.; Meganathan, C.; Joshua, B.D. Vibrational spectroscopy investigation using ab initio and density functional theory analysis on the structure of 2-amino-4,6-dimethoxypyrimidine. Spectrochim. Acta A 2006, 65, 1186–1196. [Google Scholar]
- Krishnakumar, V.; Ramasamy, R. Scaled quantum chemical studies of the structure and vibrational spectra of 2-(methylthio) benzimidazole. Spectrochim. Acta A 2005, 62, 570–577. [Google Scholar]
- Krishnakumar, V.; Xavier, R.J. Normal coordinate analysis of vibrational spectra of 2-methylindoline and 5-hydroxyindane. Indian J. Pure Ap. Phy 2003, 41, 95–99. [Google Scholar]
- Lakshmaiah, B.; Rao, G.R. Vibrational Analysis of Substituted Anisoles .1. Vibrational-Spectra and Normal Coordinate Analysis of Some Fluoro and Chloro Compounds. J. Raman Spectrosc 1989, 20, 439–448. [Google Scholar]
- Lakshmaiah, B.; Rao, G.R. Substituted Benzenes Xiv-Normal Coordinate Analysis of out-of-Plane Vibrations of Anisoles. Indian J. Pure Ap. Phy 1991, 29, 370–373. [Google Scholar]
- Reddy, B.V.; Rao, G.R. Transferable Valence Force-Fields for Substituted Benzenes .1. Monohalogenated Anisoles. Vib. Spectrosc 1994, 6, 231–250. [Google Scholar]
- Campagnaro, G.E.; Wood, J.L. The vibrational spectra and origin of torsional barriers in some aromatic systems. J. Mol. Struct 1970, 6, 117–132. [Google Scholar]
- Owen, N.L.; Hester, R.E. Vibrational spectra and torsional barriers of anisole and some monohalogen derivatives. Spectrochim. Acta A 1969, 25, 343–354. [Google Scholar]
- Babu, V.A.; Lakshmaiah, B.; Ramulu, K.S.; Rao, G.R. Substituted Benzenes. 13. Normal Coordinate Analysis of Anisoles. Indian J. Pure Ap. Phy 1987, 25, 58–65. [Google Scholar]
- Kuppens, T.; Vandyck, K.; VanderEycken, J.; Herrebout, W.; VanderVeken, B.; Bultinck, P.A. DFT conformational analysis and VCD study on methyl tetrahydrofuran-2-carboxylate. Spectrochim. Acta A 2007, 67, 402–411. [Google Scholar]
- Arslan, H.; Flörke, U.; Külcü, N. Theoretical studies of molecular structure and vibrational spectra of O-ethyl benzoylthiocarbamate. Spectrochim. Acta A 2006, 67, 936–943. [Google Scholar]
- Arslan, H.; Algül, Ö.; Dündar, Y. Structural and spectral studies on 3-(6-benzoyl-5-chloro-2-benzoxazolinon-3-yl) propanoic acid. Vib. Spectrosc 2007, 44, 248–255. [Google Scholar]
- Arslan, H.; Emen, F.M.; Kulcu, N. Structure and vibrational spectra of N,N-dimethyl-N′-(2-chlorobenzoyl)thiourea: Hartree-Fock and density functional theory studies. Asian J. Chem 2007, 19, 1888–1896. [Google Scholar]
- Arslan, H.; Algul, O. Theoretical studies of molecular structure and vibrational spectra of 2-ethyl-1H-benzo[d]imidazole. Asian J. Chem 2007, 19, 2229–2235. [Google Scholar]
- Arslan, H.; Demircan, A.; Göktürk, E. Vibrational spectroscopy investigation using ab initio and density functional theory analysis on the structure of 5-chloro-10-oxa-3-thiatricyclo[ 5.2.1.01,5]dec-8-ene-3,3-dioxide. Spectrochim. Acta A 2007. [Google Scholar] [CrossRef]
- Arslan, H.; Flörke, U.; Külcü, N.; Binzet, G. The molecular structure and vibrational spectra of 2-chloro-N-(diethylcarbamothioyl)benzamide by Hartree–Fock and density functional methods. Spectrochim. Acta A 2007. [Google Scholar] [CrossRef]
- Arslan, H.; Demircan, A. Ab initio studies on 5-bromo-10-oxa-3-thiatricyclo[5.2.1.01,5]-dec-8-ene 3,3-dioxide. Acta Chim. Slov 2007, 54, 341–353. [Google Scholar]
- Kupka, T.; Wrzalik, R.; Pasterna, G.; Pasterny, K. Theoretical DFT and experimental Raman and NMR studies on thiophene, 3-methylthiophene and selenophene. J. Mol. Struct 2002, 616, 17–32. [Google Scholar]
- Wysokinski, R.; Kuduk-Jaworska, J.; Michalska, D. Electronic structure, Raman and infrared spectra, and vibrational assignment of carboplatin. Density functional theory studies. J. Mol. Struc-Theochem 2006, 758, 169–179. [Google Scholar]
- Hanuza, J.; Sasiadek, W.; Michalski, J.; Lorenc, J.; Maczka, M.; Kaminskii, A. A.; Butashin, A. V.; Klapper, H.; Hulliger, J.; Mohmed, A. F. A. Polarized Raman and infrared spectra of the salol crystal - chemical quantum calculations of the vibrational normal modes. Vib. Spectrosc 2004, 34, 253–268. [Google Scholar]
- Rauhut, G.; Pulay, P. Transferable Scaling Factors for Density-Functional Derived Vibrational Force-Fields. J. Phys. Chem. -US 1995, 99, 3093–3100. [Google Scholar]
- Poul, S.; Gupta, M.; Gupta, R. Microwave-Induced Solvent-Free Synthesis of 2-Arylbenzothiazoles using p-TsOH. Synthetic Commun 2002, 32, 3541–3547. [Google Scholar]
- Youssef, A. M.; Mohamed, H. M.; Czezowski, C.; Ata, A.; Abd-El-Aziz, A. S. Synthesis and Biological Evaluation of Benzothiazole Derivatives of Pyrimidines, Acrylonitriles, and Coumarins. Heterocycles 2006, 68, 347–355. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr; Vreven, T.; Kudin, K.N.; Burant, J.C.; Millam, J.M.; Iyengar, S.S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G.A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J.E.; Hratchian, H.P.; Cross, J.B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A.J.; Cammi, R.; Pomelli, C.; Ochterski, J.W.; Ayala, P.Y.; Morokuma, K.; Voth, G.A.; Salvador, P.; Dannenberg, J.J.; Zakrzewski, V.G.; Dapprich, S.; Daniels, A.D.; Strain, M.C.; Farkas, O.; Malick, D.K.; Rabuck, A.D.; Raghavachari, K.; Foresman, J.B.; Ortiz, J.V.; Cui, Q.; Baboul, A.G.; Clifford, S.; Cioslowski, J.; Stefanov, B.B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R.L.; Fox, D.J.; Keith, T.; Al-Laham, M.A.; Peng, C.Y.; Nanayakkara, A.; Challacombe, M.; Gill, P.M.W.; Johnson, B.; Chen, W.; Wong, M.W.; Gonzalez, C.; Pople, J.A. Gaussian 03, Revision C.02; Gaussian, Inc: Wallingford CT, 2004. [Google Scholar]
- Foresman, J.B.; Frisch, E. Exploring Chemistry with Electronic Structure Methods: A Guide to Using Gaussian; Gaussian Pitttsburg: PA, 1993. [Google Scholar]
© 2007 by MDPI Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Arslan, H.; Algül, Ö. Synthesis and Ab Initio/DFT Studies on 2-(4-methoxyphenyl)benzo[d]thiazole. Int. J. Mol. Sci. 2007, 8, 760-776. https://0-doi-org.brum.beds.ac.uk/10.3390/i8080760
AMA Style
Arslan H, Algül Ö. Synthesis and Ab Initio/DFT Studies on 2-(4-methoxyphenyl)benzo[d]thiazole. International Journal of Molecular Sciences. 2007; 8(8):760-776. https://0-doi-org.brum.beds.ac.uk/10.3390/i8080760
Chicago/Turabian StyleArslan, Hakan, and Öztekin Algül. 2007. "Synthesis and Ab Initio/DFT Studies on 2-(4-methoxyphenyl)benzo[d]thiazole" International Journal of Molecular Sciences 8, no. 8: 760-776. https://0-doi-org.brum.beds.ac.uk/10.3390/i8080760