
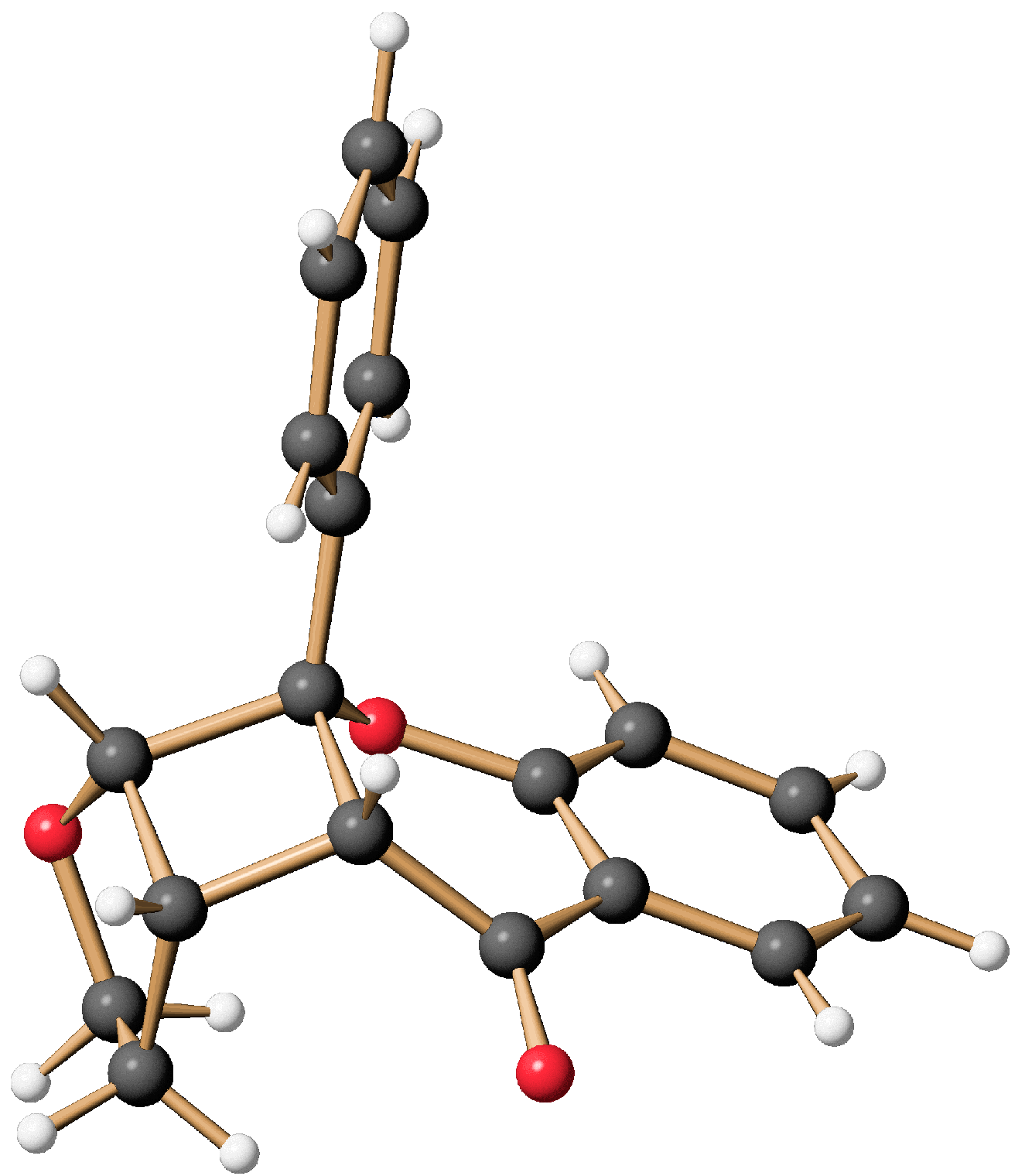
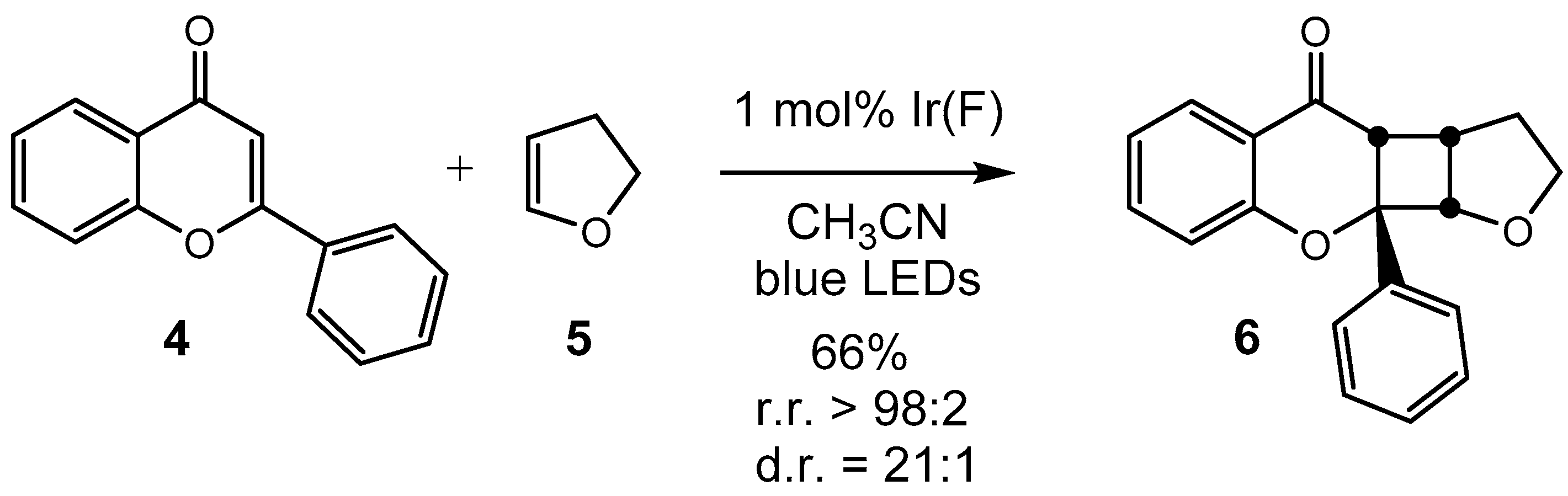
9a-Phenyl-2,3,3a,3b,9a,9b-hexahydro-4H-furo[3‘,2’:3,4]cyclobuta- [1,2-b]chromen-4-one: A Flavone-Based [2 + 2]-Photocycloadduct



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
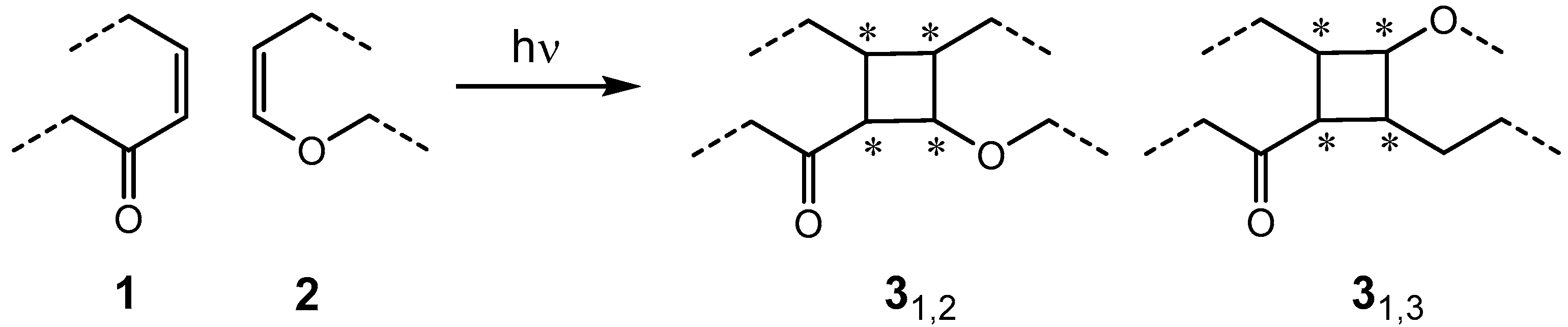
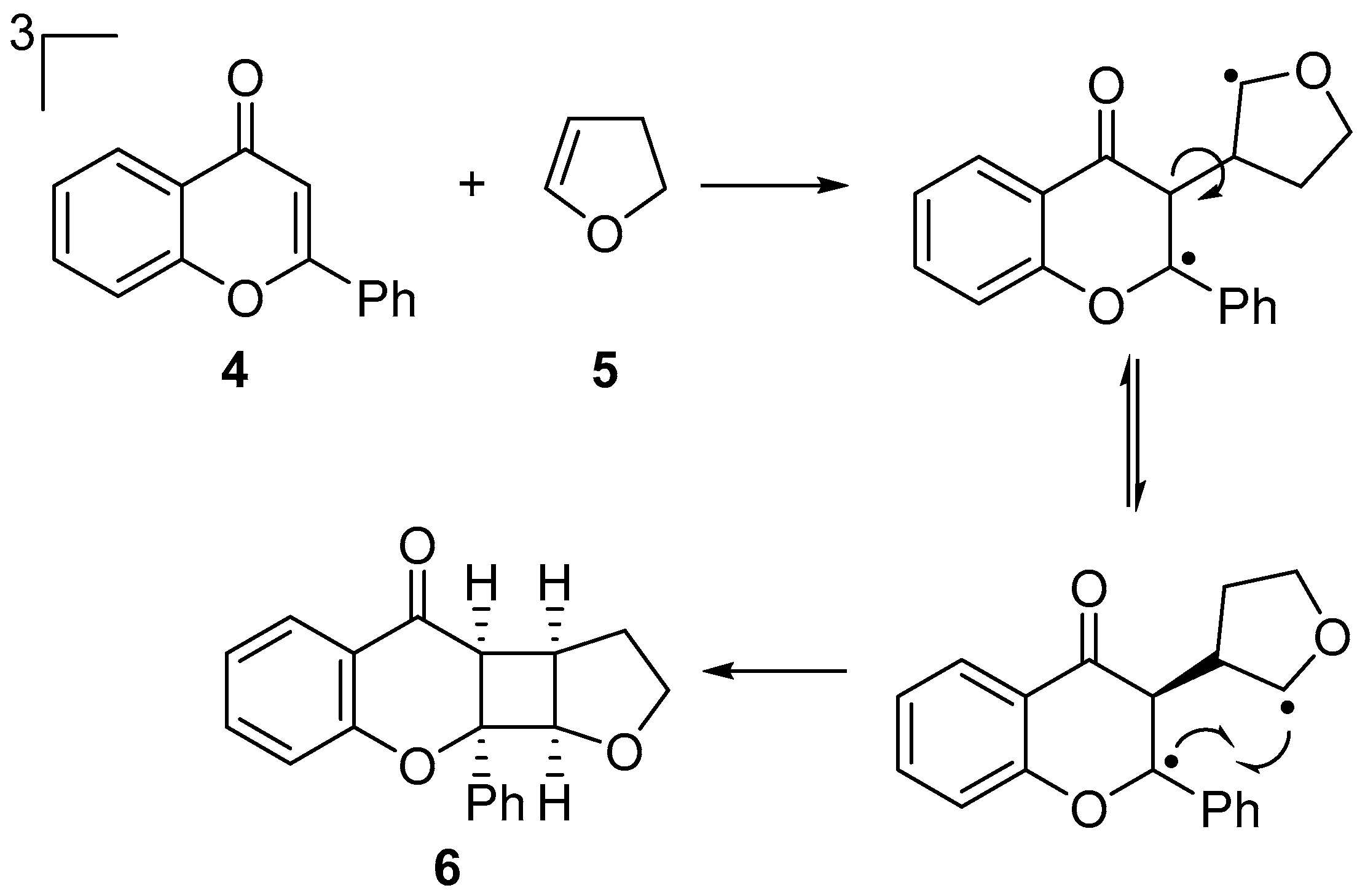
:1. Introduction
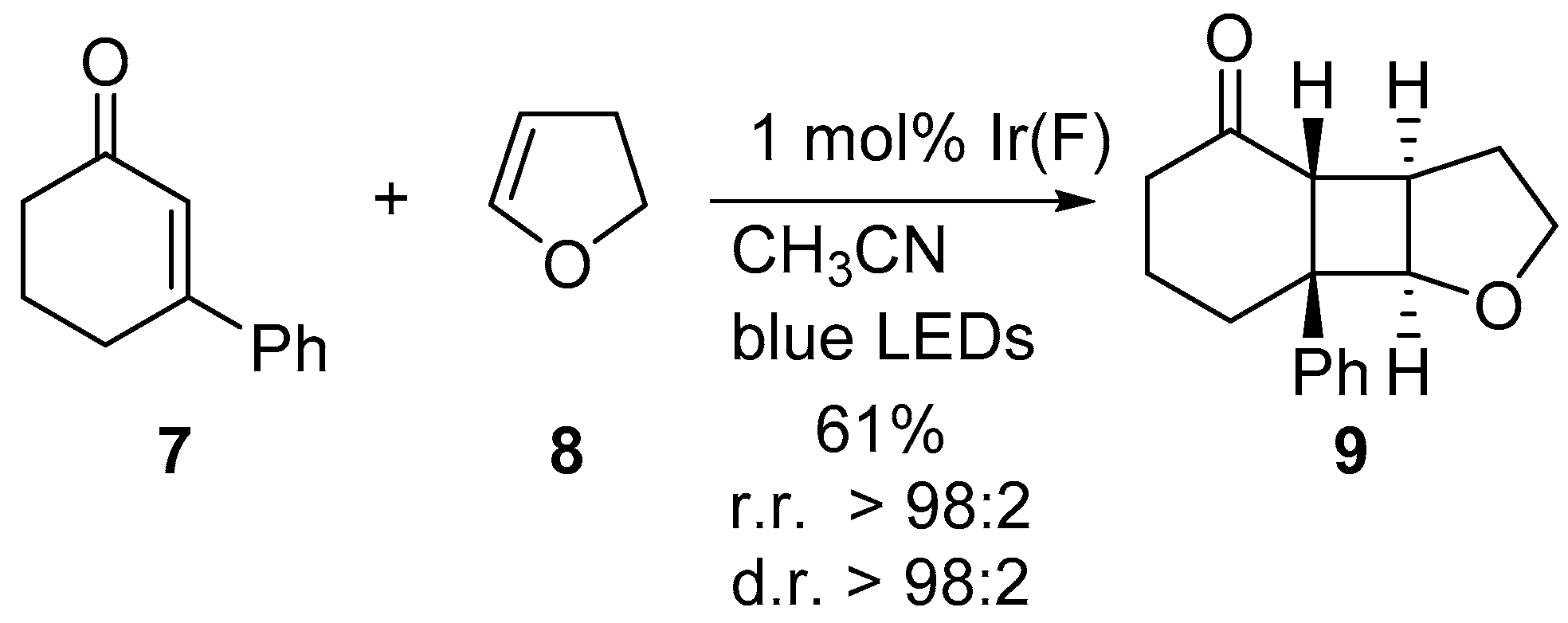
2. Results
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schuster, D.I.; Lem, G.; Kaprinidis, N.A. New insights into an old mechanism: [2 + 2] photocycloaddition of enones to alkenes. Chem. Rev. 1993, 93, 3–22. [Google Scholar] [CrossRef]
- Bach, T. Stereoselective Intermolecular [2 + 2]-Photocycloaddition Reactions and Their Application in Synthesis. Synthesis 1998, 1998, 683–703. [Google Scholar] [CrossRef]
- Poplata, S.; Tröster, A.; Zou, Y.-Q.; Bach, T. Recent Advances in the Synthesis of Cyclobutanes by Olefin [2 + 2] Photocycloaddition Reactions. Chem. Rev. 2016, 116, 9748–9815. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, D.; Bera, N.; Ghosh, S. [2 + 2] Photochemical Cycloaddition in Organic Synthesis. Eur. J. Org. Chem. 2020, 2020, 1310–1326. [Google Scholar] [CrossRef]
- Crimmins, M.T.; Wang, Z.; McKerlie, L.A. Rearrangement of cyclobutyl carbinyl radicals: Total synthesis of the spirovetivane phytoalexin (±)-lubiminol. Tetrahedron Lett. 1996, 37, 8703–8706. [Google Scholar] [CrossRef]
- Mehta, G.; Sreenivas, K. Total synthesis of the novel tricyclic sesquiterpene sulcatine G. Chem. Commun. 2001, 1892–1893. [Google Scholar] [CrossRef] [Green Version]
- Langer, K.; Mattay, J. Stereoselective Intramolecular Copper(I)-Catalyzed [2 + 2]- Photocycloadditions. Enantioselective Synthesis of (+)- and (-)-Grandisol. J. Org. Chem. 2002, 60, 7256–7266. [Google Scholar] [CrossRef]
- Lee-Ruff, E.; Mladenova, G. Enantiomerically Pure Cyclobutane Derivatives and Their Use in Organic Synthesis. Chem. Rev. 2003, 103, 1449–1483. [Google Scholar] [CrossRef]
- Iriondo-Alberdi, J.; Greaney, M.F. Photocycloaddition in Natural Product Synthesis. Eur. J. Org. Chem. 2007, 2007, 4801–4815. [Google Scholar] [CrossRef]
- Hoffmann, N. Photochemical Reactions as Key Steps in Organic Synthesis. Chem. Rev. 2008, 108, 1052–1103. [Google Scholar] [CrossRef]
- Stobbe, H.; Hensel, A. Polymere des Anisal-acetophenons und anderer Chalkone. (I. Mitteilung über Truxill-und Truxin-Ketone). Ber. Dtsch. Chem. Ges. 1926, 59, 2254–2265. [Google Scholar] [CrossRef]
- Corey, E.J.; Bass, J.D.; LeMahieu, R.; Mitra, R.B. A Study of the Photochemical Reactions of 2-Cyclohexenones with Substituted Olefins. J. Am. Chem. Soc. 1964, 86, 5570–5583. [Google Scholar] [CrossRef]
- Cantrell, T.S.; Haller, W.S.; Williams, J.C. Photocycloaddition reactions of some 3-substituted cyclohexenones. J. Org. Chem. 1969, 34, 509–519. [Google Scholar] [CrossRef]
- Sano, T.; Horiguchi, Y.; Tsuda, Y. Dioxopyrrolines. XXXVII Stereochemical pathways of dioxopyrroline-olefin photocycloaddition. Stereochemical selection rule for the photocycloaddition of enone-olefin pairs. Chem. Pharm. Bull. 1987, 35, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Lei, T.; Zhou, C.; Huang, M.Y.; Zhao, L.M.; Yang, B.; Ye, C.; Xiao, H.; Meng, Q.Y.; Ramamurthy, V.; Tung, C.H.; et al. General and Efficient Intermolecular [2 + 2] Photodimerization of Chalcones and Cinnamic Acid Derivatives in Solution through Visible-Light Catalysis. Angew. Chem. Int. Ed. 2017, 56, 15407–15410. [Google Scholar] [CrossRef]
- Riener, M.; Nicewicz, D.A. Synthesis of cyclobutane lignans via an organic single electron oxidant–electron relay system. Chem. Sci. 2013, 4, 2625–2629. [Google Scholar] [CrossRef]
- Du, J.; Yoon, T.P. Crossed Intermolecular [2 + 2] Cycloadditions of Acyclic Enones via Visible Light Photocatalysis. J. Am. Chem. Soc. 2009, 131, 14604–14605. [Google Scholar] [CrossRef] [Green Version]
- Oderinde, M.S.; Ramirez, A.; Dhar, T.G.M.; Cornelius, L.A.M.; Jorge, C.; Aulakh, D.; Sandhu, B.; Pawluczyk, J.; Sarjeant, A.A.; Meanwell, N.A.; et al. Photocatalytic Dearomative Intermolecular [2 + 2] Cycloaddition of Heterocycles for Building Molecular Complexity. J. Org. Chem. 2021, 86, 1730–1747. [Google Scholar] [CrossRef]
- Tröster, A.; Alonso, R.; Bauer, A.; Bach, T. Enantioselective Intermolecular [2 + 2] Photocycloaddition Reactions of 2(1H)-Quinolones Induced by Visible Light Irradiation. J. Am. Chem. Soc. 2016, 138, 7808–7811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pecho, F.; Zou, Y.Q.; Gramüller, J.; Mori, T.; Huber, S.M.; Bauer, A.; Gschwind, R.M.; Bach, T. A Thioxanthone Sensitizer with a Chiral Phosphoric Acid Binding Site: Properties and Applications in Visible Light-Mediated Cycloadditions. Chem. Eur. J. 2020, 26, 5190–5194. [Google Scholar] [CrossRef]
- Huang, X.; Quinn, T.R.; Harms, K.; Webster, R.D.; Zhang, L.; Wiest, O.; Meggers, E. Direct Visible-Light-Excited Asymmetric Lewis Acid Catalysis of Intermolecular [2 + 2] Photocycloadditions. J. Am. Chem. Soc. 2017, 139, 9120–9123. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Skubi, K.L.; Schultz, D.M.; Yoon, T.P. A Dual-Catalysis Approach to Enantioselective [2 + 2] Photocycloadditions Using Visible Light. Science 2014, 344, 392–396. [Google Scholar] [CrossRef] [Green Version]
- Blum, T.R.; Miller, Z.D.; Bates, D.M.; Guzei, I.A.; Yoon, T.P. Enantioselective photochemistry through Lewis acid–catalyzed triplet energy transfer. Science 2016, 354, 1391–1395. [Google Scholar] [CrossRef] [Green Version]
- Lefarth, J. Intermolecular [2 + 2]-Photocycloaddition Reactions of Complex Acceptor-Donor Systems and Development of an Enantioselective Photocatalyzed Hydroformylation Reaction. Ph.D. Thesis, University of Cologne, Köln, Germany, 2021. [Google Scholar]
- Kutateladze, A.G. Conformational Analysis of Singlet−Triplet State Mixing in Paternò−Büchi Diradicals. J. Am. Chem. Soc. 2001, 123, 9279–9282. [Google Scholar] [CrossRef]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Karak, P. Biological Activities of Flavonoids: An Overview. Int. J. Pharm. Sci. Res. 2019, 3, 1567–1574. [Google Scholar]
- Nijveldt, R.J.; van Nood, E.; van Hoorn, D.E.; Boelens, P.G.; van Norren, K.; van Leeuwen, P.A. Flavonoids: A review of probable mechanisms of action and potential applications. Am. J. Clin. Nutr. 2001, 74, 418–425. [Google Scholar] [CrossRef]
- Sisa, M.; Bonnet, S.L.; Ferreira, D.; Van der Westhuizen, J.H. Photochemistry of flavonoids. Molecules 2010, 15, 5196–5245. [Google Scholar] [CrossRef]
- Nakayama, T.; Shimizu, T.; Torii, Y.; Miki, S.; Hamanoue, K. A comparison of the photochemistry of flavanone with that of flavone originating from their lowest excited triplet states in ethanol. J. Photochem. Photobiol. A Chem. 1997, 111, 35–39. [Google Scholar] [CrossRef]
- Monici, M.; Mulinacci, N.; Baglioni, P.; Vincieri, F. Flavone photoreactivity. UV-induced reactions in organic solvents and micellar systems. J. Photochem. Photobiol. B Biol. 1993, 20, 167–172. [Google Scholar] [CrossRef]
- Chaaban, H.; Ioannou, I.; Paris, C.; Charbonnel, C.; Ghoul, M. The photostability of flavanones, flavonols and flavones and evolution of their antioxidant activity. J. Photochem. Photobiol. A Chem. 2017, 336, 131–139. [Google Scholar] [CrossRef]
- Bhattacharyya, K.; Ramaiah, D.; Das, P.K.; George, M.V. A laser flash photolysis study of 2,6-dimethyl-3,5-diphenyl-4-pyrone and related chromones. Evidence for triplet state structural relaxation from quenching behaviors. J. Phys. Chem. 1986, 90, 5984–5989. [Google Scholar] [CrossRef]
- Pownall, H.J. Solvent and substituent effects in aromatic carbonyl compounds: The triplet state of flavone. Spectrochim. Acta Part A Mol. Spectrosc. 1974, 30, 953–959. [Google Scholar] [CrossRef]
- Lowry, M.S.; Goldsmith, J.I.; Slinker, J.D.; Rohl, R.; Pascal, R.A.; Malliaras, G.; Bernhard, S. Single-Layer Electroluminescent Devices and Photoinduced Hydrogen Production from an Ionic Iridium(III) Complex. Chem. Mater. 2005, 17, 5712–5719. [Google Scholar] [CrossRef]
- Engler, G.; Nispel, M.; Marian, C.; Kleinermanns, K. Transient spectroscopy of UV excited flavone: Triplet–triplet absorption and comparison with theory. Chem. Phys. Lett. 2009, 473, 167–170. [Google Scholar] [CrossRef]
- Orlandi, G.; Monti, S.; Barigelletti, F.; Balzani, V. Triplet energy transfer to cis and trans stilbene. A quantum mechanical approach. Chem. Phys. 1980, 52, 313–319. [Google Scholar] [CrossRef]
- Kelly, J.F.D.; Kelly, J.M.; McMurry, T.B.H. Photochemistry of substituted cyclic enones. Part 12. Photocycloaddition of 3-phenylcyclopentenone and 3-phenylcyclohexenone to (E)- and (Z)-1-phenylpropene. J. Chem. Soc. Perkin Trans. 2 1999, 1933–1941. [Google Scholar] [CrossRef]
- Sicignano, M.; Rodríguez, R.I.; Alemán, J. Recent Visible Light and Metal Free Strategies in [2 + 2] and [4 + 2] Photocycloadditions. Eur. J. Org. Chem. 2021, 2021, 3303–3321. [Google Scholar] [CrossRef]
- Data from the Crystal Structure Analysis for 6 is Deposited at the Cambridge Crystallographic Data Centre (CCDC) with the Deposition Number CCDC 2089434. Available online: https://www.ccdc.cam.ac.uk/ (accessed on 21 July 2021).





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lefarth, J.; Neudörfl, J.; Griesbeck, A.G. 9a-Phenyl-2,3,3a,3b,9a,9b-hexahydro-4H-furo[3‘,2’:3,4]cyclobuta- [1,2-b]chromen-4-one: A Flavone-Based [2 + 2]-Photocycloadduct. Molbank 2021, 2021, M1256. https://0-doi-org.brum.beds.ac.uk/10.3390/M1256
Lefarth J, Neudörfl J, Griesbeck AG. 9a-Phenyl-2,3,3a,3b,9a,9b-hexahydro-4H-furo[3‘,2’:3,4]cyclobuta- [1,2-b]chromen-4-one: A Flavone-Based [2 + 2]-Photocycloadduct. Molbank. 2021; 2021(3):M1256. https://0-doi-org.brum.beds.ac.uk/10.3390/M1256
Chicago/Turabian StyleLefarth, Jens, Jörg Neudörfl, and Axel G. Griesbeck. 2021. "9a-Phenyl-2,3,3a,3b,9a,9b-hexahydro-4H-furo[3‘,2’:3,4]cyclobuta- [1,2-b]chromen-4-one: A Flavone-Based [2 + 2]-Photocycloadduct" Molbank 2021, no. 3: M1256. https://0-doi-org.brum.beds.ac.uk/10.3390/M1256