Bacterial Composition and Diversity in Deep-Sea Sediments from the Southern Colombian Caribbean Sea

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
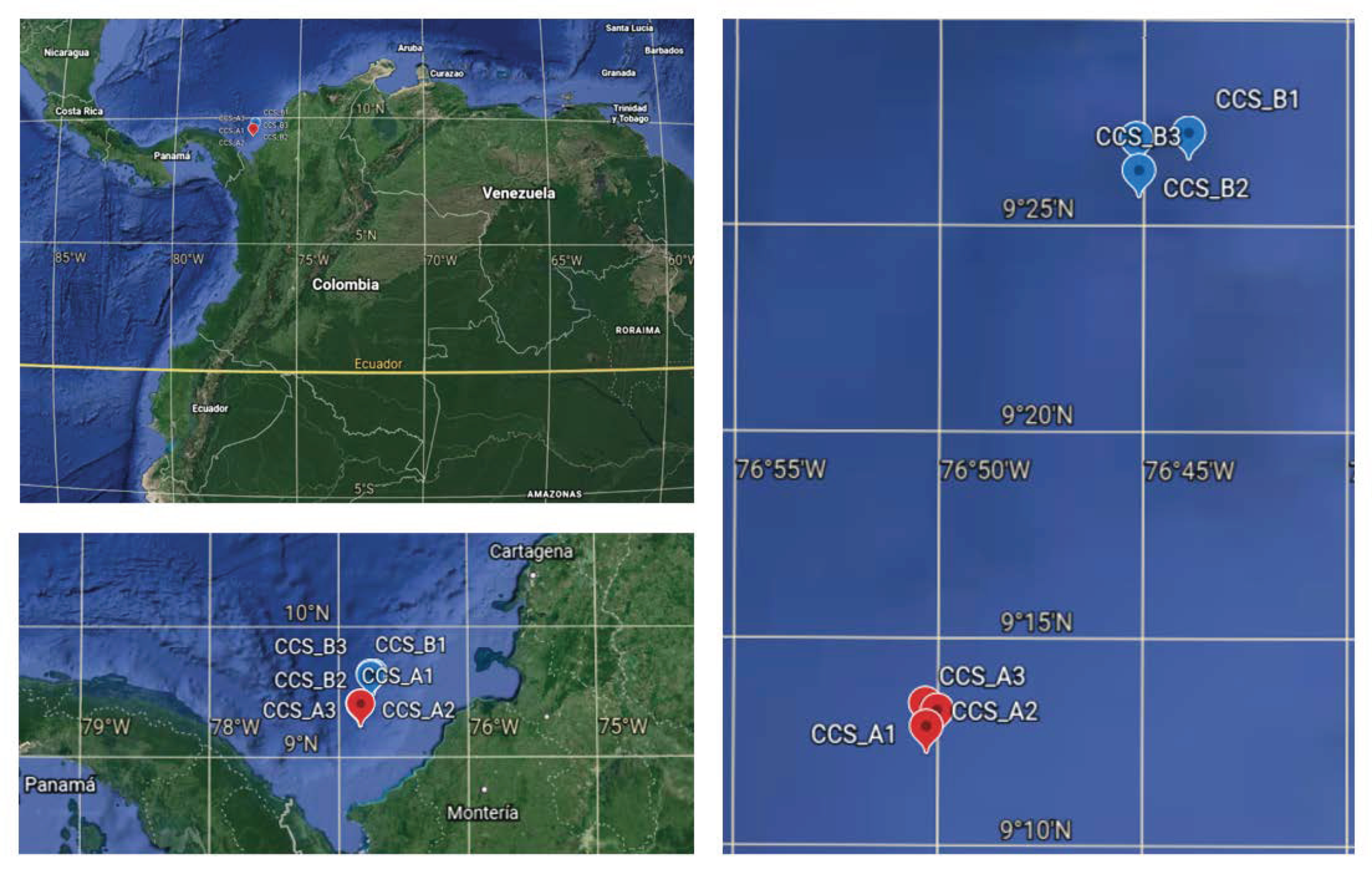
2.1. Sampling
2.2. DNA Isolation and 16S rRNA Gene Sequencing
2.3. Bioinformatics Analyses
2.4. Diversity Analysis
3. Results
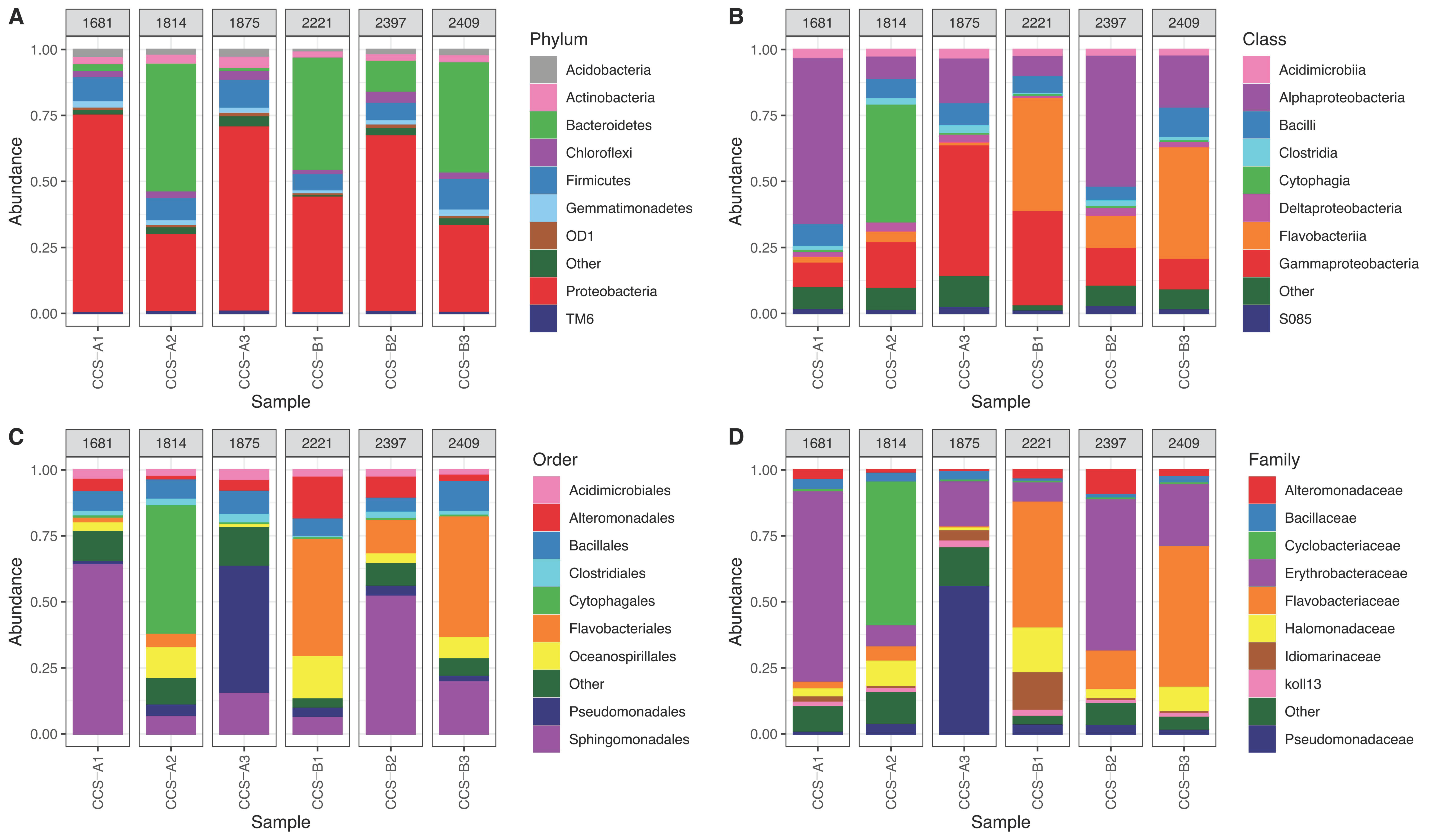
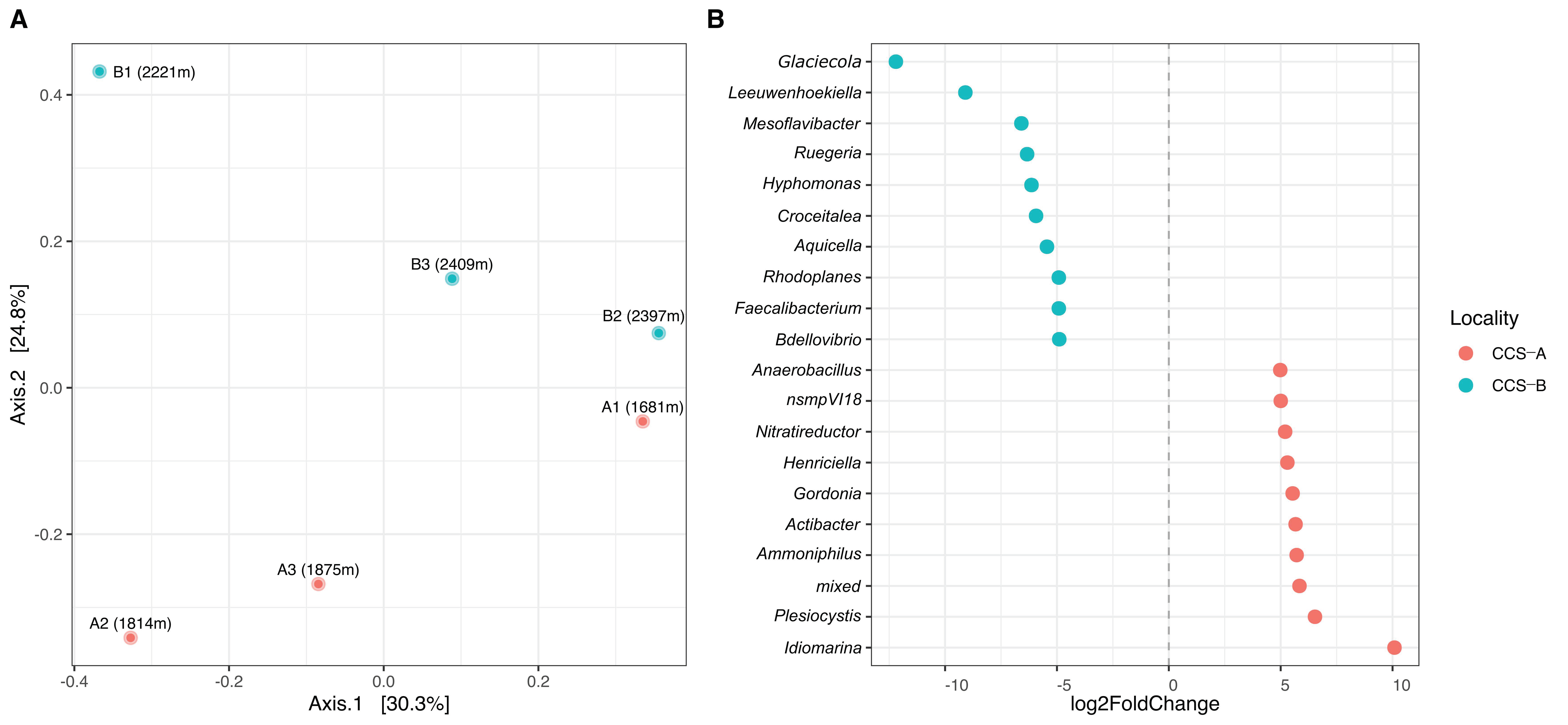
3.1. Bacterial Composition Analysis
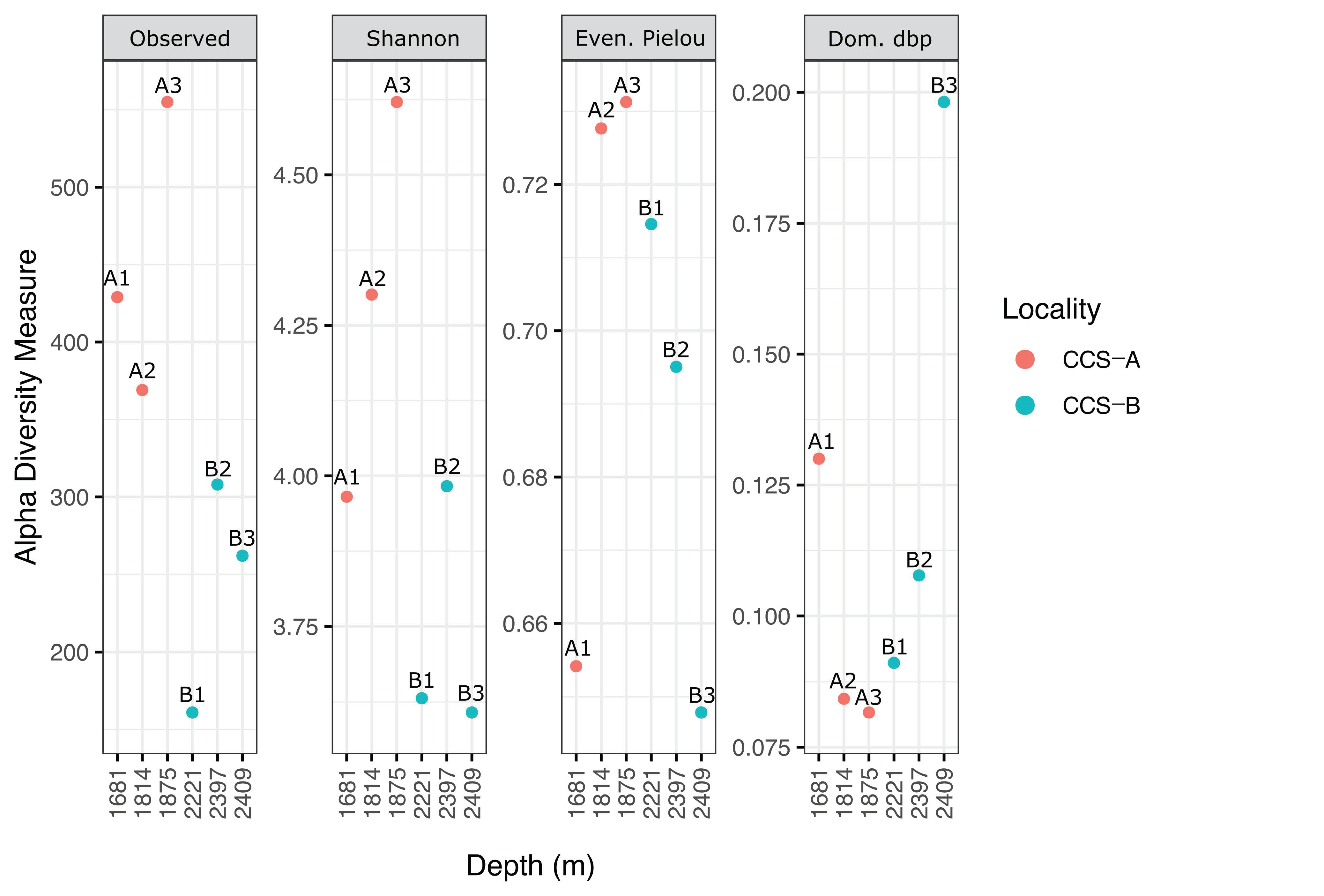
3.2. Bacterial Diversity Analysis
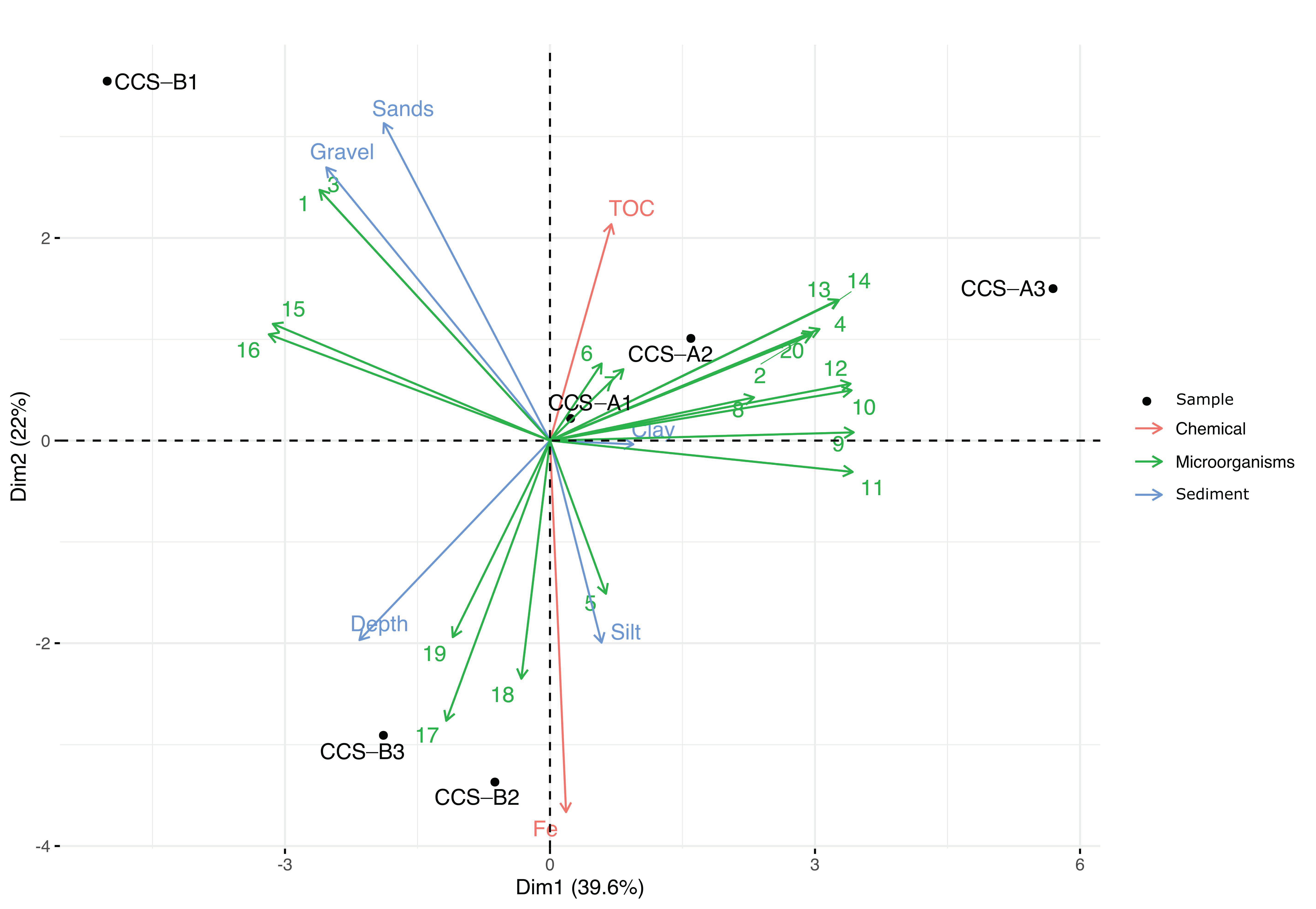
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Malek-Madani, R. Physical Oceanography: A Mathematical Introduction with MATLAB; CRC Press, Inc.: Boca Raton, FL, USA, 2012. [Google Scholar]
- Whitman, W.B.; Coleman, D.C.; Wiebe, W.J. Prokaryotes: The unseen majority. Proc. Natl. Acad. Sci. USA 1998, 95, 6578–6583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schauer, R.; Bienhold, C.; Ramette, A.; Harder, J. Bacterial diversity and biogeography in deep-sea surface sediments of the South Atlantic Ocean. ISME J. 2010, 4, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Aravindraja, C.; Viszwapriya, D.; Karutha Pandian, S. Ultradeep 16S rRNA sequencing analysis of geographically similar but diverse unexplored marine samples reveal varied bacterial community composition. PLoS ONE 2013, 8, e76724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Chun, J. 16S rRNA Gene-based identification of bacteria and archaea using the eztaxon server. In Methods in Microbiology; Academic Press: Cambridge, MA, USA, 2014; Volume 41, pp. 61–74. [Google Scholar]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [Green Version]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef]
- Salazar, G.; Cornejo-Castillo, F.M.; Benítez-Barrios, V.; Fraile-Nuez, E.; Álvarez-Salgado, X.A.; Duarte, C.M.; Gasol, J.M.; Acinas, S.G. Global diversity and biogeography of deep-sea pelagic prokaryotes. ISME J. 2016, 10, 596–608. [Google Scholar] [CrossRef] [Green Version]
- Pesant, S.; Not, F.; Picheral, M.; Kandels-Lewis, S.; Le Bescot, N.; Gorsky, G.; Iudicone, D.; Karsenti, E.; Speich, S.; Troublé, R.; et al. Open science resources for the discovery and analysis of Tara Oceans data. Sci. Data 2015, 2, 150023. [Google Scholar] [CrossRef] [Green Version]
- Kai, W.; Peisheng, Y.; Rui, M.; Wenwen, J.; Zongze, S. Diversity of culturable bacteria in deep-sea water from the South Atlantic Ocean. Bioengineered 2017, 8, 572–584. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Yu, Y.; Luo, W.; Zeng, Y.; Chen, B. Bacterial diversity in surface sediments from the Pacific Arctic Ocean. Extremophiles 2009, 13, 233–246. [Google Scholar] [CrossRef]
- Varliero, G.; Bienhold, C.; Schmid, F.; Boetius, A.; Molari, M. Microbial Diversity and Connectivity in Deep-Sea Sediments of the South Atlantic Polar Front. Front. Microbiol. 2019, 10, 665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Queiroz, L.L.; Bendia, A.G.; Duarte, R.T.D.; das Graças, D.A.; da Costa da Silva, A.L.; Nakayama, C.R.; Sumida, P.Y.; Lima, A.O.S.; Nagano, Y.; Fujikura, K.; et al. Bacterial diversity in deep-sea sediments under influence of asphalt seep at the São Paulo Plateau. Antonie Van Leeuwenhoek 2020, 113, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, D.J.; Andreote, F.D.; Chaves, D.; Montaña, J.S.; Osorio-Forero, C.; Junca, H.; Zambrano, M.M.; Baena, S. Structural and Functional Insights from the Metagenome of an Acidic Hot Spring Microbial Planktonic Community in the Colombian Andes. PLoS ONE 2012, 7, e52069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Álvarez-Yela, A.C.; Mosquera-Rendón, J.; Noreña-Puerta, A.; Cristancho, M.M.V.; Lopez-Alvarez, D. Microbial Diversity Exploration of Marine Hosts at Serrana Bank, a Coral Atoll of the Seaflower Biosphere Reserve. Front. Mar. Sci. 2019, 6. [Google Scholar] [CrossRef]
- Gonzalez-Zapata, F.L.; Bongaerts, P.; Ramírez-Portilla, C.; Adu-Oppong, B.; Walljasper, G.; Reyes, A.; Sanchez, J.A. Holobiont Diversity in a Reef-Building Coral over Its Entire Depth Range in the Mesophotic Zone. Front. Mar. Sci. 2018, 5, 29. [Google Scholar] [CrossRef] [Green Version]
- Quintanilla, E.; Ramírez-Portilla, C.; Adu-Oppong, B.; Walljasper, G.; Glaeser, S.P.; Wilke, T.; Muñoz, A.R.; Sánchez, J.A. Local confinement of disease-related microbiome facilitates recovery of gorgonian sea fans from necrotic-patch disease. Sci. Rep. 2018, 8, 14636. [Google Scholar] [CrossRef] [Green Version]
- Villegas-Plazas, M.; Wos-Oxley, M.L.; Sanchez, J.A.; Pieper, D.H.; Thomas, O.P.; Junca, H. Variations in Microbial Diversity and Metabolite Profiles of the Tropical Marine Sponge Xestospongia muta with Season and Depth. Microb. Ecol. 2019, 78, 243–256. [Google Scholar] [CrossRef]
- Naranjo-Vesga, J.; Ortiz-Karpf, A.; Wood, L.; Jobe, Z.; Paniagua-Arroyave, J.F.; Shumaker, L.; Mateus-Tarazona, D.; Galindo, P. Regional controls in the distribution and morphometry of deep-water gravitational deposits along a convergent tectonic margin. Southern Caribbean of Colombia. Mar. Pet. Geol. 2020, 121, 104639. [Google Scholar] [CrossRef]
- Correa-Ramirez, M.; Rodriguez-Santana, Á.; Ricaurte-Villota, C.; Paramo, J. The Southern Caribbean upwelling system off Colombia: Water masses and mixing processes. Deep Sea Res. Part I Oceanogr. Res. Pap. 2020, 155, 103145. [Google Scholar] [CrossRef]
- Rudzin, J.E.; Shay, L.K.; Jaimes, B.; Brewster, J.K. Upper ocean observations in eastern Caribbean Sea reveal barrier layer within a warm core eddy. J. Geophys. Res. Ocean. 2017, 122, 1057–1071. [Google Scholar] [CrossRef]
- Christ, R.D.; Wernli, R.L. (Eds.) Chapter 2—The Ocean Environment. In ROV Manual B.T.-T, 2nd ed.; Butterworth-Heinemann: Oxford, UK, 2014; pp. 21–52. ISBN 978-0-08-098288-5. [Google Scholar]
- Herlemann, D.P.R.; Labrenz, M.; Jürgens, K.; Bertilsson, S.; Waniek, J.J.; Andersson, A.F. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011, 5, 1571–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Dusa, A. Package ‘Venn’. Available online: https://cran.r-project.org/web/packages/venn/venn.pdf (accessed on 1 September 2020).
- Sinniger, F.; Pawlowski, J.; Harii, S.; Gooday, A.J.; Yamamoto, H.; Chevaldonné, P.; Cedhagen, T.; Carvalho, G.; Creer, S. Worldwide Analysis of Sedimentary DNA Reveals Major Gaps in Taxonomic Knowledge of Deep-Sea Benthos. Front. Mar. Sci. 2016, 3, 92. [Google Scholar] [CrossRef] [Green Version]
- Guardiola, M.; Wangensteen, O.S.; Taberlet, P.; Coissac, E.; Uriz, M.J.; Turon, X. Spatio-temporal monitoring of deep-sea communities using metabarcoding of sediment DNA and RNA. PeerJ 2016, 4, e2807. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, M.A.C.; Cavalett, A.; Spinner, A.; Rosa, D.C.; Jasper, R.B.; Quecine, M.C.; Bonatelli, M.L.; Pizzirani-Kleiner, A.; Corção, G.; de Lima, A.O.S. Phylogenetic identification of marine bacteria isolated from deep-sea sediments of the eastern South Atlantic Ocean. Springerplus 2013, 2, 127. [Google Scholar] [CrossRef] [Green Version]
- Quaiser, A.; Zivanovic, Y.; Moreira, D.; López-García, P. Comparative metagenomics of bathypelagic plankton and bottom sediment from the Sea of Marmara. ISME J. 2011, 5, 285–304. [Google Scholar] [CrossRef] [Green Version]
- Zinger, L.; Amaral-Zettler, L.A.; Fuhrman, J.A.; Horner-Devine, M.C.; Huse, S.M.; Welch, D.B.M.; Martiny, J.B.H.; Sogin, M.; Boetius, A.; Ramette, A. Global patterns of bacterial beta-diversity in seafloor and seawater ecosystems. PLoS ONE 2011, 6, e24570. [Google Scholar] [CrossRef]
- Hamdan, L.J.; Coffin, R.B.; Sikaroodi, M.; Greinert, J.; Treude, T.; Gillevet, P.M. Ocean currents shape the microbiome of Arctic marine sediments. ISME J. 2013, 7, 685–696. [Google Scholar] [CrossRef] [Green Version]
- Dyksma, S.; Bischof, K.; Fuchs, B.M.; Hoffmann, K.; Meier, D.; Meyerdierks, A.; Pjevac, P.; Probandt, D.; Richter, M.; Stepanauskas, R.; et al. Ubiquitous Gammaproteobacteria dominate dark carbon fixation in coastal sediments. ISME J. 2016, 10, 1939–1953. [Google Scholar] [CrossRef] [Green Version]
- Walsh, E.A.; Kirkpatrick, J.B.; Rutherford, S.D.; Smith, D.C.; Sogin, M.; D’Hondt, S. Bacterial diversity and community composition from seasurface to subseafloor. ISME J. 2016, 10, 979–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLong, E.F.; Franks, D.G.; Alldredge, A.L. Phylogenetic diversity of aggregate-attached vs. free-living marine bacterial assemblages. Limnol. Oceanogr. 1993, 38, 924–934. [Google Scholar] [CrossRef] [Green Version]
- Rath, J.; Wu, K.Y.; Herndl, G.J. High phylogenetic diversity in a marine-snow-associated bacterial assemblage. Aquat. Microb. Ecol. 1998, 14, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Fandino, L.B.; Riemann, L.; Steward, G.F.; Long, R.A. Variations in bacterial community structure during a dinoflagellate bloom analyzed by DGGE and 16S rDNA sequencing. Aquat. Microb. Ecol. 2001, 23, 119–130. [Google Scholar] [CrossRef]
- Goffredi, S.; Orphan, V. Bacterial community shifts in taxa and diversity in response to localized organic loading in the deep sea. Environ. Microbiol. 2009, 12, 344–363. [Google Scholar] [CrossRef]
- Escobar-Briones, E.; García-Villalobos, F.J. Distribución espacial del carbono orgánico total en el sedimento superficial de la planicie abisal del Golfo de México. In Carbono en Ecosistemas Acuáticos de México; Benigno Hernández de la Torre, G.G.C., Ed.; Instituto Nacional de Ecología: Mexico City, Mexico, 2007; p. 508. ISBN 9688178551. [Google Scholar]
- Bauer, M.; Kube, M.; Teeling, H.; Richter, M.; Lombardot, T.; Allers, E.; Würdemann, C.; Quast, C.; Kuhl, H.; Knaust, F.; et al. Whole genome analysis of the marine Bacteroidetes‘Gramella forsetii’ reveals adaptations to degradation of polymeric organic matter. Environ. Microbiol. 2007, 8, 2201–2213. [Google Scholar] [CrossRef]
- Panschin, I.; Becher, M.; Verbarg, S.; Spröer, C.; Rohde, M.; Schüler, M.; Amann, R.; Harder, J.; Tindall, B.; Hahnke, R. Description of Gramella forsetii sp. nov., a marine Flavobacteriaceae isolated from North Sea water, and emended description of Gramella gaetbulicola Cho et al. 2011. Int. J. Syst. Evol. Microbiol. 2016, 67. [Google Scholar] [CrossRef]
- Petro, C.; Starnawski, P.; Schramm, A. Microbial community assembly in marine sediments. Aquat. Microb. Ecol. 2017, 79, 177–195. [Google Scholar] [CrossRef] [Green Version]
- Stegen, J.C.; Lin, X.; Konopka, A.E.; Fredrickson, J.K. Stochastic and deterministic assembly processes in subsurface microbial communities. ISME J. 2012, 6, 1653–1664. [Google Scholar] [CrossRef] [Green Version]
- Bienhold, C.; Zinger, L.; Boetius, A.; Ramette, A. Diversity and Biogeography of Bathyal and Abyssal Seafloor Bacteria. PLoS ONE 2016, 11, e0148016. [Google Scholar] [CrossRef]
- Von Scheibner, M.; Sommer, U.; Jürgens, K. Tight Coupling of Glaciecola spp. and Diatoms during Cold-Water Phytoplankton Spring Blooms. Front. Microbiol. 2017, 8, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyson, R.V. Sedimentation rate, dilution, preservation and total organic carbon: Some results of a modelling study. Org. Geochem. 2001, 32, 333–339. [Google Scholar] [CrossRef]
- Walsh, E.A.; Kirkpatrick, J.B.; Pockalny, R.; Sauvage, J.; Spivack, A.J.; Murray, R.W.; Sogin, M.L.; D’Hondt, S. Relationship of Bacterial Richness to Organic Degradation Rate and Sediment Age in Subseafloor Sediment. Appl. Environ. Microbiol. 2016, 82, 4994–4999. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample-ID | Locality | Depth (m) | %Gravel | %Sand | %Silt | %Clay | %TOC | Iron (mg/Kg) |
|---|---|---|---|---|---|---|---|---|
| CCS_A1 | CCS_A | 1681 | 0.39 | 4.80 | 42.03 | 52.78 | 7.04 | 29,900 |
| CCS_A2 | CCS_A | 1814 | 0.17 | 3.55 | 40.72 | 55.56 | 7.06 | 30,600 |
| CCS_A3 | CCS_A | 1875 | 0.19 | 3.55 | 42.21 | 54.05 | 6.15 | 31,300 |
| CCS_B1 | CCS_B | 2221 | 1.50 | 7.41 | 37.85 | 53.24 | 6.35 | 26,600 |
| CCS_B2 | CCS_B | 2397 | 0.04 | 2.66 | 39.41 | 57.89 | 5.85 | 38,100 |
| CCS_B3 | CCS_B | 2409 | 0.20 | 2.40 | 48.23 | 49.17 | 5.50 | 38,600 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franco, N.R.; Giraldo, M.Á.; López-Alvarez, D.; Gallo-Franco, J.J.; Dueñas, L.F.; Puentes, V.; Castillo, A. Bacterial Composition and Diversity in Deep-Sea Sediments from the Southern Colombian Caribbean Sea. Diversity 2021, 13, 10. https://0-doi-org.brum.beds.ac.uk/10.3390/d13010010
Franco NR, Giraldo MÁ, López-Alvarez D, Gallo-Franco JJ, Dueñas LF, Puentes V, Castillo A. Bacterial Composition and Diversity in Deep-Sea Sediments from the Southern Colombian Caribbean Sea. Diversity. 2021; 13(1):10. https://0-doi-org.brum.beds.ac.uk/10.3390/d13010010
Chicago/Turabian StyleFranco, Nelson Rivera, Miguel Ángel Giraldo, Diana López-Alvarez, Jenny Johana Gallo-Franco, Luisa F. Dueñas, Vladimir Puentes, and Andrés Castillo. 2021. "Bacterial Composition and Diversity in Deep-Sea Sediments from the Southern Colombian Caribbean Sea" Diversity 13, no. 1: 10. https://0-doi-org.brum.beds.ac.uk/10.3390/d13010010