
Interface of Human/Wildlife Interactions: An Example of a Bold Coyote (Canis latrans) in Atlanta, GA, USA


1
Department of Biology, Berry College, Mount Berry, GA 30149, USA
2
Department of Biology, Emory University, Atlanta, GA 30322, USA
3
Department of Ecology and Evolutionary Biology, Princeton University, Princeton, NJ 08544, USA
*
Author to whom correspondence should be addressed.
Diversity 2021, 13(8), 372; https://0-doi-org.brum.beds.ac.uk/10.3390/d13080372
Submission received: 5 January 2021
/
Revised: 19 July 2021
/
Accepted: 5 August 2021
/
Published: 11 August 2021
(This article belongs to the Special Issue Humans and Wild Animals: Interactions in Deep Time, Recent History, and Now)
{kind=link}
{kind=link}
Abstract
:There is arguably no other North American species that better illustrates the complexities of the human-wildlife interface than the coyote. In this study, a melanistic coyote in metropolitan Atlanta, Georgia was exhibiting unusually bold behaviors that included encounters with humans, domestic dogs, and attempts to enter homes. After tracking this coyote (nicknamed Carmine) across a highly urbanized landscape with participatory science, including at least 80 publicly reported sightings, he was captured and relocated to a wildlife sanctuary. Genome-wide analyses revealed 92.8% coyote ancestry, 1.7% gray wolf ancestry, and 5.5% domestic dog ancestry. The dog alleles in Carmine’s genome were estimated to have been acquired by his ancestors 14–29 years ago. Despite his bold behavior, Carmine did not carry any mutations known to shape hypersociability in canines. He did, however, carry a single copy of the dominant mutation responsible for his melanistic coat color. This detailed study of Carmine dispels common assumptions about the reticent coyote personality and the origins of behavior. His unusual bold behavior created a higher level of human-coyote interaction. He now serves as a public ambassador for human-wildlife coexistence, urging the global community to reconsider mythologies about wildlife and promote coexistence with them in landscapes significantly altered by human activity in our rapidly changing world.
1. Introduction
The evolutionary histories and successes of humans and dogs have been intertwined for more than 15,000 years ago, with details regarding the origins of dogs highlighted by ancient DNA studies [1]. The role of dogs in society has expanded dramatically since their origins, with a dramatic focus on identifying the genes linked to their behavioral and phenotypic evolution [1,2,3,4,5,6,7,8,9,10,11]. Simultaneously, society has remained challenged by coexistence with wild canids across the globe, and particularly with coyotes (C. latrans) for North American inhabitants. Both the domestication of wolves and more recent, eradication management efforts have shaped the physiology, morphology, genetics, and behavior of several canid species through introgression and admixture. This includes the North American coyote.
Several traits have evolved in dogs throughout the course of their domestication from gray wolves. Likely the most important evolutionary change is that dogs show an increased tolerance and interest toward humans relative to wolves [12]. Recent efforts discovered a few key genetic mutations that have contributed toward the hypersocial behavioral shift in canids [8]. These mutations segregate among wolf populations and dog breeds across the globe, but appear to be rare in coyote genomes. However, these mutations follow Mendelian inheritance and could thus be introgressed into other canid genomes via hybridization. Further, selective breeding of domestic dogs has altered their pigmentation types and patterning. Melanism is an additional trait that evolved in dogs but was transferred back to wolves and coyotes through introgressive hybridization [13]. Although melanism is known to show an adaptive advantage maintained by balancing selection in gray wolf populations [14,15], coyote populations display a variable occurrence of melanistic and white coat colorations [16,17,18,19]. Although the evolutionary significance of the melanistic phenotype in coyotes remains yet to be documented, admixture and allele sharing among canids has been extensively documented [20,21,22,23,24,25,26,27].
The dynamic evolutionary history of the North American coyote has been dramatically shaped by human colonization, predatory control, and landscape conversion over the past few centuries. Historically found west of the Mississippi River, the coyote steadily expanded its geographic range during the past century and is now ubiquitous, living in nearly every major metropolitan area [28,29]. Human extirpation of congeneric red wolves (C. rufus) accelerated the southeastern range expansion of the coyote by eliminating its primary non-human competitor, while deforestation, urbanization, and the resulting increase in edge habitat were also all likely contributors [30,31,32]. In Georgia, coyotes first appeared in the central portion of the state in the 1960s and had a state-wide distribution just 20 years later [28,31,33]. Coyotes were prevalent in metropolitan Atlanta by the turn of the century and are now commonly observed throughout the region.
In this paper, we present genomic data derived from a bold coyote in Atlanta, Georgia. As we describe, this melanistic coyote lacked neophobia and exhibited proactive, exploratory, and unusually affiliative and friendly behaviors, including extensive solicitation of play from domesticated dogs [34,35]. The animal remained elusive to humans and was not overtly aggressive, nor did it attempt to copulate with dogs. Sightings of the animal, who was assumed to be a coyote and nicknamed Carmine (Figure 1A) by local residents, became commonplace over the next several weeks and its presence attracted widespread media attention due to public concern and curiosity. As a result, we initiated a plan to capture the animal in the interest of public safety, animal welfare, and scientific inquiry. Here, we provide data relevant to our goals of (1) understanding this animal’s use of an urban landscape, (2) determining its ancestry and level of admixture with other Canis species, and (3) investigating if it carries any mutations associated with canine human-directed hypersocial behavior or melanism. Investigating patterns of admixture and novel genomic traits, including those related to melanism and hypersociality, can provide insight into the origins of inter-individual variation, speciation, and radiation of diversity.
2. Materials and Methods
2.1. Tracking and Capture of Subject Animal
We received daily public sightings of the study animal through the Atlanta Coyote Project’s website (http://cs.berry.edu/coyote/report.php) and social media accounts (https://www.facebook.com/atlantacoyoteproject), which allowed us to track its movement, core areas of use, and distances traveled in the highly urbanized and fragmented landscape (Figure 1B). Concurrently, we learned that the Yellow River Wildlife Sanctuary (YRWS; Lilburn, GA, USA) was seeking a companion for a captive spayed female coyote (Wilee) that had been rehabbed but was unable to be released. With permission from the Georgia Department of Natural Resources, we targeted the study animal for capture and relocation to YRWS.
2.2. RAD Sequencing and Genomic Ancestry Analyses
We prepared genomic DNA from blood samples for restriction site-associated DNA sequencing (RADseq) following a modified protocol using the SbfI restriction enzyme and the NEBnext Ultra II DNA Library Prep Kit for paired-end (2 × 150 nt) sequencing on an Illumina NovaSeq6000 platform at Princeton University’s Lewis Sigler Genomics Institute core facility [36]. We used STACKS v2 [37,38] to process and clean sequence reads for subsequent alignment to the reference dog genome CanFam3.1 assembly [39]. Ninety publicly available canid samples previously mapped to the same reference genome assembly were included as ancestry reference genomes (Table S1). We annotated SNPs across these 91 canid genomes and filtered them to retain sites with a MAF > 3%, with an allowance of up to 20% missingness per locus. For demographic analyses of genetic structure and diversity estimates, we constructed a “statistically unlinked” dataset of SNPs through linkage disequilibrium pruning. For full protocol and bioinformatic details, see Appendix A.
We assessed the probability of population membership to the reference groups using principal component analysis (PCA) and a Bayesian posterior probability assignment method (see Appendix A). Formal assignment of ancestry across the genome was completed with the program ELAI [40], which implements a two-layer hidden Markov model to estimate the most likely ancestry proportion at every locus. We implemented ELAI in serial triplicates to explore the parameter space and averaged the results. We estimated the timing that dog alleles first appeared in the study animal’s ancestral genome through gene flow, and we followed the methods of Johnson et al. [41] to estimate the number of generations since admixture for diploid genomes (Appendix A). We utilized two estimates regarding the number of years in a canine generation (two and four years per generation) to capture possible events of canid yearling reproduction [42,43,44,45,46,47].
2.3. Genotyping Variants Associated with Canine Human-Directed Hypersocial Behavior
Due to the study animal’s propensity to be extremely proximate to human activity, we hypothesized that it carried one or more genetic variants associated with increased human-directed sociability in dogs and wolves [8]. Although coyotes rarely carry these variants, if at all, this animal’s behavior was exceedingly unusual. We used vonHoldt et al.’s [8] primers and PCR protocol to target four insertions sites for transposable elements (canine LINE and SINE transposons) on canine chromosome CFA6. Two insertions are associated with the gene WBSCR17 (referred to as Cfa6.6 and Cfa6.7), one with the gene GTF2I (Cfa6.66), and one with gene POM121 (Cfa6.83). Amplification and 2% agarose gel electrophoresis resolved the codominant status of copy number at each putative insertion site.
2.4. Sequence Assay for the Deletion Associated with K Locus Melanism in Canids
We genotyped the study animal for the 3bp indel at the gene CBD103 (also known as the K locus) on canine chromosome CFA16:58,965,448 (canfam3.1 assembly) and followed the sequencing protocol of Cubaynes et al. (T. Coulson, personal communication). We used an amplicon-sequencing approach targeting the 3bp deletion. We mapped reads (122nt in length) to the reference dog genome (CanFam3.1, GCF_000002285.3) using bwa-mem [48], followed by indel genotyping with GATK v4.0.11.0 [49,50]. Further details are provided in Appendix A.
3. Results
3.1. Behavior and Morphology
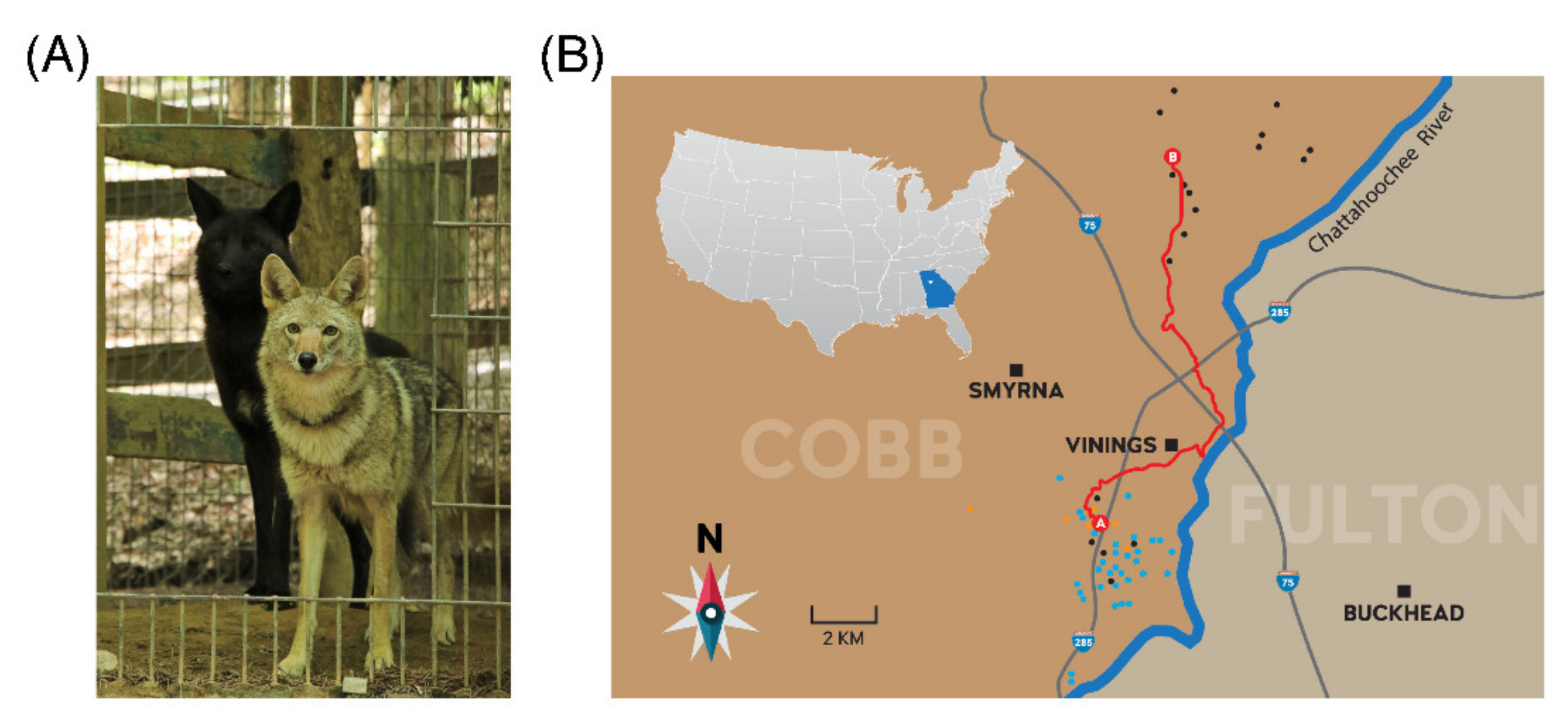
We received over 80 public sightings of the study animal between 24 December 2019 and 16 February 2020; sightings occurred at all hours of the day and night. On several occasions, Carmine was seen with at least one other coyote, although its consistent attempts to engage dogs (both male and female) in playful behavior always occurred in the absence of other coyotes (Videos S1 and S2). Based on reported sightings, the study animal spent seven weeks within a ~24 km2 area of Smryna and Vinings, GA, which meant it routinely crossed the I-285, one of metropolitan Atlanta’s busiest interstate highways. We discovered three large culverts running under the I-285 within the study animal’s core area, which likely offered the ability to avoid traffic. Two separate sightings occurred within the Smyrna, GA area on 6 February 2019, but 24 h later, the study animal was sighted and photographed 16.1 km away in the northeastern portion of Cobb County, where it utilized an ~11 km2 area over the next ten days (Figure 1B). We captured the animal on 17 February 2020 after 49 days of trapping effort and determined that it was an approximately one-year old male that weighed 17.2 kg. Despite his bold behavior in the wild, he continued to actively avoid all human approach once in captivity and had to be sedated prior to physical examination. After surgical castration, he was gradually introduced to the resident coyote (Wilee) at Yellow River Wildlife Sanctuary and they immediately engaged in playful behavior when placed in the same enclosure.
On 10 May 2020, four days after moving into his permanent exhibit with Wilee, Carmine escaped by scaling a 2 m-high enclosure fence and likely withstanding the electric shock that was delivered by the protective wire. He was monitored through a GPS collar showing that he immediately went to a nearby neighborhood and began playing with dogs, a route that had him swimming 30–35 m across the nearby Yellow River several times. Although he did not stray far, he remained at large for four days until his recapture.
3.2. Ancestry
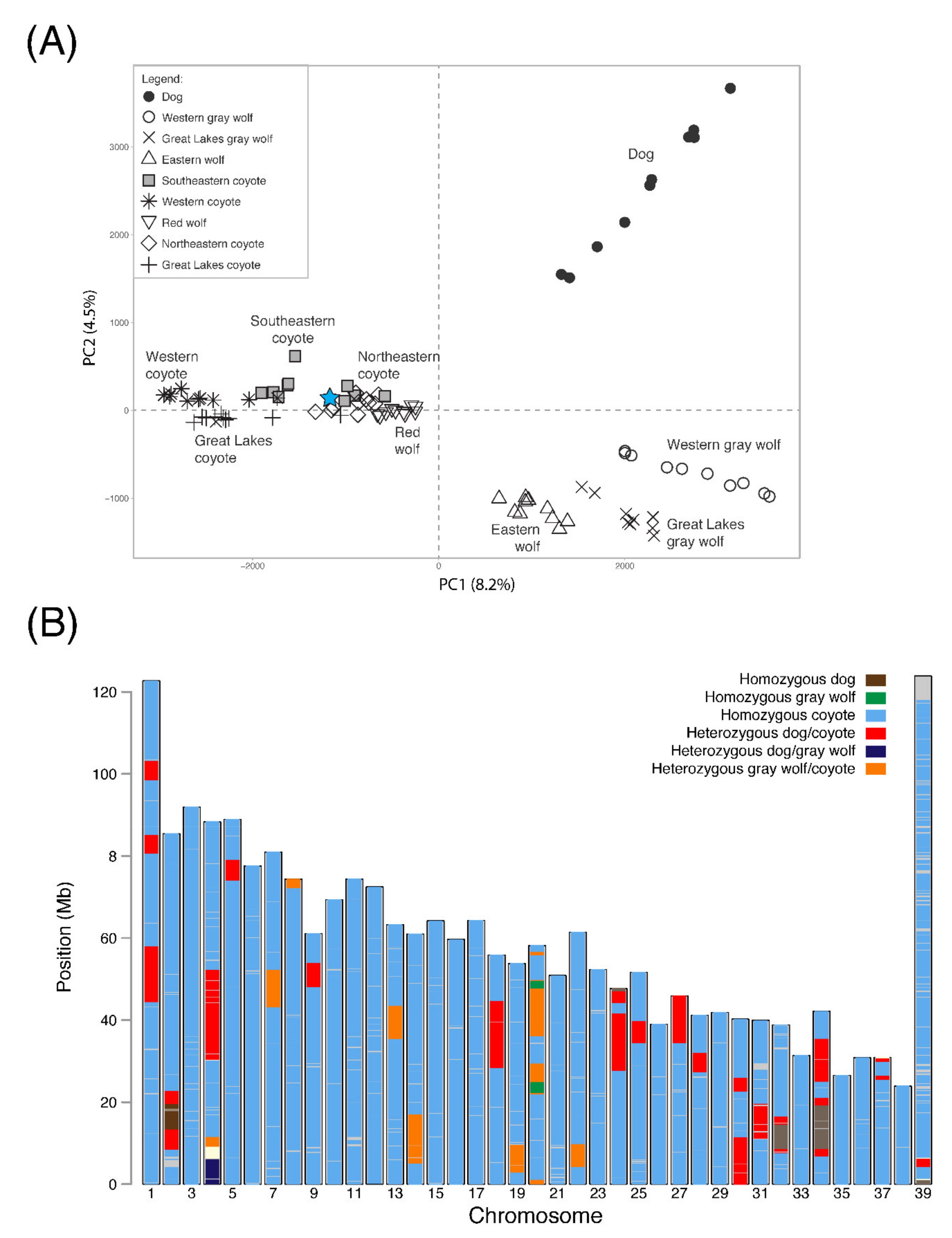
We assessed Carmine’s genomic ancestry with respect to three reference species (domestic dog, gray wolf, and coyote) across 118,201 SNP genotypes (n, autosomes = 115,623; X chromosome = 2578) with an average marker density of one SNP variant every 19.7 Kb (density: autosomes = 19 Kb, X chromosome = 48 Kb). We estimated him to carry 92.8% coyote ancestry, 1.7% gray wolf ancestry, and 5.5% domestic dog ancestry (Figure 2; Tables S1 and S2). Across these loci, 103,069 sites were homozygous for coyote ancestry, 1330 were homozygous for dog ancestry, and 255 for gray wolf ancestry. Carmine carried 13,547 SNP loci that were heterozygous for ancestry states (n sites: dog/wolf = 366; dog/coyote = 10,110; wolf/coyote = 3071). For complete details of the RADseq SNP loci and assessment of reference genomes, see Appendix B.
It was possible to estimate the number of years ago when dog alleles first appeared in Carmine’s ancestors’ genome through gene flow. The total number of dog ancestry switches (autosomal = 30, X chromosome = 3) and genome-wide ancestry proportions specific to the type of chromosome (autosomes = 0.053, X chromosome = 0.124) were used for this estimation (Table S2). The dog alleles in Carmine’s genome were estimated to have entered the ancestral genome 7.2 generations (14–29 years) ago, while the X-linked dog alleles were likely similarly acquired 6.2 generations (12–25 years) ago.
3.3. Canine Hypersociability and Melanism
Carmine lacked canine hypersocial transposons at all possible sites of integration. This is concordant with previously reported findings where such insertions are rare, at best, in coyote genomes [8]. We sequenced Carmine’s CBD103 gene to determine if his melanistic phenotype was due to the same 3bp deletion as previously reported to confer melanisms in gray wolves and some domestic dog breeds [13,51] and to determine its genotypic state. Carmine’s CBD103 gene was sequenced at an average depth of 506-fold coverage, and we determined him to be heterozygous for the deletion (coverage: wildtype allele = 232x, deletion = 236x; genotype quality (GQ) = 99).
4. Discussion
Carmine is a melanistic coyote who was exhibiting bold behavior in an urban environment at the interface of human-wildlife interactions. There is arguably no other endemic North American wildlife species that better illustrates the complexities of this interface than the coyote, and the story of Carmine’s journey across metropolitan Atlanta is a perfect example of the circumstances that result in human-wildlife conflict versus coexistence. Carmine’s ability to successfully navigate the matrix of the urban landscape prior to capture was particularly noteworthy as it demonstrated his keen awareness and understanding of his surroundings. Several recent studies have documented plasticity in coyote behavior in response to varying levels of urbanization [52,53,54], while Adducci et al. found genetic distinctiveness between urban and rural coyote populations [55]. Although urbanization has developed only relatively recently in the evolutionary history of canids, the short generation times and high fecundity rates of coyotes likely allow them to respond to such habitat selection pressures relatively quickly; this illustrates the role of diversity within the Canis genus over the past 1,000,000 years.
Carmine’s bold behavior led to much public speculation about his genetic makeup, with many people believing that a coyote would never act in this manner unless it was the product of admixture with a dog, which was not the case. Although he lacks the hypersocial mutations associated with human-directed behavior in gray wolves and dogs, there are several other candidate genetic variants that likely contribute toward Carmine’s bold personality type [56,57,58]. Previous work has quantified meaningful levels of additive heritability of the shyness-boldness spectrum of canine personality (~25% in dogs) [59]. Boldness has been linked to canine interest of playing with humans, interactions with strangers, and non-social fear [60]; such behavior is not novel for canines and has been quantitatively described in gray wolves [61]. Boldness is likely under environment-specific balancing selection where individuals who exhibit higher levels of boldness may experience benefits from resource acquisition (such as mates, food, territory). Mismatch to the environment, on the other hand, could result in deleterious outcomes (e.g., fatal interactions within urban landscapes). While urban coyotes have previously been found to be more bold and exploratory than coyotes living in rural habitats, there is little evidence that such behaviors are connected to aggression [62,63]. We are then faced with the question: can we attribute such personality to the efficiency at which eradication programs purge populations of bolder (less neophobic) individuals [62]? However, the effective population sizes of urban and rural coyotes are dramatically different, which impacts the efficacy of selection and the evolution of bold behaviors. Nonetheless, such behavioral differences serve as a reminder of the possible evolutionary options available when such standing phenotypic variation exists.
The decision to intervene in the life of a wild animal was not made lightly and it was not without some controversy. Typically, human-coyote interactions are rare and arguably the best management strategies are proactive measures that promote peaceful coexistence. Carmine’s unusual bold behavior created a higher level of human-coyote interaction, but without governmental consent and the availability of a captive sanctuary, attempts to capture him would not have been undertaken. Nevertheless, he has now made significant contributions to further our understanding of the interface of ecology and evolution while serving as a public ambassador for human-wildlife coexistence.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/d13080372/s1. Table S1: Sample information, population of origin, ancestry notes, proportion (prop.) of missing data per genome, and posterior probability (prob.) assignment to each possible reference genome. (Abbreviations: CFA, domestic dog; CLY, eastern wolf; CLU, gray wolf; CLA, coyote; CRU, red wolf). Bolded values indicate the new data collected for this study. Table S2: Posterior probability proportions (prop.) for each of the reference individuals with assignments up to two generations back. Notes are also provided to explain when admixture was detected among reference genomes and when samples were excluded from ancestry inference. (Abbreviations: gen, generation). Video S1: Carmine at the trap site. Video S2: Carmine playing with a dog.
Author Contributions
Conceptualization, C.B.M., L.A.W. and B.M.v.; methodology, C.B.M., L.A.W. and B.M.v.; formal analysis, C.B.M., L.A.W. and B.M.v.; investigation, C.B.M., L.A.W. and B.M.v.; resources, C.B.M., L.A.W. and B.M.v.; data curation, C.B.M. and B.M.v.; visualization, C.B.M., L.A.W. and B.M.v.; supervision, C.B.M., L.A.W. and B.M.v.; project administration, C.B.M.; funding acquisition, C.B.M. All authors contributed to writing and editing the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Berry College and through private donations.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and the USDA Animal Welfare Act and Animal Welfare Regulations. It was approved under permits from the USDA (#57-C-0370) and USFWS (#MB36920D-0) on 31 December 2019. Trapping of the study animal was conducted under NWCO License #1000565487.
Data Availability Statement
The processed and mapped BAM file was deposited on NCBI’s Short Read Archive (BioProject PRJNA682820). Additional data analyzed were previously published and details for their SRA information is found in references within Table S1.
Acknowledgments
We are grateful to Kris Hoffman, Brandon Sanders, Alex Taylor, Lara Shaw, Glenda Elliott, Vanessa Pryor, Jonathan Ordway, Katy Ordway, Abbey Elkert, Elizabeth Fincher, Clinton Murphy, Wayne Hubbard, Erin Brown, Bob Crabtree, Mark Johnson, and all others who contributed to the safe capture and care of Carmine. Craig Hall helped with figure preparation.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Appendix A
Appendix A.1. Detailed Materials and Methods
Appendix A.1.1. RAD Sequencing and Bioinformatic Processing
High molecular-weight genomic DNA were obtained using the Qiagen DNeasy Blood and Tissue Kit. DNA was quantified by a Qubit 2.0 fluorometry system and we subsequently standardized the DNA sample to 5 ng/µL. Genomic DNA was prepared for restriction site-associated DNA sequencing (RADseq) following a modified protocol [36]. Genomic DNA was digested with SbfI and ligated a unique 8-bp barcoded with a biotinylated adapter, which allowed for subsequent pooling and demultiplexing after sequencing. Equal amounts of up to 48 samples were pooled followed by random shearing to 300–400 bp in a Covaris LE220, which was then enriched for adapter-ligated fragments using a Dynabeads M-280 streptavidin binding assay, followed by preparation for Illumina sequencing using the NEBnext Ultra II DNA Library Prep Kit. The library was then sequenced on an Illumina NovaSeq for paired-end (2 × 150 nt) reads at Princeton University’s Lewis Sigler Genomics Institute core facility. Size selection and library purification were completed using Agencourt AMPure XP magnetic beads.
Sequence data were first processed to retain the reads (and their pairs) that contained the unique barcode and the remnant sbfI cut site. We used STACKS v2 [37,38] to demultiplex each sample using the process_radtags module, with a 2bp mismatch for barcode rescue, and retained reads with a quality score ≥ 10. PCR duplicates were removed with the filtering option in clone_filter with subsequent alignment to the reference dog genome CanFam3.1 assembly (GCF_000002285.3) [39] using STAMPY v1.0.21 [64]. A mapping quality threshold was applied to retain mapped reads (MAPQ > 96) and convert them to the BAM format in Samtools v0.1.18 [48].
Appendix A.1.2. SNP Variant Discovery
SNP discovery was conducted using all 91 samples, ensuring that they each had a minimum of 100,000 mapped reads, to obtain a catalog of all polymorphic sites possible. The gstacks and populations modules in STACKS v2 were implemented as per default guidelines, with an increase of the significance threshold in gstacks and use of the Marukilow model (flags–vt-alpha and–gt-alpha, p = 0.01) for variant discovery in the populations module. All SNPs discovered per locus were reported (we opted against using the populations flag write_single_snp) as ancestry inference is best with high-density data. Data were further filtered to retain sites with a MAF > 3% and to allow up to an 80% genotyping rate per locus (flag–geno 0.2) for the initial filtering step in PLINK v1.90b3i [65]. To determine if any individual sample was missing a significant proportion of genotypes, missingness was assessed using the PLINK function –missing, which excluded any samples with >50% missing data from all downstream analyses. For demographic analyses of genetic structure and diversity estimates, a “statistically unlinked” dataset of SNPs was constructed by excluding sites within 50-SNP windows that exceeded genotype correlations of 0.5 (with the PLINK argument–indep-pairwise 50 5 0.5). The X chromosome was analyzed independently from the autosomes to ensure the inclusion of the signal driven by the single sex chromosome captured in our sequencing and mapping efforts.
Appendix A.1.3. Population Structure Analysis
To survey genetic clustering and the structure of Carmine relative to the reference samples, two analyses were conducted with the statistically unlinked SNP set: principal component analysis (PCA) with the program flashPCA [66] and a Bayesian posterior-probability assignment test to assign Carmine to five possible genomic clusters (K) in STRUCTURE v2.3.4 [67]. The parameter flags MAPDISTANCES, POPFLAG, and USEPOPINFO were used with the others at default settings for analysis at K = 5 (i.e., dog, eastern wolf, gray wolf, red wolf, coyote) with 10,000 BURNIN and 20,000 NUMREPS.
Appendix A.1.4. Inference of Canid Ancestry
Carmine’s genomic ancestry was inferred with respect to three possible reference populations (gray wolves, coyotes, dogs) (Table S1). The full SNP set (unfiltered for statistical correlation) was analyzed with the program ELAI [40], which implemented a two-layer hidden Markov model to estimate the most likely ancestry proportion at every locus. Loci were excluded if they were lacking in any of the populations analyzed. The number of upper-layer clusters (-C) was set to 3 (i.e., red wolves and each reference coyote population) and the lower-layer clusters (-c) to 15 (5× the value of C, as recommended). To account for uncertainty in the precise timing and duration of admixture, four timepoints since admixture (-mg) were analyzed: 5, 10, 15, and 20 generations ago. ELAI was implemented in serial triplicates for each parameter setting with 30 EM steps, with the results averaged over these replicates. Only sites with allele dosages >1.5 were considered to be homozygous and all other sites were heterozygous. Sites with multiple and incompatible ancestries (e.g., three heterozygous states) were excluded. An ancestry block was defined by a minimum of three contiguous SNPs with the same ancestry assignment.
The number of dog ancestry block switches was used to infer the timing at which dog alleles first appeared in Carmine’s ancestral genome through gene flow [41] by rearranging the equation for diploid genomes:
where B is the estimated number of dog ancestry switches, T is the number of generations since admixture, L is the total genome length (2085cM for autosomes and 111cM for the X chromosome [68]), and z is the genome-wide ancestry proportion of dog ancestry across the autosomes or X chromosome.
B = (2 × 2 × 0.01) × T × L × z(1 − z)
Appendix A.1.5. Genotyping Variants Associated with Canine Human-Directed Hypersocial Behavior
Due to Carmine’s propensity to be extremely proximate to human activity, it was hypothesized that perhaps he carried one or more genetic variants that are associated with increased human-directed sociability in dogs and wolves [8]. Although coyotes rarely carry these variants, if at all, Carmine’s behavior was exceedingly unusual. The primers and protocol used were previously reported by vonHoldt et al. [8]. Briefly, PCR was used to target four insertion sites for transposable elements (canine LINE and SINE transposons) on canine chromosome 6. Two insertions are associated with the gene WBSCR17 (referred to as Cfa6.6 and Cfa6.7), one with the gene GTF2I (Cfa6.66), and one with gene POM121 (Cfa6.83). Amplification and 2% agarose gel electrophoresis resolved the codominant status of the copy number at each putative insertion site.
Appendix A.1.6. Sequence Assay for the Deletion Associated with K Locus Melanism in Canids
Carmine was genotyped for the 3bp indel at the gene CBD103 on canine chromosome CFA16:58,965,448 (canfam3.1 assembly). This is known to be a deletion of the CCC fragment, often with the preceding nucleotide also deleted (TCCC indel). Briefly, a dual-barcoding index was constructed for short-read single-end sequencing of 122 nt on an Illumina NovaSeq6000 at Princeton University’s Lewis Sigler Genomics Institute core facility. Adapter sequences were clipped (maximum error rate = 0.5, minimum overlap = 20) and a single mismatch for demultiplexing the pooled reads was allowed, which retained reads of 30nt or longer. Reads were mapped to the reference dog genome (CanFam3.1, GCF_000002285.3) using bwa-mem (arXiv: 1303.3997v2). Read groups were edited using the AddOrReplaceReadGroup function in the Picard Toolkit (Broad Institute, 2019) and we sorted/indexed the BAM file with samtools v0.1.18 [48]. The HaplotypeCaller function of GATK v4.0.11.0 [69] was implemented to genotype the 3bp deletion using the flag -ERC GVCF. Only sites with at least 10-fold sequencing coverage were considered.
Appendix B
RADseq Results
From the RADseq method, 2,091,181 SNPs were discovered across 91 canid samples. After filtering for MAF and missingness, 146,206 SNPs were retained. On average, samples contained 11.4% missing genotypes (Table S1), and all samples were included for analysis. A second dataset was constructed by removing 4025 loci that displayed a high level of inter-locus correlation. This “statistically unlinked” dataset of 105,931 loci was used for genetic clustering and structure analyses. Both PCA and a posterior probability assignment test for the current generation correctly identified a known positive control, sample #2338, that was mislabeled as a Wisconsin gray wolf. This sample correctly and accurately clustered with Great Lakes coyotes (Qcoyote = 1.0) (Figure 2; Table S2). This positive control sample was excluded from the ancestry inference. Additionally, sample #8682 (Kentucky coyote), had been assigned to ‘dog’ two generations back (Qdog = 1.0) and was also excluded from ancestry inference (Table S2). There were 11 additional samples (1 gray wolf and 10 coyotes) that were misclassified; however, their assignments were two generations back. All are consistent with previously published reports of admixture and shared demography. The gray wolf (Michigan) was assigned to eastern wolf [70]; eight northeastern coyotes were assigned to eastern wolf [70]; and two southeastern coyotes were assigned to red wolf [21,22]. These 11 samples were retained for ancestry inference due to their known demographic history and admixture, and represent crucial genetic variation found within each of the canid lineages.
References
- Frantz, L.A.F.; Bradley, D.G.; Larson, G.; Orlando, L. Animal Domestication in the Era of Ancient Genomics. Nat. Rev. Genet. 2020, 21, 449–460. [Google Scholar] [CrossRef]
- Li, Y.; vonHoldt, B.M.; Reynolds, A.; Boyko, A.R.; Wayne, R.K.; Wu, D.-D.; Zhang, Y.-P. Artificial Selection on Brain-Expressed Genes during the Domestication of Dog. Mol. Biol. Evol. 2013, 30, 1867–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akey, J.M.; Ruhe, A.L.; Akey, D.T.; Wong, A.K.; Connelly, C.F.; Madeoy, J.; Nicholas, T.J.; Neff, M.W. Tracking Footprints of Artificial Selection in the Dog Genome. Proc. Natl. Acad. Sci. USA 2010, 107, 1160–1165. [Google Scholar] [CrossRef] [Green Version]
- Axelsson, E.; Ratnakumar, A.; Arendt, M.-L.; Maqbool, K.; Webster, M.T.; Perloski, M.; Liberg, O.; Arnemo, J.M.; Hedhammar, Å.; Lindblad-Toh, K. The Genomic Signature of Dog Domestication Reveals Adaptation to a Starch-Rich Diet. Nature 2013, 495, 360–364. [Google Scholar] [CrossRef]
- vonHoldt, B.M.; Pollinger, J.P.; Lohmueller, K.E.; Han, E.; Parker, H.G.; Quignon, P.; Degenhardt, J.D.; Boyko, A.R.; Earl, D.A.; Auton, A.; et al. Genome-Wide SNP and Haplotype Analyses Reveal a Rich History Underlying Dog Domestication. Nature 2010, 464, 898–902. [Google Scholar] [CrossRef] [Green Version]
- Boyko, A.R.; Quignon, P.; Li, L.; Schoenebeck, J.J.; Degenhardt, J.D.; Lohmueller, K.E.; Zhao, K.; Brisbin, A.; Parker, H.G.; vonHoldt, B.M.; et al. A Simple Genetic Architecture Underlies Morphological Variation in Dogs. PLoS Biol. 2010, 8, e1000451. [Google Scholar] [CrossRef] [Green Version]
- Cadieu, E.; Neff, M.W.; Quignon, P.; Walsh, K.; Chase, K.; Parker, H.G.; vonHoldt, B.M.; Rhue, A.; Boyko, A.; Byers, A.; et al. Coat Variation in the Domestic Dog Is Governed by Variants in Three Genes. Science 2009, 326, 150–153. [Google Scholar] [CrossRef] [Green Version]
- vonHoldt, B.M.; Shuldiner, E.; Koch, I.J.; Kartzinel, R.Y.; Hogan, A.; Brubaker, L.; Wanser, S.; Stahler, D.; Wynne, C.D.L.; Ostrander, E.A.; et al. Structural Variants in Genes Associated with Human Williams-Beuren Syndrome Underlie Stereotypical Hypersociability in Domestic Dogs. Sci. Adv. 2017, 3, e1700398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Driscoll, C.A.; Macdonald, D.W.; O’Brien, S.J. From Wild Animals to Domestic Pets, an Evolutionary View of Domestication. Proc. Natl. Acad. Sci. USA 2009, 106, 9971–9978. [Google Scholar] [CrossRef] [Green Version]
- Price, E.O. Behavioral Aspects of Animal Domestication. Q. Rev. Biol. 1984, 59, 1–32. [Google Scholar] [CrossRef]
- Zeder, M.A. The Domestication of Animals. J. Anthropol. Res. 2012, 68, 161–190. [Google Scholar] [CrossRef] [Green Version]
- Coppinger, R.; Coppinger, L. Dogs: A New Understanding of Canine Origin, Behavior and Evolution; The University of Chicago: Chicago, IL, USA, 2002; ISBN 978-0-226-11563-4. [Google Scholar]
- Anderson, T.M.; vonHoldt, B.M.; Candille, S.I.; Musiani, M.; Greco, C.; Stahler, D.R.; Smith, D.W.; Padhukasahasram, B.; Randi, E.; Leonard, J.A.; et al. Molecular and Evolutionary History of Melanism in North American Gray Wolves. Science 2009, 323, 1339–1343. [Google Scholar] [CrossRef] [Green Version]
- Schweizer, R.M.; Durvasula, A.; Smith, J.; Vohr, S.H.; Stahler, D.R.; Galaverni, M.; Thalmann, O.; Smith, D.W.; Randi, E.; Ostrander, E.A.; et al. Natural Selection and Origin of a Melanistic Allele in North American Gray Wolves. Mol. Biol. Evol. 2018, 35, 1190–1209. [Google Scholar] [CrossRef] [Green Version]
- Hedrick, P.W. Wolf of a Different Colour. Heredity 2009, 103, 435–436. [Google Scholar] [CrossRef]
- Caudill, G.; Caudill, D. Melanism of Coyotes (Canis latrans) in Florida. AMID 2015, 174, 335–342. [Google Scholar] [CrossRef]
- Brockerville, R.M.; McGrath, M.J.; Pilgrim, B.L.; Marshall, H.D. Sequence Analysis of Three Pigmentation Genes in the Newfoundland Population of Canis latrans Links the Golden Retriever Mc1r Variant to White Coat Color in Coyotes. Mamm. Genome 2013, 24, 134–141. [Google Scholar] [CrossRef]
- Mowry, C.B.; Edge, J.L. Melanistic Coyotes in Northwest Georgia. SENA 2014, 13, 280–287. [Google Scholar] [CrossRef]
- Gipson, P.S. Melanistic Canis in Arkansas. Southwest. Nat. 1976, 21, 124–126. [Google Scholar] [CrossRef]
- vonHoldt, B.M.; Pollinger, J.P.; Earl, D.A.; Knowles, J.C.; Boyko, A.R.; Parker, H.; Geffen, E.; Pilot, M.; Jedrzejewski, W.; Jedrzejewska, B.; et al. A Genome-Wide Perspective on the Evolutionary History of Enigmatic Wolf-like Canids. Genome Res. 2011, 21, 1294–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heppenheimer, E.; Brzeski, K.; Wooten, R.; Waddell, W.; Rutledge, L.; Chamberlain, M.; Stahler, D.; Hinton, J.; vonHoldt, B. Rediscovery of Red Wolf Ghost Alleles in a Canid Population Along the American Gulf Coast. Genes 2018, 9, 618. [Google Scholar] [CrossRef] [Green Version]
- Heppenheimer, E.; Brzeski, K.E.; Hinton, J.W.; Chamberlain, M.J.; Robinson, J.; Wayne, R.K.; vonHoldt, B.M. A Genome-Wide Perspective on the Persistence of Red Wolf Ancestry in Southeastern Canids. J. Hered. 2020, 111, 277–286. [Google Scholar] [CrossRef]
- Bozarth, C.A.; Hailer, F.; Rockwood, L.L.; Edwards, C.W.; Maldonado, J.E. Coyote Colonization of Northern Virginia and Admixture with Great Lakes Wolves. J. Mammal. 2011, 92, 1070–1080. [Google Scholar] [CrossRef] [Green Version]
- Monzón, J.; Kays, R.; Dykhuizen, D.E. Assessment of Coyote-Wolf-Dog Admixture Using Ancestry-Informative Diagnostic SNPs. Mol. Ecol. 2014, 23, 182–197. [Google Scholar] [CrossRef] [Green Version]
- vonHoldt, B.M.; Cahill, J.A.; Fan, Z.; Gronau, I.; Robinson, J.; Pollinger, J.P.; Shapiro, B.; Wall, J.; Wayne, R.K. Whole-Genome Sequence Analysis Shows That Two Endemic Species of North American Wolf Are Admixtures of the Coyote and Gray Wolf. Sci. Adv. 2016, 2, e1501714. [Google Scholar] [CrossRef] [Green Version]
- Sinding, M.-H.S.; Gopalakrishan, S.; Vieira, F.G.; Samaniego Castruita, J.A.; Raundrup, K.; Heide Jørgensen, M.P.; Meldgaard, M.; Petersen, B.; Sicheritz-Ponten, T.; Mikkelsen, J.B.; et al. Population Genomics of Grey Wolves and Wolf-like Canids in North America. PLoS Genet. 2018, 14, e1007745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- vonHoldt, B.M.; Kays, R.; Pollinger, J.P.; Wayne, R.K. Admixture Mapping Identifies Introgressed Genomic Regions in North American Canids. Mol. Ecol. 2016, 25, 2443–2453. [Google Scholar] [CrossRef]
- Hody, J.W.; Kays, R. Mapping the Expansion of Coyotes (Canis latrans) across North and Central America. ZK 2018, 759, 81–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poessel, S.A.; Gese, E.M.; Young, J.K. Environmental Factors Influencing the Occurrence of Coyotes and Conflicts in Urban Areas. Landsc. Urban Plan. 2017, 259–269. [Google Scholar] [CrossRef] [Green Version]
- Gompper, M.E. Top Carnivores in the Suburbs? Ecological and Conservation Issues Raised by Colonization of North-Eastern North America by Coyotes. BioScience 2002, 52, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Parker, G.R. Eastern Coyote: The Story of Its Success; Nimbus: Halifax, NS, Canada, 1995. [Google Scholar]
- Thurber, J.M.; Peterson, R.O. Changes in Body Size Associated with Range Expansion in the Coyote (Canis latrans). J. Mammal. 1991, 72, 750–755. [Google Scholar] [CrossRef]
- Hill, E.P.; Sumner, P.W.; Wooding, J.B. Human Influences on Range Expansion of Coyotes in the Southeast. Wildl. Soc. Bull. 1987, 15, 521–524. [Google Scholar]
- Koolhaas, J.M.; Korte, S.M.; De Boer, S.F.; Van Der Vegt, B.J.; Van Reenen, C.G.; Hopster, H.; De Jong, I.C.; Ruis, M.A.; Blokhuis, H.J. Coping Styles in Animals: Current Status in Behavior and Stress-Physiology. Neurosci. Biobehav. Rev. 1999, 23, 925–935. [Google Scholar] [CrossRef]
- Sloan Wilson, D.; Clark, A.B.; Coleman, K.; Dearstyne, T. Shyness and Boldness in Humans and Other Animals. Trends Ecol. Evol. 1994, 9, 442–446. [Google Scholar] [CrossRef]
- Ali, O.A.; O’Rourke, S.M.; Amish, S.J.; Meek, M.H.; Luikart, G.; Jeffres, C.; Miller, M.R. RAD Capture (Rapture): Flexible and Efficient Sequence-Based Genotyping. Genetics 2016, 202, 389–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An Analysis Tool Set for Population Genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef] [Green Version]
- Rochette, N.C.; Rivera-Colón, A.G.; Catchen, J.M. Stacks 2: Analytical Methods for Paired—End Sequencing Improve RADseq—based Population Genomics. Mol. Ecol. 2019, 28, 4737–4754. [Google Scholar] [CrossRef] [PubMed]
- Lindblad-Toh, K.; Wade, C.M.; Mikkelsen, T.S.; Karlsson, E.K.; Jaffe, D.B.; Kamal, M.; Clamp, M.; Chang, J.L.; Kulbokas, E.J.; Zody, M.C.; et al. Genome Sequence, Comparative Analysis and Haplotype Structure of the Domestic Dog. Nature 2005, 438, 803–819. [Google Scholar] [CrossRef]
- Guan, Y. Detecting Structure of Haplotypes and Local Ancestry. Genetics 2014, 196, 625–642. [Google Scholar] [CrossRef] [Green Version]
- Johnson, N.A.; Coram, M.A.; Shriver, M.D.; Romieu, I.; Barsh, G.S.; London, S.J.; Tang, H. Ancestral Components of Admixed Genomes in a Mexican Cohort. PLoS Genet. 2011, 7, e1002410. [Google Scholar] [CrossRef] [Green Version]
- Miller, C.R.; Adams, J.R.; Waits, L.P. Pedigree—Based Assignment Tests for Reversing Coyote (Canis latrans) Introgression into the Wild Red Wolf (Canis rufus) Population. Mol. Ecol. 2003, 12, 3287–3301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vonholdt, B.M.; Stahler, D.R.; Smith, D.W.; Earl, D.A.; Pollinger, J.P.; Wayne, R.K. The Genealogy and Genetic Viability of Reintroduced Yellowstone Grey Wolves. Mol. Ecol. 2008, 17, 252–274. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, P.W.; Peterson, R.O.; Vucetich, L.M.; Adams, J.R.; Vucetich, J.A. Genetic Rescue in Isle Royale Wolves: Genetic Analysis and the Collapse of the Population. Conserv. Genet. 2014, 15, 1111–1121. [Google Scholar] [CrossRef]
- Mech, L.D.; Barber-Meyer, S.M.; Erb, J. Wolf (Canis lupus) Generation Time and Proportion of Current Breeding Females by Age. PLoS ONE 2016, 11, e0156682. [Google Scholar] [CrossRef] [PubMed]
- Albers, G.; Edwards, J.W.; Rogers, R.E.; Mastro, L.L. Natality of Yearling Coyotes in West Virginia. J. Fish Wildl. Manag. 2016, 7, 192–197. [Google Scholar] [CrossRef] [Green Version]
- Kilgo, J.C.; Shaw, C.E.; Vukovich, M.; Conroy, M.J.; Ruth, C. Reproductive Characteristics of a Coyote Population before and during Exploitation: Reproduction of Southeastern Coyotes. Jour. Wild. Mgmt. 2017, 81, 1386–1393. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling Accurate Genetic Variant Discovery to Tens of Thousands of Samples. BioRxiv 2018, 201178. [Google Scholar] [CrossRef] [Green Version]
- Candille, S.I.; Kaelin, C.B.; Cattanach, B.M.; Yu, B.; Thompson, D.A.; Nix, M.A.; Kerns, J.A.; Schmutz, S.M.; Millhauser, G.L.; Barsh, G.S. A β-Defensin Mutation Causes Black Coat Color in Domestic Dogs. Science 2007, 318, 1418. [Google Scholar] [CrossRef] [Green Version]
- Gehrt, S.D.; Brown, J.L.; Anchor, C. Is the Urban Coyote a Misanthropic Synanthrope? The Case from Chicago. Cities Environ. (CATE) 2011, 4, 3. [Google Scholar]
- Ellington, E.H.; Gehrt, S.D. Behavioral Responses by an Apex Predator to Urbanization. Behav. Ecol. 2019, 30, 821–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson, R.N.; Brown, J.L.; Karels, T.; Riley, S.P.D. Effects of Urbanization on Resource Use and Individual Specialization in Coyotes (Canis Latrans) in Southern California. PLoS ONE 2020, 15, e0228881. [Google Scholar] [CrossRef]
- Adducci, A., II; Jasperse, J.; Riley, S.; Brown, J.; Honeycutt, R.; Monzón, J. Urban Coyotes Are Genetically Distinct from Coyotes in Natural Habitats. J. Urban Ecol. 2020, 6, juaa010. [Google Scholar] [CrossRef]
- Jones, P.; Chase, K.; Martin, A.; Davern, P.; Ostrander, E.A.; Lark, K.G. Single-Nucleotide-Polymorphism-Based Association Mapping of Dog Stereotypes. Genetics 2008, 179, 1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Våge, J.; Wade, C.; Biagi, T.; Fatjó, J.; Amat, M.; Lindblad-Toh, K.; Lingaas, F. Association of Dopamine- and Serotonin-Related Genes with Canine Aggression. Genes Brain Behav. 2010, 9, 372–378. [Google Scholar] [CrossRef] [PubMed]
- MacLean, E.L.; Snyder-Mackler, N.; vonHoldt, B.M.; Serpell, J.A. Highly Heritable and Functionally Relevant Breed Differences in Dog Behaviour. Proc. R. Soc. B Biol. Sci. 2019, 286, 20190716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saetre, P.; Strandberg, E.; Sundgren, P.-E.; Pettersson, U.; Jazin, E.; Bergström, T.F. The Genetic Contribution to Canine Personality. Genes Brain Behav. 2006, 5, 240–248. [Google Scholar] [CrossRef]
- Svartberg, K. A Comparison of Behaviour in Test and in Everyday Life: Evidence of Three Consistent Boldness-Related Personality Traits in Dogs. Appl. Anim. Behav. Sci. 2005, 91, 103–128. [Google Scholar] [CrossRef]
- Fox, M.W. Socio-Ecological Implications of Individual Differences in Wolf Litters: A Developmental and Evolutionary Perspective. Behaviour 1972, 41, 298–313. [Google Scholar] [CrossRef]
- Breck, S.W.; Poessel, S.A.; Mahoney, P.; Young, J.K. The Intrepid Urban Coyote: A Comparison of Bold and Exploratory Behavior in Coyotes from Urban and Rural Environments. Sci. Rep. 2019, 9, 2104. [Google Scholar] [CrossRef]
- Young, J.K.; Mahe, M.; Breck, S. Evaluating Behavioral Syndromes in Coyotes (Canis latrans). J. Ethol. 2015, 33, 137–144. [Google Scholar] [CrossRef] [Green Version]
- Lunter, G.; Goodson, M. Stampy: A Statistical Algorithm for Sensitive and Fast Mapping of Illumina Sequence Reads. Genome Res. 2011, 21, 936–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-Generation PLINK: Rising to the Challenge of Larger and Richer Datasets. GigaScience 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Abraham, G.; Inouye, M. Fast Principal Component Analysis of Large-Scale Genome-Wide Data. PLoS ONE 2014, 9, e93766. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of Population Structure Using Multilocus Genotype Data. Genetics 2000, 155, 945. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.K.; Ruhe, A.L.; Dumont, B.L.; Robertson, K.R.; Guerrero, G.; Shull, S.M.; Ziegle, J.S.; Millon, L.V.; Broman, K.W.; Payseur, B.A.; et al. A Comprehensive Linkage Map of the Dog Genome. Genetics 2010, 184, 595–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.; Kim, J.; Kim, S.; Cook, D.E.; Evans, K.S.; Andersen, E.C.; Lee, J. Long-Read Sequencing Reveals Intra-Species Tolerance of Substantial Structural Variations and New Subtelomere Formation in C. elegans. Genome Res. 2019, 29, 1023–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heppenheimer, E.; Harrigan, R.J.; Rutledge, L.Y.; Koepfli, K.-P.; DeCandia, A.L.; Brzeski, K.E.; Benson, J.F.; Wheeldon, T.; Patterson, B.R.; Kays, R.; et al. Population Genomic Analysis of North American Eastern Wolves (Canis lycaon) Supports Their Conservation Priority Status. Genes 2018, 9, 606. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
(A) Carmine (melanistic coyote; left) and Wilee (agouti coyote; right) at the Yellow River Wildlife Sanctuary after Carmine’s capture. (B) Map of publicly reported Carmine sightings in Cobb County, GA in December 2019 (yellow) and January (blue) and February (black) 2020. The red line represents the likely route taken by Carmine between 6 February (A) and 7 February (B) 2020.
Figure 1.
(A) Carmine (melanistic coyote; left) and Wilee (agouti coyote; right) at the Yellow River Wildlife Sanctuary after Carmine’s capture. (B) Map of publicly reported Carmine sightings in Cobb County, GA in December 2019 (yellow) and January (blue) and February (black) 2020. The red line represents the likely route taken by Carmine between 6 February (A) and 7 February (B) 2020.

Figure 2.
Genomic analysis of Carmine. (A) PCA of 105,931 SNP genotypes to cluster Carmine (star) with respect to 90 reference canids of North America. The percentage of variation explained by each component is provided in parentheses. (B) Chromosomal-level visualization of the ancestry across the 118,201 SNPs in Carmine’s genome. Ancestry was assigned to three reference canids of North America: domestic dogs, gray wolves (Western and Great Lakes), and coyotes (Western, Southeastern, Northeastern, and Great Lakes). The ancestry at each SNP was designated as either homozygous or heterozygous for the ancestry-informative allele. For detailed ancestry block information, see Table S2.
Figure 2.
Genomic analysis of Carmine. (A) PCA of 105,931 SNP genotypes to cluster Carmine (star) with respect to 90 reference canids of North America. The percentage of variation explained by each component is provided in parentheses. (B) Chromosomal-level visualization of the ancestry across the 118,201 SNPs in Carmine’s genome. Ancestry was assigned to three reference canids of North America: domestic dogs, gray wolves (Western and Great Lakes), and coyotes (Western, Southeastern, Northeastern, and Great Lakes). The ancestry at each SNP was designated as either homozygous or heterozygous for the ancestry-informative allele. For detailed ancestry block information, see Table S2.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mowry, C.B.; Wilson, L.A.; vonHoldt, B.M. Interface of Human/Wildlife Interactions: An Example of a Bold Coyote (Canis latrans) in Atlanta, GA, USA. Diversity 2021, 13, 372. https://0-doi-org.brum.beds.ac.uk/10.3390/d13080372
AMA Style
Mowry CB, Wilson LA, vonHoldt BM. Interface of Human/Wildlife Interactions: An Example of a Bold Coyote (Canis latrans) in Atlanta, GA, USA. Diversity. 2021; 13(8):372. https://0-doi-org.brum.beds.ac.uk/10.3390/d13080372
Chicago/Turabian StyleMowry, Christopher B., Lawrence A. Wilson, and Bridgett M. vonHoldt. 2021. "Interface of Human/Wildlife Interactions: An Example of a Bold Coyote (Canis latrans) in Atlanta, GA, USA" Diversity 13, no. 8: 372. https://0-doi-org.brum.beds.ac.uk/10.3390/d13080372
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.