
Bacterial and Archaeal Water and Sediment Communities of Two Hot Spring Streams in Tengchong, Yunnan Province, China

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
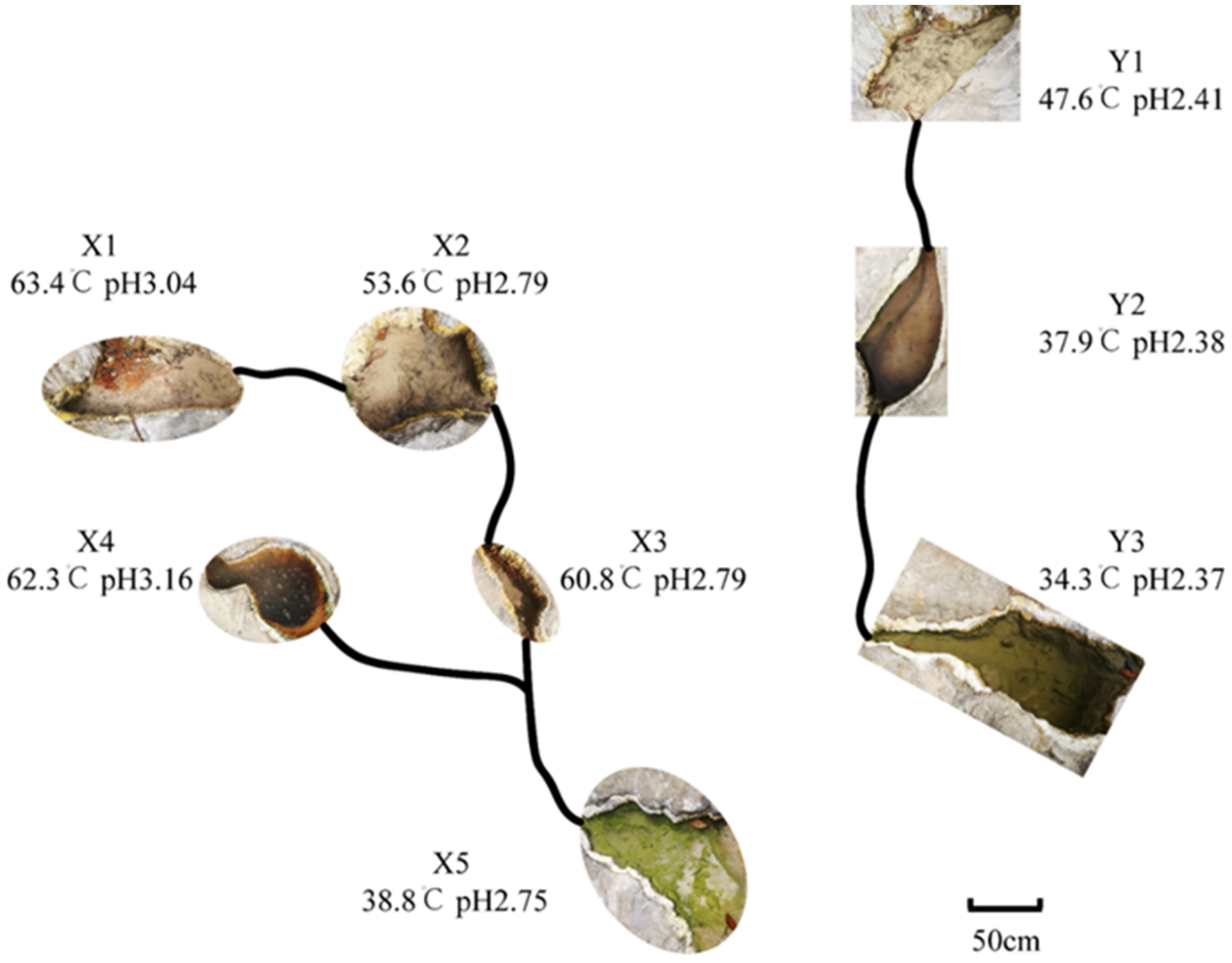
2.1. Sample Collection
2.2. DNA Extraction and Sequencing
2.3. Processing of Sequence Data
2.4. Statistical Analysis
3. Results
3.1. Sample Characteristics
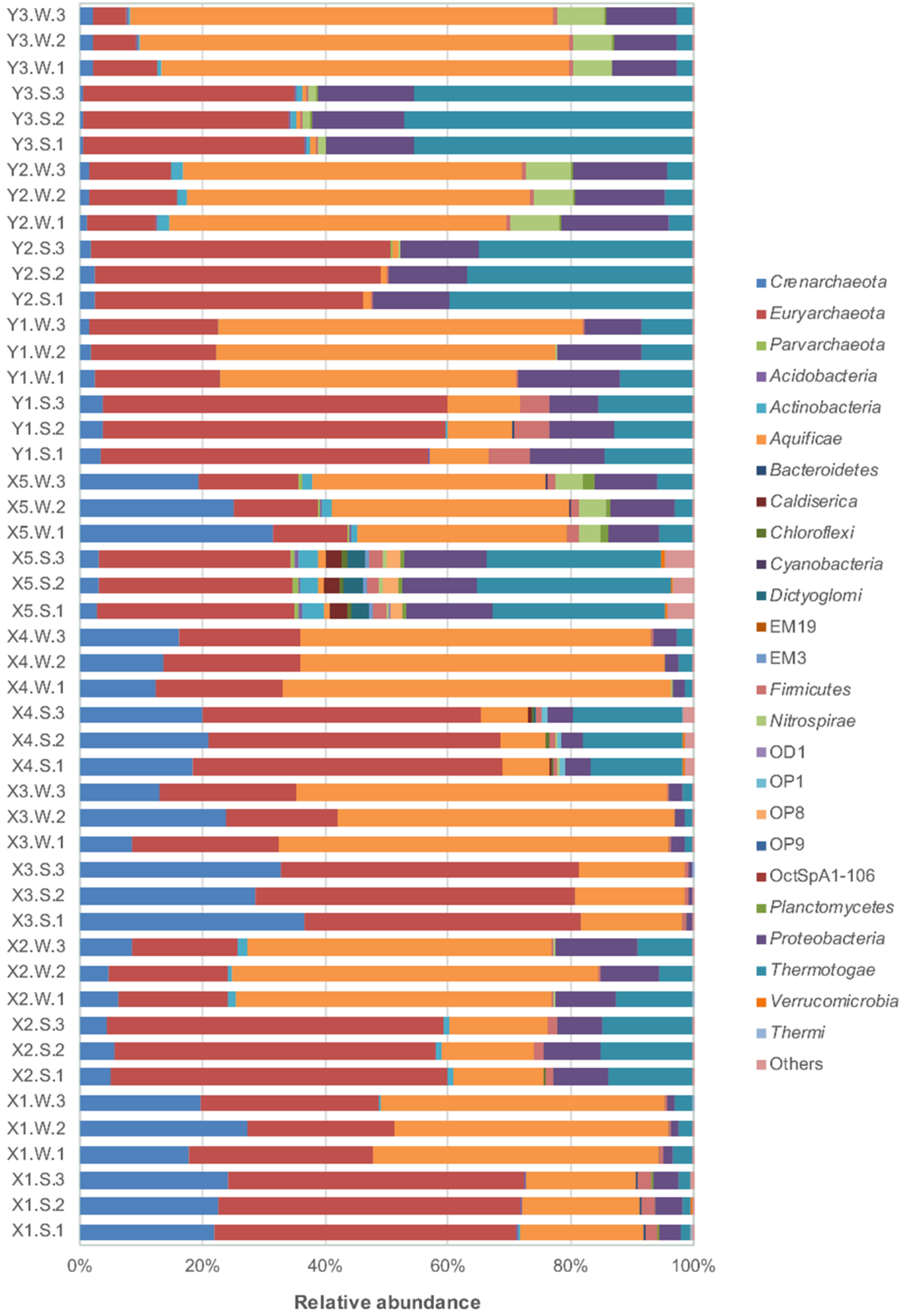
3.2. Microbial Community Composition and Diversity
3.3. Bacterial Taxonomic Composition Analysis
3.4. Archaeal Taxonomic Composition Analysis
3.5. Effect of Environmental Factors on the Microbial Communities
3.6. Effect of Different Stream Sites on the Microbial Communities
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, F.; Peng, Z.; Chen, W.; Wang, Z.; Yang, J.; Zhang, Z.; Hu, Y. High-precision thermal ionization mass spectrometry dating of young volcanic rocks by using U-series method. Chin. Sci. Bull. 2000, 45, 83–87. [Google Scholar] [CrossRef]
- Hou, W.; Wang, S.; Dong, H.; Jiang, H.; Briggs, B.R.; Peacock, J.P.; Huang, Q.; Huang, L.; Wu, G.; Zhi, X.; et al. A comprehensive census of microbial diversity in hot springs of Tengchong, Yunnan Province China using 16S rRNA gene pyrosequencing. PLoS ONE 2013, 8, e53350. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Liu, C.; Fu, B.; Ninomiya, Y.; Zhang, Y.; Wang, C.; Wang, H.; Sun, Z. Variations of geothermometry and chemical-isotopic compositions of hot spring fluids in the Rehai geothermal field, Southwestern China. J. Volcanol. Geotherm. Res. 2005, 142, 243–261. [Google Scholar] [CrossRef]
- Kearey, P.; HongBing, W. Geothermal fields of China. J. Volcanol. Geotherm. Res. 1993, 56, 415–428. [Google Scholar] [CrossRef]
- Fournier, R.O. Geochemistry and dynamics of the Yellowstone National Park hydrothermal system. Annu. Rev. Earth Planet. Sci. 1989, 17, 13–53. [Google Scholar] [CrossRef]
- Kyle, J.E.; Schroeder, P.A.; Wiegel, J. Microbial silicification in sinters from two terrestrial hot springs in the Uzon Caldera, Kamchatka, Russia. Geomicrobiol. J. 2007, 24, 627–641. [Google Scholar] [CrossRef]
- Yoshida, Y. Geochemistry of the Nigorikawa geothermal system, Southwest Hokkaido, Japan. Geochem. J. 1991, 25, 203–222. [Google Scholar] [CrossRef]
- Hedlund, B.P.; Cole, J.K.; Williams, A.J.; Hou, W.; Zhou, E.; Li, W.; Dong, H. A review of the microbiology of the Rehai geothermal field in Tengchong, Yunnan Province, China. Geosci. Front. 2012, 3, 273–288. [Google Scholar] [CrossRef] [Green Version]
- Roesch, L.F.W.; Fulthorpe, R.R.; Riva, A.; Casella, G.; Hadwin, A.K.M.; Kent, A.D.; Daroub, S.H.; Camargo, F.A.O.; Farmerie, W.G.; Triplett, E.W. Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J. 2007, 1, 283–290. [Google Scholar] [CrossRef]
- Tremblay, J.; Singh, K.; Fern, A.; Kirton, E.S.; He, S.; Woyke, T.; Lee, J.; Chen, F.; Dangl, J.L.; Tringe, S.G. Primer and platform effects on 16S rRNA tag sequencing. Front. Microbiol. 2015, 6, 771. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.K.; Herbold, C.W.; Polson, S.W.; Wommack, K.E.; Williamson, S.J.; McDonald, I.R.; Cary, S.C. Groundtruthing next-gen sequencing for microbial ecology-biases and errors in community structure estimates from PCR amplicon pyrosequencing. PLoS ONE 2012, 7, e44224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, A.J.; Raskin, L. PCR biases distort bacterial and archaeal community structure in pyrosequencing datasets. PLoS ONE 2012, 7, e43093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Zhou, B.J.; Deng, G.H.; Jiang, X.T.; Zhang, H.; Zhou, H.W. Comparison of microbial diversity determined with the same variable tag sequence extracted from two different PCR amplicons. BMC Microbiol. 2013, 13, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Dong, H.; Hou, W.; Jiang, H.; Huang, Q.; Briggs, B.R.; Huang, L. Greater temporal changes of sediment microbial community than its waterborne counterpart in Tengchong hot springs, Yunnan Province, China. Sci. Rep. 2014, 4, 7479. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Li, P.; Jiang, D.; Dai, X.; Zhang, R.; Wang, Y.; Wang, Y. Microbial community structure and arsenic biogeochemistry in an acid vapor-formed spring in Tengchong geothermal area, China. PLoS ONE 2016, 11, e0146331. [Google Scholar] [CrossRef]
- Li, F.; Leu, A.; Poff, K.; Carlson, L.T.; Ingalls, A.E.; DeLong, E.F. Planktonic archaeal ether lipid origins in surface waters of the North Pacific subtropical gyre. Front. Microbiol. 2021, 12, 610675. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- R Development Core Team R. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2012. [Google Scholar]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Vegan: Community Ecology Package; R Package Version: 2.0-3. Available online: http://cran.r-project.org/ (accessed on 24 May 2012).
- Des Marais, D.J.; Walter, M.R. Terrestrial hot spring systems: Introduction. Astrobiology 2019, 19, 1419–1432. [Google Scholar] [CrossRef]
- Kambura, A.K.; Mwirichia, R.K.; Kasili, R.W.; Karanja, E.N.; Makonde, H.M.; Boga, H.I. Bacteria and archaea diversity within the hot springs of Lake Magadi and Little Magadi in Kenya. BMC Microbiol. 2016, 16, 136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuler, C.G.; Havig, J.R.; Hamilton, T.L. Hot spring microbial community composition, morphology, and carbon fixation: Implications for interpreting the ancient rock record. Front. Earth Sci. 2017, 5, 97. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.Q.; Wang, F.P.; Zhi, X.Y.; Chen, J.Q.; Zhou, E.M.; Liang, F.; Xiao, X.; Tang, S.K.; Jiang, H.C.; Zhang, C.L.; et al. Bacterial and archaeal diversities in Yunnan and Tibetan hot springs, China. Environ. Microbiol. 2013, 15, 1160–1175. [Google Scholar] [CrossRef]
- Mohammad, B.T.; Al Daghistani, H.I.; Jaouani, A.; Abdel-Latif, S.; Kennes, C. Isolation and characterization of thermophilic bacteria from Jordanian hot springs: Bacillus licheniformis and Thermomonas hydrothermalis isolates as potential producers of thermostable enzymes. Int. J. Microbiol. 2017, 2017, 6943952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryan, C.G.; Watkin, E.L.; McCredden, T.J.; Wong, Z.R.; Harrison, S.T.L.; Kaksonen, A.H. The use of pyrite as a source of lixiviant in the bioleaching of electronic waste. Hydrometallurgy 2015, 152, 33–43. [Google Scholar] [CrossRef]
- Griffiths, E.; Gupta, R.S. Molecular signatures in protein sequences that are characteristics of the phylum Aquificae. Int. J. Syst. Evol. Microbiol. 2006, 56, 99–107. [Google Scholar] [CrossRef]
- Horiike, T.; Miyata, D.; Hamada, K.; Saruhashi, S.; Shinozawa, T.; Kumar, S.; Chakraborty, R.; Komiyama, T.; Tateno, Y. Phylogenetic construction of 17 bacterial phyla by new method and carefully selected orthologs. Gene 2009, 429, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Huber, R.; Hannig, M. Thermotogales. In The Prokaryotes: Volume 7: Proteobacteria: Delta, Epsilon Subclass; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.H., Stackebrandt, E., Eds.; Springer: New York, NY, USA, 2006; pp. 899–922. [Google Scholar]
- Reysenbach, A.L.; Banta, A.; Civello, S.; Daly, J.; Mitchel, K.; Lalonde, S.; Konhauser, K.; Rodman, A.; Rusterholtz, K.; Takacs-Vesbach, C. The Aquificales in Yellowstone National Park. In Geothermal Biology and Geochemistry in Yellowstone National Park; Inskeep, W.P., McDermott, T.R., Eds.; Montana State University: Bozeman, MT, USA, 2005; pp. 129–142. [Google Scholar]
- Spear, J.R.; Walker, J.J.; McCollom, T.M.; Pace, N.R. Hydrogen and bioenergetics in the Yellowstone geothermal ecosystem. Proc. Natl. Acad. Sci. USA 2005, 102, 2555–2560. [Google Scholar] [CrossRef] [Green Version]
- Meyer-Dombard, D.R.; Shock, E.L.; Amend, J.P. Archaeal and bacterial communities in geochemically diverse hot springs of Yellowstone National Park, USA. Geobiology 2005, 3, 211–227. [Google Scholar] [CrossRef]
- Boyd, E.S.; Leavitt, W.D.; Geesey, G.G. CO2 uptake and fixation by a thermoacidophilic microbial community attached to precipitated sulfur in a geothermal spring. Appl. Environ. Microbiol. 2009, 75, 4289–4296. [Google Scholar] [CrossRef] [Green Version]
- Macur, R.E.; Langner, H.W.; Kocar, B.D.; Inskeep, W.P. Linking geochemical processes with microbial community analysis: Successional dynamics in an arsenic-rich, acid-sulphate-chloride geothermal spring. Geobiology 2004, 2, 163–177. [Google Scholar] [CrossRef]
- Meyer-Dombard, D.R.; Swingley, W.; Raymond, J.; Havig, J.; Shock, E.L.; Summons, R.E. Hydrothermal ecotones and streamer biofilm communities in the Lower Geyser Basin, Yellowstone National Park. Environ. Microbiol. 2011, 13, 2216–2231. [Google Scholar] [CrossRef] [PubMed]
- Hügler, M.; Huber, H.; Molyneaux, S.J.; Vetriani, C.; Sievert, S.M. Autotrophic CO2 fixation via the reductive tricarboxylic acid cycle in different lineages within the phylum Aquificae: Evidence for two ways of citrate cleavage. Environ. Microbiol. 2007, 9, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Stohr, R.; Waberski, A.; Völker, H.; Tindall, B.J.; Thomm, M. Hydrogenothermus marinus gen. nov., sp. nov., a novel thermophilic hydrogen-oxidizing bacterium, recognition of Calderobacterium hydrogenophilum as a member of the genus Hydrogenobacter and proposal of the reclassification of Hydrogenobacter acidophilus as Hydrogenobaculum acidophilum gen. nov., comb. nov., in the phylum ‘Hydrogenobacter/Aquifex’. Int. J. Syst. Evol. Microbiol. 2001, 51, 1853–1862. [Google Scholar] [CrossRef]
- Donahoe-Christiansen, J.; D’Imperio, S.; Jackson, C.R.; Inskeep, W.P.; McDermott, T.R. Arsenite-oxidizing Hydrogenobaculum strain isolated from an acid-sulfate-chloride geothermal spring in Yellowstone National Park. Appl. Environ. Microbiol. 2004, 70, 1865–1868. [Google Scholar] [CrossRef] [Green Version]
- Reysenbach, A.L.; Hamamura, N.; Podar, M.; Griffiths, E.; Ferreira, S.; Hochstein, R.; Heidelberg, J.; Johnson, J.; Mead, D.; Pohorille, A.; et al. Complete and draft genome sequences of six members of the Aquificales. J. Bacteriol. 2009, 191, 1992–1993. [Google Scholar] [CrossRef] [Green Version]
- Romano, C.; D’Imperio, S.; Woyke, T.; Mavromatis, K.; Lasken, R.; Shock, E.L.; McDermott, T.R. Comparative genomic analysis of phylogenetically closely related Hydrogenobaculum sp. isolates from Yellowstone National Park. Appl. Environ. Microbiol. 2013, 79, 2932–2943. [Google Scholar] [CrossRef] [Green Version]
- Hedlund, B.P.; Reysenbach, A.L.; Huang, L.; Ong, J.C.; Liu, Z.; Dodsworth, J.A.; Ahmed, R.; Williams, A.J.; Briggs, B.R.; Liu, Y.; et al. Isolation of diverse members of the Aquificales from geothermal springs in Tengchong, China. Front. Microbiol. 2015, 6, 157. [Google Scholar] [CrossRef]
- Oren, A. Taxonomy of the family Halobacteriaceae: A paradigm for changing concepts in prokaryote systematics. Int. J. Syst. Evol. Microbiol. 2012, 62, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Schleper, C.; Puehler, G.; Holz, I.; Gambacorta, A.; Janekovic, D.; Santarius, U.; Klenk, H.P.; Zillig, W. Picrophilus gen. nov., fam. nov.: A novel aerobic, heterotrophic, thermoacidophilic genus and family comprising archaea capable of growth around pH 0. J. Bacteriol. 1995, 177, 7050–7059. [Google Scholar] [CrossRef] [Green Version]
- Itoh, T.; Yoshikawa, N.; Takashina, T. Thermogymnomonas acidicola gen. nov., sp. nov., a novel thermoacidophilic, cell wall-less archaeon in the order Thermoplasmatales, isolated from a solfataric soil in Hakone, Japan. Int. J. Syst. Evol. Microbiol. 2007, 57, 2557–2561. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, R.J.; Grogan, D.W.; Taylor, J.W. Geographic barriers isolate endemic populations of hyperthermophilic archaea. Science 2003, 301, 976–978. [Google Scholar] [CrossRef] [PubMed]
- Huber, G.; Spinnler, C.; Gambacorta, A.; Stetter, K.O. Metallosphaera sedula gen, and sp. nov. represents a new genus of aerobic, metal-mobilizing, thermoacidophilic archaebacteria. Syst. Appl. Microbiol. 1989, 12, 38–47. [Google Scholar] [CrossRef] [Green Version]
- Auernik, K.S.; Maezato, Y.; Blum, P.H.; Kelly, R.M. The genome sequence of the metal-mobilizing, extremely thermoacidophilic archaeon Metallosphaera sedula provides insights into bioleaching-associated metabolism. Appl. Environ. Microbiol. 2008, 74, 682–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, T.; Huber, H.; Teiner, K.; Burggraf, S.; Stetter, K.O. Metallosphaera prunae, sp. nov., a novel metal-mobilizing, thermoacidophilic Archaeum, isolated from a uranium mine in Germany. Syst. Appl. Microbiol. 1995, 18, 560–566. [Google Scholar] [CrossRef]
- Liu, L.J.; You, X.Y.; Guo, X.; Liu, S.J.; Jiang, C.Y. Metallosphaera cuprina sp. nov., an acidothermophilic, metal-mobilizing archaeon. Int. J. Syst. Evol. Microbiol. 2011, 61, 2395–2400. [Google Scholar] [CrossRef] [PubMed]
- Kurosawa, N.; Itoh, Y.H.; Itoh, T. Reclassification of Sulfolobus hakonensis Takayanagi et al., 1996 as Metallosphaera hakonensis comb. nov. based on phylogenetic evidence and DNA G+C content. Int. J. Syst. Evol. Microbiol. 2003, 53, 1607–1608. [Google Scholar] [CrossRef]
- Peng, T.J.; Liu, L.J.; Liu, C.; Yang, Z.F.; Liu, S.J.; Jiang, C.Y. Metallosphaera tengchongensis sp. nov., an acidothermophilic archaeon isolated from a hot spring. Int. J. Syst. Evol. Microbiol. 2015, 65, 537–542. [Google Scholar] [CrossRef]
- Dodsworth, J.A.; Hungate, B.A.; Hedlund, B.P. Ammonia oxidation, denitrification and dissimilatory nitrate reduction to ammonium in two US Great Basin hot springs with abundant ammonia-oxidizing archaea. Environ. Microbiol. 2011, 13, 2371–2386. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Zhang, Z.; Liu, T.; Xiong, H.; Zhao, S.; Mei, Y.; Peng, N.; Liang, Y. Bacterial and Archaeal Water and Sediment Communities of Two Hot Spring Streams in Tengchong, Yunnan Province, China. Diversity 2022, 14, 381. https://0-doi-org.brum.beds.ac.uk/10.3390/d14050381
Li J, Zhang Z, Liu T, Xiong H, Zhao S, Mei Y, Peng N, Liang Y. Bacterial and Archaeal Water and Sediment Communities of Two Hot Spring Streams in Tengchong, Yunnan Province, China. Diversity. 2022; 14(5):381. https://0-doi-org.brum.beds.ac.uk/10.3390/d14050381
Chicago/Turabian StyleLi, Jinshan, Zhufeng Zhang, Tao Liu, Hui Xiong, Shumiao Zhao, Yuxia Mei, Nan Peng, and Yunxiang Liang. 2022. "Bacterial and Archaeal Water and Sediment Communities of Two Hot Spring Streams in Tengchong, Yunnan Province, China" Diversity 14, no. 5: 381. https://0-doi-org.brum.beds.ac.uk/10.3390/d14050381