Towards the Development of a 3-D Biochip for the Detection of Hepatitis C Virus

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Supplements
2.2. Instrumentation
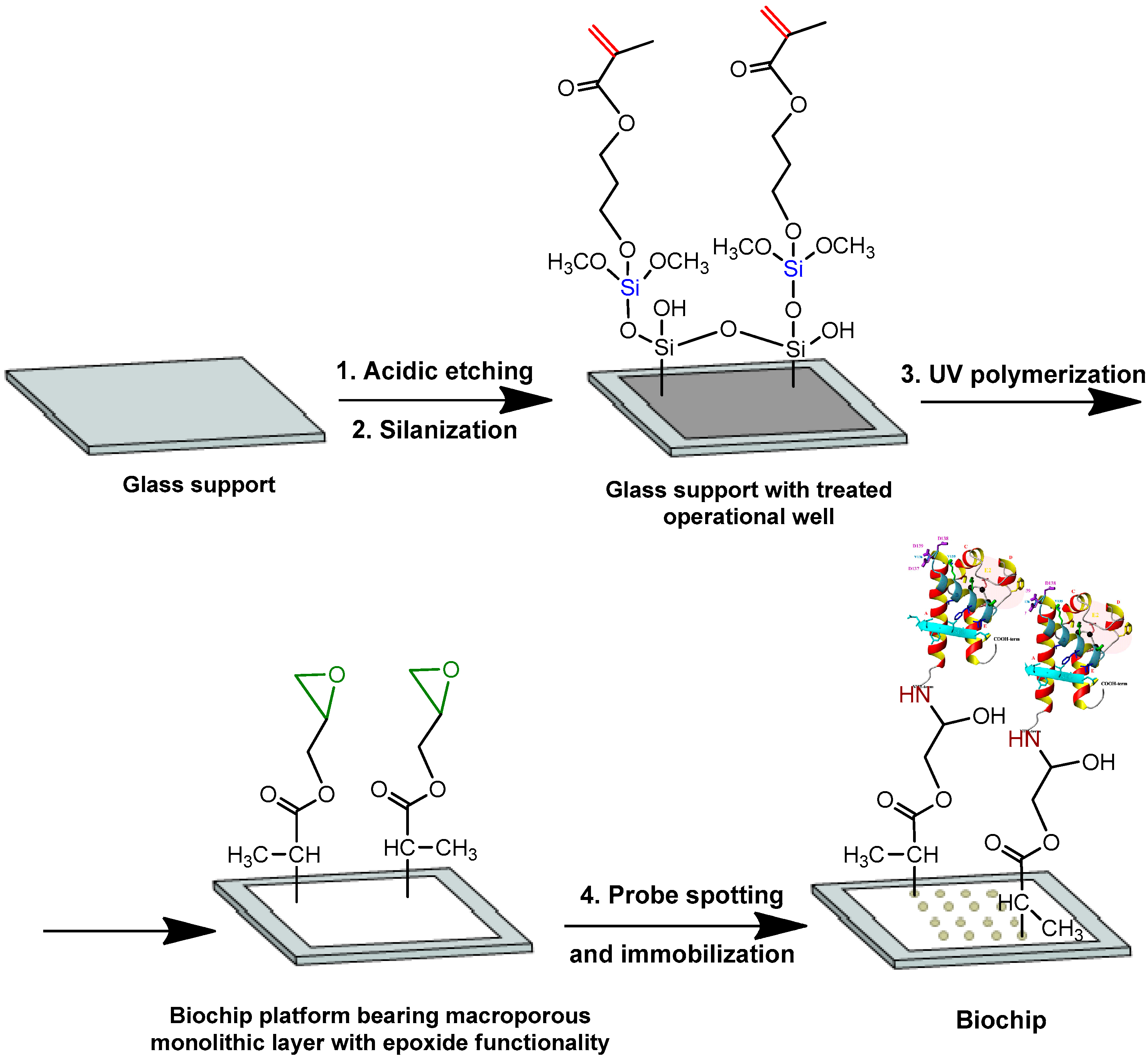
2.3. Synthesis and Characterization of Monolithic Layers
2.4. Biochip Printing
2.5. E2 Production
2.6. Labeling of Antibodies
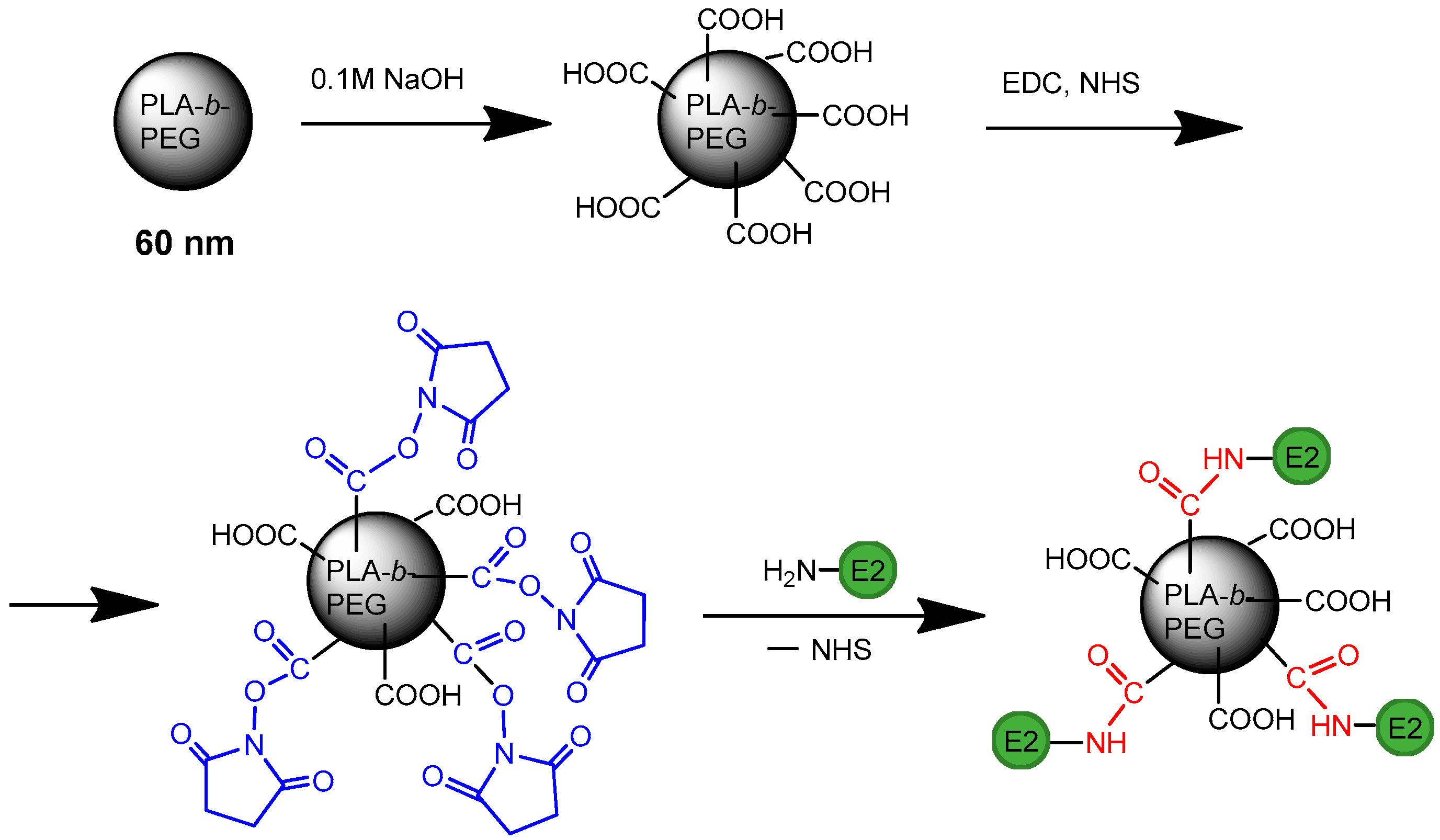
2.7. Preparation HC VMPs
2.7.1. Preparation of Nanoparticles
2.7.2. E2 Immobilization
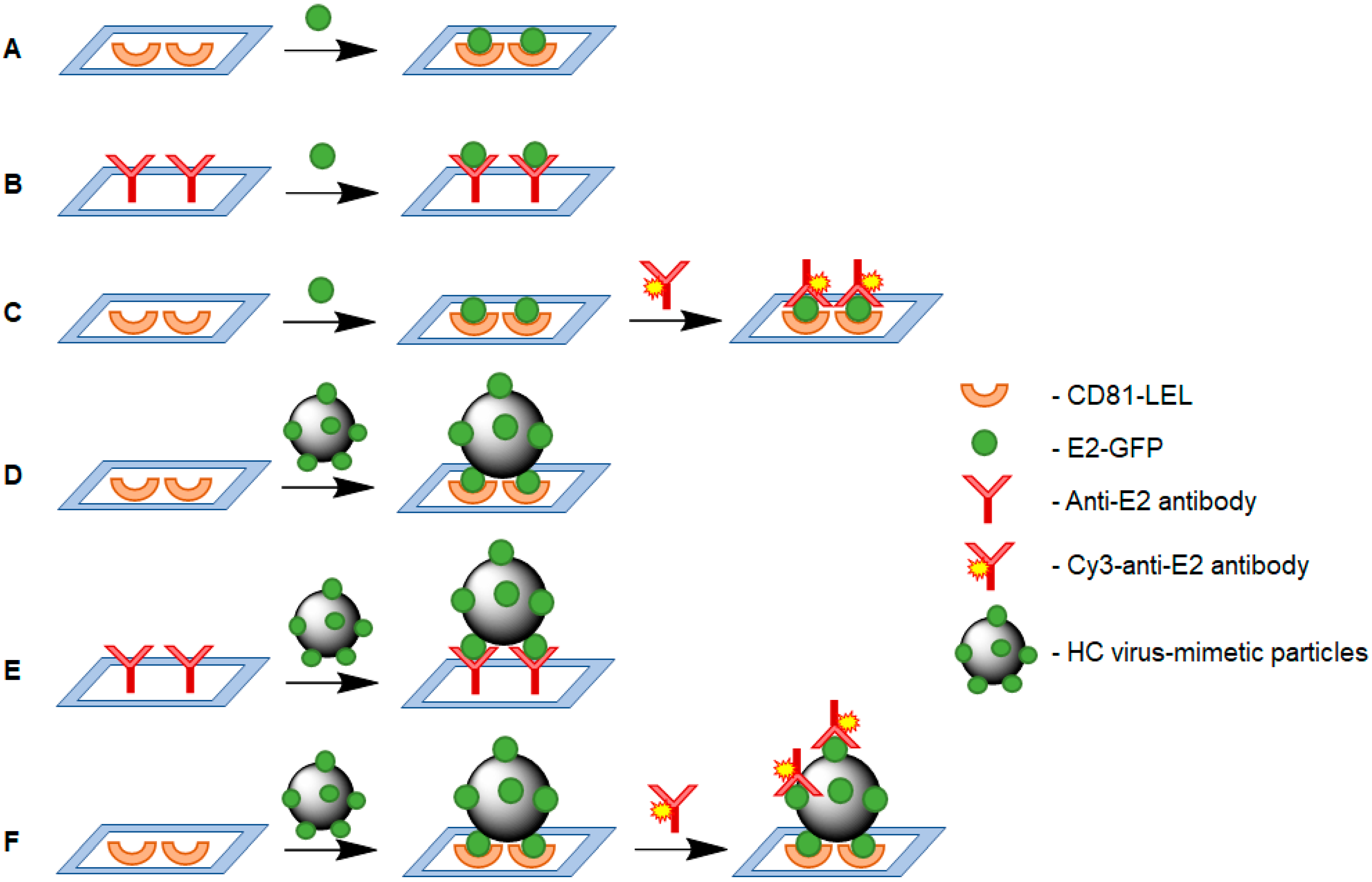
2.8. Bioanalisys
2.8.1. Direct Analysis
2.8.2. Sandwich-Analysis
2.9. Statistics and Reproducibility, LOD and LOQ
3. Results and Discussion
3.1. Preparation of Biochips
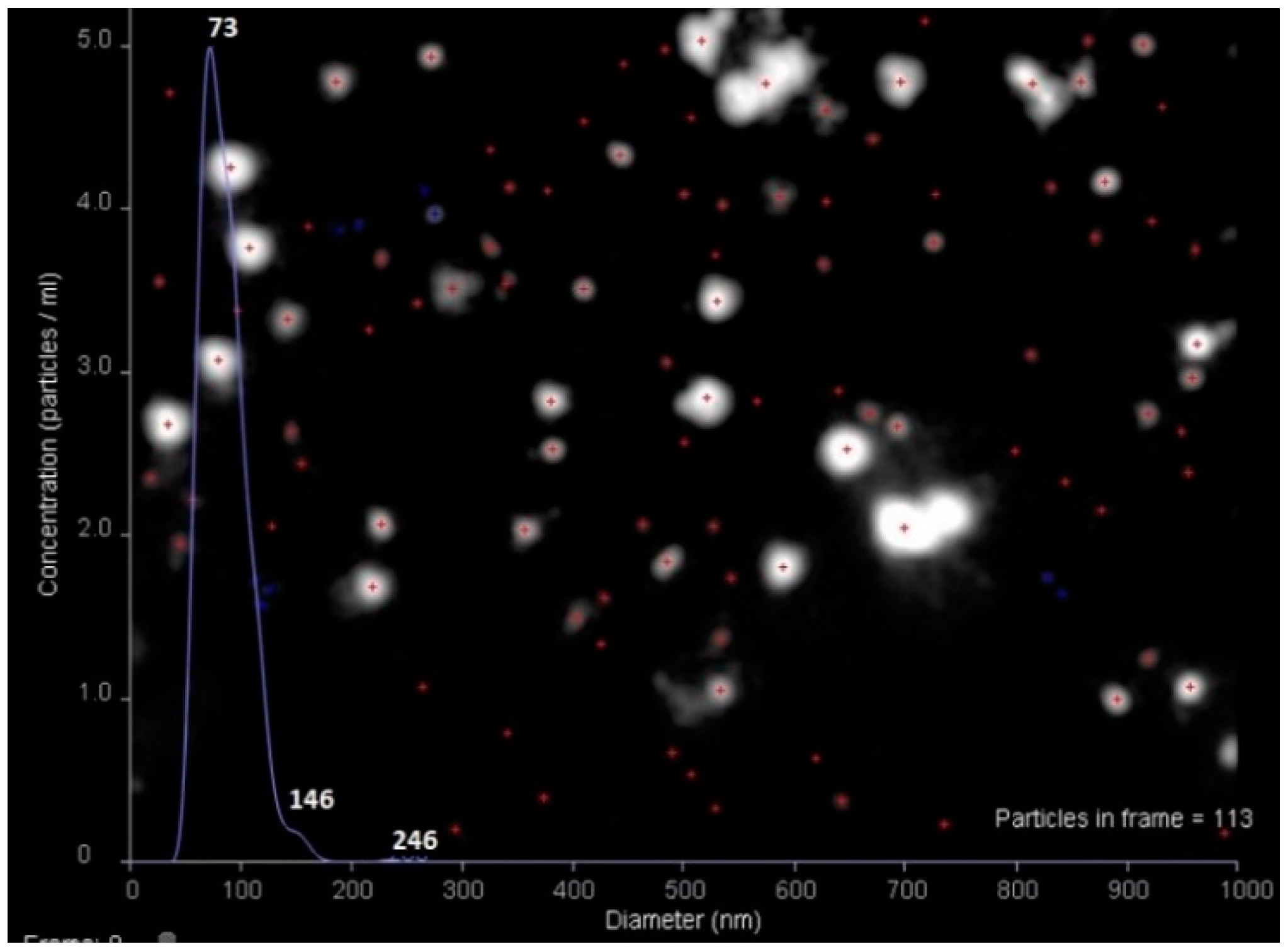
3.2. Preparation of HC VMPs
3.3. Analysis on Biochips
3.4. Optimization of Analytical Procedure
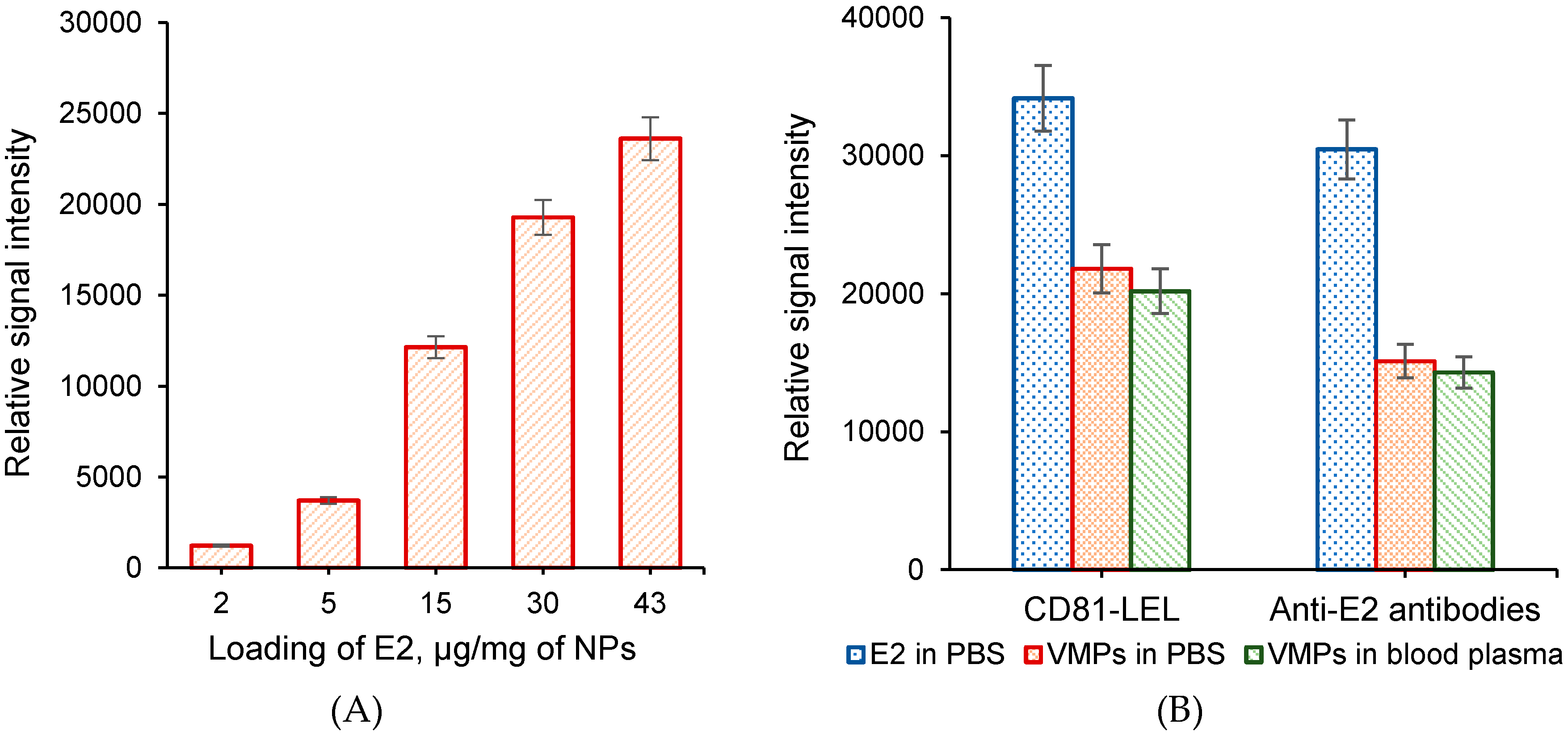
3.4.1. Probe Amount
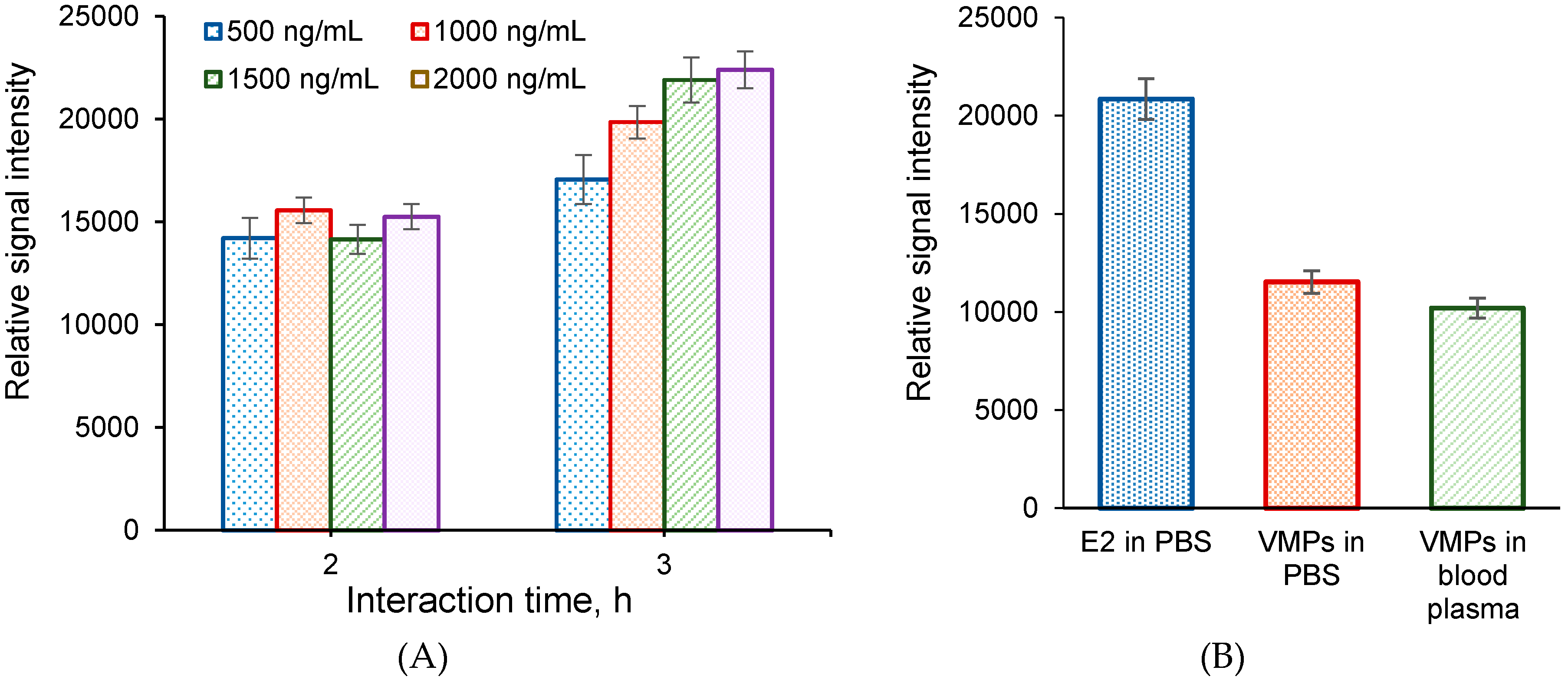
3.4.2. Interaction Time
3.4.3. Analyte Concentration
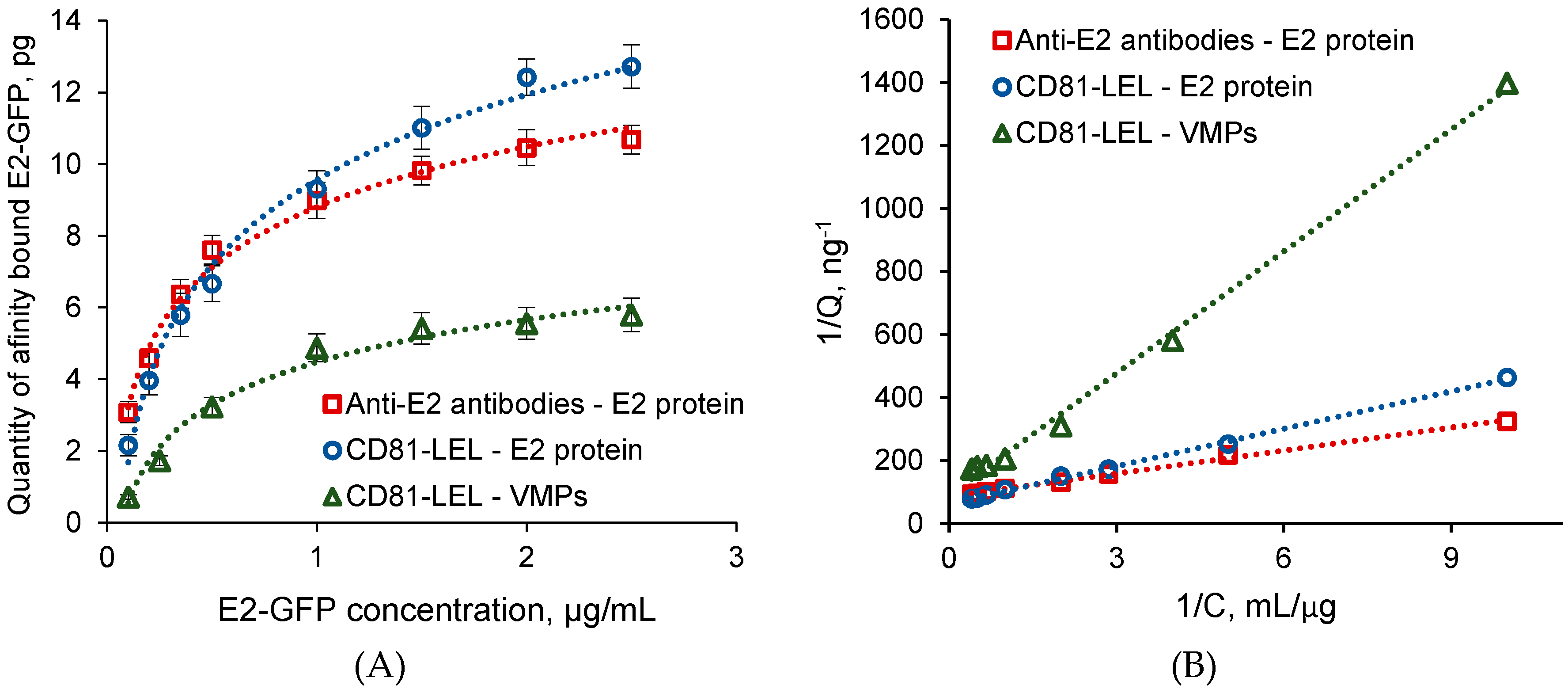
3.5. Selection of VMPs and Comparison of Their Detection to Free Protein
3.6. “Sandwich” Analysis
3.7. LOD, LOQ and Reproducibility of Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mysore, K.R.; Leung, D.H. Hepatitis B and C. Clin. Liver Dis. 2018, 22, 703–722. [Google Scholar] [CrossRef]
- Espinosa, C.M.; Jhaveri, R.; Barritt, A.S. Unique challenges of hepatitis c in infants, children, and adolescents. Clin. Ther. 2018, 40, 1299–1307. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Qiao, M.; Atanasov, I.; Hu, Z.; Kato, T.; Liang, T.J.; Zhou, Z.H. Cryo-electron microscopy and three-dimensional reconstructions of hepatitis C virus particles. Virology 2007, 367, 126–134. [Google Scholar] [CrossRef] [Green Version]
- Catanese, M.T.; Uryu, K.; Kopp, M.; Edwards, T.J.; Andrus, L.; Rice, W.J.; Silvestry, M.; Kuhn, R.J.; Rice, C.M. Ultrastructural analysis of hepatitis C virus particles. Proc. Natl. Acad. Sci. USA 2013, 110, 9505–9510. [Google Scholar] [CrossRef] [Green Version]
- Dubuisson, J.D.; Cosset, F.-L. Virology and cell biology of the hepatitis C virus life cycle—An update. J. Hepatol. 2014, 61, S3–S13. [Google Scholar] [CrossRef] [Green Version]
- Kabashima, T.; Tonooka, K.; Takada, M.; Kai, M.; Shibata, T. Simultaneous assay for protease activities of hepatitis C virus and human immunodeficiency virus based on fluorescence detection. Sci. Rep. 2019, 9, 9150. [Google Scholar] [CrossRef]
- Warkad, S.D.; Song, K.; Pal, D. Developments in the HCV screening technologies. Sensors 2019, 19, 4257. [Google Scholar] [CrossRef] [Green Version]
- Arca-Lafuente, S.; Martínez-Román, P.; Mate-Cano, I.; Madrid, R.; Briz, V. Nanotechnology: A reality for diagnosis of HCV infectious disease. J. Infect. 2020, 80, 8–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevaliez, S.; Pawlotsky, J.-M. New virological tools for screening, diagnosis and monitoring of hepatitis B and C in resource-limited settings. J. Hepatol. 2018, 69, 916–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Kardos, K.W.; Schiff, E.; Berne, C.A.; Mounzer, K.; Banks, A.T.; Tatum, H.A.; Friel, T.J.; Demicco, M.P.; Lee, W.M.; et al. Evaluation of a new, rapid test for detecting HCV infection, suitable for use with blood or oral fluid. J. Virol. Methods 2011, 172, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Kemmler, M.; Sauer, U.; Schleicher, E.; Preininger, C.; Brandenburg, A. Biochip point-of-care device for sepsis diagnostics. Sens. Actuators B Chem. 2014, 192, 205–215. [Google Scholar] [CrossRef]
- Feyzkhanova, G.; Filippova, M.; Talibov, V.; Dementieva, E.; Maslennikov, V.; Reznikov, Y.; Offermann, N.; Zasedatelev, A.; Rubina, A.; Fooke-Achterrath, M. Development of hydrogel biochip for in vitro allergy diagnostics. J. Immunol. Methods 2014, 406, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Kenyon, F.; O’Looney, N.; Ross, A.J.; Kwan, M.C.; Beattie, J.S.; Petrik, J.; Ghazal, P.; Campbell, C.J. A multiplexed protein microarray for the simultaneous serodiagnosis of human immunodeficiency virus/hepatitis C virus infection and typing of whole blood. Anal. Biochem. 2008, 382, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, J.-A.; Lee, H.; Lee, K.N.; Chae, K.; Lee, S.; Lee, D.-K.; Kim, S. High diagnostic accuracy of antigen microarray for sensitive detection of hepatitis C virus infection. Clin. Chem. 2008, 54, 424–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talha, S.M.; Saviranta, P.; Hattara, L.; Vuorinen, T.; Hytönen, J.; Khanna, N.; Pettersson, K. Array-in-well platform–based multiplex assay for the simultaneous detection of anti-HIV- and treponemal-antibodies, and Hepatitis B surface antigen. J. Immunol. Methods 2016, 429, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, S.; Ibrahim, F.; Rothan, H.A.; Yusof, R.; van der Marel, C.; Djordjevic, I.; Koole, L.H. Aging effect and antibody immobilization on COOH exposed surfaces designed for dengue virus detection. Biochem. Eng. J. 2015, 99, 183–192. [Google Scholar] [CrossRef]
- Jang, H.; Kim, J.; Choi, J.-J.; Son, Y.; Park, H. Peptide nucleic acid array for detection of point mutations in hepatitis B virus associated with antiviral resistance. J. Clin. Microbiol. 2010, 48, 3127–3131. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Kang, H.S.; Hur, S.-S.; Sim, S.; Ahn, S.H.; Park, Y.K.; Park, E.-S.; Lee, A.R.; Park, S.; Kwon, S.Y.; et al. Direct detection of drug-resistant hepatitis B virus in serum using a dendron-modified microarray. Gut Liver 2017, 12, 331–341. [Google Scholar] [CrossRef] [Green Version]
- Yuk, C.-S.; Lee, H.K.; Kim, H.T.; Choi, Y.K.; Lee, B.C.; Chun, B.-H.; Chung, N. Development and evaluation of a protein microarray chip for diagnosis of hepatitis C virus. Biotechnol. Lett. 2004, 26, 1563–1568. [Google Scholar] [CrossRef]
- Xu, R.; Gan, X.; Fang, Y.; Zheng, S.; Dong, Q. A simple, rapid, and sensitive integrated protein microarray for simultaneous detection of multiple antigens and antibodies of five human hepatitis viruses (HBV, HCV, HDV, HEV, and HGV). Anal. Biochem. 2007, 362, 69–75. [Google Scholar] [CrossRef]
- Yun, S.G.; Jang, J.W.; Lee, J.H.; Lim, C.S.; Kim, J.; Ki, Y.; Jo, M.; Kim, S. Evaluation of novel multiplex antibody kit for human immunodeficiency virus 1/2 and hepatitis C virus using sol-gel based microarray. BioMed Res. Int. 2015, 2015, 837296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Oidovsambuu, O.; Liu, P.; Grosely, R.; Elazar, M.; Winn, V.D.; Fram, B.; Boa, Z.; Dai, H.; Dashtseren, B.; et al. A novel quantitative microarray antibody capture assay identifies an extremely high hepatitis delta virus prevalence among hepatitis B virus-infected mongolians. Hepatology 2017, 66, 1739–1749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Hu, W.; Lu, Z.; Li, C.M. Photografted poly(methyl methacrylate)-based high performance protein microarray for hepatitis B virus biomarker detection in human serum. MedChemComm 2010, 1, 132. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Kuo, C.-H.; Hsiao, W.-T.; Huang, K.-C.; Tseng, S.-F.; Chou, C.-P. Glass biochip fabrication by laser micromachining and glass-molding process. J. Mater. Process. Technol. 2012, 212, 633–639. [Google Scholar] [CrossRef]
- Shang, J.; Yan, J.; Zhang, Z.; Huang, X.; Maturavongsadit, P.; Song, B.; Jia, Y.; Ma, T.; Li, D.; Xu, K.; et al. A hydrogel-based glucose affinity microsensor. Sens. Actuators B Chem. 2016, 237, 992–998. [Google Scholar] [CrossRef] [Green Version]
- Le Goff, G.C.; Corgier, B.P.; Mandon, C.; De Crozals, G.; Chaix, C.; Blum, L.J.; Marquette, C.A. Impact of immobilization support on colorimetric microarrays performances. Biosens. Bioelectron. 2012, 35, 94–100. [Google Scholar] [CrossRef]
- Rober, M.; Walter, J.; Vlakh, E.; Stahl, F.; Kasper, C.; Tennikova, T. New 3-D microarray platform based on macroporous polymer monoliths. Anal. Chim. Acta 2009, 644, 95–103. [Google Scholar] [CrossRef]
- Svec, F.; Tennikova, T.B.; Deyl, Z. Monolithic Materials: Preparation, Properties and Applications; Elsevier: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Cretich, M.; Sedini, V.; Damin, F.; Pelliccia, M.; Sola, L.; Chiari, M. Coating of nitrocellulose for colorimetric DNA microarrays. Anal. Biochem. 2010, 397, 84–88. [Google Scholar] [CrossRef]
- Groarke, R.; Brabazon, D. Methacrylate polymer monoliths for separation applications. Materials 2016, 9, 446. [Google Scholar] [CrossRef] [Green Version]
- Volokitina, M.V.; Nikitina, A.V.; Tennikova, T.B.; Korzhikova-Vlakh, E.G. Immobilized enzyme reactors based on monoliths: Effect of pore size and enzyme loading on biocatalytic process. Electrophoresis 2017, 38, 2931–2939. [Google Scholar] [CrossRef]
- Kalashnikova, I.; Ivanova, N.; Tennikova, T.B. Macroporous monolithic layers as efficient 3-D microarrays for quantitative detection of virus-like particles. Anal. Chem. 2007, 79, 5173–5180. [Google Scholar] [CrossRef] [PubMed]
- Slabospitskaya, M.Y.; Vlakh, E.G.; Saprykina, N.N.; Tennikova, T.B. Synthesis and investigation of a new macroporous monolithic material based on anN-hydroxyphthalimide ester of acrylic acid-co-glycidyl methacrylate-co-ethylene dimethacrylate terpolymer. J. Appl. Polym. Sci. 2008, 111, 692–700. [Google Scholar] [CrossRef]
- Sinitsyna, E.S.; Walter, J.G.; Vlakh, E.G.; Stahl, F.; Kasper, C.; Tennikova, T.B. Macroporous methacrylate-based monoliths as platforms for DNA microarrays. Talanta 2012, 93, 139–146. [Google Scholar] [CrossRef]
- Glotov, A.S.; Sinitsyna, E.; Danilova, M.; Vashukova, E.; Walter, J.; Stahl, F.; Baranov, V.; Korzhikova-Vlakh, E.G.; Tennikova, T.B. Detection of human genome mutations associated with pregnancy complications using 3-D microarray based on macroporous polymer monoliths. Talanta 2016, 147, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Volokitina, M.; Krutyakova, M.; Sirotov, V.; Larionov, M.; Tennikova, T.B.; Korzhikova-Vlakh, E.G. Protein biochips based on macroporous polymer supports: Material properties and analytical potential. J. Pharm. Biomed. Anal. 2019, 165, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Kramberger, P.; Peterka, M.; Boben, J.; Ravnikar, M.; Štrancar, A. Short monolithic columns—A breakthrough in purification and fast quantification of tomato mosaic virus. J. Chromatogr. A 2007, 1144, 143–149. [Google Scholar] [CrossRef]
- Zaveckas, M.; Snipaitis, S.; Pesliakas, H.; Nainys, J.; Gedvilaite, A. Purification of recombinant virus-like particles of porcine circovirus type 2 capsid protein using ion-exchange monolith chromatography. J. Chromatogr. B 2015, 991, 21–28. [Google Scholar] [CrossRef]
- Kalashnikova, I.; Ivanova, N.; Tennikova, T.B. Development of a strategy of influenza virus separation based on pseudoaffinity chromatography on short monolithic columns. Anal. Chem. 2008, 80, 2188–2198. [Google Scholar] [CrossRef]
- Kalashnikova, I.V.; Ivanova, N.D.; Evseeva, T.G.; Menshikova, A.Y.; Vlakh, E.G.; Tennikova, T.B. Study of dynamic adsorption behavior of large-size protein-bearing particles. J. Chromatogr. A 2007, 1144, 40–47. [Google Scholar] [CrossRef]
- Figueroa, S.M.; Veser, A.; Abstiens, K.; Fleischmann, D.; Beck, S.; Goepferich, A. Influenza A virus mimetic nanoparticles trigger selective cell uptake. Proc. Natl. Acad. Sci. USA 2019, 116, 9831–9836. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Hwang, A.; Jose, L.; Park, J.H.; Jang, S.; Song, J.K.; Kim, Y.; Cho, Y.; Jeon, H.B.; Jin, J.O.; et al. RAFT/PISA based Ni-NTA polymeric particles for virus-mimetic influenza vaccines. J. Ind. Eng. Chem. 2020, 86, 35–38. [Google Scholar] [CrossRef]
- Kong, L.; Giang, E.; Nieusma, T.; Kadam, R.U.; Cogburn, K.E.; Hua, Y.; Dai, X.; Stanfield, R.L.; Burton, D.R.; Ward, A.B.; et al. Hepatitis C virus E2 envelope glycoprotein core structure. Science 2013, 342, 1090–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fénéant, L.; Levy, S.; Cocquerel, L. CD81 and hepatitis C virus (HCV) infection. Viruses 2014, 6, 535–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solovyov, K.V.; Polyakov, D.S.; Grudinina, N.A.; Egorov, V.V.; Morozova, I.V.; Aleynikova, T.D.; Shavlovsky, M.M. Expression in E. coli and purification of the fibrillogenic fusion proteins TTR-sfGFP and.2M-sfGFP. Prep. Biochem. Biotechnol. 2011, 41, 337–349. [Google Scholar] [CrossRef]
- Polyakov, D.; Sakhabeyev, R.G.; Shavlovsky, M.M. Partial denaturation of recombinant protein for affinity purification. Appl. Biochem. Microbiol. 2016, 52, 105–109. [Google Scholar] [CrossRef]
- Ayankojo, A.G.; Reut, J.; Ciocan, V.; Öpik, A.; Syritski, V. Molecularly imprinted polymer-based sensor for electrochemical detection of erythromycin. Talanta 2020, 209, 120502. [Google Scholar] [CrossRef]
- Daly, T.M.; Dumaual, C.M.; Dotson, C.A.; Farmen, M.W.; Kadam, S.K.; Hockett, R.D. Precision profiling and components of variability analysis for affymetrix microarray assays run in a clinical context. J. Mol. Diagn. 2005, 7, 404–412. [Google Scholar] [CrossRef] [Green Version]
- Yuen, T.; Wurmbach, E.; Pfeffer, R.L.; Ebersole, B.J.; Sealfon, S.C. Accuracy and calibration of commercial oligonucleotide and custom cDNA microarrays. Nucleic Acids Res. 2002, 30, e48. [Google Scholar] [CrossRef] [Green Version]
- Selvarajah, S.; Negm, O.; Hamed, M.R.; Tubby, C.; Todd, I.; Tighe, P.; Harrison, T.; Fairclough, L.C. Development and validation of protein microarray technology for simultaneous inflammatory mediator detection in human sera. Mediat. Inflamm. 2014, 2014, 820304. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Probe | Analyte | Analysis | Sx/y | b | R2 | LOD, ng/mL | LOQ, ng/mL |
|---|---|---|---|---|---|---|---|
| Anti-E2 antibodies | E2-GFP | direct, in PBS | 1.95 | 0.047 | 0.99151 | 124 | 414 |
| CD81-LEL | E2-GFP | direct, in PBS | 3.87 | 0.050 | 0.98482 | 232 | 774 |
| Anti-E2 antibodies | VMPs | direct, in PBS | 0.93 | 0.037 | 0.99361 | 75 | 251 |
| CD81-LEL | VMPs | direct, in PBS | 1.38 | 0.038 | 0.99093 | 108 | 363 |
| CD81-LEL | VMPs | sandwich, in PBS | 0.63 | 0.027 | 0.99248 | 70 | 233 |
| CD81-LEL | VMPs | sandwich, in human blood plasma | 0.69 | 0.028 | 0.99142 | 73 | 246 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antipchik, M.; Polyakov, D.; Sinitsyna, E.; Dzhuzha, A.; Shavlovsky, M.; Korzhikova-Vlakh, E.; Tennikova, T. Towards the Development of a 3-D Biochip for the Detection of Hepatitis C Virus. Sensors 2020, 20, 2719. https://0-doi-org.brum.beds.ac.uk/10.3390/s20092719
Antipchik M, Polyakov D, Sinitsyna E, Dzhuzha A, Shavlovsky M, Korzhikova-Vlakh E, Tennikova T. Towards the Development of a 3-D Biochip for the Detection of Hepatitis C Virus. Sensors. 2020; 20(9):2719. https://0-doi-org.brum.beds.ac.uk/10.3390/s20092719
Chicago/Turabian StyleAntipchik, Mariia, Dmitry Polyakov, Ekaterina Sinitsyna, Apollinariia Dzhuzha, Mikhail Shavlovsky, Evgenia Korzhikova-Vlakh, and Tatiana Tennikova. 2020. "Towards the Development of a 3-D Biochip for the Detection of Hepatitis C Virus" Sensors 20, no. 9: 2719. https://0-doi-org.brum.beds.ac.uk/10.3390/s20092719