
A Novel Coupled Reaction-Diffusion System for Explainable Gene Expression Profiling


 , , , ,
, , , ,
Abstract
:1. Introduction
- An innovative mathematical explanation of potential biomarkers denoting tumorigenesis in non-small cell lung cancer.
- A detailed formulation of the regulators of gene expression of non-thermal plasma treatment of non-small cell lung cancer: regulators of mRNA, the regulators of ncRNA, and the coupled mRNA–ncRNA regulators.
- An approximation of the temporal expression-profile of genes in a two-dimensional spatial domain using a system comprised of coupled-reaction partial differential equations.
- Various simulation experiments conducted to validate the proposed mathematical gene-expression profile.
2. Diffusion Equation
3. Proposed Computational and Qualitative Analysis: Two-Species Diffusion Equation
- Intercellular ncRNA mobility
- Intercellular mRNA mobility
- Intercellular ncRNA and mRNA mobility
- Intercellular ncRNA mobility
- Intercellular mRNA mobility
- Intercellular ncRNA and mRNA mobility
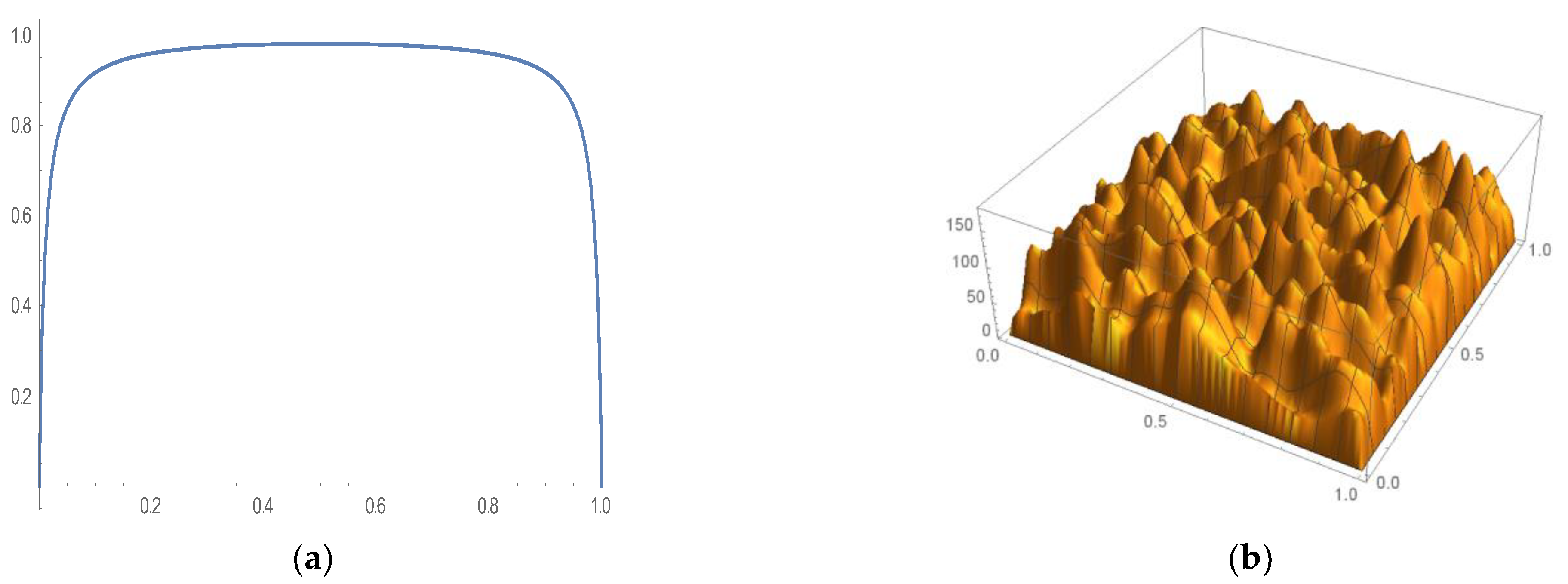
3.1. Two-Species Model in Dirichlet Initial Boundary Value Problem
3.2. Consistency Correction for Initial Condition and Dirichlet Boundary Conditions
3.3. Two-Species Model in Neumann Initial Boundary Value Problem
4. Experiments
4.1. Dataset
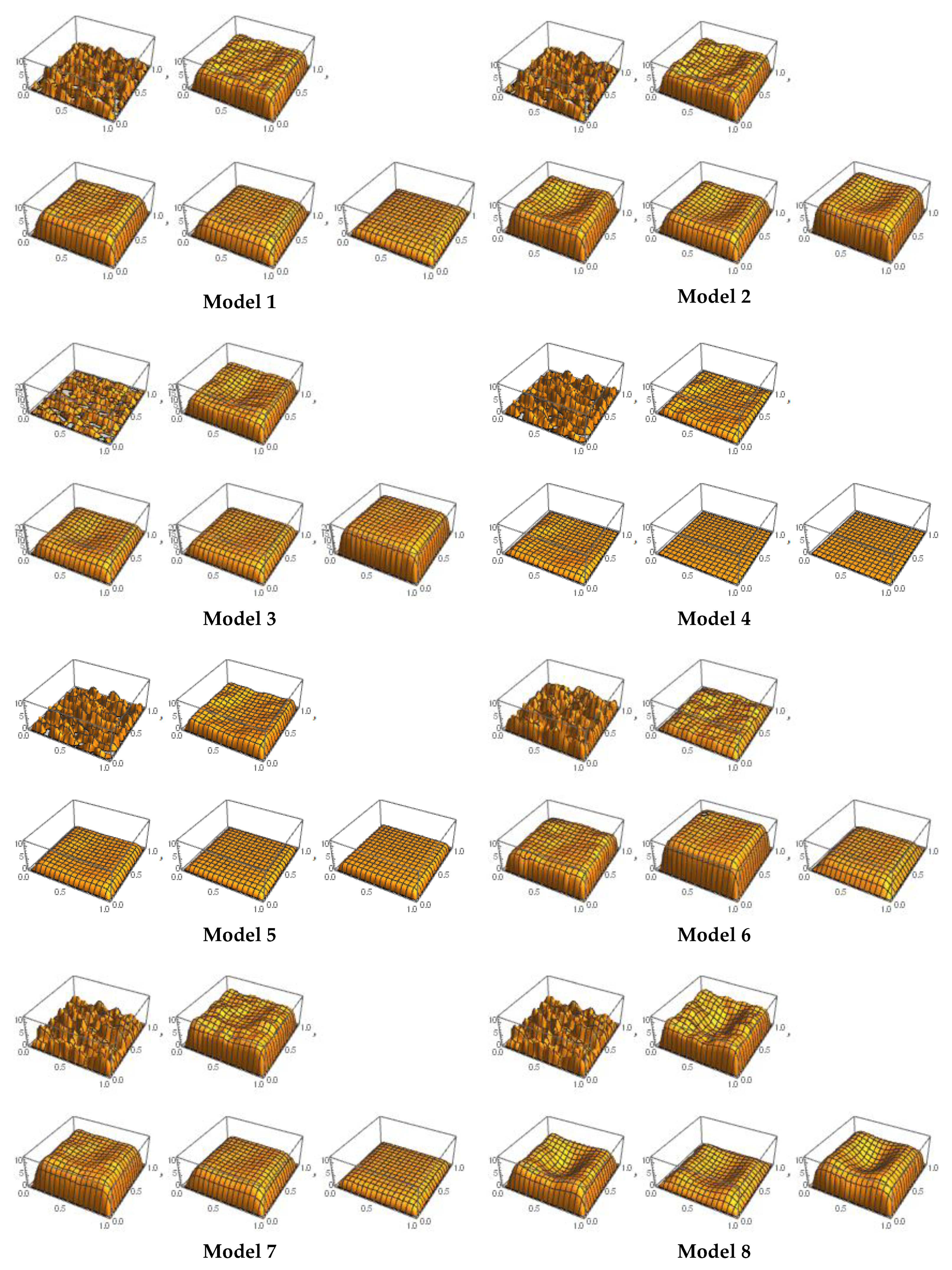
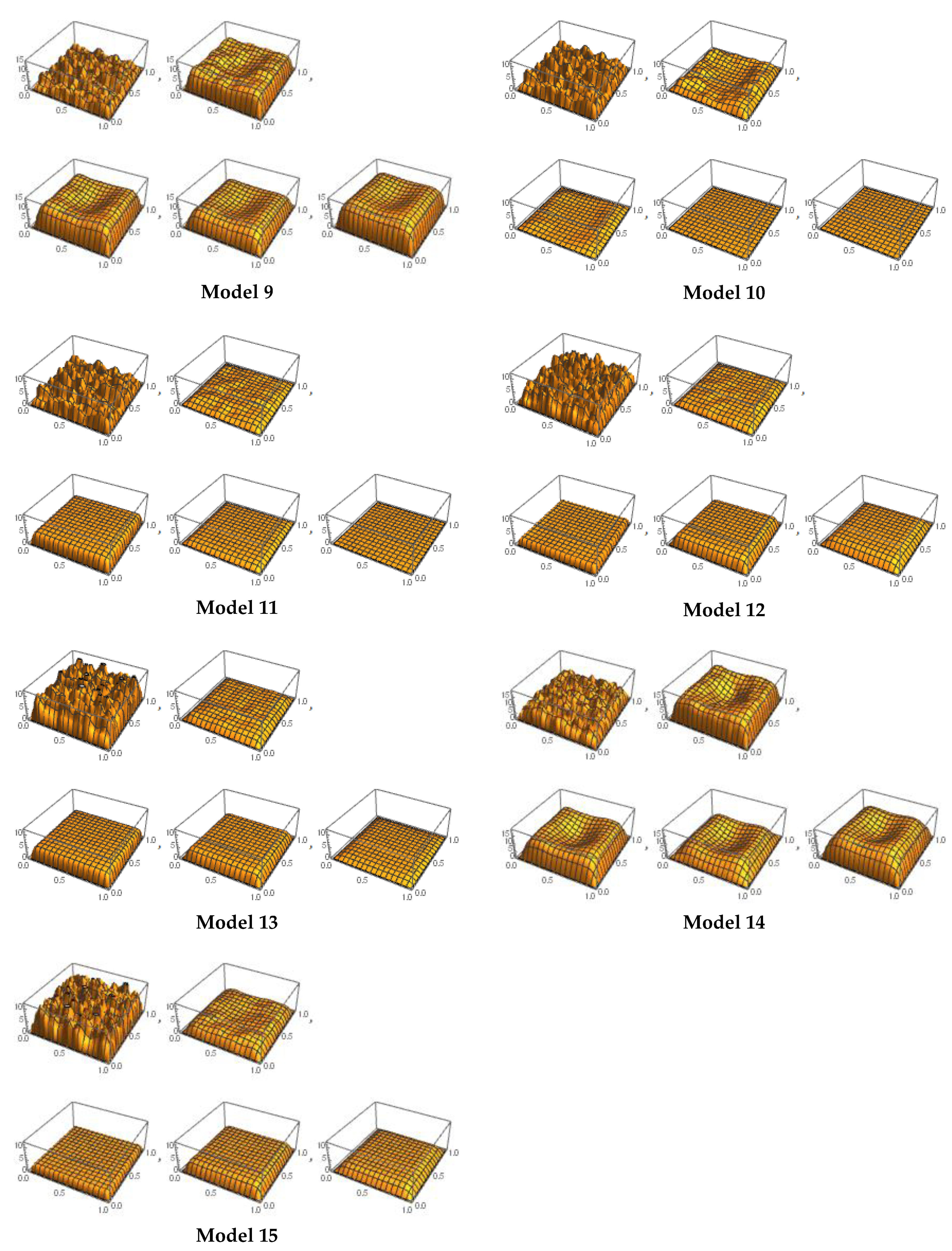
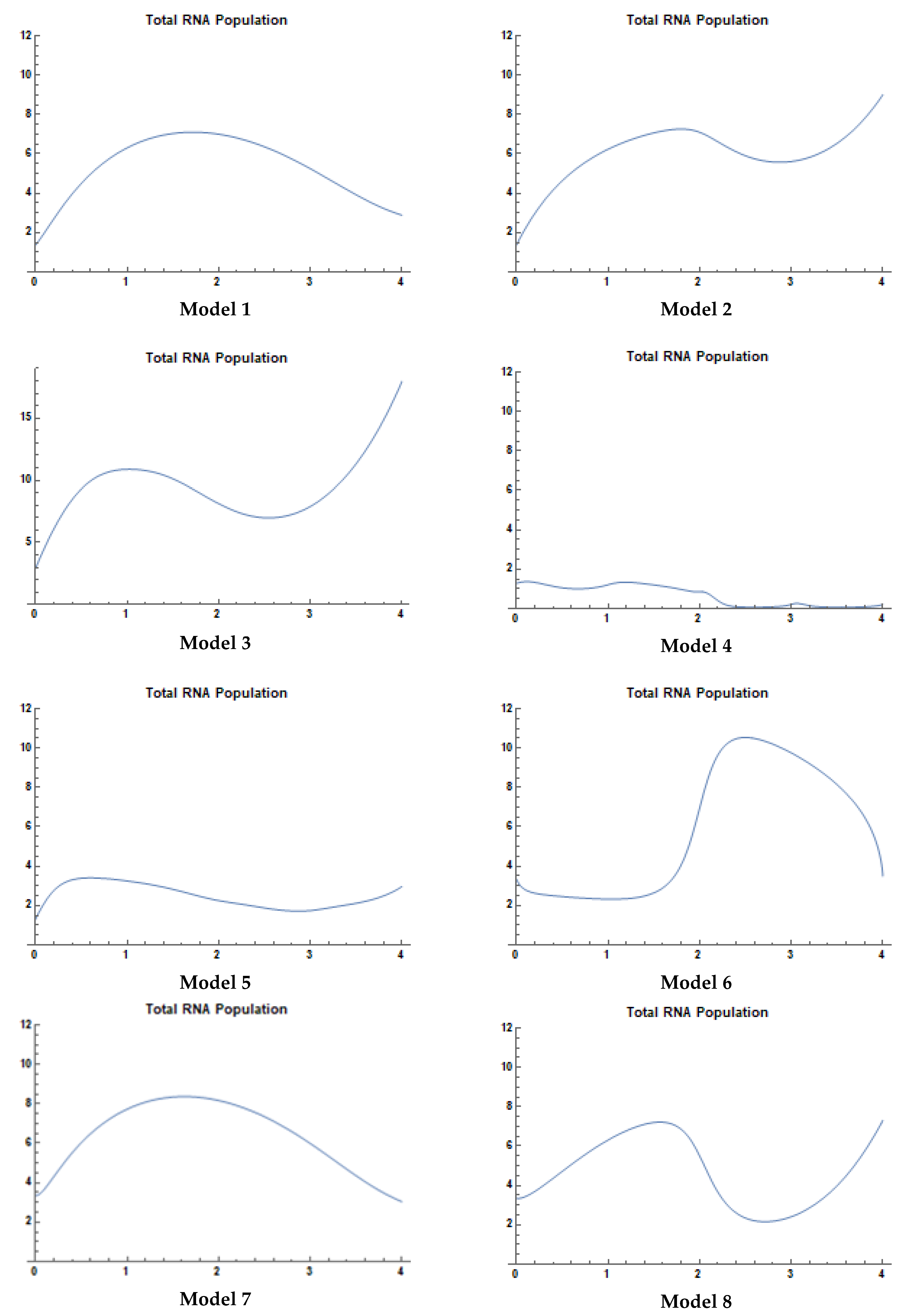
4.2. Experimental Results
- Model 1 is the best fit for MET1, KRAS1, NRAS2, EGFR1, and PIK3CA3 patterns.
- Model 2 is the complete fit for MET4 pattern.
- Model 3 is the complete fit for MET2 pattern.
- Model 4 is the complete fit for AKT2, EGFR7, and PIK3CA2 patterns.
- Model 5 is the best fit for KRAS3, EGFR4, and BRAF patterns.
- Model 6 provides the best fit for RET1 patterns.
- Model 7 is the complete fit for ALK2 pattern.
- Model 8 is the best fit for AKT3, EGFR5, RET2, and RET3 patterns.
- Model 9 is the complete fit for ALK1 pattern.
- Model 10 is the complete fit for EGFR8 pattern.
- Model 11 is the best fit for ROS1 patterns.
- Model 12 is the best fit for EGFR6 patterns.
- Model 13 is the best fit for AKT1, NRAS1, and EGFR3 patterns.
- Model 14 is the complete fit for MET3, KRAS4, and EGFR2 patterns.
- Model 15 is the complete fit for SMEK1 pattern.
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brazma, A.; Parkinson, H.; Schlitt, T.; Shojatalab, M. A Quick Introduction to Elements of Biology-Cells, Molecules, Genes, Functional Genomics, Microarrays; EMBL-EBI: Cambridge, UK, 2001. [Google Scholar]
- Widłak, W. Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Epigenetics: Fundamentals. 2016. Available online: https://www.whatisepigenetics.com/fundamentals/ (accessed on 24 September 2019).
- Phillips, T. Small Non-coding RNA and Gene Expression. Nat. Educ. 2008, 1, 115. [Google Scholar]
- King, M.W. Regulation of Gene Expression, The Medical Biochemistry Page. 2016. Available online: http://themedicalbiochemistrypage.org/gene-regulation.php (accessed on 24 September 2019).
- Liu, Y. Active learning with support vector machine applied to gene expression data for cancer classification. J. Chem. Inf. Comput. Sci. 2004, 44, 1936–1941. [Google Scholar] [CrossRef] [PubMed]
- Glaab, E.; Bacardit, J.; Garibaldi, J.M.; Krasnogor, N. Using rule-based machine learning for candidate disease gene prioritization and sample classification of cancer gene expression data. PLoS ONE 2012, 7, e39932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wajid, S.K.; Hussain, A.; Huang, K.; Boulila, W. Lung cancer detection using Local Energy-based Shape Histogram (LESH) feature extraction and cognitive machine learning techniques. In Proceedings of the 2016 IEEE 15th International Conference on Cognitive Informatics & Cognitive Computing (ICCI* CC), Palo Alto, CA, USA, 22–23 August 2016; pp. 359–366. [Google Scholar]
- Salem, H.; Attiya, G.; El-Fishawy, N. Classification of human cancer diseases by gene expression profiles. Appl. Soft Comput. 2017, 50, 124–134. [Google Scholar] [CrossRef]
- Jiang, Z.; Li, T.; Min, W.; Qi, Z.; Rao, Y. Fuzzy c-means clustering based on weights and gene expression programming. Pattern Recognit. Lett. 2017, 90, 1–7. [Google Scholar] [CrossRef]
- Matsubara, T.; Ochiai, T.; Hayashida, M.; Akutsu, T.; Nacher, J.C. Convolutional neural network approach to lung cancer classification integrating protein interaction network and gene expression profiles. J. Bioinform. Comput. Biol. 2019, 17, 1940007. [Google Scholar] [CrossRef]
- Farouq, M.W.; Boulila, W.; Abdel-aal, M.; Hussain, A.; Salem, A. Novel Multi-Stage Fusion based Approach for Gene Expression Profiling in Non-Small Cell Lung Cancer. IEEE Access 2019, 7, 37141–37150. [Google Scholar] [CrossRef]
- Janizek, J.D.; Celik, S.; Lee, S.I. Explainable machine learning prediction of synergistic drug combinations for precision cancer medicine. bioRxiv 2018, 331769. [Google Scholar] [CrossRef]
- Lamy, J.-B.; Sekar, B.; Guezennec, G.; Bouaud, J.; Séroussi, B. Explainable artificial intelligence for breast cancer: A visual case-based reasoning approach. Artif. Intell. Med. 2019, 94, 42–53. [Google Scholar] [CrossRef]
- Pintelas, E.; Liaskos, M.; Livieris, I.E.; Kotsiantis, S.; Pintelas, P. Explainable Machine Learning Framework for Image Classification Problems: Case Study on Glioma Cancer Prediction. J. Imaging 2020, 6, 37. [Google Scholar] [CrossRef]
- Sabol, P.; Sinčák, P.; Hartono, P.; Kočan, P.; Benetinová, Z.; Blichárová, A.; Verbóová, Ľ.; Štammová, E.; Sabolová-Fabianová, A.; Jašková, A. Explainable classifier for improving the accountability in decision-making for colorectal cancer diagnosis from histopathological images. J. Biomed. Inform. 2020, 109, 103523. [Google Scholar] [CrossRef] [PubMed]
- Gadgil, C.; Yeckel, A.; Derby, J.J.; Hu, W.S. A diffusion-reaction model for DNA microarray assays. J. Biotechnol. 2004, 114, 31–45. [Google Scholar] [CrossRef]
- Wang, L.; Chu, F.; Xie, W. Accurate Cancer Classification Using Expressions of Very Few Genes. IEEE/ACM Trans. Computational Biol. Bioinform. 2007, 4, 40–53. [Google Scholar] [CrossRef] [Green Version]
- Mahmud, M.; Kaiser, M.S.; Hussain, A.; Vassanelli, S. Applications of Deep Learning and Reinforcement Learning to Biological Data. IEEE Trans. Neural Netw. Learn. Syst. 2018, 29, 2063–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, L.; Ng, M.K.; Liu, Y. Construction of Gene Networks with Hybrid Approach From Expression Profile and Gene Ontology. IEEE Trans. Inf. Technol. Biomed. 2010, 14, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Levy, D. Modeling the chemotherapy-induced selection of drug-resistant traits during tumor growth. J. Theor. Biol. 2018, 436, 120–134. [Google Scholar] [CrossRef]
- Zhang, X.; Han, Y.; Wu, L.; Wang, Y. State Estimation for Delayed Genetic Regulatory Networks With Reaction–Diffusion Terms. IEEE Trans. Neural Netw. Learn. Syst. 2018, 29, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Wang, M.; Song, S.; Ahn, C.K. Sampled-Data State Estimation of Reaction Diffusion Genetic Regulatory Networks via Space-Dividing Approaches. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019. [Google Scholar] [CrossRef]
- Jamieson, N.B.; Maker, A.V. Gene-expression profiling to predict responsiveness to immunotherapy. Cancer Gene Ther. 2017, 24, 134–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Atitallah, S.; Driss, M.; Boulila, W.; Ben Ghézala, H. Leveraging Deep Learning and IoT big data analytics to support the smart cities development: Review and future directions. Comput. Sci. Rev. 2020, 38, 100303. [Google Scholar] [CrossRef]
- Hajjaji, Y.; Boulila, W.; Farah, I.R.; Romdhani, I.; Hussain, A. Big data and IoT-based applications in smart environments: A systematic review. Comput. Sci. Rev. 2021, 39, 100318. [Google Scholar] [CrossRef]
- Boulila, W. A top-down approach for semantic segmentation of big remote sensing images. Earth Sci. Inform. 2019, 12, 295–306. [Google Scholar] [CrossRef]
- Boulila, A.; Sellami, M.; Driss, M.; Al-Sarem, M.; Safai, M.; Ghaleb, F.A. RS-DCNN: A Novel Distributed Convolutional-Neural-Networks based-Approach for Big Remote-Sensing Image Classification. Comput. Electron. Agric. 2021, 182, 106014. [Google Scholar] [CrossRef]
- Bellman, R.; Adomian, G. Partial Differential Equations New Methods for Their Treatment and Solution; Media, D.R., Ed.; Springer Science & Business: Berlin, Germany, 1985. [Google Scholar]
- Chirikjian, G.S. Stochastic Models, Information Theory, and Lie Groups; Birkhäuser: Basel, Switzerland, 2009. [Google Scholar]
- Salsa, S. Partial Differential Equations in Action; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Tveito, A.; Langtangen, H.P.; Nielsen, B.F.; Cai, X. Elements of Scientific Computing; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Fangohr, H. Solving Partial Differential Equations (PDEs). 2012, pp. 1–47. Available online: https://www.southampton.ac.uk/~fangohr/teaching/comp6024/comp6024-pdes.pdf (accessed on 24 September 2019).
- Chasnov, J.R. Introduction to Differential Equations. 2016, pp. 1–151. Available online: http://www.ms.uky.edu/~ghly222/teaching/Summer2018/Lecture%20Notes%20-%20Chasnov (accessed on 24 September 2019).
- Grossmann, C.; Roos, H.; Stynes, M. Numerical Treatment of Partial Differential Equations; Springer: Berlin/Heidelberg, Germany, 2007. [Google Scholar]
- Mohamad, A.A. Lattice Boltzmann Method; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Kreiss, H.-O.; Ortiz, O.E.; Anders Petersson, N. Initial-Boundary Value Problems for Second Order Systems of Partial Differential Equations. ESAIM 2012, 46, 559–593. [Google Scholar] [CrossRef] [Green Version]
- Kuttler, K. Notes for Partial Differential Equations. 2003. Available online: http://www.math.byu.edu/~klkuttle/547notesB.pdf (accessed on 24 September 2019).
- Grigoryan, V. Partial Differential Equations. 2010, pp. 1–96. Available online: http://www.math.ucsb.edu/~grigoryan/124A.pdf (accessed on 24 September 2019).
- Dhar, D. States of Matter. 2009. Available online: http://www.tifr.res.in/~alumni/States_of_matter_Deepak_Dhar.pdf (accessed on 24 September 2019).
- Hou, J.; Ma, J.; Yu, K.N.; Li, W.; Cheng, C.; Bao, L.; Han, W. Non-thermal plasma treatment altered gene expression profiling in non-small-cell lung cancer A549 cells. BMC Genom. 2015, 16, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Levine, E.; Zhang, Z.; Kuhlman, T.; Hwa, T. Quantitative characteristics of gene regulation by small RNA. PLoS Biology 2007, 5, 2007. [Google Scholar] [CrossRef]
- Levine, E.; McHale, P.; Levine, H. Small Regulatory RNAs May Sharpen Spatial Expression Patterns. PLoS Comput. Biol. 2007, 3, e233. [Google Scholar] [CrossRef] [Green Version]
- Hohn, M.E. Partial Differential Equation Models and Numerical Simulations of RNA Interactions and Gene Expression. Ph.D. Thesis, University of California, San Diego, CA, USA, 2013. [Google Scholar]
- Wyler, E.; Mösbauer, K.; Franke, V.; Diag, A.; Gottula, L.T.; Arsie, R.; Klironomos, F.; Koppstein, D.; Ayoub, S.; Buccitelli, C.; et al. Bulk and single-cell gene expression profiling of SARS-CoV-2 infected human cell lines identifies molecular targets for therapeutic intervention. bioRxiv 2020. [Google Scholar] [CrossRef]
- Bar-Sinai, Y.; Hoyer, S.; Hickey, J.; Brenner, M.P. Learning data-driven discretizations for partial differential equations. Proc. Natl. Acad. Sci. USA 2019, 116, 15344–15349. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Xie, Z.; Jiang, X.; Li, Z.; Shen, Y.; Wang, B.; Liu, J. The Applications of Promoter-gene-Engineered Biosensors. Sensors 2018, 18, 2823. [Google Scholar] [CrossRef] [Green Version]
- Sierpe, R.; Kogan, M.J.; Bollo, S. Label-Free Oligonucleotide-Based SPR Biosensor for the Detection of the Gene Mutation Causing Prothrombin-Related Thrombophilia. Sensors 2020, 20, 6240. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Notation | Description |
|---|---|
| Messenger RNA (mRNA) Population | |
| Non-coding small interfering RNA (siRNA) Population | |
| Production rate of mRNA | |
| Self-degradation rate of mRNA | |
| Coupled Degradation rate of mRNA due to siRNA | |
| Production rate of siRNA | |
| Self-degradation rate of siRNA | |
| Diffusion coefficient of mRNA | |
| Diffusion coefficient of siRNA |
| Boundaries of IC | Dirichlet B.Cs |
|---|---|
| NT | Non-Treatment or Control Group |
| SE | Short-exposure non-thermal plasma treatment, measured post 1 h of treatment |
| LE post 1 h | Long-exposure non-thermal plasma treatment, measured post 1 h of treatment |
| LE post 2 h | Long-exposure non-thermal plasma treatment, measured post 2 h of treatment |
| LE post 4 h | Long-exposure non-thermal plasma treatment, measured post 4 h of treatment |
| Constant | Exponential | Log |
|---|---|---|
1 10 | ||
| Epigenetic | ||
| Experimental | ||
| Model | |||||
|---|---|---|---|---|---|
| 1 | ) | 10 | 1 | ||
| 2 | ) | ||||
| 3 | |||||
| 4 | |||||
| 5 | ) | ||||
| 6 | 10 | 1 | |||
| 7 | ) | 10 | 1 | ||
| 8 | ) | 1 | |||
| 9 | ) | ||||
| 10 | |||||
| 11 | |||||
| 12 | |||||
| 13 | 1 | ||||
| 14 | ) | 1 | |||
| 15 |
| Normalized Scale | ||
|---|---|---|
| C1 | 0 | 2.265458 |
| C2 | 2.267602 | 4.383277 |
| C3 | 4.383667 | 7.001413 |
| C4 | 7.002388 | 12 |
| Fusion NT | Fusion LE Post 1 h | Fusion LE Post 2 h | Fusion LE Post 4 h | Model | |
|---|---|---|---|---|---|
| AKT1(1) | 4 | 1 | 1 | 1 | 13 |
| AKT1(2) | 1 | 1 | 1 | 1 | 4 |
| AKT1(3) | 2 | 3 | 3 | 3 | 8 |
| MET(1) | 1 | 3 | 3 | 3 | 1 |
| MET(2) | 1 | 4 | 4 | 4 | 3 |
| MET(3) | 4 | 4 | 4 | 4 | 14 |
| MET(4) | 1 | 3 | 4 | 4 | 2 |
| KRAS(1) | 1 | 3 | 3 | 3 | 1 |
| KRAS(2) | 4 | 1 | 1 | 2 | NA |
| KRAS(3) | 1 | 2 | 2 | 2 | 5 |
| KRAS(4) | 4 | 4 | 4 | 4 | 14 |
| NRAS(1) | 4 | 1 | 1 | 1 | 13 |
| NRAS(2) | 1 | 3 | 3 | 3 | 1 |
| EGFR(1) | 1 | 3 | 3 | 3 | 1 |
| EGFR(2) | 4 | 4 | 4 | 4 | 14 |
| EGFR(3) | 4 | 1 | 1 | 1 | 13 |
| EGFR(4) | 1 | 2 | 2 | 2 | 5 |
| EGFR(5) | 2 | 3 | 3 | 3 | 8 |
| EGFR(6) | 3 | 2 | 2 | 2 | 12 |
| EGFR(7) | 1 | 1 | 1 | 1 | 4 |
| EGFR(8) | 2 | 1 | 1 | 1 | 10 |
| BRAF | 1 | 2 | 2 | 2 | 5 |
| ALK(1) | 2 | 4 | 4 | 4 | 9 |
| ALK(2) | 2 | 4 | 4 | 2 | 7 |
| PIK3CA(1) | 1 | 3 | 3 | 3 | 1 |
| PIK3CA(2) | 1 | 1 | 1 | 1 | 4 |
| SMEK1 | 4 | 2 | 2 | 2 | 15 |
| RET(1) | 2 | 2 | 2 | 2 | 6 |
| RET(2) | 2 | 3 | 3 | 3 | 8 |
| RET(3) | 2 | 3 | 3 | 3 | 8 |
| ROS1 | 2 | 3 | 3 | 1 | 11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farouq, M.W.; Boulila, W.; Hussain, Z.; Rashid, A.; Shah, M.; Hussain, S.; Ng, N.; Ng, D.; Hanif, H.; Shaikh, M.G.; et al. A Novel Coupled Reaction-Diffusion System for Explainable Gene Expression Profiling. Sensors 2021, 21, 2190. https://0-doi-org.brum.beds.ac.uk/10.3390/s21062190
Farouq MW, Boulila W, Hussain Z, Rashid A, Shah M, Hussain S, Ng N, Ng D, Hanif H, Shaikh MG, et al. A Novel Coupled Reaction-Diffusion System for Explainable Gene Expression Profiling. Sensors. 2021; 21(6):2190. https://0-doi-org.brum.beds.ac.uk/10.3390/s21062190
Chicago/Turabian StyleFarouq, Muhamed Wael, Wadii Boulila, Zain Hussain, Asrar Rashid, Moiz Shah, Sajid Hussain, Nathan Ng, Dominic Ng, Haris Hanif, Mohamad Guftar Shaikh, and et al. 2021. "A Novel Coupled Reaction-Diffusion System for Explainable Gene Expression Profiling" Sensors 21, no. 6: 2190. https://0-doi-org.brum.beds.ac.uk/10.3390/s21062190