A Novel Protocol Using Small-Scale Spray-Drying for the Efficient Screening of Solid Dispersions in Early Drug Development and Formulation, as a Straight Pathway from Screening to Manufacturing Stages

 ,
,
Abstract
:1. Introduction
2. Material and Methods
2.1. Materials
2.2. Methods
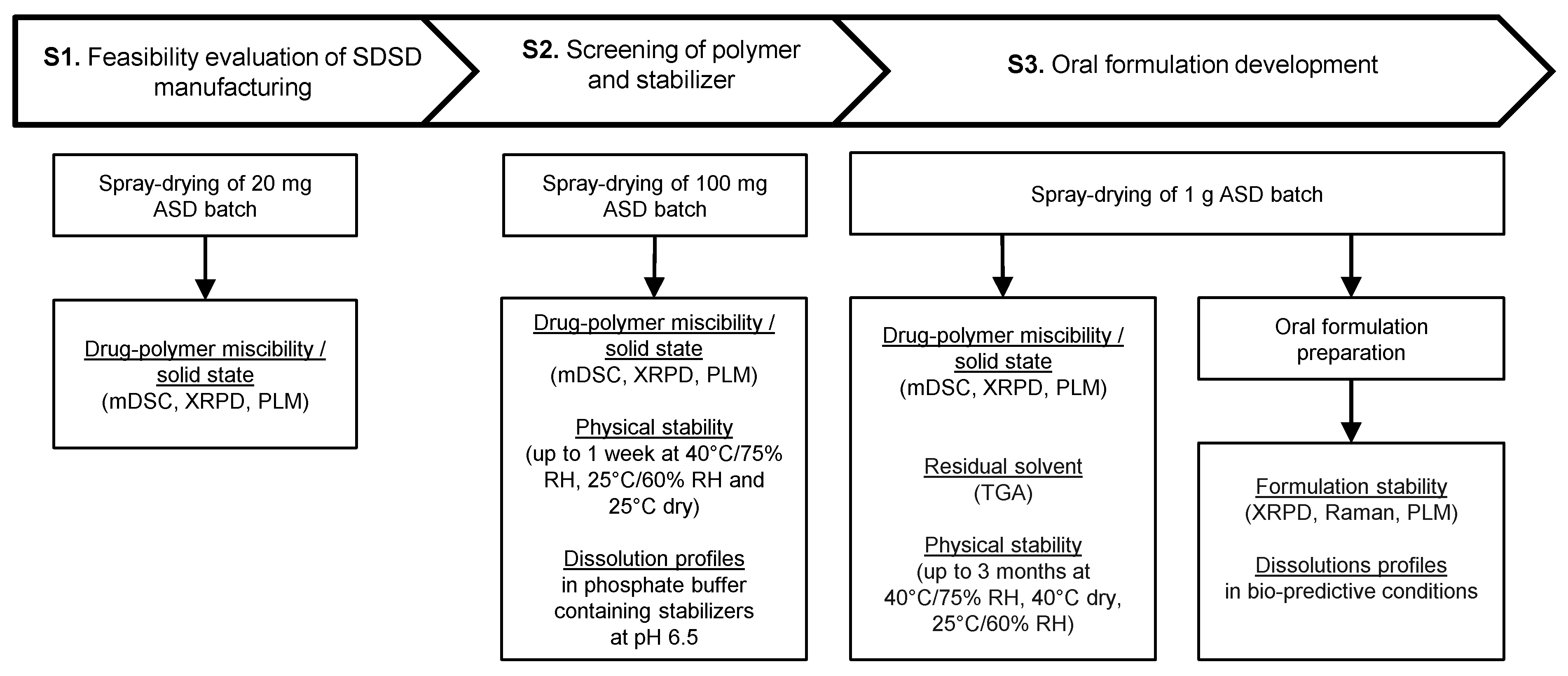
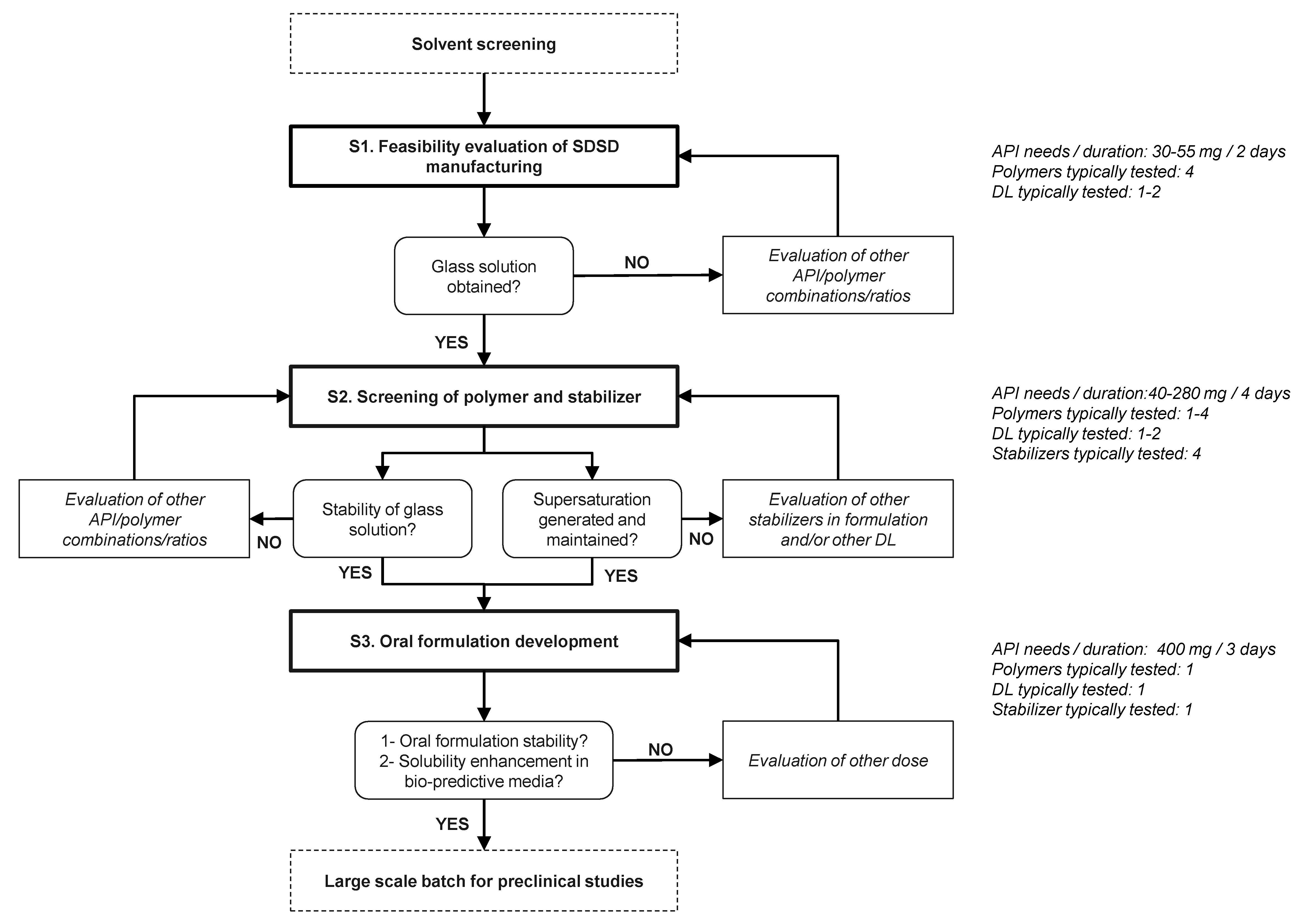
2.2.1. Screening Strategy
General Considerations
- As a preliminary step of screening protocol, a common solvent or binary solvent mixture of interest that allows dissolving both drug and polymer, needs to be identified. A solute concentration higher than 2% (w/v) is defined as acceptance criteria for spray-drying development to achieve a reasonable yield, process time and solvent consumption to comply with HSE considerations [8,23].
- In the first Stage (S1), the potential of screened carriers was evaluated based on their ability to generate glass solutions by spray-drying. The evaluation of drug–polymer miscibility in the early stage of drug development is known to offer a reliable assessment of the ASD potential [24,25,26]. Specifically, the formation of glass solution system where amorphous drug is molecularly dispersed in the carrier, combines the best performance in terms of physical stability and solubility improvement [27]. On the contrary, semi-crystalline and phase-separated ASDs are known to provide limited potential of solubility enhancement and higher tendency for drug recrystallization during both dissolution and upon storage [28,29]. Commonly used excipients for the preparation of solid dispersions include cellulose, polyvinlylpyrrolidone, poloxamer, polyethylene glycol or polymethacrylate derivatives [30]. These polymers are recognized as “generally regarded as safe” (GRAS) excipients. Additional criteria such as Tg, hygroscopicity, solubility in organic solvents, viscosifying properties, pH of hydration in water and solid solution capacity need to be considered regarding the carrier selection for the manufacturing of solid dispersions by spray-drying [31].
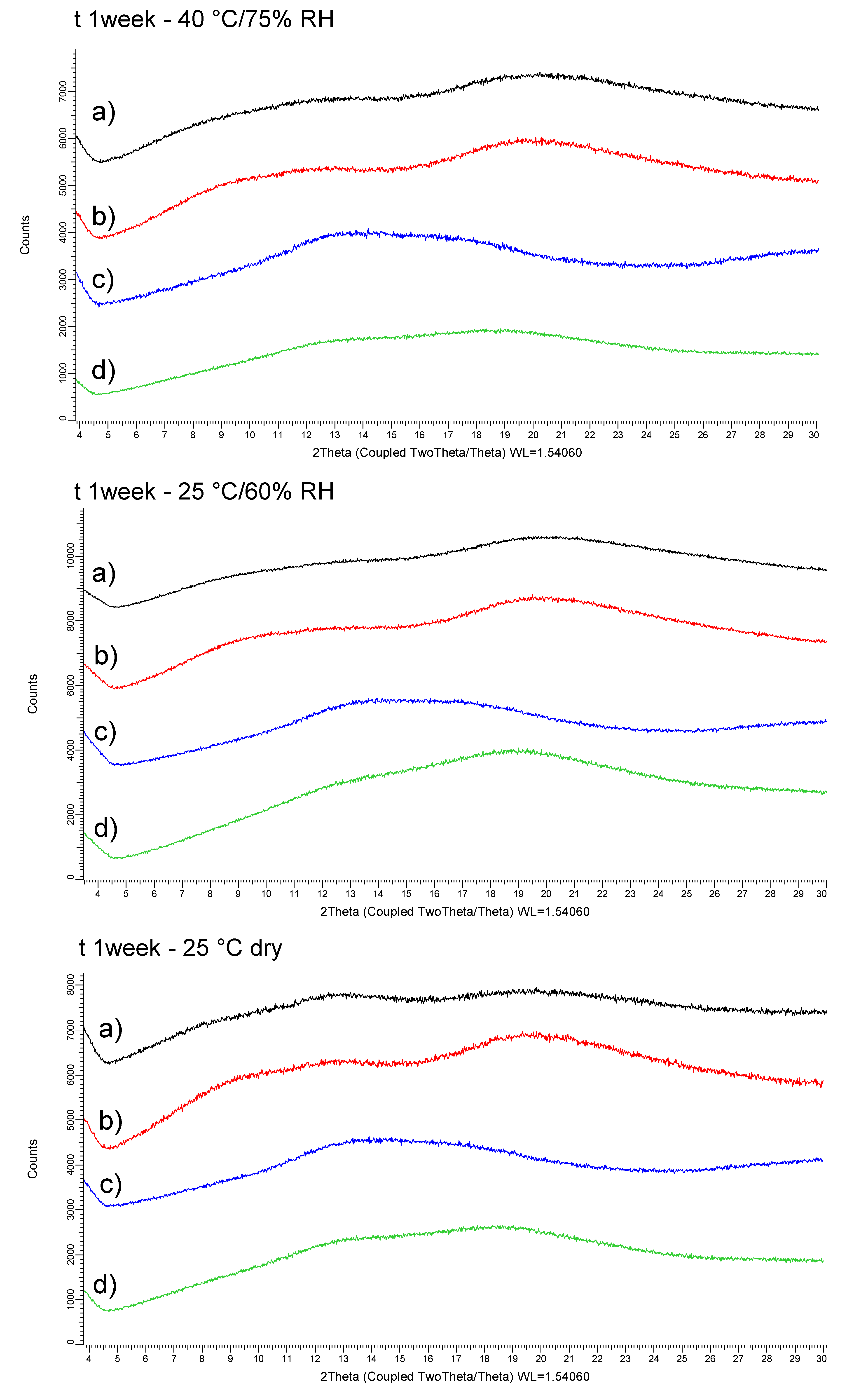
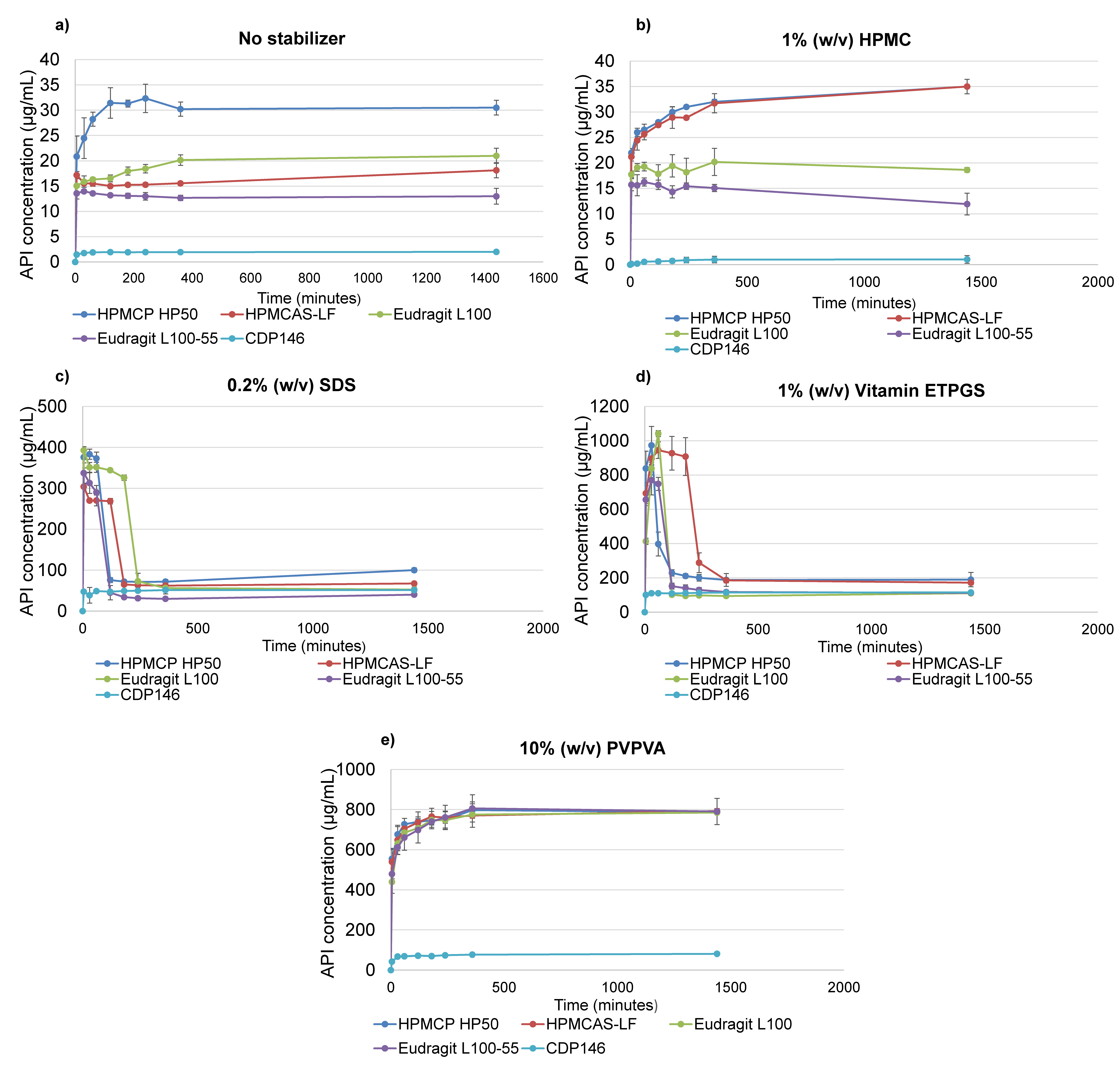
- In the second Stage (S2), the physical stability of API-polymer systems identified as glass solutions was assessed up to one week. In parallel, the dissolution properties of these ASDs was examined with and without stabilizers dissolved in aqueous medium. Therefore, the influence of the stabilizer in the dosing vehicle of the oral formulation and its ability to maintain drug supersaturation and parachute effect during dissolution tests were investigated. The use of stabilizer in the dosing vehicle of suspensions allows converting the API into suitable dosage form for administration to non-clinical species. The wide variety of stabilizers commonly used during non-clinical studies allows overcoming the diversity of molecule specific exposure limitations so that the formulation maintains stability, homogeneity and dosability within the range of doses tested [5].
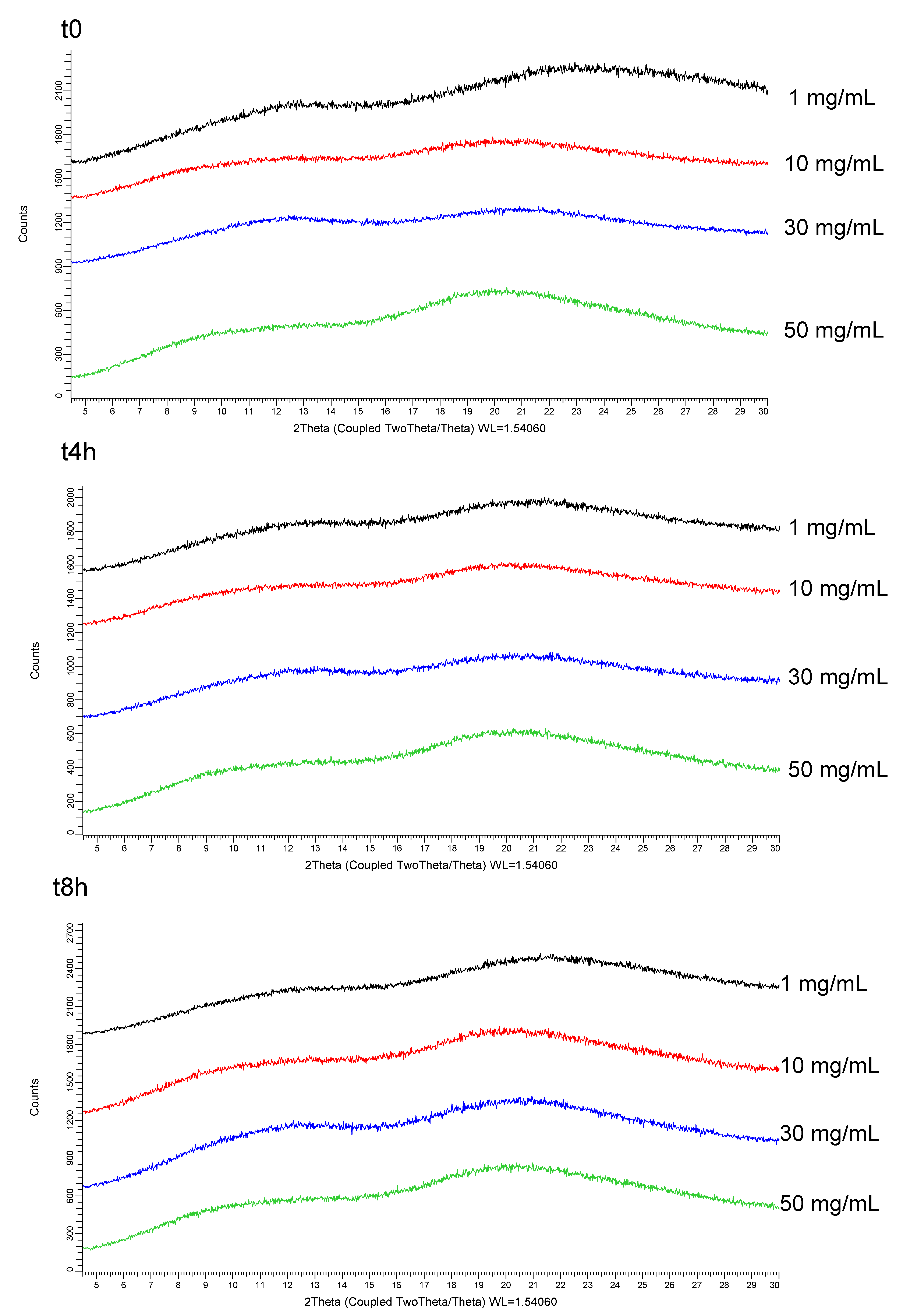
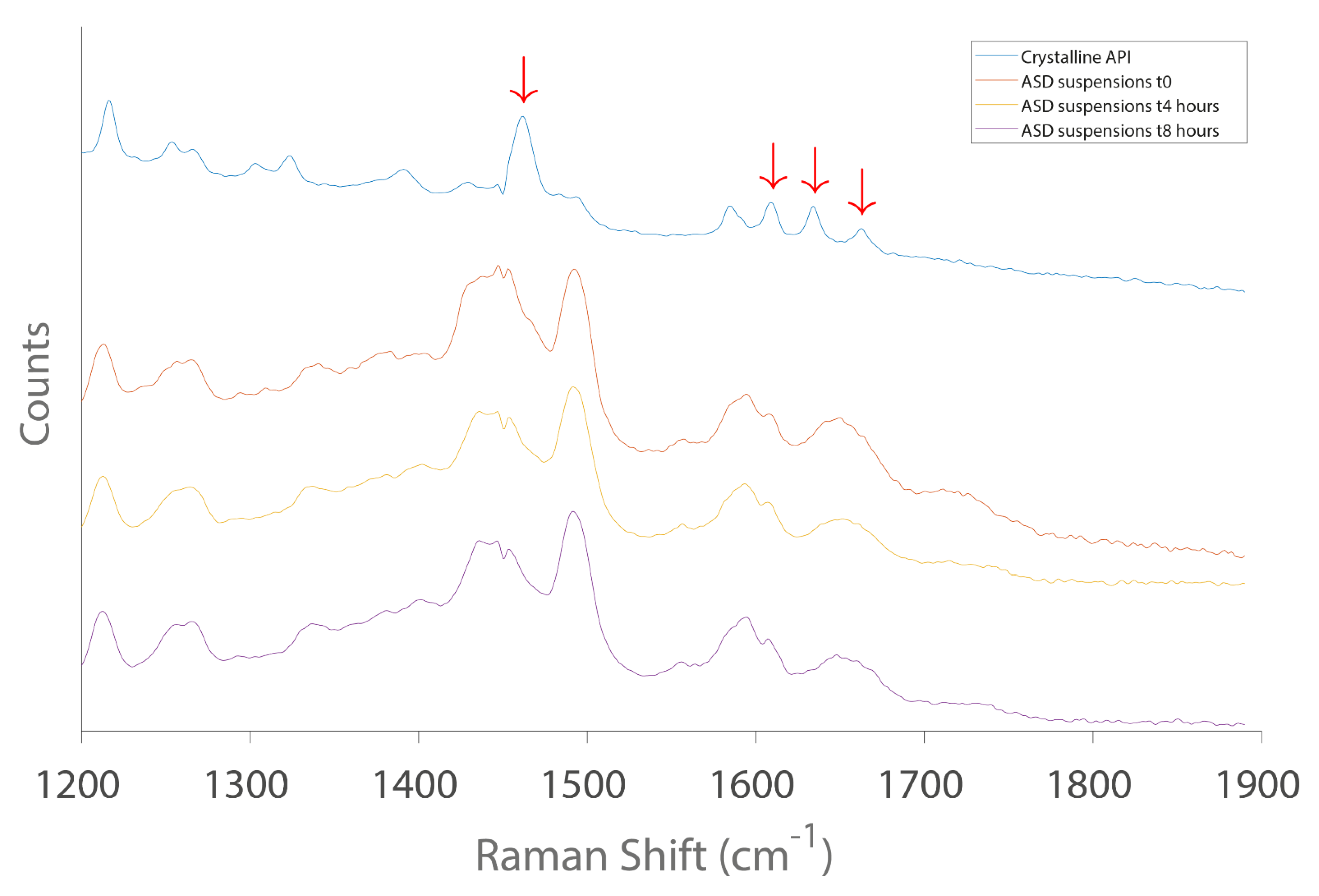
- In the last Stage (S3), the long-term stability of the lead SDSD identified during Stage 2 was investigated for up to 3 months. Moreover, the lead SDSD was prepared as non-clinical suspension formulation in the vehicle containing the stabilizer of interest. Then, the oral formulation was prepared at various doses generally tested during non-clinical studies. Its stability prior to administration and dissolution performance in bio-predictive conditions were assessed.
Application to CDP146 ASD Screening
2.2.2. Analytical or Characterization Methods
Modulated Differential Scanning Calorimetry
Thermogravimetric Analysis
X-ray Powder Diffraction
Polarized Light Microscopy
Raman Spectroscopy
HPLC
3. Results and Discussion
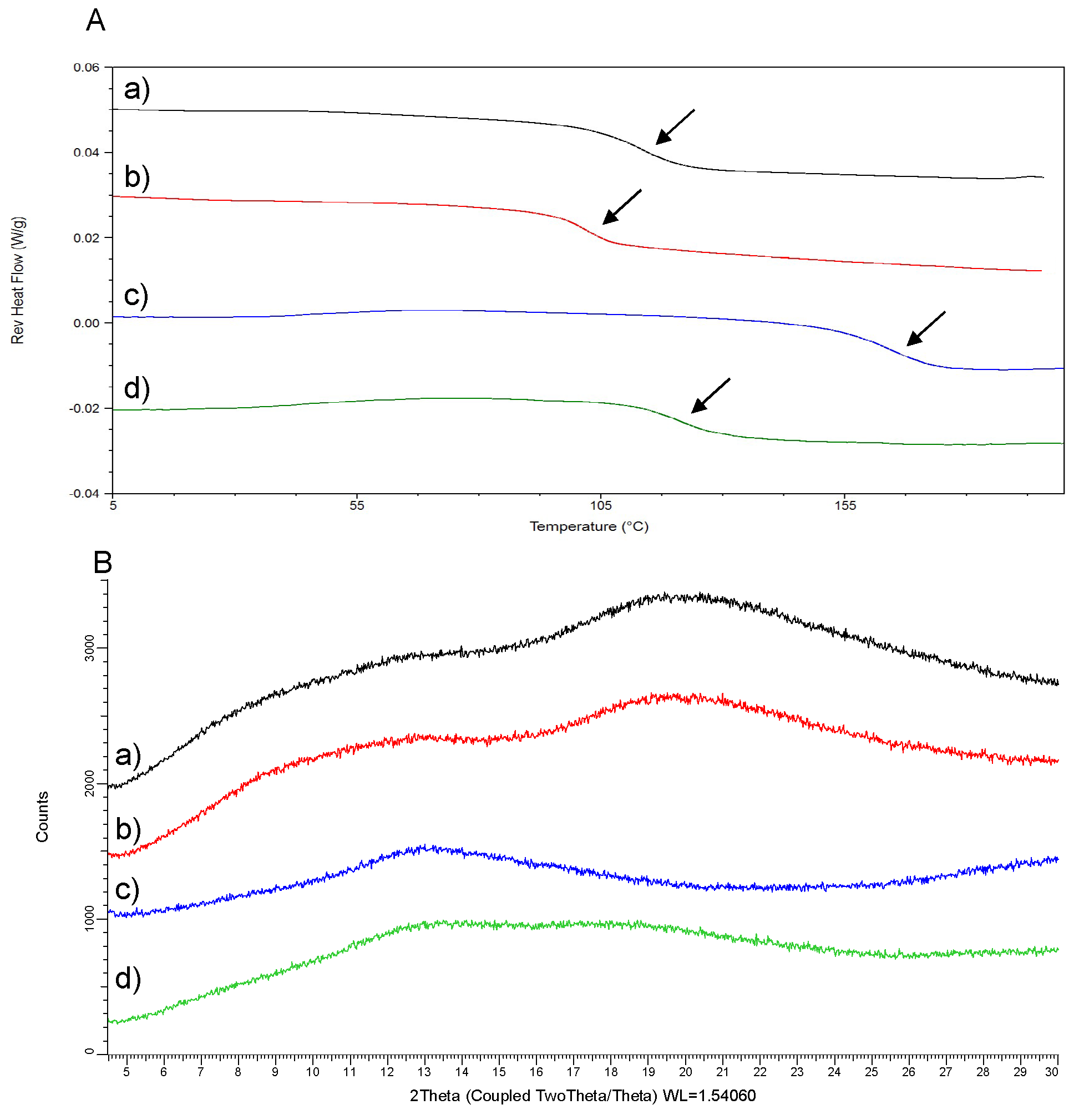
3.1. Feasibility Evaluation of SDSD Manufacturing (Stage 1)
3.2. Screening of Polymer and Stabilizer (Stage 2)
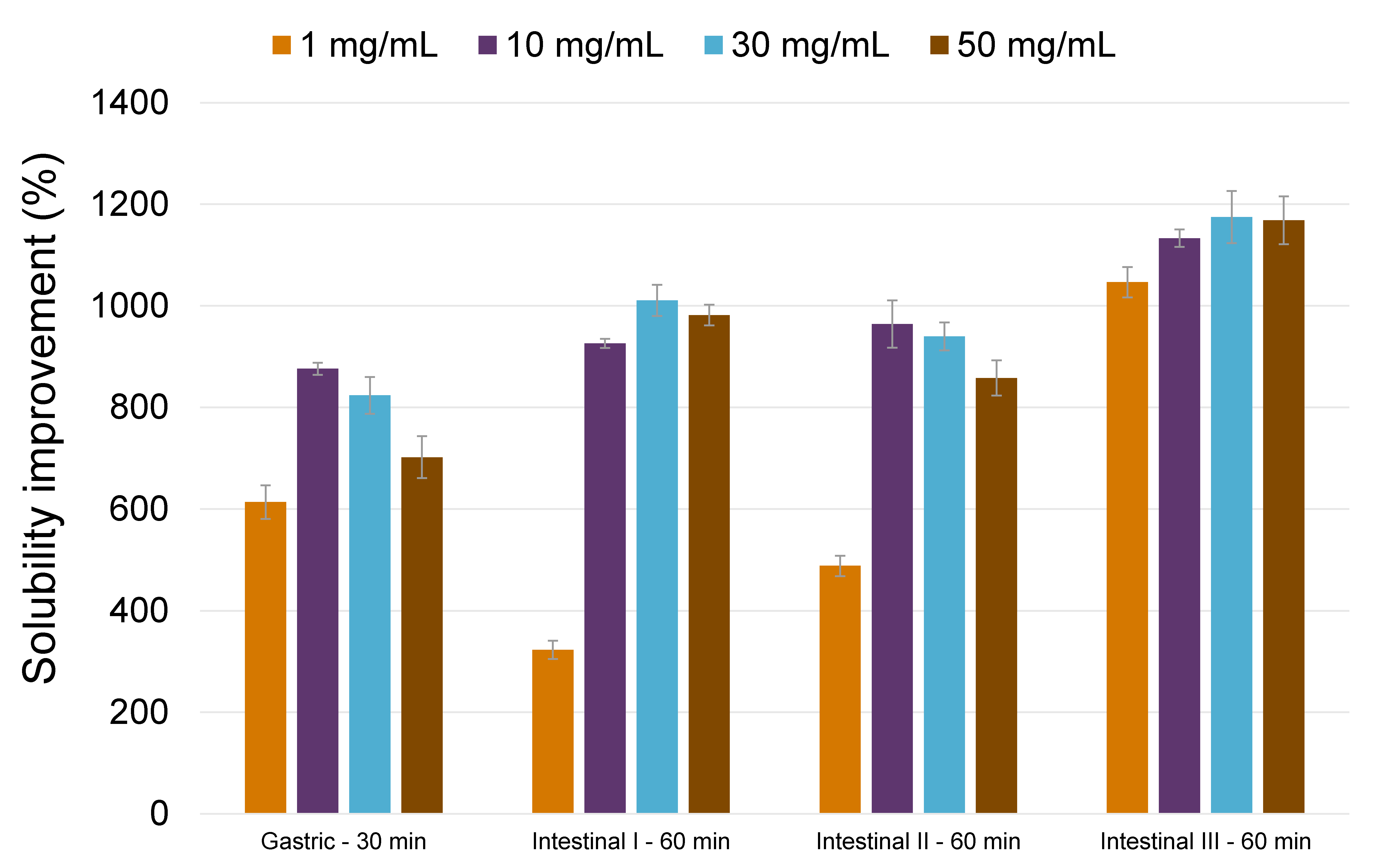
3.3. Oral Formulation Development (Stage 3)
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACN | Acetonitrile |
| AFM | Atomic force microscopy |
| API | Active pharmaceutical ingredient |
| ASD | amorphous solid dispersions |
| DCM | Dichloromethane |
| DL | Drug-loading |
| EtOH | Ethanol |
| HPLC | High performance liquid chromatography |
| mDSC | Modulated differential scanning calorimetry |
| NCE | New chemical entity |
| PLM | Polarized light microscopy |
| RH | Relative humidity |
| SDSD | Spray-dried solid dispersion |
| SIF | Simulated intestinal fluids |
| Tg | Glass transition temperature |
| TGA | Thermogravimetric analysis |
| Tm | Melting temperature |
| XRPD | X-ray powder diffraction |
References
- Nieto-Gutierrez, M. Non-clinical assessment requirements. In Safety and Efficacy of Medicines/Human Medicines Development and Evaluation; European Medicines Agency: London, UK, 2013. [Google Scholar]
- Gad, S.C.; Cassidy, C.D.; Aubert, N.; Spainhour, B.; Robbe, H. Nonclinical vehicle use in studies by multiple routes in multiple species. Int. J. Toxicol. 2006, 25, 499–521. [Google Scholar] [CrossRef] [PubMed]
- Kalepu, S.; Nekkanti, V. Insoluble drug delivery strategies: Review of recent advances and business prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Thackaberry, E.A. Vehicle selection for nonclinical oral safety studies. Expert Opin. Drug Metab. Toxicol. 2013, 9, 1635–1646. [Google Scholar] [CrossRef] [PubMed]
- Turner, P.V.; Pekow, C.; Vasbinder, M.A.; Brabb, T. Administration of substances to laboratory animals: Equipment considerations, vehicle selection, and solute preparation. J. Am. Assoc. Lab. Anim. 2011, 50, 614–627. [Google Scholar]
- Kawabata, Y.; Wada, K.; Nakatani, M.; Yamada, S.; Onoue, S. Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: Basic approaches and practical applications. Int. J. Pharm. 2011, 420, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Vo, C.L.N.; Park, C.; Lee, B.J. Current trends and future perspectives of solid dispersions containing poorly water-soluble drugs. Eur. J. Pharm. Biopharm. 2013, 85, 799–813. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Ho, C. Amorphous solid dispersions: Utilization and challenges in drug discovery and development. J. Pharm. Sci. 2015, 104, 3237–3258. [Google Scholar] [CrossRef] [PubMed]
- Teja, S.B.; Patil, S.P.; Shete, G.; Patel, S.; Bansal, A.K. Drug-excipient behavior in polymeric amorphous solid dispersions. J. Excip. Food Chem. 2013, 4, 70–93. [Google Scholar]
- Singh, A.; Van den Mooter, G. Spray drying formulation of amorphous solid dispersions. Adv. Drug Deliv. Rev. 2016, 100, 27–50. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Q.; Stefanski, K.; Shen, H.; Huang, C.; Caporuscio, C.; Yang, W.; Lam, P.; Su, C.; Gudmundsson, O.; Hageman, M. Oral delivery of highly lipophilic poorly water-soluble drugs: Spray-dried dispersions to improve oral absorption and enable high-dose toxicology studies of a p2y1 antagonist. J. Pharm. Sci. 2014, 103, 3924–3931. [Google Scholar] [CrossRef] [PubMed]
- Chiang, P.C.; Ran, Y.; Chou, K.J.; Cui, Y.; Sambrone, A.; Chan, C.; Hart, R. Evaluation of drug load and polymer by using a 96-well plate vacuum dry system for amorphous solid dispersion drug delivery. AAPS Pharm. Sci. Tech. 2012, 13, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.G.; Pollock-Dove, C.; Dong, L.C.; Li, S. Advanced screening assays to rapidly identify solubility-enhancing formulations: High-throughput, miniaturization and automation. Adv. Drug. Deliv. Rev. 2008, 60, 657–672. [Google Scholar] [CrossRef] [PubMed]
- Janssens, S.; De Zeure, A.; Paudel, A.; Van Humbeeck, J.; Rombaut, P.; Van den Mooter, G. Influence of preparation methods on solid state supersaturation of amorphous solid dispersions: A case study with itraconazole and eudragit e100. Pharm. Res. 2010, 27, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Shanbhag, A.; Rabel, S.; Nauka, E.; Casadevall, G.; Shivanand, P.; Eichenbaum, G.; Mansky, P. Method for screening of solid dispersion formulations of low-solubility compounds--miniaturization and automation of solvent casting and dissolution testing. Int. J. Pharm. 2008, 351, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Ousset, A.; Meeus, J.; Robin, F.; Schubert, M.A.; Somville, P.; Dodou, K. Comparison of a novel miniaturized screening device with büchi b290 mini spray-dryer for the development of spray-dried solid dispersions (SDSDs). Processes 2018, 6, 129. [Google Scholar] [CrossRef]
- Ousset, A.; Chavez, P.F.; Meeus, J.; Robin, F.; Schubert, M.A.; Somville, P.; Dodou, K. Prediction of phase behavior of spray-dried amorphous solid dispersions: Assessment of thermodynamic models, standard screening methods and a novel atomization screening device with regard to prediction accuracy. Pharmaceutics 2018, 10, 29. [Google Scholar] [CrossRef] [PubMed]
- Wyttenbach, N.; Janas, C.; Siam, M.; Lauer, M.E.; Jacob, L.; Scheubel, E.; Page, S. Miniaturized screening of polymers for amorphous drug stabilization (spads): Rapid assessment of solid dispersion systems. Eur. J. Pharm. Biopharm. 2013, 84, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Parikh, T.; Gupta, S.S.; Meena, A.K.; Vitez, I.; Mahajan, N.; Serajuddin, A.T. Application of film-casting technique to investigate drug-polymer miscibility in solid dispersion and hot-melt extrudate. J. Pharm. Sci. 2015, 104, 2142–2152. [Google Scholar] [CrossRef] [PubMed]
- Overhoff, K.A.; Moreno, A.; Miller, D.A.; Johnston, K.P.; Williams, R.O. Solid dispersions of itraconazole and enteric polymers made by ultra-rapid freezing. Int. J. Pharm. 2007, 336, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Higashi, K.; Yamamoto, K.; Moribe, K. The effect of hpmcas functional groups on drug crystallization from the supersaturated state and dissolution improvement. Int. J. Pharm. 2014, 464, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Rowe, R.C.; Sheskey, P.J.; Owen, S.C. Handbook of Pharmaceutical Excipients, 5th ed.; Pharmaceutical Press: London, UK, 2006; Volume 5. [Google Scholar]
- Patel, R.P.; Patel, M.P.; Suthar, A.M. Spray drying technology: An overview. Indian J. Sci. Technol. 2009, 2, 44–47. [Google Scholar]
- Albers, J.; Matthee, K.; Knop, K.; Kleinebudde, P. Evaluation of predictive models for stable solid solution formation. J. Pharm. Sci. 2011, 100, 667–680. [Google Scholar] [CrossRef] [PubMed]
- Engers, D.; Teng, J.; Jimenez-Novoa, J.; Gent, P.; Hossack, S.; Campbell, C.; Thomson, J.; Ivanisevic, I.; Templeton, A.; Byrn, S.; et al. A solid-state approach to enable early development compounds: Selection and animal bioavailability studies of an itraconazole amorphous solid dispersion. J. Pharm. Sci. 2010, 99, 3901–3922. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Trivino, A.; Prasad, D.; Chauhan, H. Investigation and correlation of drug polymer miscibility and molecular interactions by various approaches for the preparation of amorphous solid dispersions. Eur. J. Pharm. Sci. 2015, 71, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Janssens, S.; Van den Mooter, G. Review: Physical chemistry of solid dispersions. J. Pharm. Pharmacol. 2009, 61, 1571–1586. [Google Scholar] [CrossRef] [PubMed]
- Rumondor, A.C.; Stanford, L.A.; Taylor, L.S. Effects of polymer type and storage relative humidity on the kinetics of felodipine crystallization from amorphous solid dispersions. Pharm. Res. 2009, 26, 2599–2606. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Dai, W.G. Fundamental aspects of solid dispersion technology for poorly soluble drugs. Acta Pharm. Sin. B 2014, 4, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Paudel, A.; Worku, Z.A.; Meeus, J.; Guns, S.; Van den Mooter, G. Manufacturing of solid dispersions of poorly water soluble drugs by spray drying: Formulation and process considerations. Int. J. Pharm. 2013, 453, 253–284. [Google Scholar] [CrossRef] [PubMed]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Polymeric amorphous solid dispersions: A review of amorphization, crystallization, stabilization, solid-state characterization, and aqueous solubilization of biopharmaceutical classification system class ii drugs. J. Pharm. Sci. 2016, 105, 2527–2544. [Google Scholar] [CrossRef] [PubMed]
- Ormes, J.D.; Zhang, D.; Chen, A.M.; Hou, S.; Krueger, D.; Nelson, T.; Templeton, A. Design of experiments utilization to map the processing capabilities of a micro-spray dryer: Particle design and throughput optimization in support of drug discovery. Pharm. Dev. Technol. 2013, 18, 121–129. [Google Scholar] [CrossRef] [PubMed]
- ProCept. Procept information brochure spray dryer. Available online: http://www.procept.be/spray-dryer-chiller (accessed on 27 August 2018).
- Sun, D.D.; Wen, H.; Taylor, L.S. Non-sink dissolution conditions for predicting product quality and in vivo performance of supersaturating drug delivery systems. J. Pharm. Sci. 2016, 105, 2477–2488. [Google Scholar] [CrossRef] [PubMed]
- Baird, J.A.; Van Eerdenbrugh, B.; Taylor, L.S. A classification system to assess the crystallization tendency of organic molecules from undercooled melts. J. Pharm. Sci. 2010, 99, 3787–3806. [Google Scholar] [CrossRef] [PubMed]
- Paudel, A.; Van den Mooter, G. Influence of solvent composition on the miscibility and physical stability of naproxen/pvp k 25 solid dispersions prepared by cosolvent spray-drying. Pharm. Res. 2012, 29, 251–270. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Hu, Y.; Liu, L.; Su, L.; Li, N.; Yu, J.; Tang, B.; Yang, Z. Physical stability of amorphous solid dispersions: A physicochemical perspective with thermodynamic, kinetic and environmental aspects. Pharm. Res. 2018, 35, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Konecke, S.; Wegiel, L.A.; Taylor, L.S.; Edgar, K.J. Both solubility and chemical stability of curcumin are enhanced by solid dispersion in cellulose derivative matrices. Carbohydr. Polym. 2013, 98, 1108–1116. [Google Scholar] [CrossRef] [PubMed]
- Lauer, M.E.; Siam, M.; Tardio, J.; Page, S.; Kindt, J.H.; Grassmann, O. Rapid assessment of homogeneity and stability of amorphous solid dispersions by atomic force microscopy--from bench to batch. Pharm. Res. 2013, 30, 2010–2022. [Google Scholar] [CrossRef] [PubMed]
- Baghel, S.; Cathcart, H.; O'Reilly, N.J. Theoretical and experimental investigation of drug-polymer interaction and miscibility and its impact on drug supersaturation in aqueous medium. Eur. J. Pharm. Biopharm. 2016, 107, 16–31. [Google Scholar] [CrossRef] [PubMed]
- Prasad, D.; Chauhan, H.; Atef, E. Amorphous stabilization and dissolution enhancement of amorphous ternary solid dispersions: Combination of polymers showing drug-polymer interaction for synergistic effects. J. Pharm. Sci. 2014, 103, 3511–3523. [Google Scholar] [CrossRef] [PubMed]
- Ziaee, A.; Albadarin, A.B.; Padrela, L.; Faucher, A.; O'Reilly, E.; Walker, G. Spray drying ternary amorphous solid dispersions of ibuprofen—An investigation into critical formulation and processing parameters. Eur. J. Pharm. Biopharm. 2017, 120, 43–51. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Mw (g/mol) a | Dissolution pH a | Tg (°C) b | T Degradation (°C) c |
|---|---|---|---|---|
| HPMCP-HP50 | 78,000 | >5.0 | 140 | 160 |
| HPMCAS-LF | 18,167 | >5.5 | 122 | 170 |
| Eudragit L100 | 125,000 | >6.0 | 192 | 165 |
| Eudragit L100-55 | 320,000 | >5.5 | 122 | 165 |
| ASD Composition | Yield | Miscibility/Solid State | |||
|---|---|---|---|---|---|
| Polymer | DL (w/w) | Yield (%) | Tg (°C) | Tm (°C) | XRPD Pattern/PLM |
| HPMCP HP50 | 40% | 68.1 | 113.5 | - | A |
| HPMCAS-LF | 40% | 70.9 | 102.5 | - | A |
| Eudragit L100 | 40% | 71.8 | 163.4 | - | A |
| Eudragit L100-55 | 40% | 71.7 | 120.9 | - | A |
| Lead ASD | Process Considerations | Miscibility/Solid State | Residual Solvent | Physical Stability | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Polymer | DL (w/w) | Yield (%) | Tg (°C) | Tm (°C) | XRPDPattern/PLM | Weight Loss (%) | 25 °C/60% RH | 40 °C Dry | 40 °C/75% RH | ||||||
| 1 m | 2 m | 3 m | 1 m | 2 m | 3 m | 1 m | 2 m | 3 m | |||||||
| HPMCAS-LF | 40% | 88.2 | 102.5 | - | A | 0.2 | A | A | A | A | A | A | A | A | A |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ousset, A.; Chirico, R.; Robin, F.; Schubert, M.A.; Somville, P.; Dodou, K. A Novel Protocol Using Small-Scale Spray-Drying for the Efficient Screening of Solid Dispersions in Early Drug Development and Formulation, as a Straight Pathway from Screening to Manufacturing Stages. Pharmaceuticals 2018, 11, 81. https://0-doi-org.brum.beds.ac.uk/10.3390/ph11030081
Ousset A, Chirico R, Robin F, Schubert MA, Somville P, Dodou K. A Novel Protocol Using Small-Scale Spray-Drying for the Efficient Screening of Solid Dispersions in Early Drug Development and Formulation, as a Straight Pathway from Screening to Manufacturing Stages. Pharmaceuticals. 2018; 11(3):81. https://0-doi-org.brum.beds.ac.uk/10.3390/ph11030081
Chicago/Turabian StyleOusset, Aymeric, Rosanna Chirico, Florent Robin, Martin Alexander Schubert, Pascal Somville, and Kalliopi Dodou. 2018. "A Novel Protocol Using Small-Scale Spray-Drying for the Efficient Screening of Solid Dispersions in Early Drug Development and Formulation, as a Straight Pathway from Screening to Manufacturing Stages" Pharmaceuticals 11, no. 3: 81. https://0-doi-org.brum.beds.ac.uk/10.3390/ph11030081