Development of a Microwave-assisted Chemoselective Synthesis of Oxime-linked Sugar Linkers and Trivalent Glycoclusters


Abstract
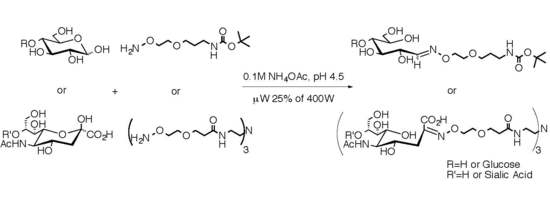
:
1. Introduction
2. Results
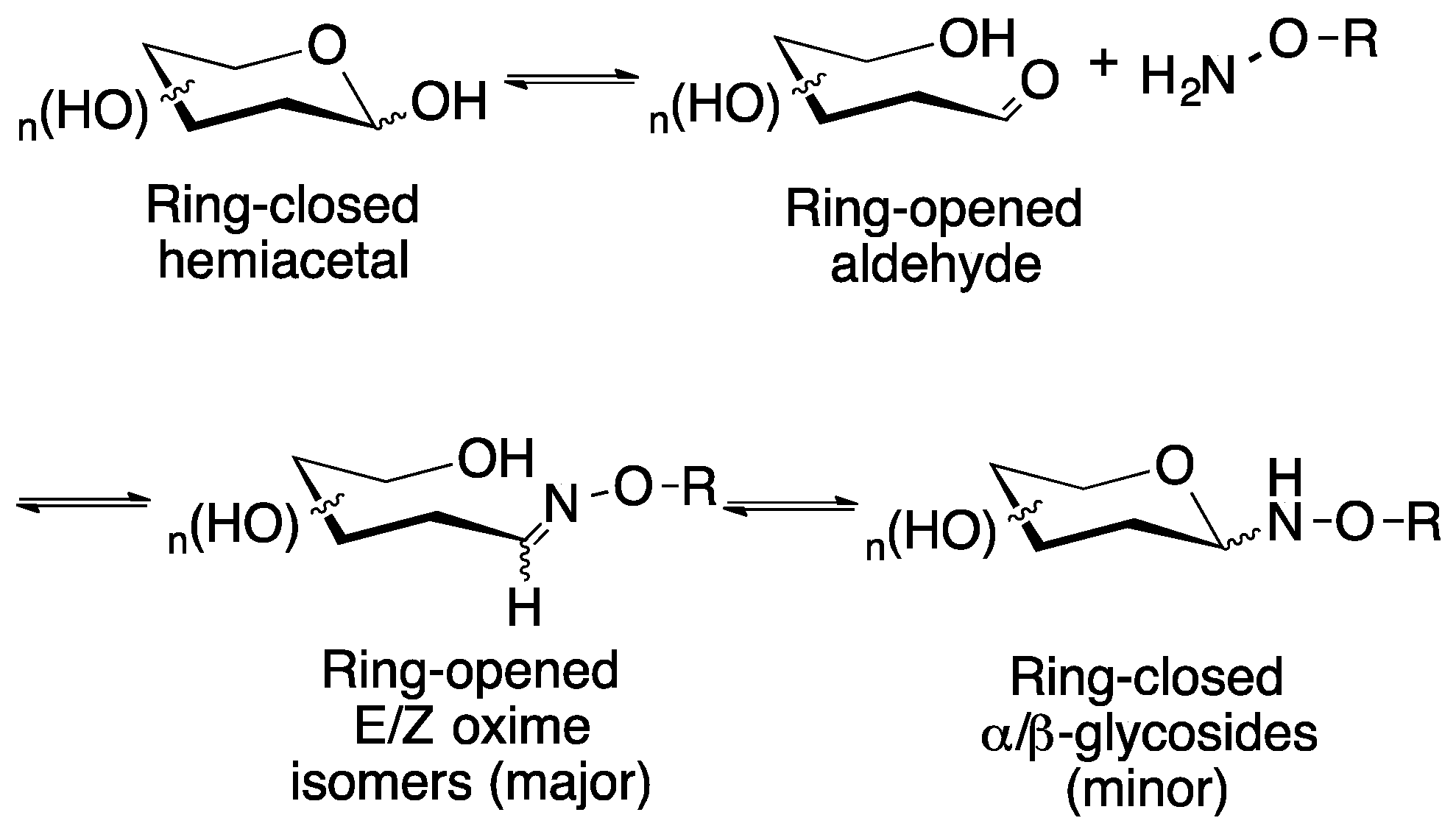
2.1. Medium Scale Microwave-mediated Synthesis of Monovalent Sugar-Linkers
2.2. Large Scale Microwave-mediated Synthesis of Monovalent Sugar-Linkers
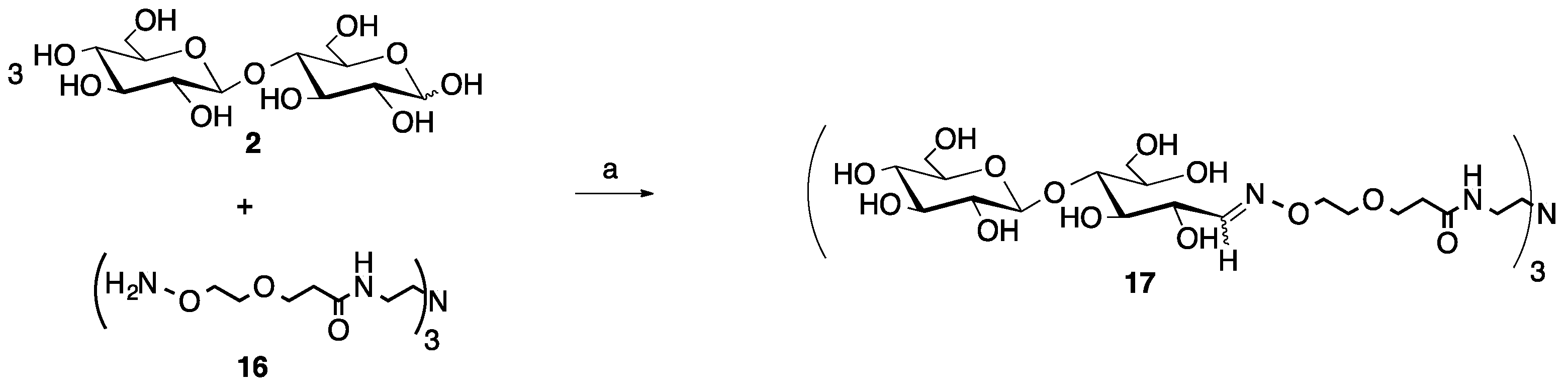
2.3. Multivalent Glycocluster Microwave-mediated Synthesis
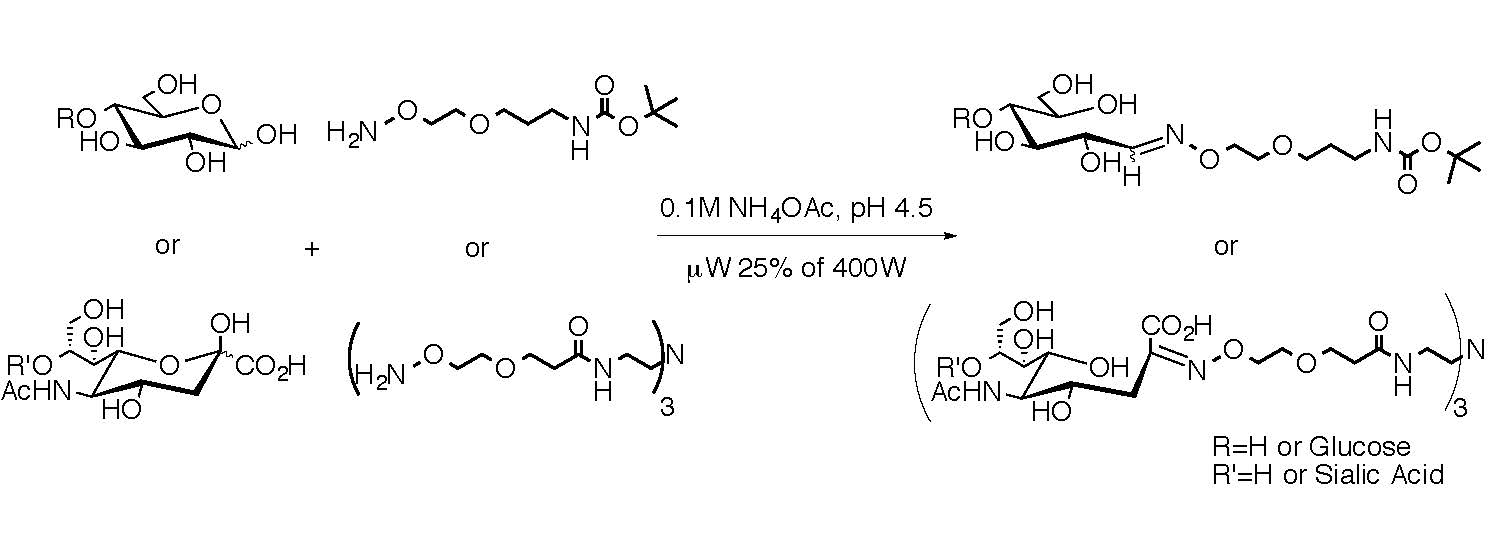
3. Discussion
4. Materials and Methods
4.1. General Methods
4.2. Synthesis of Monovalent Sugar-Linkers
4.2.1. General Procedure for the Medium-Scale (0.187–0.250 mmol) Synthesis of Sugar-Linkers
Without Aniline-microwave or Traditional Heating
With Aniline
4.2.2. General Procedure for the Large-scale (≥0.800 mmol) Synthesis of Sugar-linkers:
4.3. Synthesis of Trivalent Glycoclusters
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kottari, N.; Chabre, Y.M.; Sharma, R.; Roy, R. Applications of glyconanoparticles as “sweet” glycobiological therapeutics and diagnostics. Adv. Polym. Sci. 2013, 254, 297–342. [Google Scholar]
- Mellet, C.O.; Mendez-Ardoy, A.; Ferenandez, J.M.G. Click multivalent glycomaterials: Glycoclusters, glycodendrimers, glycopolymers, hybrid glycomaterials and glycosurfaces. In Click Chemistry in Glycoscience: New Developments and Strategies, 1st ed.; Witczak, Z.J., Bielski, R., Eds.; John Wiley and Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 143–182. [Google Scholar]
- Adak, A.K.; Yu, C.-C.; Lin, C.-C. Synthesis and applications of glyconanoparticles, glycodendrimers, and glycoclusters in biological systems. In Glycochemical Synthesis: Strategies and Applications; Hung, S.-C., Zulueta, M.M.L., Eds.; John Wiley and Sons, Inc.: Hoboken, NJ, USA, 2016; pp. 425–454. [Google Scholar]
- Jebali, A.; Nayeri, E.K.; Roohana, S.; Aghaei, S.; Ghaffari, M.; Daliri, K.; Fuente, G. Nano-carbohydrates: Synthesis and application in genetics, biotechnology, and medicine. Adv. Colloid Interface 2017, 240, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ramella, D.; Polito, L.; Mazzini, S.; Ronchi, S.; Scaglioni, L.; Marelli, M.; Lay, L. A strategy for multivalent presentation of carba analogues from N. meningitidis a capsular polysaccharide. Eur. J. Org. Chem. 2014, 2014, 5915–5924. [Google Scholar] [CrossRef]
- Lee, Y.-C.; Lee, R.T. Carbohydrate-protein interactions: Basis of glycobiology. Acc. Chem. Res. 1995, 28, 321–327. [Google Scholar] [CrossRef]
- Clayton, R.; Hardman, J.; LaBranche, C.C.; McReynolds, K.D. Evaluation of the synthesis of sialic acid-PAMAM glycodendrimers without the use of sugar protecting groups, and the anti-HIV-1 properties of these compounds. Bioconj. Chem. 2011, 22, 2186–2197. [Google Scholar] [CrossRef] [PubMed]
- Cecioni, S.; Praly, J.-P.; Matthews, S.E.; Wimmerova, M.; Imberty, A.; Vidal, S. Rational design and synthesis of optimized glycoclusters for multivalent lectin-carbohydrate interactions: Influence of the linker arm. Chem. Eur. J. 2012, 18, 6250–6263. [Google Scholar] [CrossRef]
- Yang, Z.-L.; Zeng, X.-F.; Liu, H.-P.; Yu, Q.; Meng, X.; Yan, Z.-L.; Fan, Z.-C.; Xiao, S.S.; Yang, Y.; Yu, P. Synthesis of multivalent difluorinated zanamivir analogs as potent antiviral inhibitors. Tetrahedron Lett. 2016, 57, 2579–2582. [Google Scholar] [CrossRef]
- Bagul, R.S.; Hosseini, M.; Shiao, T.C.; Saadeh, N.K.; Roy, R. Heterolayered hybrid dendrimers with optimized sugar head groups for enhancing carbohydrate-protein interactions. Polym. Chem. 2017, 8, 5354–5366. [Google Scholar] [CrossRef]
- Villadson, K.; Martos-Maldonado, M.C.; Jensen, K.J.; Thygesen, M.B. Chemoselective reactions for the synthesis of glycoconjugates from unprotected carbohydrates. ChemBioChem 2017, 18, 574–612. [Google Scholar] [CrossRef]
- Kolmel, D.K.; Kool, E.T. Oximes and hydrazones in bioconjugation: Mechanism and catalysis. Chem. Rev. 2017, 117, 10358–10376. [Google Scholar] [CrossRef]
- Kalia, J.; Raines, R.T. Hydrolytic stability of hydrazones and oximes. Angew. Chem. Int. Ed. Engl. 2008, 47, 7523–7526. [Google Scholar] [CrossRef]
- Gudmundsdottir, A.V.; Paul, C.E.; Nitz, M. Stability studies of hydrazide and hydroxylamine-based glycoconjugates in aqueous solution. Carbohydr. Res. 2009, 344, 278–284. [Google Scholar] [CrossRef]
- Iqbal, A.; Chibli, H.; Hamilton, C.J. Stability of aminooxy glycosides to glycosidase catalyzed hydrolysis. Carbohydr. Res. 2013, 377, 1–3. [Google Scholar] [CrossRef]
- McReynolds, K.D.; Dimas, D.; Le, H. Synthesis of hydrophilic aminooxy linkers and multivalent cores for chemoselective aldehyde/ketone conjugation. Tetrahedron Lett. 2014, 55, 2270–2273. [Google Scholar] [CrossRef]
- Chen, N.; Xie, J. N-O linkage in carbohydrates and glycoconjugates. Org. Biomol. Chem. 2016, 14, 11028–11047. [Google Scholar] [CrossRef]
- Pifferi, C.; Daskhan, G.C.; Fiore, M.; Shiao, T.C.; Roy, R. Aminooxylated carbohydrates: Synthesis and applications. Chem. Rev. 2017, 117, 9839–9873. [Google Scholar] [CrossRef]
- Thygesen, M.B.; Sauer, J.; Jensen, K.J. Chemoselective capture of glycans for analysis on gold nanoparticles: Carbohydrate oxime tautomers provide functional recognition by proteins. Chem. Eur. J. 2009, 15, 1649–1660. [Google Scholar] [CrossRef]
- Rathi, A.K.; Gawande, M.B.; Zboril, R.; Varma, R.S. Microwave-assisted synthesis-catalytic applications in aqueous media. Coord. Chem. Rev. 2015, 291, 68–94. [Google Scholar] [CrossRef]
- Corsaro, A.; Pistaria, V.; Chiacchio, M.A.; Romeo, G. A journey into recent microwave-assisted carbohydrate chemistry. In Microwaves in Organic Synthesis, 3rd ed.; de la Hoz, A., Loupy, A., Eds.; Wiley-VCH Verlag GmbH and Co.: Weinheim, Germany, 2012; pp. 961–1011. [Google Scholar]
- Joosten, J.A.F.; Tholen, N.T.H.; El Maate, F.A.; Brouwer, A.J.; van Esse, G.W.; Rijkers, D.T.S.; Liskamp, R.M.J.; Pieters, R.J. High-yielding microwave-assisted synthesis of triazole-linked glycodendrimers by copper-catalyzed [3+2] cycloaddition. Eur. J. Org. Chem. 2005, 2005, 3182–3185. [Google Scholar] [CrossRef]
- Brun, M.A.; Disney, M.D.; Seeberger, P.H. Miniaturization of microwave assisted carbohydrate functionalization to create oligosaccharide microarrays. ChemBioChem 2006, 7, 421–424. [Google Scholar] [CrossRef]
- Cecioni, S.; Faure, S.; Darbost, U.; Bonnamour, I.; Parrot-Lopez, H.; Roy, O.; Taillefumier, C.; Wimmerova, M.; Praly, J.-P.; Imberty, A.; et al. Selectivity among two lectins: Probing the effect of topology, multivalency and flexibility of “clicked” multivalent glycoclusters. Chem. Eur. J. 2011, 17, 2146–2159. [Google Scholar] [CrossRef]
- Cagnoni, A.J.; Varela, O.; Gouin, S.G.; Kovensky, J.; Uhrig, M.L. Synthesis of multivalent glycoclusters from 1-thio-β-d-galactose and their inhibitory activity agains the β-galactosidase from E. coli. J. Org. Chem. 2011, 76, 3064–3077. [Google Scholar] [CrossRef]
- Cecioni, S.; Matthews, S.E.; Blanchard, H.; Praly, J.-P.; Imberty, A.; Vidal, S. Synthesis of lactosylated glycoclusters and inhibition studies with plant and human lectins. Carbohydr. Res. 2012, 356, 132–141. [Google Scholar] [CrossRef]
- Cagnoni, A.J.; Kovensky, J.; Uhrig, M.L. Design and synthesis of hydrolytically stable multivalent ligands bearing thiodigalactoside analogues for peanut lectin and human galectin-3 binding. J. Org. Chem. 2014, 79, 6456–6467. [Google Scholar] [CrossRef]
- Ding, F.; Ji, L.; William, R.; Chai, H.; Liu, X.-W. Design and synthesis of multivalent neoglycoconjugates by click conjugations. Beilstein J. Org. Chem. 2014, 10, 1325–1332. [Google Scholar] [CrossRef] [Green Version]
- Chuang, Y.-J.; Zhou, X.; Pan, Z.; Turchi, C. A convenient method for synthesis of glyconanoparticles for calorimetric measuring carbohydrate-protein interaction. Biochem. Biophys. Res. Commun. 2009, 389, 22–27. [Google Scholar] [CrossRef]
- Seo, J.; Michaelian, N.; Owens, S.C.; Dashner, S.T.; Wong, A.J.; Barron, A.E.; Carrasco, M.R. Chemoselective and microwave-assisted synthesis of glycopeptoids. Org. Lett. 2009, 11, 5210–5213. [Google Scholar] [CrossRef]
- Thygesen, M.B.; Munch, H.; Sauer, J.; Cló, E.; Jorgensen, M.R.; Hindsgaul, O.; Jensen, K.J. Nucleophilic catalysis of carbohydrate oxime formation by anilines. J. Org. Chem. 2010, 75, 1752–1755. [Google Scholar] [CrossRef]
- Ray, G.J.; Siekman, J.; Scheinecker, R.; Zhang, Z.; Gerasimov, M.V.; Szabo, C.M.; Kosma, P. Reaction of oxidized polysialic acid and a diaminooxy linker: Characterization and process optimization using nuclear magnetic resonance spectroscopy. Bioconj. Chem. 2016, 27, 2071–2080. [Google Scholar] [CrossRef]
- Berthet, N.; Thomas, B.; Bossu, I.; DuFour, E.; Gillon, E.; Garcia, J.; Spinelli, N.; Imberty, A.; Dumy, P.; Renaudet, O. High affinity glycodendrimers for the lectin LecB from Pseudomonas aeruginosa. Bioconj. Chem. 2013, 24, 1598–1611. [Google Scholar] [CrossRef]
- Dirksen, A.; Dawson, P.E. Rapid oxime and hydrazone ligations with aromatic aldehydes for biomolecular labeling. Bioconj. Chem. 2008, 19, 2543–2548. [Google Scholar] [CrossRef]
- Patane, J.; Trapani, V.; Villavert, J.; McReynolds, K.D. Preparative production of colominic acid oligomers via a facile microwave hydrolysis. Carbohydr. Res. 2009, 344, 820–824. [Google Scholar] [CrossRef] [Green Version]
- Sato, C.; Kitajima, K. Disialic, oligosialic and polysialic acids: Distribution, functions and related disease. J. Biochem. 2013, 154, 115–136. [Google Scholar] [CrossRef]
- Bhide, G.P.; Colley, K.J. Sialylation of N-glycans: Mechanism, cellular compartmentalization and function. Histochem. Cell Biol. 2017, 147, 149–174. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sugar | Additive | % Yield | % Yield |
|---|---|---|---|
| Medium Scale (≤0.250 mmol) | |||
| Non-Microwave Conditions: | |||
| Cellobiose | 0.1 M Aniline | 65 | |
| N/A | 56 | −9 | |
| Microwave-Mediated Conditions: | |||
| GlcNAc | 0.1 M Aniline | 78 | |
| N/A | 68 | −10 | |
| Cellobiose | 0.1 M Aniline | 76 | |
| N/A | 63 | −13 | |
| Gentiobiose | 0.1 M Aniline | 73 | |
| N/A | 60 | −13 | |
| Lactose | 0.1 M Aniline | 93 | |
| N/A | 65 | −28 | |
| Maltose | 0.1 M Aniline | 74 | |
| N/A | 65 | −9 | |
| Maltotriose | 0.1 M Aniline | 92 | |
| N/A | 68 | −24 | |
| Melibiose | 0.1 M Aniline | 80 | |
| N/A | 68 | −12 | |
| Large Scale (≥0.800 mmol) | |||
| GlcNAc | 0.1 M Aniline | 79 | 1 |
| Cellobiose | 0.1 M Aniline | 75 | −1 |
| Gentiobiose | 0.1 M Aniline | 65 | −8 |
| Lactose | 0.1 M Aniline | 63 | −30 |
| Maltose | 0.1 M Aniline | 62 | −12 |
| Maltotriose | 0.1 M Aniline | 64 | −28 |
| Melibiose | 0.1 M Aniline | 60 | −20 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McReynolds, K.; Dimas, D.; Floyd, G.; Zeman, K. Development of a Microwave-assisted Chemoselective Synthesis of Oxime-linked Sugar Linkers and Trivalent Glycoclusters. Pharmaceuticals 2019, 12, 39. https://0-doi-org.brum.beds.ac.uk/10.3390/ph12010039
McReynolds K, Dimas D, Floyd G, Zeman K. Development of a Microwave-assisted Chemoselective Synthesis of Oxime-linked Sugar Linkers and Trivalent Glycoclusters. Pharmaceuticals. 2019; 12(1):39. https://0-doi-org.brum.beds.ac.uk/10.3390/ph12010039
Chicago/Turabian StyleMcReynolds, Katherine, Dustin Dimas, Grace Floyd, and Kara Zeman. 2019. "Development of a Microwave-assisted Chemoselective Synthesis of Oxime-linked Sugar Linkers and Trivalent Glycoclusters" Pharmaceuticals 12, no. 1: 39. https://0-doi-org.brum.beds.ac.uk/10.3390/ph12010039