Twenty Years of Ferroportin Disease: A Review or An Update of Published Clinical, Biochemical, Molecular, and Functional Features

 , ,
, ,
Abstract
:1. Introduction
2. Methods
2.1. Data Collection and Selection
Identification, Demographic Characteristics, and Iron Parameters
2.2. Variant Classification Based on Functional In Vitro Studies
2.3. Bioinformatics Prediction Software Analysis
3. Evaluation and Analysis
3.1. Description of the Phenotype of Patients (Including Related Family Members) in Relation Findings in Functional Studies
3.1.1. Patients Characteristics
Ferroportin Disease Occurs in Different Parts of the World and Has no Specific Symptoms
Modification by Additional Hereditary and Acquired Conditions
3.2. Relation between In Vitro Functional Studies of Ferroportin Variants on Clinical Features
3.2.1. Clear Distinct Phenotypical Features in Patients with GOF and LOF Variants
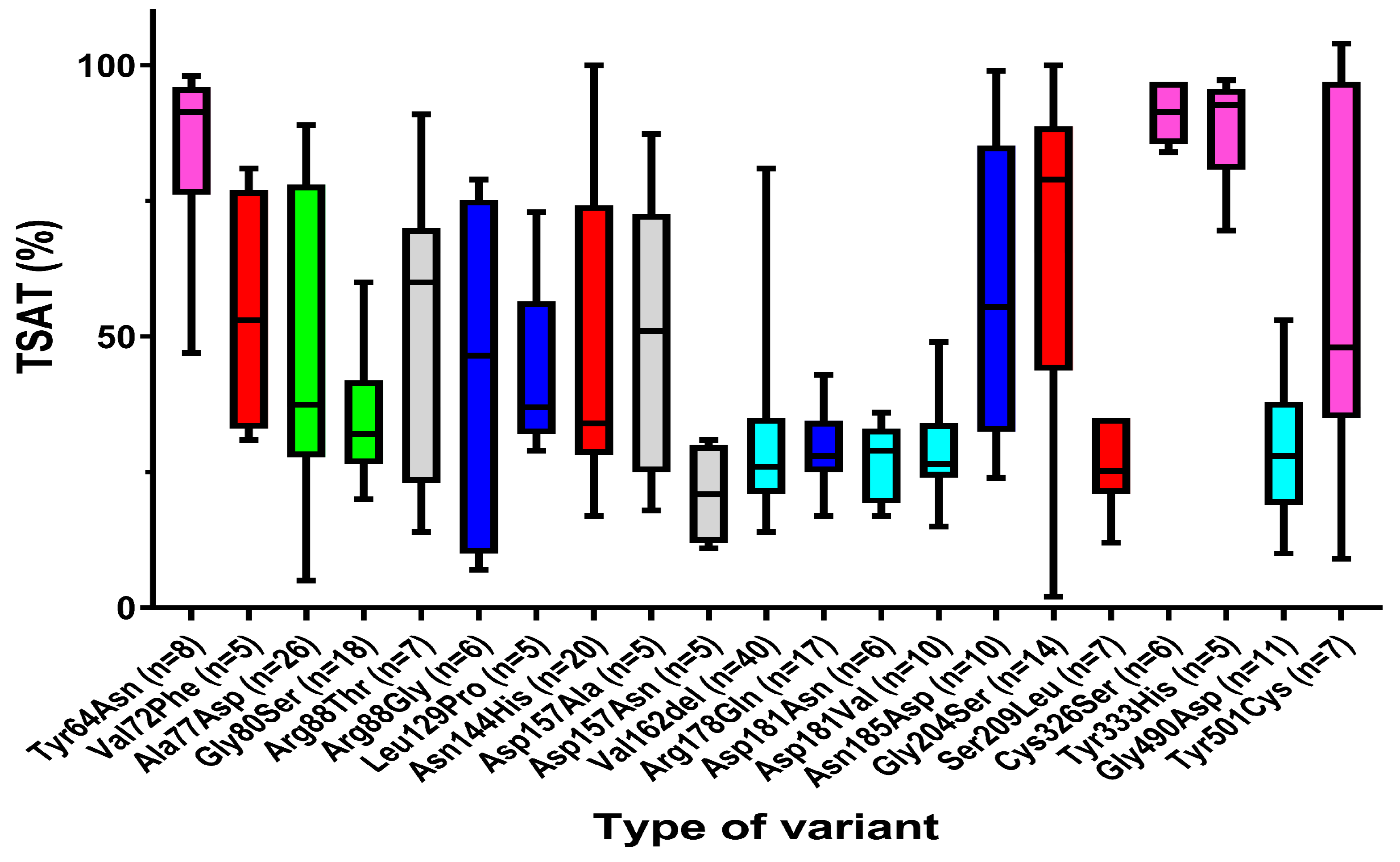
3.2.2. Strong Association of Hepcidin Sensitivity of Ferroportin Functional Gene Variants with Serum Iron Parameters
3.3. Therapeutic Considerations
4. Critical Annotations on Pathogenicity of Variants
4.1. Shortcomings in Description of Clinical Phenotypes
4.2. Shortcomings in Establishment of Pathogenicity
4.2.1. Co-Segregation with Disease in the Family
4.2.2. Variant Occurrence and Allele Frequency
4.2.3. Limitations of Functional In Vitro Tests
4.2.4. Lack of Concordance between Various In Silico Prediction Models
4.3. Assessment of Pathogenicity of Ferroportin Variants on Molecular Level in Context of Current Knowledge on the Function and Structure of Ferroportin
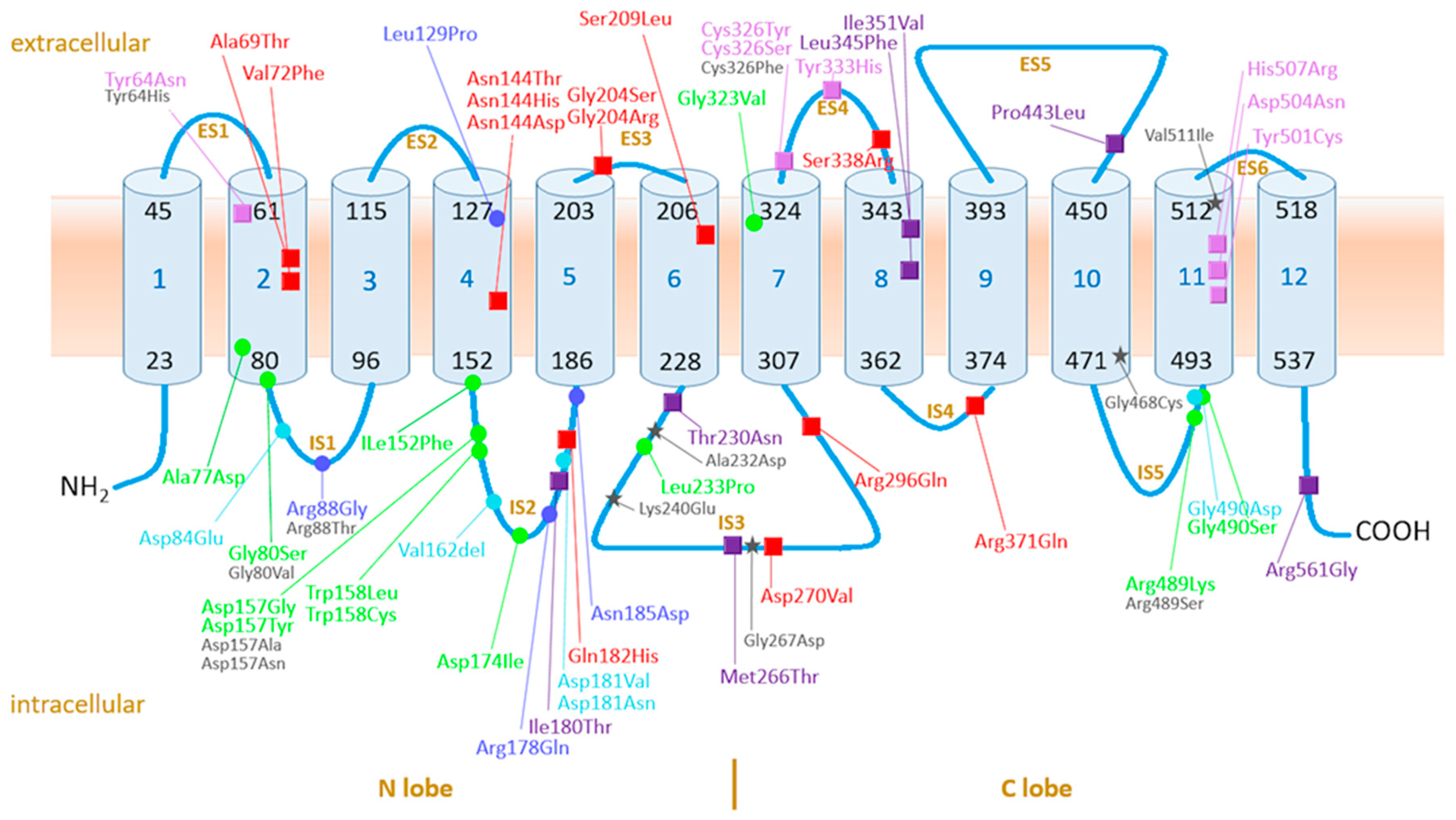
4.3.1. Ferroportin, the Protein, Its Variants, and Function: Current Knowledge
4.3.2. GOF Variants
4.3.3. LOF Variants
4.4. Is the Mode of Iron Overload in LOF Variants Related to a Difference in Ferroportin Expression and Activation between Enterocytes and Macrophages?
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pietrangelo, A.; Montosi, G.; Totaro, A.; Garuti, C.; Fraquelli, M.; Sardini, C.; Vasta, F.; Pietrangelo, A.; Conte, D.; Cassanelli, S.; et al. Hereditary hemochromatosis in adults without pathogenic mutations in the hemochromatosis gene. N. Engl. J. Med. 1999, 341, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Montosi, G.; Donovan, A.; Totaro, A.; Garuti, C.; Pignatti, E.; Cassanelli, S.; Trenor, C.C.; Gasparini, P.; Andrews, N.C.; Pietrangelo, A. Autosomal-dominant hemochromatosis is associated with a mutation in the ferroportin (SLC11A3) gene. J. Clin. Investig. 2001, 108, 619–623. [Google Scholar] [CrossRef] [PubMed]
- Njajou, O.T.; Vaessen, N.; Joosse, M.; Berghuis, B.; Van Dongen, J.W.; Breuning, M.H.; Snijders, P.J.; Rutten, W.P.; Sandkuijl, L.A.; Oostra, B.A.; et al. A mutation in SLC11A3 is associated with autosomal dominant hemochromatosis. Nat. Genet. 2001, 28, 213–214. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E. Ferroportin mutations: A tale of two phenotypes. Blood 2005, 105, 3763–3764. [Google Scholar] [CrossRef]
- Fleming, R.E.; Sly, W.S. Ferroportin mutation in autosomal dominant hemochromatosis: Loss of function, gain in understanding. J. Clin. Investig. 2001, 108, 521–522. [Google Scholar] [CrossRef]
- Wallace, D.F.; Harris, J.M.; Subramaniam, V.N. Functional analysis and theoretical modeling of ferroportin reveals clustering of mutations according to phenotype. Am. J. Physiol. Cell Physiol. 2010, 298, C75–C84. [Google Scholar] [CrossRef]
- Mayr, R.; Janecke, A.R.; Schranz, M.; Griffiths, W.J.; Vogel, W.; Pietrangelo, A.; Zoller, H. Ferroportin disease: A systematic meta-analysis of clinical and molecular findings. J. Hepatol. 2010, 53, 941–949. [Google Scholar] [CrossRef] [Green Version]
- Callebaut, I.; Joubrel, R.; Pissard, S.; Kannengiesser, C.; Gérolami, V.; Ged, C.; Cadet, E.; Cartault, F.; Ka, C.; Gourlaouen, I.; et al. Comprehensive functional annotation of 18 missense mutations found in suspected hemochromatosis type 4 patients. Hum. Mol. Genet. 2014, 23, 4479–4490. [Google Scholar] [CrossRef] [Green Version]
- Le Lan, C.; Mosser, A.; Ropert, M.; Détivaud, L.; Loustaud–Ratti, V.; Vital–Durand, D.; Roget, L.; Bardou–Jacquet, E.; Turlin, B.; David, V.; et al. Sex and acquired cofactors determine phenotypes of ferroportin disease. Gastroenterology 2011, 140, 1199–1207. [Google Scholar] [CrossRef]
- Pietrangelo, A.; Caleffi, A.; Corradini, E. Non-HFE hepatic iron overload. Semin. Liver Dis. 2011, 31, 302–318. [Google Scholar] [CrossRef]
- Detivaud, L.; Island, M.L.; Jouanolle, A.M.; Ropert, M.; Bardou-Jacquet, E.; Le Lan, C.; Mosser, A.; Leroyer, P.; Deugnier, Y.; David, V. Ferroportin diseases: Functional studies, a link between genetic and clinical phenotype. Hum. Mutat. 2013, 34, 1529–1536. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Yano, M.; Urawa, N.; Mizutani, A.; Hamaoka, S.; Araki, J.; Kojima, Y.; Naito, Y.; Kato, A.; Tatsumi, Y.; et al. A 10-year follow-up study of a Japanese family with ferroportin disease A: Mild iron overload with mild hyperferritinemia co-occurring with hyperhepcidinemia may be benign. Intern. Med. 2018, 57, 2865–2871. [Google Scholar] [CrossRef] [PubMed]
- Altes, A.; Bach, V.; Ruiz, A.; Esteve, A.; Remacha, A.F.; Sarda, M.P.; Felez, J.; Baiget, M. Does the SLC40A1 gene modify HFE-related haemochromatosis phenotypes? Annu. Hematol. 2009, 88, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Zaahl, M.G.; Merryweather-Clarke, A.T.; Kotze, M.J.; van der Merwe, S.; Warnich, L.; Robson, K.J. Analysis of genes implicated in iron regulation in individuals presenting with primary iron overload. Hum. Genet. 2004, 115, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Santos, P.C.; Cancado, R.D.; Pereira, A.C.; Schettert, I.T.; Soares, R.A.; Pagliusi, R.A.; Hirata, R.D.; Hirata, M.H.; Teixeira, A.C.; Figueiredo, M.S.; et al. Hereditary hemochromatosis: Mutations in genes involved in iron homeostasis in Brazilian patients. Blood Cells Mol. Dis. 2011, 46, 302–307. [Google Scholar] [CrossRef]
- Barton, J.C.; LaFreniere, S.A.; Li, H.; Acton, R.T.; Press, R.D.; Eckfeldt, J.H.; Leiendecker-Foster, C.; Leiendecker-Foster, C. HFE, SLC40A1, HAMP, HJV, TFR2, and FTL mutations detected by denaturing high-performance liquid chromatography after iron phenotyping and HFE C282Y and H63D genotyping in 785 HEIRS study participants. Am. J. Hematol. 2009, 84, 710–714. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.L.; Gelbart, T.; West, C.; Halloran, C.; Felitti, V.; Beutler, E. A study of genes that may modulate the expression of hereditary hemochromatosis: Transferrin receptor-1, ferroportin, ceruloplasmin, ferritin light and heavy chains, iron regulatory proteins (IRP)-1 and -2, and hepcidin. Blood Cells Mol. Dis. 2001, 27, 783–802. [Google Scholar] [CrossRef]
- McNamara, L.; Gordeuk, V.R.; MacPhail, A.P. Ferroportin (Q248H) mutations in African families with dietary iron overload. J. Gastroenterol. Hepatol. 2005, 20, 1855–1858. [Google Scholar] [CrossRef]
- Rivers, C.A.; Barton, J.C.; Gordeuk, V.R.; Acton, R.T.; Speechley, M.R.; Snively, B.M.; Leiendecker-Foster, C.; Press, R.D.; Adams, P.C.; McLaren, G.D.; et al. Association of ferroportin Q248H polymorphism with elevated levels of serum ferritin in African Americans in the hemochromatosis and iron overload screening (HEIRS) study. Blood Cells Mol. Dis. 2007, 38, 247–252. [Google Scholar] [CrossRef]
- Albuquerque, D.; Manco, L.; Loua, K.M.; Arez, A.P.; Trovoada, M.D.J.; Relvas, L.; Milimono, T.S.; Rath, S.L.; Lopes, D.; Nogueira, F.; et al. SLC40A1 Q248H allele frequencies and associated SLC40A1 haplotypes in three West African population samples. Ann. Hum. Biol. 2011, 38, 378–381. [Google Scholar] [CrossRef]
- Beutler, E.; Barton, J.C.; Felitti, V.J.; Gelbart, T.; West, C.; Lee, P.L.; Waalen, J.; Vulpe, C. Ferroportin 1 (SCL40A1) variant associated with iron overload in African-Americans. Blood Cells Mol. Dis. 2003, 31, 305–309. [Google Scholar] [CrossRef]
- Jarvik, G.P.; Browning, B.L. Consideration of cosegregation in the pathogenicity classification of Genomic variants. Am. J. Hum. Gen. 2016, 98, 1077–1781. [Google Scholar] [CrossRef] [PubMed]
- Moller, P.; Clark, N.; Maehle, L. A simplified method for segregation analysis (SISA) to determine penetrance and expression of a genetic variant in a family. Hum. Mutat. 2011, 32, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the Association for molecular pathology. Off. J. Am. Coll. Med. Gen. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Broad Institute. Available online: http://gnomad.broadinstitute.org (accessed on 12 July 2019).
- PolyPhen-2 Prediction of Functional Effects of Human nsSNPs. Available online: http://genetics.bwh.harvard.edu/pph2 (accessed on 12 July 2019).
- Sorting Intolerant from Tolerant. Available online: http://www.blocks.fhcrc.org/sift/SIFT.html (accessed on 12 July 2019).
- Align-GVGD. Available online: http://agvgd.hci.utah.edu (accessed on 12 July 2019).
- Duca, L.; Delbini, P.; Nava, I.; Vaja, V.; Fiorelli, G.; Cappellini, M.D. Mutation analysis of hepcidin and ferroportin genes in Italian prospective blood donors with iron overload. Am. J. Hematol. 2009, 84, 592–593. [Google Scholar] [CrossRef] [PubMed]
- Aschemeyer, S.; Qiao, B.; Stefanova, D.; Valore, E.V.; Sek, A.C.; Ruwe, T.A.; Vieth, K.R.; Jung, G.; Casu, C.; Rivella, S.; et al. Structure-function analysis of ferroportin defines the binding site and an alternative mechanism of action of hepcidin. Blood 2018, 131, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Drakesmith, H.; Schimanski, L.M.; Ormerod, E.; Merryweather-Clarke, A.T.; Viprakasit, V.; Edwards, J.P.; Sweetland, E.; Bastin, J.M.; Cowley, D.; Chinthammitr, Y.; et al. Resistance to hepcidin is conferred by hemochromatosis-associated mutations of ferroportin. Blood 2005, 106, 1092–1097. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.; Preza, G.C.; Phung, Y.; De Domenico, I.; Kaplan, J.; Ganz, T.; Nemeth, E. The molecular basis of hepcidin-resistant hereditary hemochromatosis. Blood 2009, 114, 437–443. [Google Scholar] [CrossRef] [Green Version]
- Letocart, E.; Le Gac, G.; Majore, S.; Ka, C.; Radio, F.C.; Gourlaouen, I.; De Bernardo, C.; Ferec, C.; Grammatico, P. A novel missense mutation in SLC40A1 results in resistance to hepcidin and confirms the existence of two ferroportin-associated iron overload diseases. Br. J. Haematol. 2009, 147, 379–385. [Google Scholar] [CrossRef]
- Rice, A.E.; Mendez, M.J.; Hokanson, C.A.; Rees, D.C.; Bjorkman, P.J. Investigation of the biophysical and cell biological properties of ferroportin, a multipass integral membrane protein iron exporter. J. Mol. Biol. 2009, 386, 717–732. [Google Scholar] [CrossRef]
- Schimanski, L.M.; Drakesmith, H.; Merryweather-Clarke, A.T.; Viprakasit, V.; Edwards, J.P.; Sweetland, E.; Bastin, J.M.; Cowley, D.; Chinthammitr, Y.; Robson, K.J.; et al. In vitro functional analysis of human ferroportin (FPN) and hemochromatosis-associated FPN mutations. Blood 2005, 105, 4096–4102. [Google Scholar] [CrossRef] [PubMed]
- Rivard, S.R.; Lanzara, C.; Grimard, D.; Carella, M.; Simard, H.; Ficarella, R.; Simard, R.; D’Adamo, A.P.; De Braekeleer, M.; Gasparini, P. Autosomal dominant reticuloendothelial iron overload (HFE type 4) due to a new missense mutation in the FERROPORTIN 1 gene (SLC11A3) in a large French-Canadian family. Haematologica 2003, 88, 824–826. [Google Scholar] [PubMed]
- Sham, R.L.; Phatak, P.D.; Nemeth, E.; Ganz, T. Hereditary hemochromatosis due to resistance to hepcidin: high hepcidin concentrations in a family with C326S ferroportin mutation. Blood 2009, 114, 493–494. [Google Scholar] [CrossRef] [PubMed]
- Sham, R.L.; Phatak, P.D.; West, C.; Lee, P.; Andrews, C.; Beutler, E. Autosomal dominant hereditary hemochromatosis associated with a novel ferroportin mutation and unique clinical features. Blood Cells Mol. Dis. 2005, 34, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Le Gac, G.; Ka, C.; Joubrel, R.; Gourlaouen, I.; Lehn, P.; Mornon, J.P.; Ferec, C.; Callebaut, I. Structure-function analysis of the human ferroportin iron exporter (SLC40A1): Effect of hemochromatosis type 4 disease mutations and identification of critical residues. Hum. Mutat. 2013, 34, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, W.J.; Mayr, R.; McFarlane, I.; Hermann, M.; Halsall, D.J.; Zoller, H.; Cox, T.M. Clinical presentation and molecular pathophysiology of autosomal dominant hemochromatosis caused by a novel ferroportin mutation. Hepatology 2010, 51, 788–795. [Google Scholar] [CrossRef] [PubMed]
- Mayr, R.; Griffiths, W.J.; Hermann, M.; McFarlane, I.; Halsall, D.J.; Finkenstedt, A.; Douds, A.; Davies, S.E.; Janecke, A.R.; Vogel, W.; et al. Identification of mutations in SLC40A1 that affect ferroportin function and phenotype of human ferroportin iron overload. Gastroenterology 2011, 140, 2056–2063.e1. [Google Scholar] [CrossRef]
- Praschberger, R.; Schranz, M.; Griffiths, W.J.; Baumgartner, N.; Hermann, M.; Lomas, D.J.; Pietrangelo, A.; Cox, T.M.; Vogel, W.; Zoller, H. Impact of D181V and A69T on the function of ferroportin as an iron export pump and hepcidin receptor. Biochimica et Biophysica Acta 2014, 1842, 1406–1412. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.F.; McDonald, C.J.; Ostini, L.; Iser, D.; Tuckfield, A.; Subramaniam, V.N. The dynamics of hepcidin-ferroportin internalization and consequences of a novel ferroportin disease mutation. Am. J. Hematol. 2017, 92, 1052–1061. [Google Scholar] [CrossRef] [Green Version]
- Lok, C.Y.; Merryweather-Clarke, A.T.; Viprakasit, V.; Chinthammitr, Y.; Srichairatanakool, S.; Limwongse, C.; Oleesky, D.; Robins, A.J.; Hudson, J.; Wai, P.; et al. Iron overload in the Asian community. Blood 2009, 114, 20–25. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Xu, A.; Li, Y.; Zhao, S.; Zhou, D.; Wu, L.; Zhang, B.; Zhao, X.; Wang, Y.; Wang, X.; et al. A novel SLC40A1 p.Y333H mutation with gain of function of ferroportin: A recurrent cause of haemochromatosis in China. Liver Int. Off. J. Int. Assoc. Study Liver 2019, 39, 1120–1127. [Google Scholar] [CrossRef] [PubMed]
- Bonaccorsi di Patti, M.C.; Polticelli, F.; Cece, G.; Cutone, A.; Felici, F.; Persichini, T.; Musci, G. A structural model of human ferroportin and of its iron binding site. FEBS J. 2014, 281, 2851–2860. [Google Scholar] [CrossRef]
- Yamakawa, N.; Oe, K.; Yukawa, N.; Murakami, K.; Nakashima, R.; Imura, Y.; Yoshifuji, H.; Ohmura, K.; Miura, Y.; Tomosugi, N.; et al. A Novel Phenotype of a Hereditary Hemochromatosis Type 4 with Ferroportin-1 Mutation, Presenting with Juvenile Cataracts. Intern. Med. 2016, 55, 2697–2701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bach, V.; Remacha, A.; Altes, A.; Barcelo, M.J.; Molina, M.A.; Baiget, M. Autosomal dominant hereditary hemochromatosis associated with two novel Ferroportin 1 mutations in Spain. Blood Cells Mol. Dis. 2006, 36, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Del-Castillo-Rueda, A.; Moreno-Carralero, M.I.; Cuadrado-Grande, N.; Alvarez-Sala-Walther, L.A.; Enriquez-de-Salamanca, R.; Mendez, M.; Moran-Jimenez, M.J. Mutations in the HFE, TFR2, and SLC40A1 genes in patients with hemochromatosis. Gene 2012, 508, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Sussman, N.L.; Lee, P.L.; Dries, A.M.; Schwartz, M.R.; Barton, J.C. Multi-organ iron overload in an African-American man with ALAS2 R452S and SLC40A1 R561G. Acta Haematol. 2008, 120, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Majore, S.; Bonaccorsi di Patti, M.C.; Valiante, M.; Polticelli, F.; Cortese, A.; Di Bartolomeo, S.; De Bernardo, C.; De Muro, M.; Faienza, F.; Radio, F.C.; et al. Characterization of three novel pathogenic SLC40A1 mutations and genotype/phenotype correlations in 7 Italian families with type 4 hereditary hemochromatosis. Biochim. Biophys. Acta. Mol. Basis Dis. 2018, 1864, 464–470. [Google Scholar] [CrossRef]
- Ferbo, L.; Manzini, P.M.; Badar, S.; Campostrini, N.; Ferrarini, A.; Delledonne, M.; Francisci, T.; Tassi, V.; Valfre, A.; Dall’omo, A.M.; et al. Detection of a rare mutation in the ferroportin gene through targeted next generation sequencing. Blood Transfus. 2016, 1–4. [Google Scholar]
- Pelucchi, S.; Mariani, R.; Salvioni, A.; Bonfadini, S.; Riva, A.; Bertola, F.; Trombini, P.; Piperno, A. Novel mutations of the ferroportin gene (SLC40A1): analysis of 56 consecutive patients with unexplained iron overload. Clin Genet 2008, 73, 171–178. [Google Scholar] [CrossRef]
- Wallace, D.F.; Clark, R.M.; Harley, H.A.; Subramaniam, V.N. Autosomal dominant iron overload due to a novel mutation of ferroportin1 associated with parenchymal iron loading and cirrhosis. J. Hepatol. 2004, 40, 710–713. [Google Scholar] [CrossRef]
- De Domenico, I.; Ward, D.M.; Nemeth, E.; Vaughn, M.B.; Musci, G.; Ganz, T.; Kaplan, J. The molecular basis of ferroportin-linked hemochromatosis. Proc. Natl. Acad. Sci. USA 2005, 102, 8955–8960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.B.; Yang, F.; Haile, D.J. Functional consequences of ferroportin 1 mutations. Blood Cells Mol. Dis. 2005, 35, 33–46. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.J.; Wallace, D.F.; Ostini, L.; Bell, S.J.; Demediuk, B.; Subramaniam, V.N. G80S-linked ferroportin disease: classical ferroportin disease in an Asian family and reclassification of the mutant as iron transport defective. J. Hepatol. 2011, 54, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Njajou, O.T.; de Jong, G.; Berghuis, B.; Vaessen, N.; Snijders, P.J.; Goossens, J.P.; Wilson, J.H.; Breuning, M.H.; Oostra, B.A.; Heutink, P.; et al. Dominant hemochromatosis due to N144H mutation of SLC11A3: Clinical and biological characteristics. Blood Cells Mol. Dis. 2002, 29, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Rosmorduc, O.; Wendum, D.; Arrive, L.; Elnaggar, A.; Ennibi, K.; Hannoun, L.; Charlotte, F.; Grange, J.D.; Poupon, R. Phenotypic expression of ferroportin disease in a family with the N144H mutation. Gastroenterol. Clin. Biol. 2008, 32, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Arden, K.E.; Wallace, D.F.; Dixon, J.L.; Summerville, L.; Searle, J.W.; Anderson, G.J.; Ramm, G.A.; Powell, L.W.; Subramaniam, V.N. A novel mutation in ferroportin1 is associated with haemochromatosis in a Solomon Islands patient. Gut 2003, 52, 1215–1217. [Google Scholar] [CrossRef] [PubMed]
- Hetet, G.; Devaux, I.; Soufir, N.; Grandchamp, B.; Beaumont, C. Molecular analyses of patients with hyperferritinemia and normal serum iron values reveal both L ferritin IRE and 3 new ferroportin (slc11A3) mutations. Blood 2003, 102, 1904–1910. [Google Scholar] [CrossRef] [Green Version]
- An, P.; Jiang, L.; Guan, Y.; Wang, H.; Wang, J.; Tian, Y.; Yang, W.; Shi, Y.; Xue, J.; Min, J.; et al. Identification of hereditary hemochromatosis pedigrees and a novel SLC40A1 mutation in Chinese population. Blood Cells Mol. Dis. 2017, 63, 34–36. [Google Scholar] [CrossRef]
- Wang, Y.; Du, Y.; Liu, G.; Guo, S.; Hou, B.; Jiang, X.; Han, B.; Chang, Y.; Nie, G. Identification of novel mutations in HFE, HFE2, TfR2, and SLC40A1 genes in Chinese patients affected by hereditary hemochromatosis. Int. J. Hematol. 2016, 105, 521–525. [Google Scholar] [CrossRef]
- Lee, P.L.; Gaasterland, T.; Barton, J.C. Mild iron overload in an African American man with SLC40A1 D270V. Acta Haematol. 2012, 128, 28–32. [Google Scholar] [CrossRef]
- Wallace, D.F.; Dixon, J.L.; Ramm, G.A.; Anderson, G.J.; Powell, L.W.; Subramaniam, V.N. A novel mutation in ferroportin implicated in iron overload. J. Hepatol. 2007, 46, 921–926. [Google Scholar] [CrossRef] [PubMed]
- McGregor, J.A.; Shayeghi, M.; Vulpe, C.D.; Anderson, G.J.; Pietrangelo, A.; Simpson, R.J.; McKie, A.T. Impaired iron transport activity of ferroportin 1 in hereditary iron overload. J. Membr. Biol. 2005, 206, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Akoum, R. Point mutations in ferroportin disease: genotype/phenotype correlation. In Point Mutation; Logie, C., Ed.; InTech: London, UK, 2012; pp. 285–300. [Google Scholar]
- Cazzola, M.; Cremonesi, L.; Papaioannou, M.; Soriani, N.; Kioumi, A.; Charalambidou, A.; Paroni, R.; Romtsou, K.; Levi, S.; Ferrari, M.; et al. Genetic hyperferritinaemia and reticuloendothelial iron overload associated with a three base pair deletion in the coding region of the ferroportin gene (SLC11A3). Br. J. Haematol. 2002, 119, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Galicia-Poblet, G.; Cid-Paris, E.; Lopez-Andres, N.; Losada-Pajares, A.; Jurado-Lopez, J.C.; Moreno-Carralero, M.I.; Moran-Jimenez, M.J. A Pediatric Case Report of Ferroportin Disease. J. Pediatr. Gastroenterol. Nutr. 2015, 63, e205–e207. [Google Scholar] [CrossRef]
- Ikuta, K.; Hatayama, M.; Addo, L.; Toki, Y.; Sasaki, K.; Tatsumi, Y.; Hattori, A.; Kato, A.; Kato, K.; Hayashi, H.; et al. Iron overload patients with unknown etiology from national survey in Japan. Int. J. Hematol. 2017, 105, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Lim, F.L.; Dooley, J.S.; Roques, A.W.; Grellier, L.; Dhillon, A.P.; Walker, A.P. Hepatic iron concentration, fibrosis and response to venesection associated with the A77D and V162del “loss of function” mutations in ferroportin disease. Blood Cells Mol. Dis. 2008, 40, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Papanikolaou, G.; Tzilianos, M.; Christakis, J.I.; Bogdanos, D.; Tsimirika, K.; MacFarlane, J.; Goldberg, Y.P.; Sakellaropoulos, N.; Ganz, T.; Nemeth, E. Hepcidin in iron overload disorders. Blood 2005, 105, 4103–4105. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A.; Corradini, E.; Ferrara, F.; Vegetti, A.; De Jong, G.; Luca Abbati, G.; Paolo Arcuri, P.; Martinelli, S.; Cerofolini, E. Magnetic resonance imaging to identify classic and nonclassic forms of ferroportin disease. Blood Cells Mol. Dis. 2006, 37, 192–196. [Google Scholar] [CrossRef]
- Roetto, A.; Merryweather-Clarke, A.T.; Daraio, F.; Livesey, K.; Pointon, J.J.; Barbabietola, G.; Piga, A.; Mackie, P.H.; Robson, K.J.; Camaschella, C. A valine deletion of ferroportin 1: A common mutation in hemochromastosis type 4. Blood 2002, 100, 733–734. [Google Scholar] [CrossRef]
- Speletas, M.; Kioumi, A.; Loules, G.; Hytiroglou, P.; Tsitouridis, J.; Christakis, J.; Germenis, A.E. Analysis of SLC40A1 gene at the mRNA level reveals rapidly the causative mutations in patients with hereditary hemochromatosis type IV. Blood Cells Mol. Dis. 2008, 40, 353–359. [Google Scholar] [CrossRef]
- Unal, S.; Piperno, A.; Gumruk, F. Iron chelation with deferasirox in a patient with de-novo ferroportin mutation. J. Trace Elem. Med. Biol. 2015, 30, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.F.; Browett, P.; Wong, P.; Kua, H.; Ameratunga, R.; Subramaniam, V.N. Identification of ferroportin disease in the Indian subcontinent. Gut 2005, 54, 567–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, D.F.; Pedersen, P.; Dixon, J.L.; Stephenson, P.; Searle, J.W.; Powell, L.W.; Subramaniam, V.N. Novel mutation in ferroportin1 is associated with autosomal dominant hemochromatosis. Blood 2002, 100, 692–694. [Google Scholar] [CrossRef] [PubMed]
- Zoller, H.; McFarlane, I.; Theurl, I.; Stadlmann, S.; Nemeth, E.; Oxley, D.; Ganz, T.; Halsall, D.J.; Cox, T.M.; Vogel, W. Primary iron overload with inappropriate hepcidin expression in V162del ferroportin disease. Hepatology 2005, 42, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Cremonesi, L.; Forni, G.L.; Soriani, N.; Lamagna, M.; Fermo, I.; Daraio, F.; Galli, A.; Pietra, D.; Malcovati, L.; Ferrari, M.; et al. Genetic and clinical heterogeneity of ferroportin disease. Br. J. Haematol. 2005, 131, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Jouanolle, A.M.; Douabin-Gicquel, V.; Halimi, C.; Loreal, O.; Fergelot, P.; Delacour, T.; de Lajarte-Thirouard, A.S.; Turlin, B.; Le Gall, J.Y.; Cadet, E.; et al. Novel mutation in ferroportin 1 gene is associated with autosomal dominant iron overload. J. Hepatol. 2003, 39, 286–289. [Google Scholar] [CrossRef]
- Cunat, S.; Giansily-Blaizot, M.; Bismuth, M.; Blanc, F.; Dereure, O.; Larrey, D.; Quellec, A.L.; Pouderoux, P.; Rose, C.; Raingeard, I.; et al. Global sequencing approach for characterizing the molecular background of hereditary iron disorders. Clin. Chem. 2007, 53, 2060–2069. [Google Scholar] [CrossRef]
- Moreno-Carralero, M.I.; Munoz-Munoz, J.A.; Cuadrado-Grande, N.; Lopez-Rodriguez, R.; Jose Hernandez-Alfaro, M.; del-Castillo-Rueda, A.; Enriquez-de-Salamanca, R.; Mendez, M.; Moran-Jimenez, M.J. A novel mutation in the SLC40A1 gene associated with reduced iron export in vitro. Am. J. Hematol. 2014, 89, 689–694. [Google Scholar] [CrossRef]
- Ka, C.; Guellec, J.; Perpermans, X.; Kannengiesser, C.; Ged, C.; Wuyts, W.; Cassiman, D.; de Ledinghen, V.; Varet, B.; de Kerguenec, C.; et al. The SLC40A1 R178Q mutation is a recurrent cause of hemochromatosis and is associated with a novel pathogenic mechanism. Haematologica 2018, 103, 1796–1805. [Google Scholar] [CrossRef] [Green Version]
- Speletas, M.; Kioumi, A.; Germenis, A.E. Reply to: “SLC40A1-R178G or R178Q and ferroportin disease? A call for vigilance in mutation reporting”. J. Hepatol. 2013, 59, 397. [Google Scholar] [CrossRef]
- Morris, T.J.; Litvinova, M.M.; Ralston, D.; Mattman, A.; Holmes, D.; Lockitch, G. A novel ferroportin mutation in a Canadian family with autosomal dominant hemochromatosis. Blood Cells Mol. Dis. 2005, 35, 309–314. [Google Scholar] [CrossRef] [PubMed]
- De Domenico, I.; McVey Ward, D.; Nemeth, E.; Ganz, T.; Corradini, E.; Ferrara, F.; Musci, G.; Pietrangelo, A.; Kaplan, J. Molecular and clinical correlates in iron overload associated with mutations in ferroportin. Haematologica 2006, 91, 1092–1095. [Google Scholar] [PubMed]
- Corradini, E.; Montosi, G.; Ferrara, F.; Caleffi, A.; Pignatti, E.; Barelli, S.; Garuti, C.; Pietrangelo, A. Lack of enterocyte iron accumulation in the ferroportin disease. Blood Cells Mol. Dis. 2005, 35, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Relvas, L.C.M.; Bento, M.C.; Ribeiro, L. Novel human pathological mutation: Gene Symbol: SLC40A1; Disease: Haemochromatosis, type IV. Hum. Genet. 2009, 125, 338. [Google Scholar] [PubMed]
- Subramaniam, V.N.; Wallace, D.F.; Dixon, J.L.; Fletcher, L.M.; Crawford, D.H. Ferroportin disease due to the A77D mutation in Australia. Gut 2005, 54, 1048–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mougiou, A.; Pietrangelo, A.; Caleffi, A.; Kourakli, A.; Karakantza, M.; Zoumbos, N. G80S-linked ferroportin disease: the first clinical description in a Greek family. Blood Cells Mol. Dis. 2008, 41, 138–139. [Google Scholar] [CrossRef] [PubMed]
- Wolff, F.; Bailly, B.; Gulbis, B.; Cotton, F. Monitoring of hepcidin levels in a patient with G80S-linked ferroportin disease undergoing iron depletion by phlebotomy. Clin. Chim. Acta 2014, 430, 20–21. [Google Scholar] [CrossRef]
- Girelli, D.; De Domenico, I.; Bozzini, C.; Campostrini, N.; Busti, F.; Castagna, A.; Soriani, N.; Cremonesi, L.; Ferrari, M.; Colombari, R.; et al. Clinical, pathological, and molecular correlates in ferroportin disease: A study of two novel mutations. J. Hepatol. 2008, 49, 664–671. [Google Scholar] [CrossRef] [Green Version]
- Hattori, A.; Miyajima, H.; Tomosugi, N.; Tatsumi, Y.; Hayashi, H.; Wakusawa, S. Clinicopathological study of Japanese patients with genetic iron overload syndromes. Pathol. Int. 2012, 62, 612–618. [Google Scholar] [CrossRef]
- Raszeja-Wyszomirska, J.; Caleffi, A.; Milkiewicz, P.; Pietrangelo, A. Ferroportin-related haemochromatosis associated with novel Y64H mutation of the SCL40A1 gene. Prz. Gastroenterol. 2014, 9, 307–309. [Google Scholar] [CrossRef]
- Muehlenberg, K.; Faltermeier, N.; Lohse, P.; Tannapfel, A.; Pech, O. [Family with marked hyperferritinemia as a result of hemochromatosis type 4 (ferroportin disease)]. Z. Gastroenterol. 2014, 52, 1075–1080. [Google Scholar] [PubMed]
- Saja, K.; Bignell, P.; Robson, K.; Provan, D. A novel missense mutation c.470 A>C (p.D157A) in the SLC40A1 gene as a cause of ferroportin disease in a family with hyperferritinaemia. Br. J. Haematol. 2010, 149, 914–916. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Morotomi, N.; Sohda, T.; Hayashi, H.; Yoshida, N.; Ochi, K.; Ohkura, I.; Karita, M.; Fujiwara, H.; Yamashita, H.; et al. A male patient with ferroportin disease B and a female patient with iron overload similar to ferroportin disease B. Clin. J. Gastroenterol. 2014, 7, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Caetano, G.; Relvas, L.; Bento, C.; Silveira, M.P.; Ribeiro, L. Atypical iron deficiency anemia—Association of two new mutations in ferroportin and TMPRSS6 genes. In Proceedings of the International BioIron Society Annual Meeting, Porto, Portugal, 7–11 June 2009. [Google Scholar]
- Del-Castillo-Rueda, A.; Moreno-Carralero, M.I.; Alvarez-Sala-Walther, L.A.; Cuadrado-Grande, N.; Enriquez-de-Salamanca, R.; Mendez, M.; Moran-Jimenez, M.J. Two novel mutations in the SLC40A1 and HFE genes implicated in iron overload in a Spanish man. Eur. J. Haematol. 2011, 86, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.R.; Yang, L.Q.; Chong, Y.T.; Jie, Y.S.; Wu, Y.K.; Yang, J.; Lin, G.L.; Li, X.H. Novel gain of function mutation in the SLC40A1 gene associated with hereditary haemochromatosis type 4. Intern. Med. J. 2015, 45, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.L.; Gelbart, T.; West, C.; Barton, J.C. SLC40A1 c.1402G-->a results in aberrant splicing, ferroportin truncation after glycine 330, and an autosomal dominant hemochromatosis phenotype. Acta Haematol. 2007, 118, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Koyama, C.; Wakusawa, S.; Hayashi, H.; Ueno, T.; Suzuki, R.; Yano, M.; Saito, H.; Okazaki, T. A Japanese family with ferroportin disease caused by a novel mutation of SLC40A1 gene: hyperferritinemia associated with a relatively low transferrin saturation of iron. Intern. Med. 2005, 44, 990–993. [Google Scholar] [CrossRef]
- Nekhai, S.; Xu, M.; Foster, A.; Kasvosve, I.; Diaz, S.; Machado, R.F.; Castro, O.L.; Kato, G.J.; Taylor, J.G.; Gordeuk, V.R. Reduced sensitivity of the ferroportin Q248H mutant to physiological concentrations of hepcidin. Haematologica 2013, 98, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A. The ferroportin disease. Blood Cells Mol. Dis. 2004, 32, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Corradini, E.; Ferrara, F.; Pollicino, T.; Vegetti, A.; Abbati, G.L.; Losi, L.; Raimondo, G.; Pietrangelo, A. Disease progression and liver cancer in the ferroportin disease. Gut 2007, 56, 1030–1032. [Google Scholar] [CrossRef] [Green Version]
- Devalia, V.; Carter, K.; Walker, A.P.; Perkins, S.J.; Worwood, M.; May, A.; Dooley, J.S. Autosomal dominant reticuloendothelial iron overload associated with a 3-base pair deletion in the ferroportin 1 gene (SLC11A3). Blood 2002, 100, 695–697. [Google Scholar] [CrossRef] [PubMed]
- Abboud, S.; Haile, D.J. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J. Biol. Chem. 2000, 275, 19906–19912. [Google Scholar] [CrossRef] [PubMed]
- McKie, A.T.; Marciani, P.; Rolfs, A.; Brennan, K.; Wehr, K.; Barrow, D.; Miret, S.; Bomford, A.; Peters, T.J.; Farzaneh, F.; et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol. Cell 2000, 5, 299–309. [Google Scholar] [CrossRef]
- Donovan, A.; Brownlie, A.; Zhou, Y.; Shepard, J.; Pratt, S.J.; Moynihan, J.; Paw, B.H.; Drejer, A.; Barut, B.; Zapata, A.; et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 2000, 403, 776–781. [Google Scholar] [CrossRef] [PubMed]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, C.; Oates, P.S. Ferroportin/IREG-1/MTP-1/SLC40A1 modulates the uptake of iron at the apical membrane of enterocytes. Gut 2004, 53, 44–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drakesmith, H.; Nemeth, E.; Ganz, T. Ironing out Ferroportin. Cell Metab. 2015, 22, 777–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, D.M.; Kaplan, J. Ferroportin-mediated iron transport: expression and regulation. Biochimica Biophysica Acta 2012, 1823, 1426–1433. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, R.; Kato, H.E.; Font, J.; Deshpande, C.N.; Wada, M.; Ito, K.; Ishitani, R.; Jormakka, M.; Nureki, O. Outward- and inward-facing structures of a putative bacterial transition-metal transporter with homology to ferroportin. Nat. Commun. 2015, 6, 8545. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A. The ferroportin disease: pathogenesis, diagnosis and treatment. Haematologica 2017. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.L.; Tran, L.; Winters, A.; Lee, K.J.; Plewa, C.; Foltz, I.; King, C.; Miranda, L.P.; Allen, J.; Beckman, H.; et al. Molecular mechanism of hepcidin-mediated ferroportin internalization requires ferroportin lysines, not tyrosines or JAK-STAT. Cell Metab. 2012, 15, 905–917. [Google Scholar] [CrossRef] [PubMed]
- De Domenico, I.; Lo, E.; Ward, D.M.; Kaplan, J. Hepcidin-induced internalization of ferroportin requires binding and cooperative interaction with Jak2. Proc. Natl. Acad. Sci. USA 2009, 106, 3800–3805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Tertre, M.; Ka, C.; Guellec, J.; Gourlaouen, I.; Ferec, C.; Callebaut, I.; Le Gac, G. Deciphering the molecular basis of ferroportin resistance to hepcidin: Structure/function analysis of rare SLC40A1 missense mutations found in suspected hemochromatosis type 4 patients. Transfusion Clinique et Biologique: Journal de la Societe Francaise de Transfusion Sanguine 2017, 24, 462–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.J.; Tan, C.C.; Preza, G.C.; Nemeth, E.; Ganz, T.; Craik, D.J. Understanding the structure/activity relationships of the iron regulatory peptide hepcidin. Chem. Biol. 2011, 18, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Qiao, B.; Sugianto, P.; Fung, E.; Del-Castillo-Rueda, A.; Moran-Jimenez, M.J.; Ganz, T.; Nemeth, E. Hepcidin-induced endocytosis of ferroportin is dependent on ferroportin ubiquitination. Cell Metab. 2012, 15, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA--a self-parameterizing force field. Proteins 2002, 47, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Vriend, G. WHAT IF: a molecular modeling and drug design program. J. Mol. Graph. 1990, 8, 52–56. [Google Scholar] [CrossRef]
- Altamura, S.; Kessler, R.; Grone, H.J.; Gretz, N.; Hentze, M.W.; Galy, B.; Muckenthaler, M.U. Resistance of ferroportin to hepcidin binding causes exocrine pancreatic failure and fatal iron overload. Cell Metab. 2014, 20, 359–367. [Google Scholar] [CrossRef]
- Folgueras, A.R.; de Lara, F.M.; Pendas, A.M.; Garabaya, C.; Rodriguez, F.; Astudillo, A.; Bernal, T.; Cabanillas, R.; Lopez-Otin, C.; Velasco, G. Membrane-bound serine protease matriptase-2 (Tmprss6) is an essential regulator of iron homeostasis. Blood 2008, 112, 2539–2545. [Google Scholar] [CrossRef] [Green Version]
- De Falco, L.; Sanchez, M.; Silvestri, L.; Kannengiesser, C.; Muckenthaler, M.U.; Iolascon, A.; Gouya, L.; Camaschella, C.; Beaumont, C. Iron refractory iron deficiency anemia. Haematologica 2013, 98, 845–853. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.J.; Ostini, L.; Bennett, N.; Subramaniam, N.; Hooper, J.; Velasco, G.; Wallace, D.F.; Subramaniam, V.N. Functional analysis of matriptase-2 mutations and domains: insights into the molecular basis of iron-refractory iron deficiency anemia. Am. J. Physiol. Cell Physiol. 2015, 308, C539–C547. [Google Scholar] [CrossRef] [PubMed]
- Donker, A.E.; Raymakers, R.A.; Vlasveld, L.T.; van Barneveld, T.; Terink, R.; Dors, N.; Brons, P.P.; Knoers, N.V.; Swinkels, D.W. Practice guidelines for the diagnosis and management of microcytic anemias due to genetic disorders of iron metabolism or heme synthesis. Blood 2014, 123, 3873–3886, quiz 4005. [Google Scholar] [CrossRef] [PubMed]
- Sabelli, M.; Montosi, G.; Garuti, C.; Caleffi, A.; Oliveto, S.; Biffo, S.; Pietrangelo, A. Human macrophage ferroportin biology and the basis for the ferroportin disease. Hepatology 2017, 65, 1512–1525. [Google Scholar] [CrossRef] [PubMed]
- Delaby, C.; Pilard, N.; Goncalves, A.S.; Beaumont, C.; Canonne-Hergaux, F. Presence of the iron exporter ferroportin at the plasma membrane of macrophages is enhanced by iron loading and down-regulated by hepcidin. Blood 2005, 106, 3979–3984. [Google Scholar] [CrossRef] [PubMed]
- Nairz, M.; Schroll, A.; Demetz, E.; Tancevski, I.; Theurl, I.; Weiss, G. ‘Ride on the ferrous wheel’--the cycle of iron in macrophages in health and disease. Immunobiology 2015, 220, 280–294. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.; Qu, A.; Anderson, E.R.; Matsubara, T.; Martin, A.; Gonzalez, F.J.; Shah, Y.M. Hypoxia-inducible factor-2alpha mediates the adaptive increase of intestinal ferroportin during iron deficiency in mice. Gastroenterology 2011, 140, 2044–2055. [Google Scholar] [CrossRef]
- Muckenthaler, M.U.; Galy, B.; Hentze, M.W. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu. Rev. Nutr. 2008, 28, 197–213. [Google Scholar] [CrossRef]
- Wilkinson, N.; Pantopoulos, K. The IRP/IRE system in vivo: insights from mouse models. Front. Pharmacol. 2014, 5, 176. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.L.; Ghosh, M.C.; Rouault, T.A. The physiological functions of iron regulatory proteins in iron homeostasis—An update. Front. Pharmacol. 2014, 5, 124. [Google Scholar] [CrossRef]
- Lymboussaki, A.; Pignatti, E.; Montosi, G.; Garuti, C.; Haile, D.J.; Pietrangelo, A. The role of the iron responsive element in the control of ferroportin1/IREG1/MTP1 gene expression. J. Hepatol. 2003, 39, 710–715. [Google Scholar] [CrossRef]
- Zhang, D.L.; Hughes, R.M.; Ollivierre-Wilson, H.; Ghosh, M.C.; Rouault, T.A. A ferroportin transcript that lacks an iron-responsive element enables duodenal and erythroid precursor cells to evade translational repression. Cell Metab. 2009, 9, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Han, O.; Kim, E.Y. Colocalization of ferroportin-1 with hephaestin on the basolateral membrane of human intestinal absorptive cells. J. Cell. Biochem. 2007, 101, 1000–1010. [Google Scholar] [CrossRef] [PubMed]
- Musci, G.; Polticelli, F.; Bonaccorsi di Patti, M.C. Ceruloplasmin-ferroportin system of iron traffic in vertebrates. World J. Biol. Chem. 2014, 5, 204–215. [Google Scholar] [PubMed]
- Canonne-Hergaux, F.; Donovan, A.; Delaby, C.; Wang, H.J.; Gros, P. Comparative studies of duodenal and macrophage ferroportin proteins. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G156–G163. [Google Scholar] [CrossRef] [PubMed]
- Theurl, I.; Aigner, E.; Theurl, M.; Nairz, M.; Seifert, M.; Schroll, A.; Sonnweber, T.; Eberwein, L.; Witcher, D.R.; Murphy, A.T.; et al. Regulation of iron homeostasis in anemia of chronic disease and iron deficiency anemia: Diagnostic and therapeutic implications. Blood 2009, 113, 5277–5286. [Google Scholar] [CrossRef]
- Jacolot, S.; Ferec, C.; Mura, C. Iron responses in hepatic, intestinal and macrophage/monocyte cell lines under different culture conditions. Blood Cells Mol. Dis. 2008, 41, 100–108. [Google Scholar] [CrossRef]
- Mena, N.P.; Esparza, A.; Tapia, V.; Valdes, P.; Nunez, M.T. Hepcidin inhibits apical iron uptake in intestinal cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G192–G198. [Google Scholar] [CrossRef] [Green Version]
- Yamaji, S.; Sharp, P.; Ramesh, B.; Srai, S.K. Inhibition of iron transport across human intestinal epithelial cells by hepcidin. Blood 2004, 104, 2178–2180. [Google Scholar] [CrossRef]
- Chaston, T.; Chung, B.; Mascarenhas, M.; Marks, J.; Patel, B.; Srai, S.K.; Sharp, P. Evidence for differential effects of hepcidin in macrophages and intestinal epithelial cells. Gut 2008, 57, 374–382. [Google Scholar] [CrossRef]
- Bergamaschi, G.; Di Sabatino, A.; Pasini, A.; Ubezio, C.; Costanzo, F.; Grataroli, D.; Masotti, M.; Alvisi, C.; Corazza, G.R. Intestinal expression of genes implicated in iron absorption and their regulation by hepcidin. Clin. Nutr. 2016. [Google Scholar] [CrossRef] [PubMed]
- Guida, C.; Altamura, S.; Klein, F.A.; Galy, B.; Boutros, M.; Ulmer, A.J.; Hentze, M.W.; Muckenthaler, M.U. A novel inflammatory pathway mediating rapid hepcidin-independent hypoferremia. Blood 2015, 125, 2265–2275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enculescu, M.; Metzendorf, C.; Sparla, R.; Hahnel, M.; Bode, J.; Muckenthaler, M.U.; Legewie, S. Modelling Systemic Iron Regulation during Dietary Iron Overload and Acute Inflammation: Role of Hepcidin-Independent Mechanisms. PLoS Comput. Biol. 2017, 13, e1005322. [Google Scholar] [CrossRef] [PubMed]
- Deschemin, J.C.; Vaulont, S. Role of hepcidin in the setting of hypoferremia during acute inflammation. PLoS ONE 2013, 8, e61050. [Google Scholar] [CrossRef] [PubMed]
- Agoro, R.; Mura, C. Inflammation-induced up-regulation of hepcidin and down-regulation of ferroportin transcription are dependent on macrophage polarization. Blood Cells Mol. Dis. 2016, 61, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Mantovani, A. Orchestration of metabolism by macrophages. Cell Metab. 2012, 15, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Vlasveld, L.T.; Swinkels, D.W. Loss-of-function ferroportin disease: novel mechanistic insights and unanswered questions. Haematologica 2018, 103, 1753–1755. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Knovich, M.A.; Coffman, L.G.; Torti, F.M.; Torti, S.V. Serum ferritin: Past, present and future. Biochimica et Biophysica Acta 2010, 1800, 760–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NON-HFE Registry. Available online: http://non-hfe.com/ (accessed on 12 July 2019).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | n | Value (Median) | Range |
|---|---|---|---|
| Patients | 359 | ||
| Probands | 68 | ||
| Relatives from probands | 191 | ||
| Individual patients | 88 | ||
| Gender | 342 | ||
| Female | 131 | ||
| Male | 211 | ||
| Age (years) | 322 | 41 | 2–87 |
| Continent | 352 | ||
| Europe | 258 | ||
| North–South America | 37 | ||
| Asia | 44 | ||
| Australia/Oceania | 13 | ||
| Presenting clinical symptoms | 71 | ||
| Fatigue | 34 | ||
| Elevated liver enzymes / hepatomegaly | 30 | ||
| Joint complaints/arthralgia | 36 | ||
| Miscellaneous | 47 | ||
| Referral because of high ferritin level | 131 | ||
| Hematological Parameters | |||
| Hemoglobin (g/dL)# | |||
| Female | 60 | 13.1 | 7.5–16.3 |
| Male | 92 | 15.0 | 6.5–18.4 |
| MCV (fl) | |||
| Female | 38 | 91 | 77–107 |
| Male | 46 | 91 | 70–108 |
| Serum iron (µmol/L) | |||
| Female | 34 | 20.9 | 6.7–94.0 |
| Male | 56 | 26.3 | 6.2–86.0 |
| TSAT (%) | |||
| Female | 116 | 31.5 | 2.0–100.0 |
| Male | 180 | 38.0 | 7.0–104.0 |
| Ferritin (µg/L) | |||
| Female | 122 | 1026 | 4–8943 |
| Male | 201 | 1514 | 12–18695 |
| Protein | Iron Export | Hepcidin Effect on | TSAT | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Variant | Expression | Capacity | Expression | Export Capacity | Fate of Hepcidin | Patients (n) | Normal | High | Reference% |
| Tyr64Asn | PM | = | no | no | ↓↓ubiquitination/uptake | 8 | - | 8 | [11,30,31,32,33,34,35,36] |
| Cys326Ser | PM | = | no | no | ↓binding/no uptake | 7 | - | 6 | [30,32,34,37,38] |
| Cys326Tyr | PM | = | no | no | ↓/no uptake | 4 | - | 4 | [8,31,34,35,39,40,41,42,43,44] |
| Tyr333His | PM | = | no | no | 5 | - | 5 | [45] | |
| Tyr501Cys | PM | = | no | no | ↓binding | 7 | 2 | 5 | [30,33,46] |
| Asp504Asn | PM | = | no | no | ↓binding/uptake | 2 | - | 2 | [8,30] |
| His507Arg | PM | = | no | no | ↓↓ubiquitination | 4 | - | 4 | [30,41,47] |
| Sum: 7 | GOF | Hepcidin Resistant | 37 | 2 | 34 | ||||
| ILe180Thr | PM | = | yes | -- | normal uptake | 3 | 2 | 1 | [8,48,49] |
| Thr230Asn | PM | = | yes | -- | normal uptake | 1 | 1 | - | [8] |
| Met266Thr | PM | = | yes | -- | normal uptake | 1 | 1 | - | [8] |
| Leu345Phe | PM | = | yes | -- | normal uptake | 1 | 1 | - | [8] |
| Ile351Val | PM | = | yes | -- | ↑uptake | 1 | 1 | - | [8] |
| Pro443Leu | PM | = | yes | -- | ↑uptake | 1 | - | - | [8] |
| Arg561Gly | PM | = | yes | -- | normal uptake | 4 | 1 | 2 | [8,29,50] |
| Sum: 7 | GOF | Hepcidin Sensitive: Neutral | 12 | 7 | 3 | ||||
| Ala69Thr | PM | = | -- | ↓! | -- | 4 | 2 | 2 | [42,51,52] |
| Val72Phe | PM | = | yes | ↓! | ↓binding | 5 | 2 | 3 | [30,53] |
| Asn144Asp | PM | =# | conflicting | no# | ↓binding/uptake | 2 | - | 1 | [30,31,32,35,54] |
| Asn144His | PM | = | conflicting | yes | ↓uptake | 20 | 13 | 7 | [3,6,31,34,35,39,40,42,55,56,57,58,59] |
| Asn144Thr | PM | = | conflicting | yes# | ↓uptake | 1 | - | 1 | [6,31,32,34,60] |
| Gln182His | PM | = | conflicting | yes | normal uptake | 2 | 1 | - | [55,56,61] |
| Gly204Arg~ | PM | = | no$ | no$ | -- | 1 | - | 1 | [51] |
| Gly204Ser | PM | = | conflicting | no# | ↓ubiquitination | 14 | 4 | 10 | [9,11,15,30] |
| Ser209Leu | PM | = | no$ | no$ | -- | 7 | 7 | - | [62,63] |
| Asp270Val | PM | =# | yes | ↓! | ⇟ubiquitination | 2 | 1 | 1 | [14,30,64] |
| Arg296Gln~ | PM | = | no$ | ⇟ | -- | 1 | 1 | - | [51] |
| Ser338Arg | PM | = | yes | ↓! | ⇟ubiquitination | 1 | - | 1 | [6,30,65] |
| Arg371Gln | IC/PM | = | no | yes | -- | 1 | 1 | - | [11] |
| Sum: 13 | GOF | Uncertain/Conflicting/Unknown | 61 | 32 | 27 | ||||
| Asp84Glu | -- | ↓ | no | -- | -- | 1 | - | - | [43] |
| Val162del | IC/↓PM | ↓ | no | no | ↓↓uptake | 50 | 39 | 2 | [6,8,35,43,51,55,56,66,67,68,69,70,71,72,73,74,75,76,77,78,79] |
| Asp181Asn | PM | ↓ | no | no | -- | 6 | 6 | - | [51] |
| Asp181Val | ↓PM | ↓ | no | no | -- | 10 | 10 | - | [8,42,46,80] |
| Gly490Asp | IC/PM | ↓ | no | no | no uptake | 12 | 9 | 3 | [8,34,55,81] |
| Sum: 5 | LOF | Hepcidin Resistant | 79 | 64 | 5 | ||||
| Arg88Gly | IC/PM | ↓ | yes | -- | -- | 7 | 3 | 3 | [8,11,82] |
| Leu129Pro | PM | ↓ | yes | ⇟yes | -- | 5 | 3 | 2 | [83] |
| Arg178Gln | PM | ↓ | yes | -- | -- | 26 | 17 | - | [75,82,84,85] |
| Asn185Asp | PM | ↓ | yes | -- | -- | 19 | 4 | 6 | [9,11,86] |
| Sum: 4 | LOF | Hepcidin Sensitive | 57 | 27 | 11 | ||||
| Ala77Asp | IC/PM | ↓ | conflicting | not reliable@ | no uptake | 26 | 15 | 11 | [1,2,8,11,34,35,39,41,42,56,57,66,71,73,87,88,89,90] |
| Gly80Ser | ↓PM | ↓& | not reliable@ | not reliable@ | normal uptake& | 24 | 15 | 3 | [8,57,73,87,88,91,92] |
| Ile152Phe | PM | ↓ | ↓ | ↓ | -- | 2 | - | - | [39,93] |
| Asp157Gly | IC/PM | ↓ | conflicting | no& | -- | 4 | 3 | 1 | [8,34,55,56,61] |
| Asp157Tyr | ↓PM | ↓ | -- | -- | -- | 2 | 1 | 1 | [8,94] |
| Trp158Cys | IC | ↓ | not reliable@ | not reliable@ | -- | 4 | 4 | - | [41,62] |
| Trp158Leu | IC | ↓ | not reliable@ | not reliable@ | -- | 2 | 2 | - | [11] |
| Asn174Ile | IC/PM | ↓ | conflicting | not reliable@ | ↓uptake | 3 | 1 | 2 | [34,39,46,87,88] |
| Leu233Pro | IC/↓PM | ↓ | not reliable@ | -- | -- | 3 | 1 | 2 | [8,55,93] |
| Gly323Val | IC/PM | ↓ | conflicting | not reliable@ | ↓↓uptake | 1 | - | - | [34,55,56,61] |
| Arg489Lys | IC | ↓ | -- | -- | -- | 6 | 4 | - | [40] |
| Gly490Ser | ↓PM | ↓ | -- | -- | -- | 3 | 2 | 1 | [8,35,82] |
| Sum: 12 | LOF | Uncertain/Conflicting/Unknown | 80 | 48 | 21 | ||||
| Tyr64His | 1 | - | 1 | [95] | |||||
| Gly80Val | 2 | 2 | - | [80] | |||||
| Arg88Thr | 7 | 3 | 4 | [48] | |||||
| Asp157Ala | 5 | 2 | 3 | [96,97,98] | |||||
| Asp157Asn | 5 | 5 | - | [45,53] | |||||
| Ala232Asp | 2 | 2 | - | [99] | |||||
| Lys240Glu | 1 | - | 1 | [100] | |||||
| Gly267Asp | 1 | 1 | - | [80] | |||||
| Cys326Phe | 1 | - | 1 | [101] | |||||
| Gly468Ser | 3 | 3 | - | [102] | |||||
| Arg489Ser | 4 | 2 | 2 | [94,103] | |||||
| Val511Ile | 1 | - | 1 | [45] | |||||
| Sum: 12 | Non-Classified | 33 | 20 | 13 | |||||
| Sum: 60 | 359 | 200 | 114 | ||||||
| Phenotypic Features | Gain-of-Function (n = 110) | Loss-of-Function (n = 216) | p |
|---|---|---|---|
| Age (years) | n = 93 | n = 203 | 0.032& |
| median (range) | 46 (2–80) | 36 (6–87) | 0.055&& |
| Gender (n) | |||
| Female | 35 | 82 | |
| Male | 66 | 126 | ns |
| Hb (g/dL) | n = 39 | n = 104 | |
| median (range) | 14.4 (9.5–16.5) | 14.4 (10.1–18.4) | ns |
| Anemia | |||
| Yes | 4 | 14 | |
| No | 35 | 90 | ns |
| MCV (fl) | n = 29 | n = 50 | |
| median (range) | 93 (70–108) | 91 (73–98) | ns |
| Serum iron (µmol/L) | n = 45 | n = 51 | 0.0002& |
| median (range) | 36.0 (8.0–74.0) | 15.7 (6.2–94.0) | 0.00017&& |
| TSAT (%) | n = 105 | n = 174 | < 0.0001& |
| median (range) | 62 (2–104) | 32 (5–99) | < 0.0001&& |
| Ferritin (µg/L) | n = 100 | n = 208 | < 0.0001& |
| median (range) | 755 (4–15000) | 1595 (24–21665) | 0.0013&& |
| Iron Removed (g) | n = 10 | n = 31 | |
| median (range) | 10.2 (2–24.4) | 8.0 (1.6–80.0) | ns |
| Tolerance to Phlebotomy (n) | |||
| Good | 16 | 38 | |
| Poor | 2 | 13 | ns |
| HIC(µg/g) | n = 19 | n = 53 | |
| median (range) | 11718 (925–38665) | 10052 (307–58590) | ns |
| Grade of fibrosis | |||
| 0–2 | 14 | 20 | |
| 3–4 | 10 | 2 | 0.012 |
| Hepatic Iron distribution | |||
| Hepatic (predominant) | 18 | 7 | < 0.00001@ |
| Macrophagic (predominant) | 0 | 28 | |
| Mixed | 10 | 16 | < 0.00001@@ |
| ALT (IU/L) | n = 31 | n = 48 | 0.0026& |
| median (range) | 52.0 (14.0–304.0) | 29.5 (6.0–98.0) | 0.004&& |
| In silico Prediction | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Variant | Pheno-Type | Co-Segregation | VO/Controls | gnomAD | Functional | Molecular | Poly-phen2 | SIFT | Align-GVGD |
| Tyr64Asn | +++ | +++ | 0/100 | ++++ | ++ | ++ | 0 | 0 | |
| Tyr64His | +++& | ++ | ++ | 0 | 0 | ||||
| Ala69Thr | +++& | 0 | ++ | 0 | ++ | + | 0 | ||
| Val72Phe | + | + | +++ | 0 | ++ | 0 | 0 | ||
| Ala77Asp | +++ | ++,+++# | 0/100 | +++ | 0 | ++ | + | 0 | |
| Gly80Ser | ++ | 0/734 | +++ | + | ++ | + | 0 | ||
| Gly80Val | ++ | 0/100 | + | ++ | + | 0 | |||
| Asp84Glu | +++& | ++ | + | ++ | + | 0 | |||
| Arg88Gly | +++ | 0/734 | ++ | + | ++ | + | 0 | ||
| Arg88Thr | ++ | +++ | 0/60 | + | ++ | + | 0 | ||
| Leu129Pro | 0 | + | ++++ | 0 | ++ | + | +++ | ||
| Asn144Asp | +++ | +++ | + | ++ | 0 | 0 | |||
| Asn144His | 0 | +++ | 0/200 | +++ | + | + | 0 | 0 | |
| Asn144Thr | 0& | 0/100 | +++ | + | ++ | 0 | 0 | ||
| Ile152Phe | +++ | + | +++ | + | ++ | 0 | 0 | ||
| Asp157Ala | ++ | ++ | + | ++ | + | 0 | |||
| Asp157Asn | ++ | 0 | + | ++ | 0 | 0 | |||
| Asp157Gly | +++ | 0/80, 0/734 | +++ | + | + | 0 | 0 | ||
| Asp157Tyr | +++ | 0/734 | + | + | ++ | + | + | ||
| Trp158Cys | +++ | 0,+ | 0/50 | +++ | 0 | ++ | + | ++ | |
| Trp158Leu | +++ | +++ | 0 | + | 0 | 0 | |||
| Val162del | ++ | +,+++ | 0/100, 0/103, 0/734 | ++++ | 0 | @ | @ | @ | |
| Asn174Ile | +++ | +++ | + | ++ | + | +++ | |||
| Arg178Gln | +++ | 0-+++## | ++ | +++ | ++ | 0 | 0 | ||
| Ile180Thr | 0 | 0 | 0/100 | 7.076−5 | ++ | 0 | ++ | + | +++ |
| Asp181Asn | ++ | ++ | + | ++ | + | + | |||
| Asp181Val | ++ | +,++ | 0/100, 0/734 | ++ | + | ++ | + | +++ | |
| Gln182His | +++ | 0/80 | +++ | 0 | ++ | + | + | ||
| Asn185Asp | +++ | +,+++ | 0/50 | + | 0 | ++ | + | + | |
| Gly204Arg | +++& | ++ | 0 | ++ | + | +++ | |||
| Gly204Ser | + | +++ | 2/100 | 7.955−6 | +++ | 0 | ++ | 0 | 0 |
| Ser209Leu | 0 | ++ | 1.485−4 | +++ | 0 | 0 | 0 | + | |
| Thr230Asn | 0& | 0/734 | 1.096−4 | ++ | 0 | + | 0 | 0 | |
| Ala232Asp | 0 | 4.95−5 | 0 | + | 0 | + | |||
| Leu233Pro | +++ | 0 | 0/734 | ++ | 0 | ++ | + | +++ | |
| Lys240Glu | +++& | 0/50 | 0 | 0 | + | 0 | |||
| Met266Thr | 0& | 0/734 | ++ | 0 | 0 | + | 0 | ||
| Gly267Asp | ++& | 0/100 | 4.382−5 | 0 | + | 0 | 0 | ||
| Asp270Val | + | 4/100, 1/516 | 7.09−5 | +++ | 0 | 0 | 0 | 0 | |
| Arg296Gln | 0& | ++ | 0 | + | 0 | ++ | |||
| Gly323Val | +++& | 0/80 | +++ | 0 | ++ | + | +++ | ||
| Cys326Phe | +++& | 0 | ++ | ++ | 0 | + | |||
| Cys326Ser | +++ | +++ | ++++ | +++ | + | 0 | 0 | ||
| Cys326Tyr | +++ | 0/800 | ++++ | ++ | + | 0 | 0 | ||
| Tyr333His | +++ | 0/40 | ++ | + | ++ | + | +++ | ||
| Ser338Arg | +++& | +++ | 0 | 0 | 0 | 0 | |||
| Leu345Phe | 0& | 0/734 | ++ | 0 | ++ | 0 | 0 | ||
| Ile351Val | 0& | 0/734 | 2.125−5 | ++ | 0 | 0 | 0 | 0 | |
| Arg371Gln | 0& | 2.388−5 | +++ | 0 | 0 | 0 | 0 | ||
| Pro443Leu | 0& | 0/734 | 5.87−4 | ++ | 0 | 0 | 0 | 0 | |
| Gly468Ser | ++ | 0 | 4.005−6 | 0 | ++ | 0 | + | ||
| Arg489Lys | +++ | +++ | 0/50 | + | + | ++ | + | +++ | |
| Arg489Ser | + | 0/734 | + | ++ | + | +++ | |||
| Gly490Asp | +++ | 0 | 0/734 | ++++ | 0 | ++ | 0 | 0 | |
| Gly490Ser | +++ | 0/734 | + | 0 | ++ | 0 | 0 | ||
| Tyr501Cys | + | 0/200 | 3.184−5 | ++++ | +++ | + | 0 | + | |
| Asp504Asn | +++ | 0/734 | ++++ | +++ | ++ | 0 | 0 | ||
| His507Arg | +++ | + | 0/50 | ++++ | ++ | ++ | 0 | 0 | |
| Val511Ile | +++& | 0 | ++ | + | ++ | ||||
| Arg561Gly | + | 0/734 | 1.638−3 | ++ | 0 | 0 | 0 | 0 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vlasveld, L.T.; Janssen, R.; Bardou-Jacquet, E.; Venselaar, H.; Hamdi-Roze, H.; Drakesmith, H.; Swinkels, D.W. Twenty Years of Ferroportin Disease: A Review or An Update of Published Clinical, Biochemical, Molecular, and Functional Features. Pharmaceuticals 2019, 12, 132. https://0-doi-org.brum.beds.ac.uk/10.3390/ph12030132
Vlasveld LT, Janssen R, Bardou-Jacquet E, Venselaar H, Hamdi-Roze H, Drakesmith H, Swinkels DW. Twenty Years of Ferroportin Disease: A Review or An Update of Published Clinical, Biochemical, Molecular, and Functional Features. Pharmaceuticals. 2019; 12(3):132. https://0-doi-org.brum.beds.ac.uk/10.3390/ph12030132
Chicago/Turabian StyleVlasveld, L. Tom, Roel Janssen, Edouard Bardou-Jacquet, Hanka Venselaar, Houda Hamdi-Roze, Hal Drakesmith, and Dorine W. Swinkels. 2019. "Twenty Years of Ferroportin Disease: A Review or An Update of Published Clinical, Biochemical, Molecular, and Functional Features" Pharmaceuticals 12, no. 3: 132. https://0-doi-org.brum.beds.ac.uk/10.3390/ph12030132