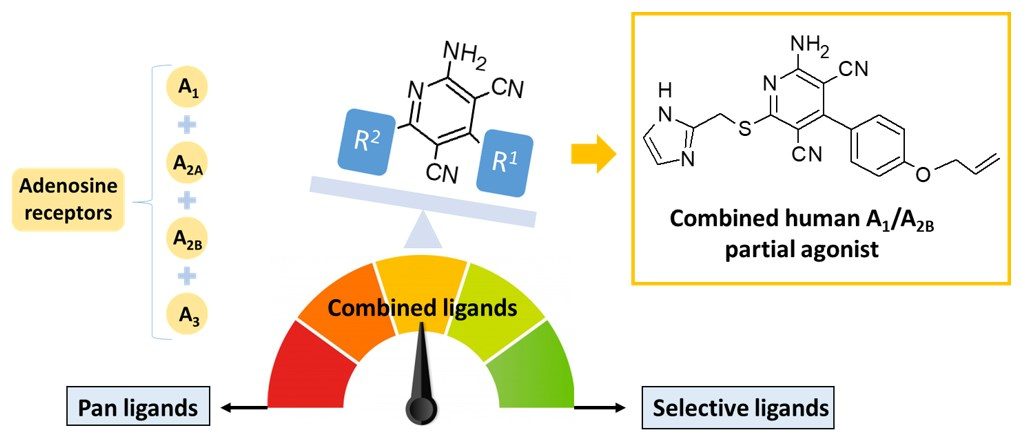
Amino-3,5-Dicyanopyridines Targeting the Adenosine Receptors. Ranging from Pan Ligands to Combined A1/A2B Partial Agonists

 , , , and
, , , and
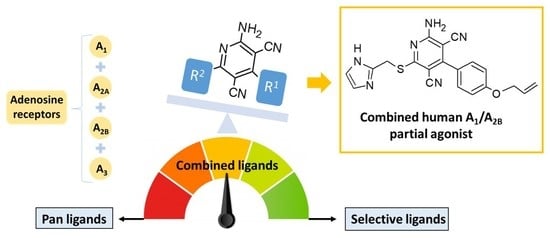
Abstract
:
1. Introduction
2. Results
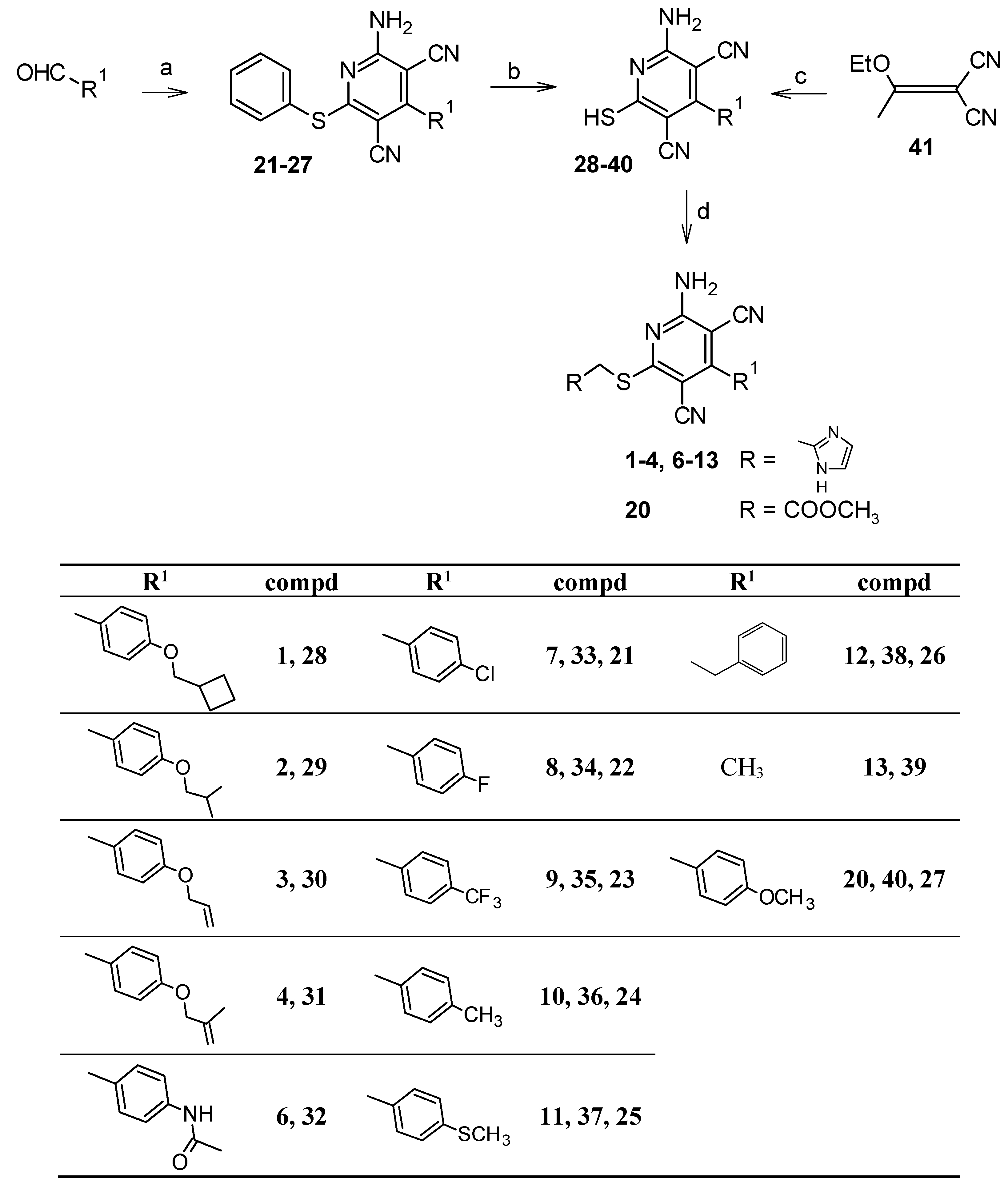
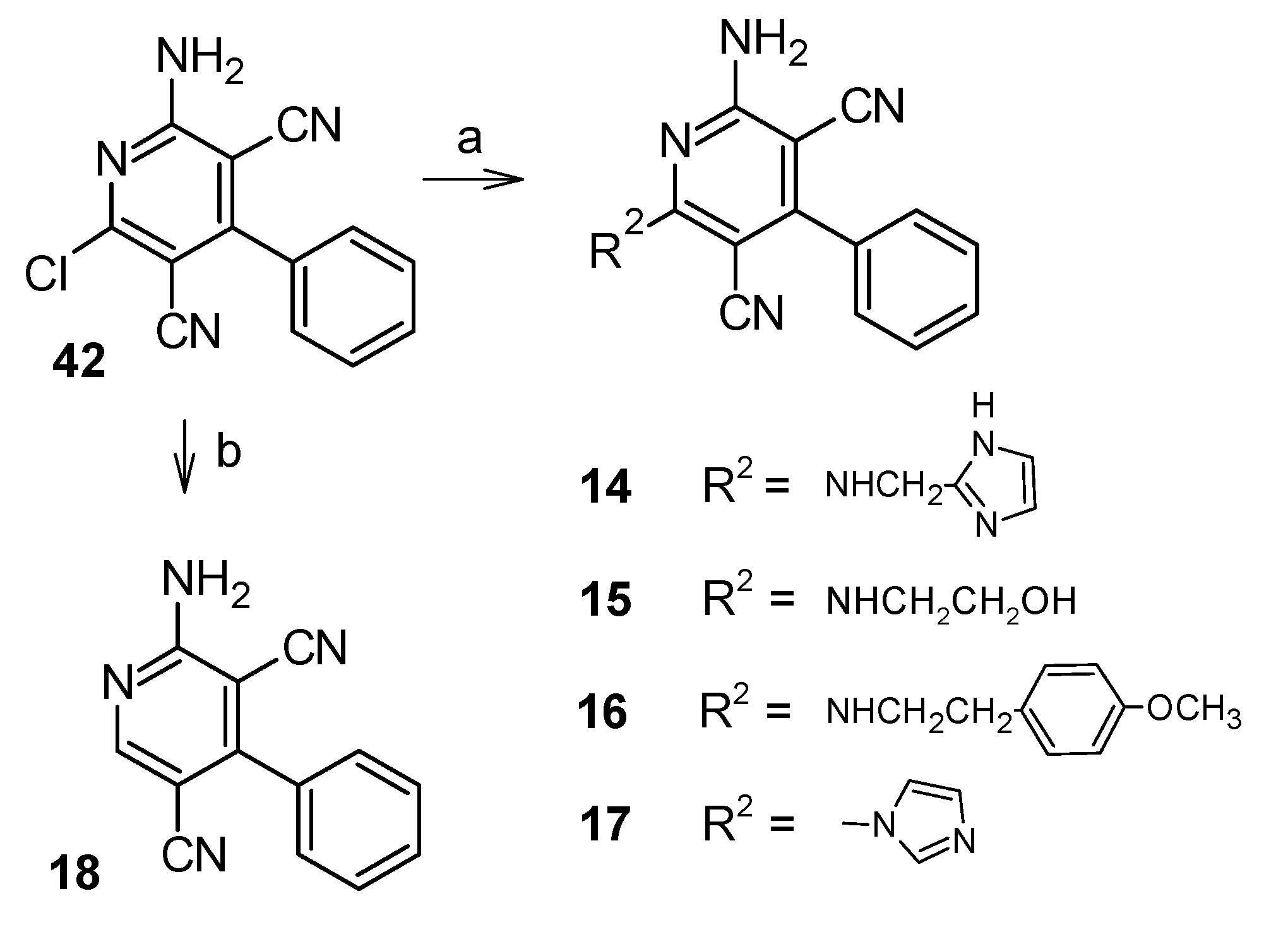
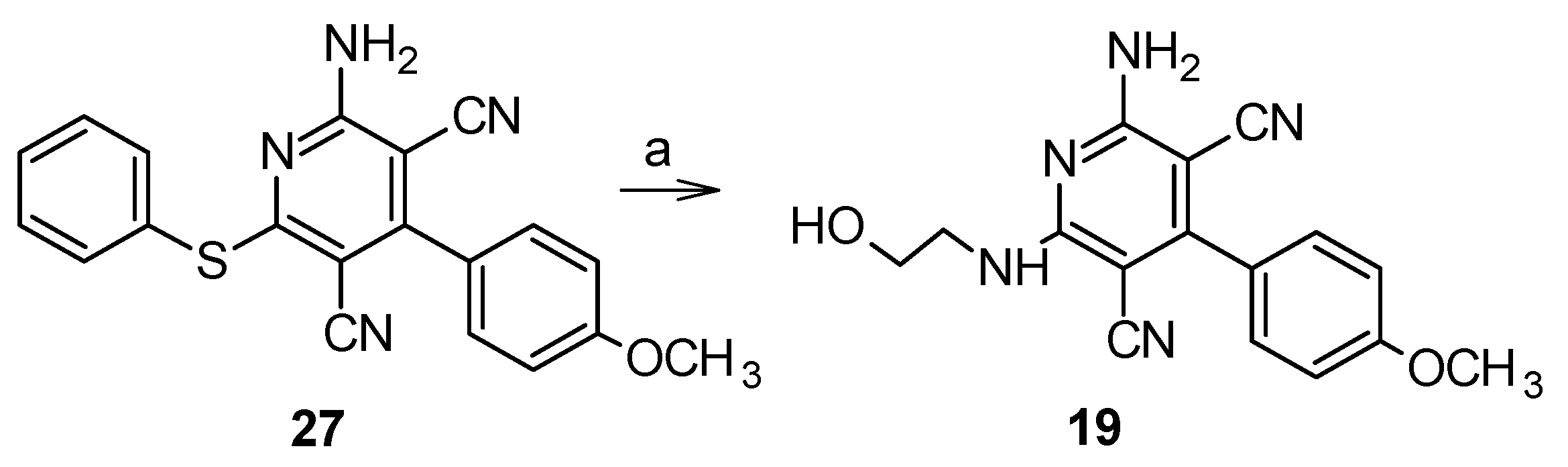
2.1. Chemistry
2.2. Pharmacological Assays
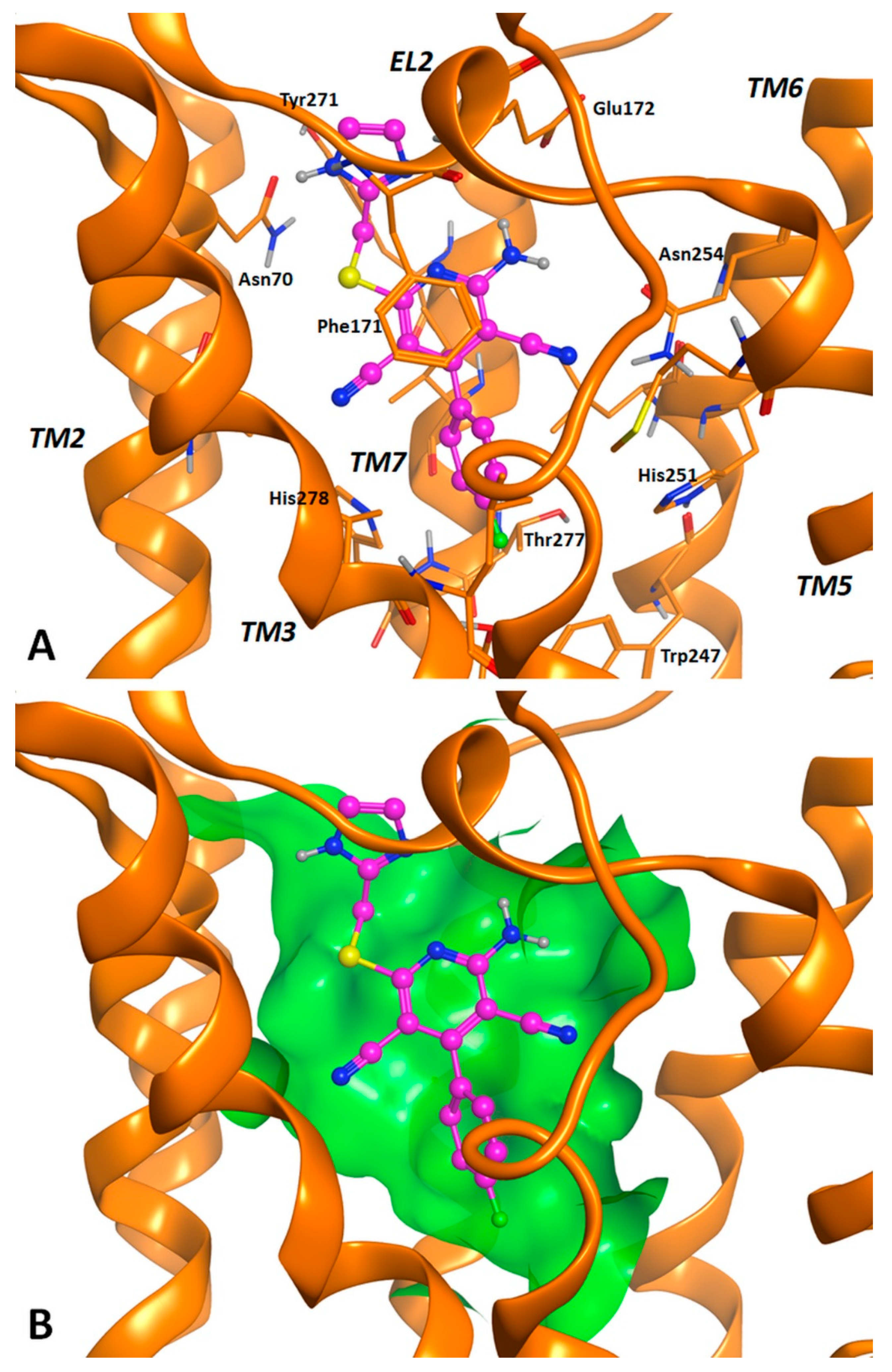
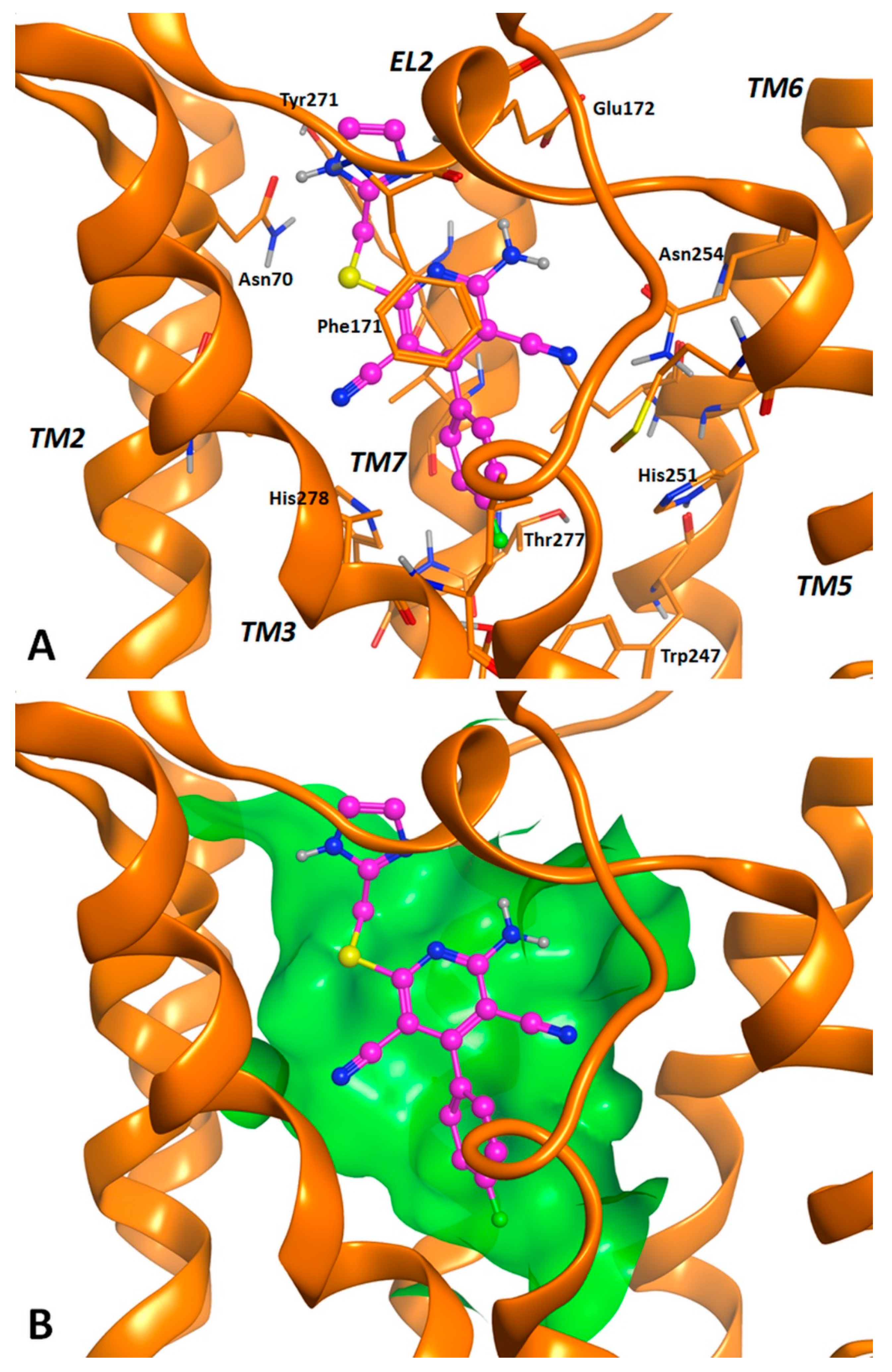
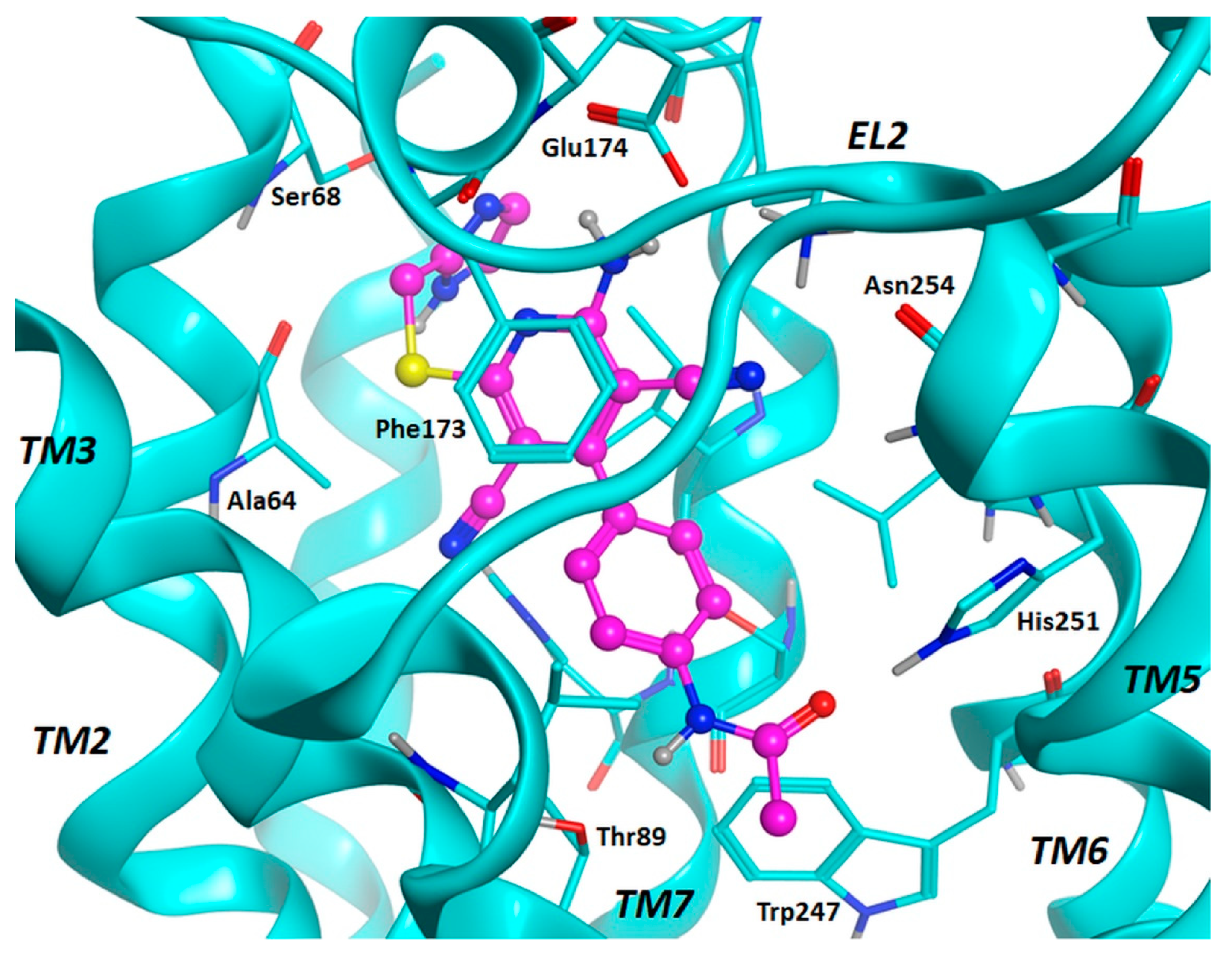
2.3. Molecular Docking Studies
3. Discussion
3.1. Structure–Activity Relationships
3.2. Molecular Docking Investigation and In Silico ADMET Prediction.
4. Conclusions
5. Materials and Methods
5.1. Chemistry
5.1.1. General Methods
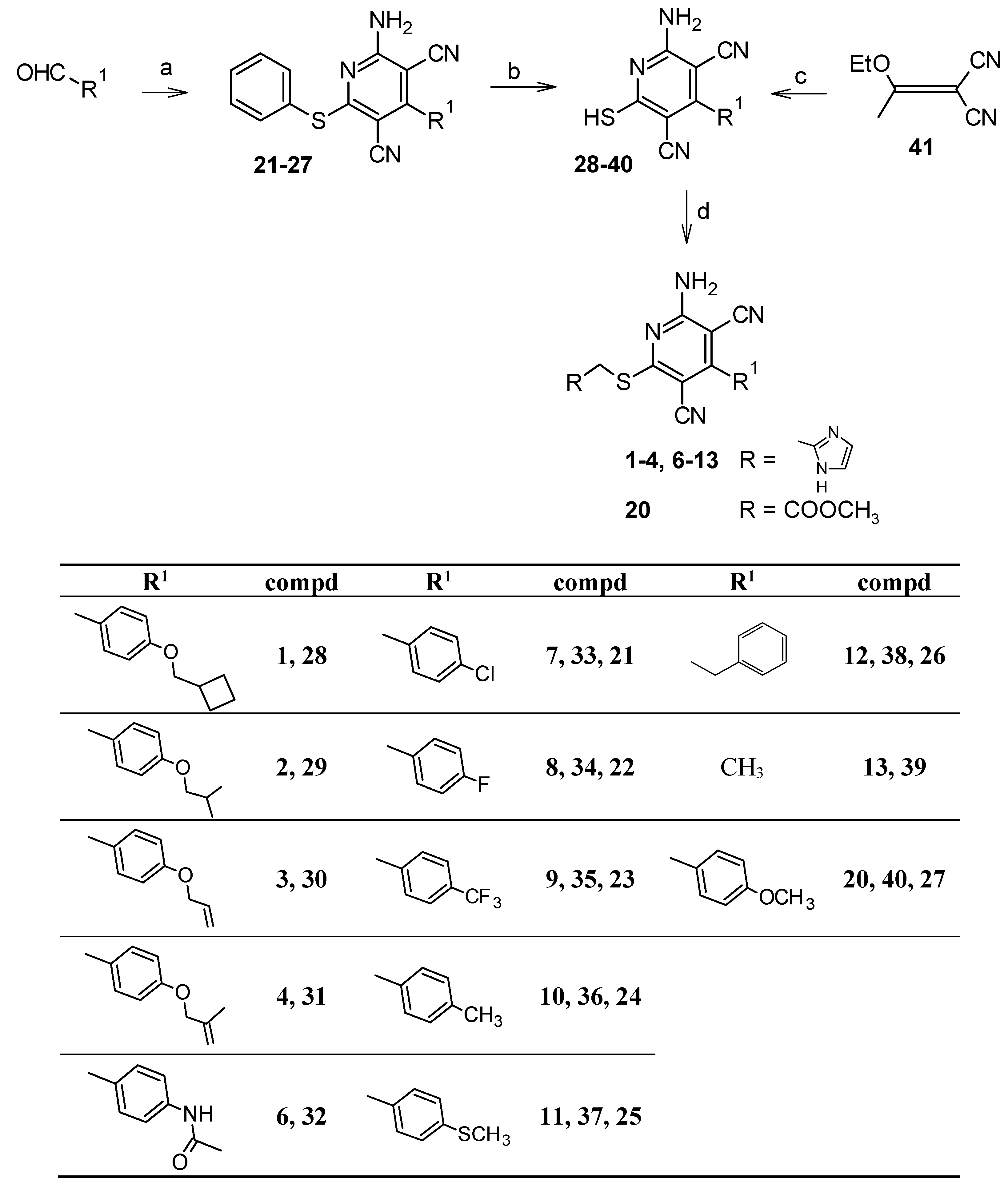
5.1.2. Synthesis of the target compounds 1–4, 6–13, and 20
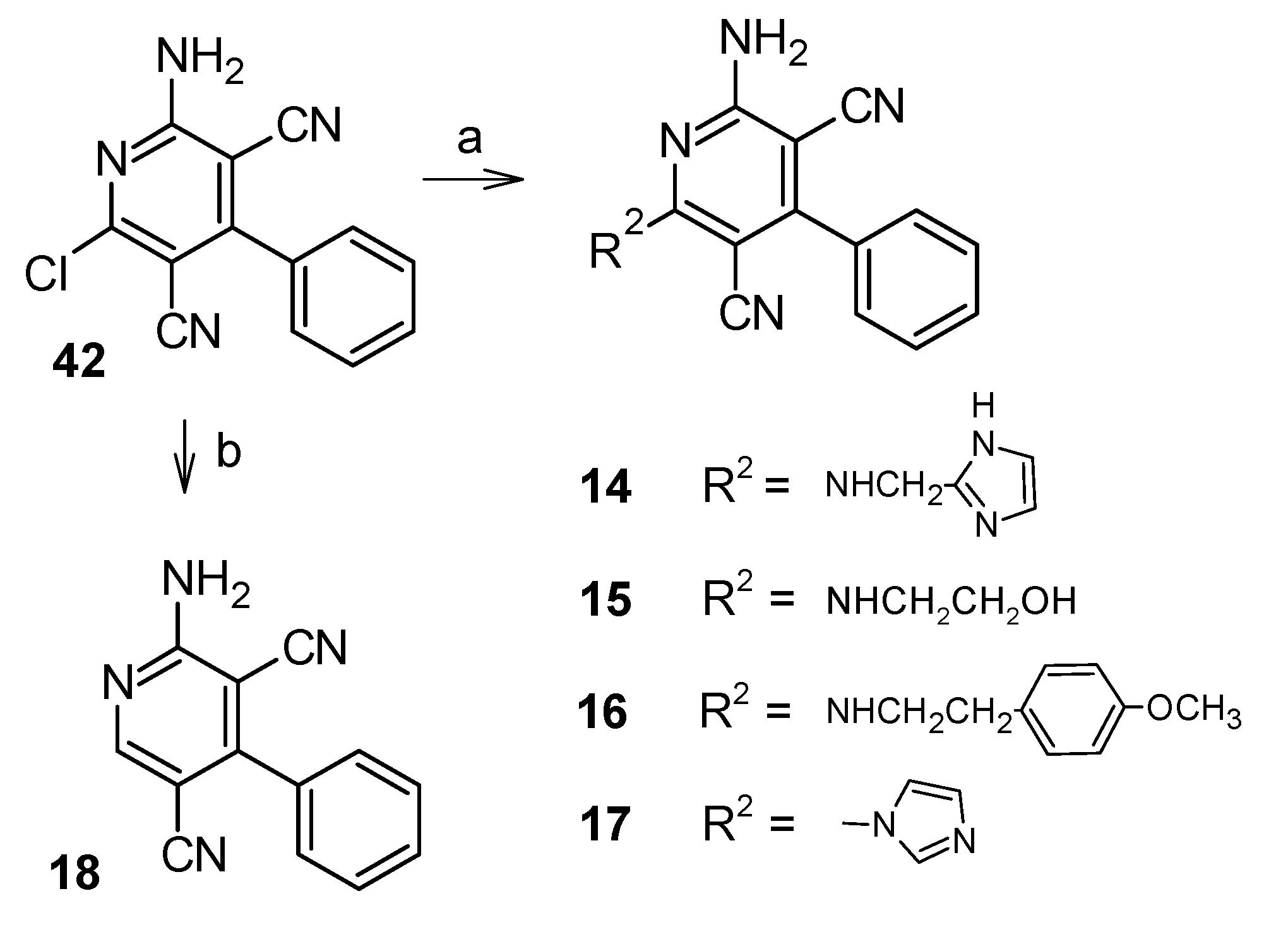
5.1.3. Synthesis of the Target Compounds 14–17
5.1.4. Synthesis of 2-amino-4-phenylpyridine-3,5-dicarbonitrile (18)
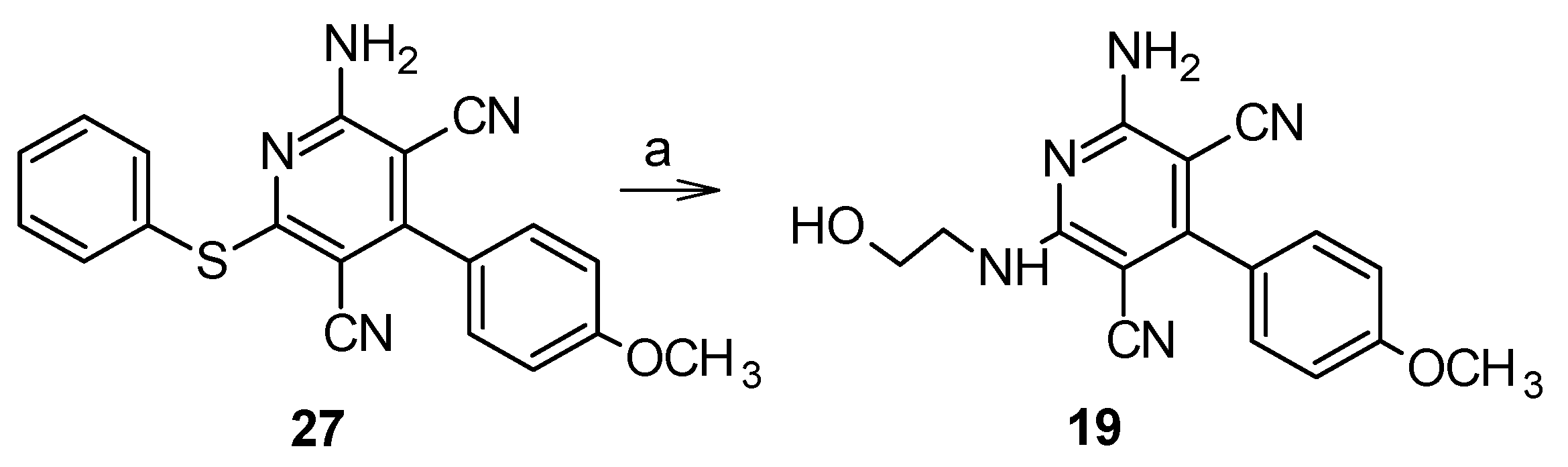
5.1.5. 2-Amino-6-[(2-hydroxyethyl)amino]-4-(4-methoxyphenyl)pyridine-3,5-dicarbonitrile (19)
5.1.6. Synthesis of 4-substituted-2-amino-6-(phenylsulfanyl)pyridine-3,5-dicarbonitriles 21–27 [18,24–26]
5.1.7. Synthesis of 2-amino-4-substituted-6-sulfanylpyridine-3,5-dicarbonitriles 33–40 [14,18,20,21]
5.1.8. Synthesis of 2-amino-4-methyl-6-sulfanylpyridine-3,5-dicarbonitrile (39) [22,23]
5.1.9. Synthesis of 2-amino-6-chloro-4-phenylpyridine-3,5-dicarbonitrile (42) [27]
5.2. Pharmacological Assays
5.2.1. Cell Culture and Membrane Preparation.
5.2.2. Competition Binding Experiments
5.2.3. Cyclic AMP Assays.
5.2.4. Data Analysis
5.3. Molecular Modelling
- Conformational Analysis of ligands. The algorithm generated conformations from a single 3D conformation by conducting a systematic search. In this way, all combinations of angles were created for each ligand.
- Placement. A collection of poses was generated from the pool of ligand conformations using Alpha Triangle placement method. Poses were generated by superposition of ligand atom triplets and triplet points in the receptor binding site. The receptor site points are alpha sphere centres which represent locations of tight packing. At each iteration, a random conformation was selected, a random triplet of ligand atoms and a random triplet of alpha sphere centres were used to determine the pose.
- Scoring. Poses generated by the placement methodology were scored using the Alpha HB scoring function, which combines a term measuring the geometric fit of the ligand to the binding site and a term measuring hydrogen bonding effects.
- Induced Fit. The generated docking conformations were subjected to energy minimization within the binding site and the protein sidechains are included in the refinement stage. In detail, the protein backbone is set as rigid while the side chains are not set to “free to move” but are set to “tethered”, where an atom tether is a distance restraint that restrains the distance not between two atoms but between an atom and a fixed point in space.
- Rescoring. Complexes generated by the Induced Fit methodology stage were scored using the Alpha HB scoring function. Gold tool was used with default efficiency settings through MOE interface, by selecting GoldScore as scoring function [31].
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABMECA | N6-(4-Aminobenzyl)-N-methylcarboxamidoadenosine |
| AC | Adenylate Cyclase |
| ADMET | Absorption, Distribution, Metabolism, Excretion, Toxicity |
| Ado | Adenosine |
| AR | Adenosine Receptor |
| BSA | Bovine Serum Albumin |
| CCPA | 2-Chloro-N6-cyclopentyladenosine |
| CHO | Chinese Hamster Ovary |
| CPA | N6-Cyclopentyladenosine |
| DPCPX | 8-Cyclopentyl-1,3-dipropylxanthine |
| EL | Extracellular loop |
| EtOAc | Ethyl Acetate |
| DMF | Dimethylformamide |
| DMSO | Dimethylsulfoxide |
| EDTA | Ethylendiaminotetracetic Acid |
| HEPES | 4-(2-Hydroxyethyl)-1-piperazine-1-ethane sulfonic Acid |
| MOE | Molecular operating environment; |
| NECA | 5′-(N-Ethylcarboxamido)adenosine |
| rt | room temperature |
| SARs | Structure-Activity Relationships |
| THF | Tetrahydrofuran |
| TM | Transmembrane |
References
- Borea, P.A.; Gessi, S.; Merighi, S.; Varani, K. Adenosine as a multisignalling guardian angel in human diseases: When, where and how does it exert its protective effects? Trends Pharmacol. Sci. 2016, 37, 419–434. [Google Scholar] [CrossRef]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of adenosine receptors: The state of the art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef]
- Chen, J.F.; Eltzschig, H.K.; Fredholm, B.B. Adenosine receptors as drug targets—what are the challenges? Nature Rev. Drug Discov. 2013, 12, 265–286. [Google Scholar] [CrossRef]
- Müller, C.E.; Jacobson, K.A. Recent developments in adenosine receptor ligands ad their potential as novel drugs. Biochim. Biophys. Acta 2011, 1808, 1290–1308. [Google Scholar] [CrossRef] [PubMed]
- Dhalla, A.K.; Chisholm, J.W.; Reaven, G.M.; Belardinelli, L. A1 adenosine receptor: Role in diabetes and obesity. Handb. Exp. Pharmacol. 2009, 193, 271–295. [Google Scholar]
- Koupenova, M.; Ravid, K. Adenosine, adenosine receptors and their role in glucose homeostasis and lipid metabolism. J. Cell. Physiol. 2013, 228, 1703–1712. [Google Scholar] [CrossRef] [PubMed]
- Eisenstein, A.; Patterson, S.; Ravid, K. The many faces of the A2B adenosine receptor in cardiovascular and metabolic diseases. J. Cell. Physiol. 2015, 230, 2891–2897. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, M.; Zieleniak, A.; Wozniak, L.A. New insight into A1 adenosine receptors in diabetes treatment. Curr. Pharm. Design 2010, 16, 4237–4242. [Google Scholar] [CrossRef] [PubMed]
- Gessi, S.; Merighi, S.; Fazzi, D.; Stefanelli, A.; Varani, K.; Borea, P.A. Adenosine receptor targeting in health and disease. Expert Opin. Investig. Drug. 2011, 20, 1591–1609. [Google Scholar] [CrossRef] [PubMed]
- Johansson, S.M.; Lindgren, E.; Yang, J.N.; Herling, A.W.; Fredholm, B.B. Adenosine A1 receptors regulate lipolysis and lipogenesis in mouse adipose tissue-interactions with insulin. Eur. J. Pharmacol. 2008, 597, 92–101. [Google Scholar] [CrossRef]
- Rosentreter, U.; Henning, R.; Bauser, M.; Krämer, T.; Vaupel, A.; Hübsch, W.; Dembowsky, K.; Salcher-Schraufstätter, O.; Stasch, J.P.; Krahn, T.; et al. Substituted 2-Thio-3,5-Dicyano-4-Aryl-6-Aminopyridines and the use Thereof as Adenosine Receptor Ligands. WO/2001/025210. 2001. Available online: https://worldwide.espacenet.com/ (accessed on 15 October 2019).
- Betti, M.; Catarzi, D.; Varano, F.; Falsini, M.; Varani, K.; Vincenzi, F.; Dal Ben, D.; Lambertucci, C.; Colotta, V. The aminopyridine-3,5-dicarbonitrile core for the design of new non-nucleoside-like agonists of the human adenosine A2B receptor. Eur. J. Med. Chem. 2018, 150, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Fusco, I.; Cherchi, F.; Catarzi, D.; Colotta, V.; Varano, F.; Pedata, F.; Pugliese, A.M.; Coppi, E. Functional characterization of a novel adenosine A2B receptor agonist on short-term plasticity and synaptic inhibition during oxygen and glucose deprivation in the rat CA1 hippocampus. Brain Res. Bull. 2019, 151, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Beukers, M.W.; Chang, L.C.W.; von Frijtag Drabbe Kunzel, J.K.; Mulder-Krieger, T.; Spanjersberg, R.F.; Brussee, J.; IJzerman, A.P. New, non-adenosine, high-potency agonists for the human adenosine A2B receptor with an improved selectivity profile compared to the reference agonist N-ethylcarboxamidoadenosine. J. Med. Chem. 2004, 47, 3707–3709. [Google Scholar] [CrossRef] [PubMed]
- Betti, M.; Catarzi, D.; Varano, F.; Falsini, M.; Varani, K.; Vincenzi, F.; Pasquini, S.; di Cesare Mannelli, L.; Ghelardini, C.; Lucarini, E.; et al. Modifications on the amino-3,5-dicyanopyridine core to obtain multifaceted adenosine receptor ligands with antineuropathic activity. J. Med. Chem. 2019, 62, 6894–6912. [Google Scholar] [CrossRef] [PubMed]
- Baraldi, P.G.; Tabrizi, M.A.; Fruttarolo, F.; Romagnoli, R.; Preti, D. Recent improvement in the development of A2B adenosine receptor agonists. Purinergic Signal. 2009, 5, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Krahn, T.; Krämer, T.; Rosentreter, U.; Downey, J.M.; Solenkova, N. Use of substituted 2-thio-3,5-dicyano-4-phenyl-6-aminopyridines for the treatment of reperfusion injury and reperfusion damage. WO 2006099958 A1. 2006. Available online: https://worldwide.espacenet.com/ (accessed on 15 October 2019).
- Chang, L.C.W.; von Frijtag Drabbe Künzel, J.K.; Mulder-Krieger, T.; Spanjersberg, R.F.; Roerink, S.F.; van den Hout, G.; Beukers, M.W.; Brussee, J.; IJzerman, A.P. A series of ligand displaying a remarkable agonistic-antagonistic profile at the adenosine A1 receptor. J. Med. Chem. 2005, 48, 2045–2053. [Google Scholar] [CrossRef]
- Meibom, D.; Albrecht-Küpper, B.; Diedrichs, N.; Hübsch, W.; Kast, R.; Krämer, T.; Krenz, U.; Lerchen, H.-J.; Mittendorf, J.; Nell, P.G.; et al. Neladenoson bialanate hydrochloride: A prodrug of a partial adenosine A1 receptor agonist for the chronic treatment of heart disease. ChemMedChem. 2017, 12, 728–737. [Google Scholar] [CrossRef]
- El-Torgoman, A.M.; El-Kousy, S.M.; El-Shahat, K.Z. Nitriles in heterocyclic synthesis: The reaction of 2-(thiocarbamoyl)cinnamonitriles with active methylene reagents. Z. Naturforsch. B Chem. Sci. 1987, 42, 107–111. [Google Scholar] [CrossRef]
- Alinaghizadeh, F.; Zahedifar, M.; Seifi, M.; Sheibani, H. Cascade synthesis of thieno[2,3-b]pyridines by using intramolecular cyclization reactions of 3-cyano-2-(organylmethylthio)pyridines. J. Braz. Chem. Soc. 2016, 27, 663–669. [Google Scholar] [CrossRef]
- Ibrahim, D.A.; Ismail, N.S.M. Design, synthesis and biological study of novel pyrido[2,3-d]pyrimidine as anti-proliferative CDK2 inhibitors. Eur. J. Med. Chem. 2011, 46, 5825–5832. [Google Scholar] [CrossRef]
- Geies, A.A.; Kamal El-Dean, A.M.; Abdel Monem, M.I. Reinvestigation of the reaction of arylidenemalonitriles with cyanothioacetamide: New approach for the synthesis of pyridine derivatives. Z. Naturforsch. B. 1992, 47, 1438–1440. [Google Scholar] [CrossRef]
- Kambe, S.; Saito, K.; Sakurai, A.; Midorikawa, H. Synthetic studies using α,β-unsaturated nitriles: Facile synthesis of pyridine derivatives. Synthesis 1981, 7, 531–533. [Google Scholar] [CrossRef]
- Guo, K.; Thompson, M.J.; Reddy, T.R.K.; Mutter, R.; Chen, B. Mechanistic studies leading to a new procedure for rapid, microwave assisted generation of pyridine-3,5-dicarbonitrile libraries. Tetrahedron 2007, 63, 5300–5311. [Google Scholar] [CrossRef]
- Ranu, B.C.; Jana, R.; Sowmiah, S. An improved procedure for the three-component synthesis of highly substituted pyridines using ionic liquid. J. Org.Chem. 2007, 72, 3152–3154. [Google Scholar] [CrossRef]
- Murray, T.J.; Zimmerman, S.C.; Kolotuchin, S.V. Synthesis of heterocyclic compounds containing three contiguous hydrogen bonding sites in all possible arrangements. Tetrahedron 1995, 51, 635–648. [Google Scholar] [CrossRef]
- Draper-Joyce, C.J.; Khoshouei, M.; Thal, D.M.; Liang, Y.L.; Nguyen, A.T.N.; Furness, S.G.B.; Venugopal, H.; Baltos, J.A.; Plitzko, J.M.; Danev, R.; et al. Structure of the adenosine-bound human adenosine A1 receptor-Gi complex. Nature 2018, 558, 559–563. [Google Scholar] [CrossRef]
- Lebon, G.; Warne, T.; Edwards, P.C.; Bennett, K.; Langmead, C.J.; Leslie, A.G.; Tate, C.G. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 2011, 474, 521–525. [Google Scholar] [CrossRef]
- Molecular Operating Environment, C.C.G., Inc.: Montreal, QC, Canada, H3B 3X3.
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Rosentreter, U.; Krämer, T.; Shimada, M.; Hübsch, W.; Diedrichs, N.; Krahn, T.; Henninger, K.; Stasch, J.P. Substituted 2-thio-3,5-dicyano-4-phenyl-6-aminopyridines and their use as adenosine receptor-selective ligands. WO/2003/008384. 2003. Available online: https://worldwide.espacenet.com/ (accessed on 15 October 2019).
- Thimm, D.; Schiedel, A.C.; Sherbiny, F.F.; Hinz, S.; Hochheiser, K.; Bertarelli, D.C.G.; Maass, A.; Müller, C.E. Ligand-Specific Binding and Activation of the Human Adenosine A2B Receptor. Biochemistry 2013, 52, 726–740. [Google Scholar] [CrossRef]
- Dal Ben, D.; Buccioni, M.; Lambertucci, C.; Thomas, A.; Volpini, R. Simulation and comparative analysis of binding modes of nucleoside and non-nucleoside agonists at the A2B adenosine receptor. In Silico Pharmacol. 2013, 1, 24. [Google Scholar] [CrossRef]
- Dal Ben, D.; Buccioni, M.; Lambertucci, C.; Marucci, G.; Santinelli, C.; Spinaci, A.; Thomas, A.; Volpini, R. Simulation and Comparative Analysis of Different Binding Modes of Non-nucleoside Agonists at the A2A Adenosine Receptor. Mol. Inform. 2016, 35, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, D.; Gao, Z.G.; Moss, S.M.; Jacobson, K.A.; Carlsson, J. Molecular docking screening using agonist-bound GPCR structures: Probing the A2A adenosine receptor. J. Chem. Inf. Model. 2015, 55, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Wu, H.; Katritch, V.; Han, G.W.; Jacobson, K.A.; Gao, Z.G.; Cherezov, V.; Stevens, R.C. Structure of an agonist-bound human A2A adenosine receptor. Science 2011, 332, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Lebon, G.; Bennett, K.; Jazayeri, A.; Tate, C.G. Thermostabilisation of an agonist-bound conformation of the human adenosine A2A receptor. J. Mol. Biol. 2011, 409, 298–310. [Google Scholar] [CrossRef]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Pereira, A.S.P.; den Haan, H.; Peña-García, J.; Moreno, M.M.; Pérez-Sánchez, H.; Apostolides, Z. Exploring african medicinal plants for potential anti-diabetic compounds with the DIA-DB inverse virtual screening web server. Molecules 2019, 24, 2002. [Google Scholar] [CrossRef]
- Vincenzi, F.; Targa, M.; Romagnoli, R.; Merighi, S.; Gessi, S.; Baraldi, P.G.; Borea, P.A.; Varani, K. TRR469, a potent A1 adenosine receptor allosteric modulator, exhibits anti-nociceptive properties in acute and neuropathic pain models in mice. Neuropharmacology 2014, 81, 6–14. [Google Scholar] [CrossRef]
- Varani, K.; Massara, A.; Vincenzi, F.; Tosi, A.; Padovan, M.; Trotta, F.; Borea, P.A. Normalization of A2A and A3 adenosine receptor up-regulation in rheumatoid arthritis patients by treatment with anti-tumor necrosis factor alpha but not methotrexate. Arthritis Rheum. 2009, 60, 2880–2891. [Google Scholar] [CrossRef]
- Varani, K.; Merighi, S.; Gessi, S.; Klotz, K.-N.; Leung, E.; Baraldi, P.G.; Cacciari, B.; Romagnoli, R.; Spalluto, G.; Borea, P.A. [3H]MRE 3008F20: A novel antagonist radioligand for the pharmacological and biochemical characterization of human A3 adenosine receptors. Mol. Pharmacol. 2000, 57, 968–975. [Google Scholar]
- Ravani, A.; Vincenzi, F.; Bortoluzzi, A.; Padovan, M.; Pasquini, S.; Gessi, S.; Merighi, S.; Borea, P.A.; Govoni, M.; Varani, K. Role and function of A2A and A3 adenosine receptors in patients with ankylosing spondylitis, psoriatic arthritis and rheumatoid arthritis. Int. J. Mol. Sci. 2017, 18, 697. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| cAMP Assays | Binding Experiments a | |||||
|---|---|---|---|---|---|---|
| EC50 (nM) b Efficacy, c % | Ki (nM) or I% | |||||
| hA2B | hA1 d | hA2A e | hA3 f | |||
| R1 | R2 | |||||
| 1 |  |  | ˃1000 (12%) | 25% | 4% | 574 ± 46 |
| 2 |  |  | 227 ± 21 (36%) | 26% | 1% | 1% |
| 3 |  |  | 27 ± 3 (56%) | 16 ± 2 | 17% | 8% |
| 4 |  |  | ˃1000 (14%) | 77 ± 6 | 1% | 8% |
| 5g |  |  | 11.7 ± 1.2 (62%) | 8.21 ± 0.73 | 221 ± 19 | 85 ± 6 |
| 6 |  |  | 12 ± 1 (100%) | 83 ± 7 | 38 ± 3 | 48 ± 4 |
| 7 |  |  | 109 ± 12 (32%) | 30 ± 4 | 865 ± 68 | 850 ± 62 |
| 8 |  |  | 18 ± 2 (76%) | 0.78 ± 0.08 | 139 ± 12 | 410 ± 38 |
| 9 |  |  | ˃1000 (4%) | 419 ± 32 | 32% | 21% |
| 10 |  |  | 78 ± 6 (78%) | 5.32 ± 0.42 | 47 ± 4 | 212 ± 19 |
| 11 |  |  | 36 ± 3 (30%) | 8.32 ± 0.78 | 227 ± 21 | 388 ± 29 |
| 12 |  |  | 260 ± 24 (44%) | 31% | 8% | 1% |
| 13 | CH3 |  | ˃1000 (1%) | 74 ± 8 | 331 ± 32 | 510 ± 53 |
| 14 |  |  | ˃1000 (1%) | 140 ± 12 | 115 ± 10 | 19% |
| 15 |  | NHCH2CH2OH | ˃1000 (1%) | 943 ± 87 | 1% | 1% |
| 16 |  |  | ˃1000 (1%) | 1% | 1% | 1% |
| 17 |  |  | ˃1000 (1%) | 27% | 1% | 25% |
| 18 |  | H | ˃1000 (1%) | 15% | 16% | 5% |
| 19 |  | NHCH2CH2OH | ˃1000 (1%) | 14% | 2% | 15% |
| 20 |  | SCH2COOCH3 | ˃1000 (1%) | 987 ± 81 | 1% | 5% |
| P453 g |  |  | 9.52 ± 0.91 (70%) | 235 ± 24 | 764 ± 72 | 474 ± 45 |
| LUF5833 h |  |  | 19 ± 7 (81%) | 2.4 ± 1 | 28 ± 1 | 171 ± 109 |
| compd | Efficacy, b % | EC50 (nM) c |
|---|---|---|
| 3 | 39 ± 4 | 19.6 ± 1.7 |
| 8 | 34 ± 3 | 1.34 ± 0.11 |
| 11 | 52 ± 4 | 8.87 ± 0.64 |
| DPCPX | 100 ± 9 | 1.52 ± 0.12 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Catarzi, D.; Varano, F.; Varani, K.; Vincenzi, F.; Pasquini, S.; Dal Ben, D.; Volpini, R.; Colotta, V. Amino-3,5-Dicyanopyridines Targeting the Adenosine Receptors. Ranging from Pan Ligands to Combined A1/A2B Partial Agonists. Pharmaceuticals 2019, 12, 159. https://0-doi-org.brum.beds.ac.uk/10.3390/ph12040159
Catarzi D, Varano F, Varani K, Vincenzi F, Pasquini S, Dal Ben D, Volpini R, Colotta V. Amino-3,5-Dicyanopyridines Targeting the Adenosine Receptors. Ranging from Pan Ligands to Combined A1/A2B Partial Agonists. Pharmaceuticals. 2019; 12(4):159. https://0-doi-org.brum.beds.ac.uk/10.3390/ph12040159
Chicago/Turabian StyleCatarzi, Daniela, Flavia Varano, Katia Varani, Fabrizio Vincenzi, Silvia Pasquini, Diego Dal Ben, Rosaria Volpini, and Vittoria Colotta. 2019. "Amino-3,5-Dicyanopyridines Targeting the Adenosine Receptors. Ranging from Pan Ligands to Combined A1/A2B Partial Agonists" Pharmaceuticals 12, no. 4: 159. https://0-doi-org.brum.beds.ac.uk/10.3390/ph12040159