Chemical Probes for the Adenosine Receptors

Department of Chemical and Pharmaceutical Sciences, University of Trieste, Via Licio Giorgeri 1, 34127 Trieste, Italy
*
Author to whom correspondence should be addressed.
Pharmaceuticals 2019, 12(4), 168; https://0-doi-org.brum.beds.ac.uk/10.3390/ph12040168
Submission received: 26 September 2019
/
Revised: 4 November 2019
/
Accepted: 7 November 2019
/
Published: 12 November 2019
(This article belongs to the Special Issue Adenosine Receptors as Attractive Targets in Human Diseases)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Research on the adenosine receptors has been supported by the continuous discovery of new chemical probes characterized by more and more affinity and selectivity for the single adenosine receptor subtypes (A1, A2A, A2B and A3 adenosine receptors). Furthermore, the development of new techniques for the detection of G protein-coupled receptors (GPCR) requires new specific probes. In fact, if in the past radioligands were the most important GPCR probes for detection, compound screening and diagnostic purposes, nowadays, increasing importance is given to fluorescent and covalent ligands. In fact, advances in techniques such as fluorescence resonance energy transfer (FRET) and fluorescent polarization, as well as new applications in flow cytometry and different fluorescence-based microscopic techniques, are at the origin of the extensive research of new fluorescent ligands for these receptors. The resurgence of covalent ligands is due in part to a change in the common thinking in the medicinal chemistry community that a covalent drug is necessarily more toxic than a reversible one, and in part to the useful application of covalent ligands in GPCR structural biology. In this review, an updated collection of available chemical probes targeting adenosine receptors is reported.
1. Introduction
Since their discovery in the mid seventies [1,2], adenosine receptors (ARs) have attracted research interest for their implication in a wide range of physiological and pathological processes (i.e., asthma, ischemia, cancer, Parkinson’s disease, etc.) [3]. As a consequence, at the same time research commenced on specific receptor probes that are essential tools for receptor characterization [4,5]. ARs exist as four different subtypes: A1, A2A, A2B and A3 ARs [6,7]. Due to the advancement in techniques for detection and characterization of receptors, and in particular of G protein-coupled receptors (GPCRs) [8,9,10,11,12], the availability of suitable probes is a constant need. In particular, this review covers three specific chemical probe families for ARs: radioactive, covalent and fluorescent ligands. Radioactive ligands, properly called radioligands, are the oldest class of AR probes, and still represent the principal tool in drug discovery since their use in binding assays [7]. Recently, the broad interest in radioactive ligands is due to their development as radiotracers in positron emission tomography (PET), leading to new diagnostic possibilities [13,14]. On the other hand, covalent ligands for GPCRs, which were in the past used as tools to purify, isolate or pharmacologically characterize receptors, have recently attracted the interest of the scientific community for their ability to stabilize their target protein, increasing the probability of obtaining X-ray crystal structures [10]. This strategy was successfully applied for the A1 AR subtype [15,16]. Since A2B and A3 AR crystal structures are still lacking, it is easy to imagine that several works will focus on development of covalent ligands for these receptor subtypes in the near future. Finally, the last few years have been characterized by the application of a variety of fluorescence-based methods for GPCR structure biology and drug discovery [17]. These techniques involve the introduction of a fluorescent tag on a GPCR or on a GPCR ligand, leading to fluorescent ligands, which are discussed here [9,18,19,20]. The aim of this review is to give a panorama of the available chemical probes for the ARs to researchers working in this field or medicinal chemists working on ARs or other GPCR targets.
2. Radioligands and Radiotracers
It is well known, that radioligand probes are useful for studying both the distribution and functions of receptors. In this class of compounds, two families of derivatives should be considered: i) radioligands, generally tritiated or iodinated, for binding studies; ii) radioligands used for imaging, in general probes including isotopes such as 11C, 18F and 15O.
In the first class of compounds, in the last decades, several examples of radioligands for all AR subtypes, both agonists and antagonists, with different degrees of potency and selectivity have been reported and extensively reviewed [7,21,22,23,24]. Our purpose is to give a brief update of the work developed in this field in this review.
Considering labeled derivatives for binding studies only an agonist for A2B AR named [3H]-BAY60-6583 (1) was recently reported by the group of Prof. C.A. Müller (Figure 1) [25].
This partial agonist in its tritiated form (the position of tritium is not reported) failed to be a good probe for binding studies. This is probably due to its moderate affinity at the human A2B receptor and high level of non-specific binding. The only results obtained using this radioligand indicate that nucleoside and non-nucleoside agonists most probably bind the receptor in different conformations [25].
In contrast to the development of tritiated or 125I radioligands, in the last few years great efforts have been made in the field of radiotracers for imaging [14]. In particular, several examples of 11C or 18F derivatives for the different AR subtypes have been reported.
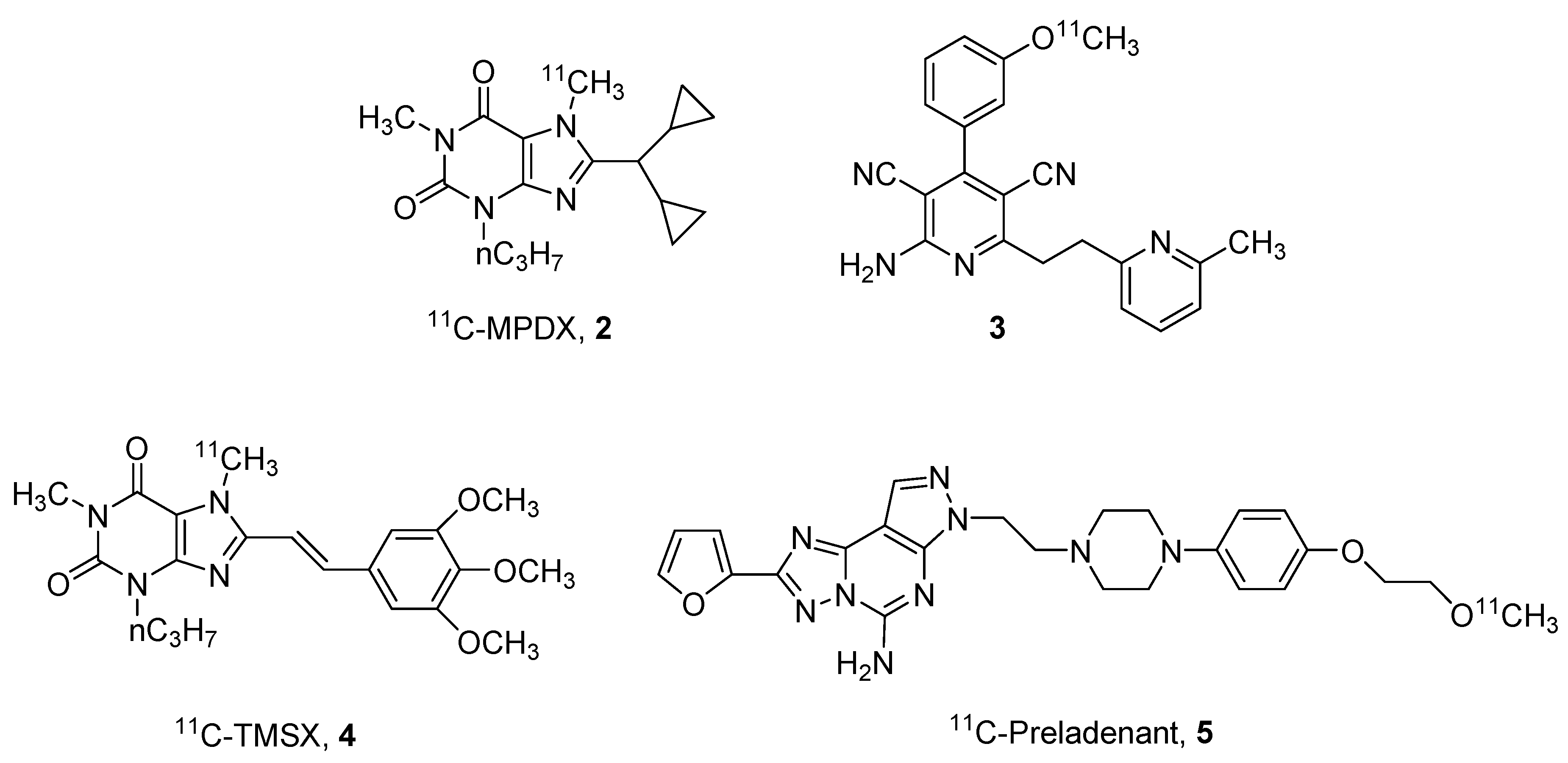
Regarding the 11C derivatives, some recent examples (compounds 2–5) have been reported in Figure 2, in particular, regarding the A1 and A2A ARs.
One of the most studied derivatives is xanthine derivative [1-methyl-11C]8-dicyclopropylmethyl-1-methyl-3-propylxanthine (11C-MPDX, 2). This compound has been utilized for various studies [26,27,28]. For example, Paul and coworkers performed in vivo binding studies in rats hypothesizing that agonists and antagonists may bind in different sites at the A1 receptor [26]. The same derivative was also utilized to evaluate A1 AR alterations in patients with chronic diffuse axonal injury [27] or to investigate the density of A1 ARs in patients with early-stage Parkinson’s disease [28].
Another interesting ligand for the A1 AR, is the pyridine derivative 3 reported by Guo et al. This compound could be considered the first brain blood barrier (BBB)-permeable A1 partial agonist with promising properties of detection of endogenous adenosine fluctuations [29].
Also in the field of A2A AR antagonists, some labeled compounds have been recently reported, in particular the styryl-xanthine derivative [7-methyl-11C]-(E)-8-(3,4,5-trimethoxystyryl)-1,3,7-trimethylxanthine (11C-TMSX, 4) that has been utilized for demonstrating that A2A receptors could be a future target to enhance brown adipose tissue (BAT) metabolism [30]. In addition, the non-xanthine labeled derivative 11C-preladenant (5) has been reported to be an interesting probe for investigating cerebral distribution of A2A ARs and other kinetic aspects such as its extensive distribution in the striatum [31,32].
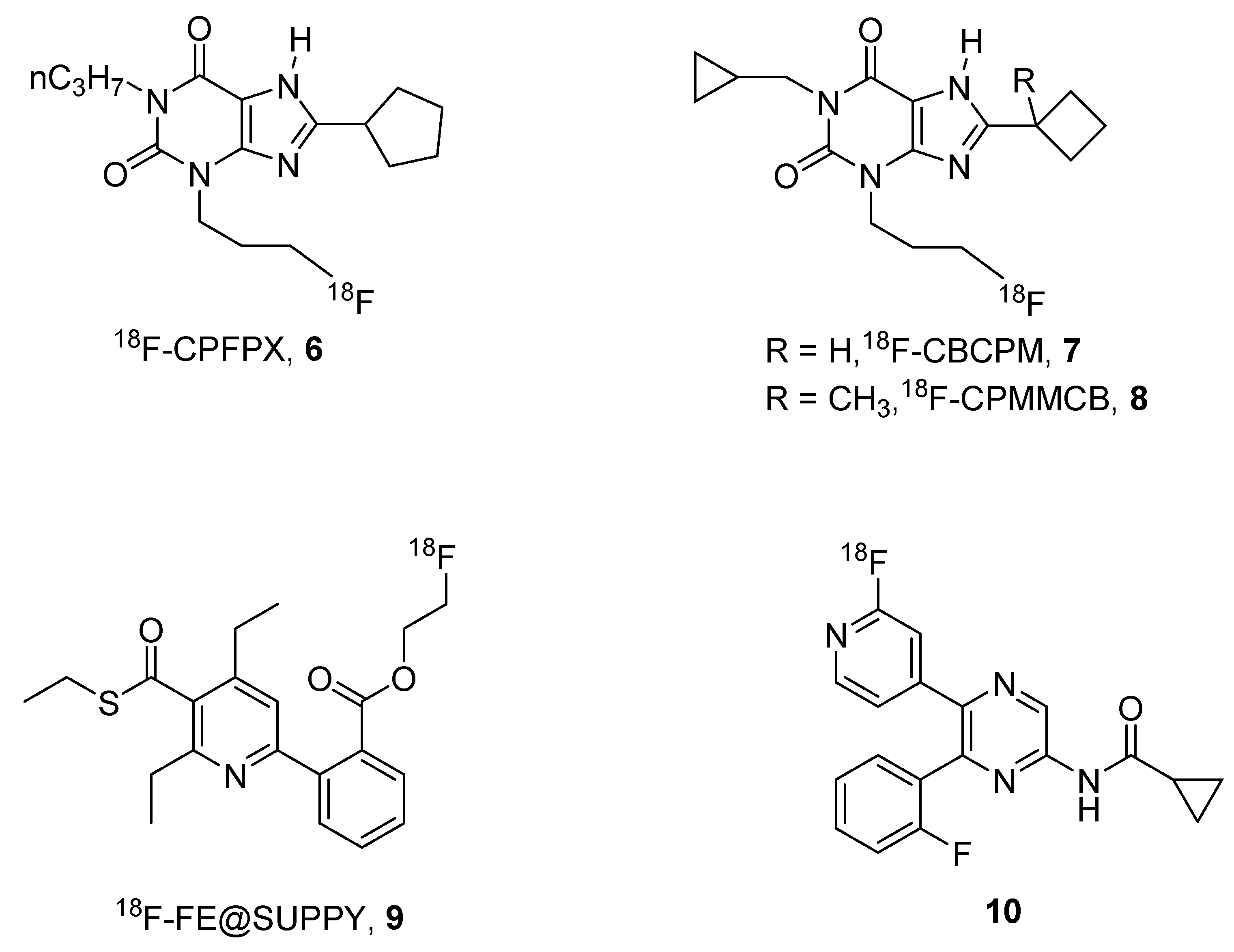
Nevertheless, one of the major problems of the use of 11C-labeled compounds is the short half-life of this isotope (20 min), this means that the compound has to be prepared and immediately utilized before its inactivation. For this reason, other isotopes are preferred to 11C such as 18F, which has a half-life of 109 min. In this field, some promising agents for A1, A2B and A3 receptors (6–10) have been reported (Figure 3).
One of the most used 18F-labeled compounds could be considered to be the xanthine derivative 18F-8-cyclopentyl-3-(3-fluoropropyl)-1-propylxanthine (18F-CPFPX, 6). This compound has been extensively used for developing suitable pharmacokinetic models for the quantification of cerebral A1 ARs [33].
It has also been utilized to visualize and quantify the in vivo occupancy of the human cerebral adenosine A1 receptor by caffeine [34], and demonstrated that its chronic use did not lead to persistent changes in functional availability of A1 ARs [35].
Taking into account the results obtained with these derivatives, Kreft and coworkers developed other 18F-CPFPX structurally-related derivatives named CBCPM (8-cyclobutyl-1-cyclopropymethyl-3-(3-fluoropropyl)xanthine, 7) and CPMMCB (1-cyclopropylmethyl-3-(3-fluoropropyl)-8-(1-methylcyclobutyl)xanthine, 8), respectively, which showed similar behavior to the reference compound in preliminary PET studies [36].
In addition, a promising labeled derivative, named 18F-FE@SUPPY (5-(2-fluoroethyl)2,4-diethyl-3-(ethylsulfanylcarbonyl)-6-phenylpyridine-5-carboxylate, 9), has been developed for the A3 AR, which has been shown to be a tracer for studying pharmacokinetic aspects and mapping A3 ARs in rats [37,38].
Very recently, a 18F-labeled form of a pyrazine A2B AR antagonists (10) has been reported by Lindemann et al. [39]. Preliminary studies on this compound, which showed binding affinity in the nanomolar range towards the human A2B AR, clearly indicated the potential of this derivative as a tracer for studying this receptor subtype and provided an important basis for the development of new tracers for A2B AR [39].
3. Covalent Ligands
It is well known that covalent ligands may be considered important pharmacological tools for the study of the receptors; in fact, the irreversible blockade of the receptors could reveal details of both structural binding interactions and the patho-physiological role of the examined receptor.
The strategy utilized for obtaining irreversible ligands is generally based on the introduction of chemoreactive moieties on the scaffold of well-known agonists and/or antagonists. The limiting step in preparing this kind of probe is the position on which the chemoreactive group should be introduced in order to obtain irreversible block of the receptor without losing both affinity and selectivity. For this reason, several structure–activity relationship studies (SAR) have been performed in order to optimize the structural requirements that are indispensable for obtaining irreversible probes that retain affinity and selectivity.
In the field of ARs, several examples of covalent agonists and antagonists have been reported in the last decades. We wish, for this reason, to briefly summarize the results obtained in this research area.
Regarding agonists for ARs, several examples of A1, A2A and A3 receptor subtypes are reported and depicted in Figure 4 and Figure 5.
The strategy to obtain irreversible agonists for ARs is based on the core modification of the natural nucleoside adenosine.
Concerning the A1 AR subtype, examples of covalent agents are reported and summarized in Figure 4 [40,41,42].
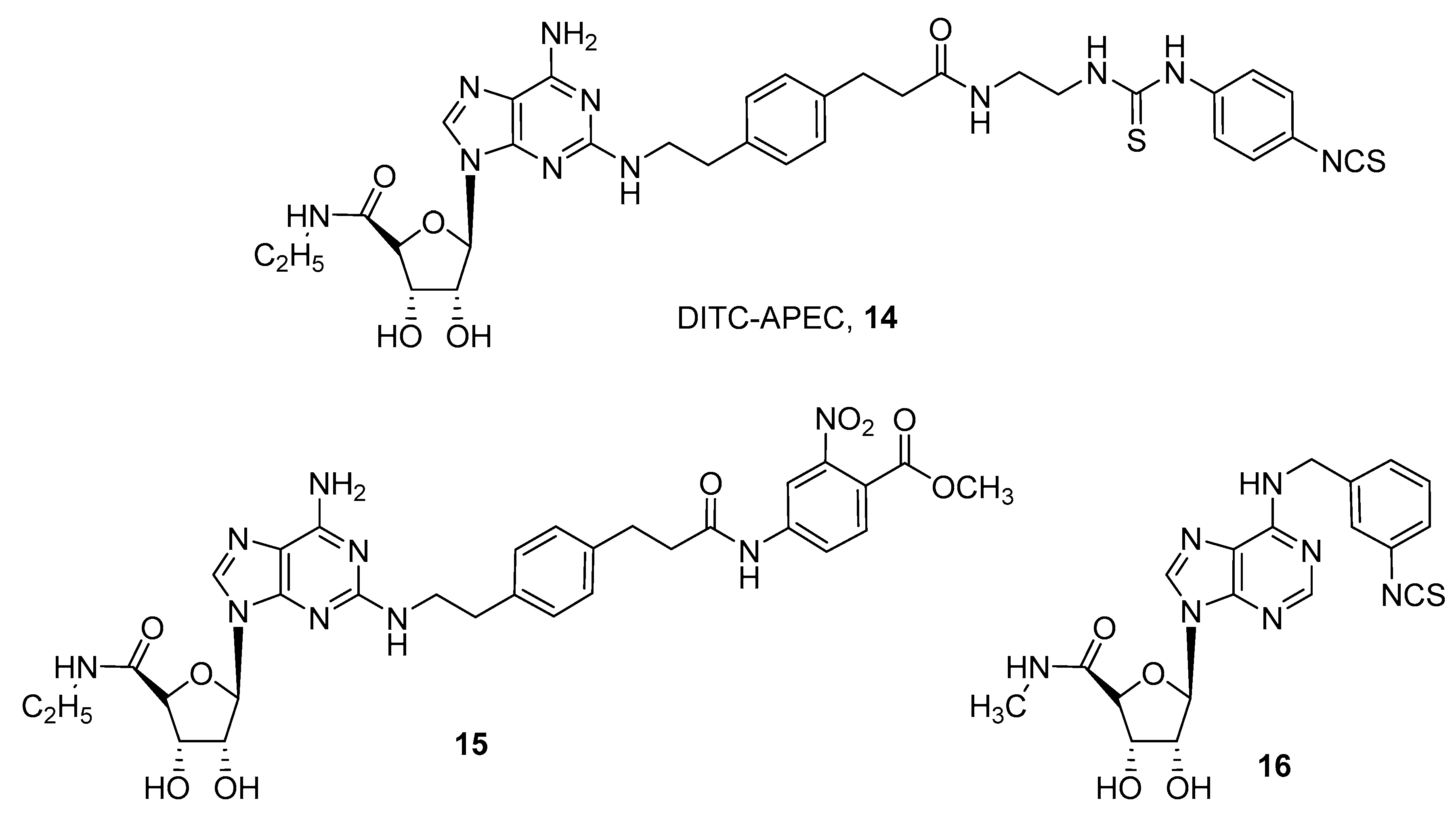
In particular the N6 substituted derivative of adenosine, named m-DITC-ADAC (4-isothiocyanatophenylaminothiocarbonyl, DITC; adenosine amine congener or N6-[4-[[[4-[[[(2-Aminoethyl)amino]carbonyl]methyl]-anilino]carbonyl]methyl]phenyl]adenosine, ADAC) (11), proved to be a good irreversible agonist for A1 AR. It was able to mimic ischemic preconditioning in rabbits [40] and also prolong the stimulus of the His bundle (SA) interval by 2.1 fold in guinea pig isolated hearts [41].
Another interesting covalent agonist, R-AHPIA (2-azido-N-(2-(4-hydroxyphenyl)-1-methylethyl)-adenosine, 12) was reported by Klotz and coworkers [42]. This ligand was also labeled with 125I and, after UV irradiation, it irreversibly binds to the rat receptor, which was isolated for the first time as a protein of about 35 KDa [42].
A quite extensive SAR study on the adenosine nucleus for obtaining other covalent ligands for human ARs was performed by Jorg and coworkers [16], who obtained a series of compounds of general formula 13. Nevertheless no extensive pharmacological studies for their characterization were reported [16].
Development of covalent A2A agonists was performed by an extensive SAR study on the CGS21680 (2-p-(2-carboxyethyl)phenethylamino-5′-N-ethylcarboxamidoadenosine) scaffold, by introduction of several chemoreactive groups on its carboxylic acid moiety (Figure 5) [24,43,44].
These studies led to the discovery of a compound named DITC-APEC (2-[(2-aminoethyl-aminocarbonylethyl)phenylethylamino]-5’-N-ethyl-carboxamidoadenosine, APEC) (14), which was able to induce concentration dependent coronary vasodilation that persisted for a long time, even after washout. This suggests that compound 14 irreversibly bound A2A AR in guinea pig coronary arteries [44].
A quite similar compound (15) was obtained by introducing a reactive ester on the CGS21680 nucleus as an acylating agent. This derivative was found to be an irreversible agonist for the human A2A AR, and docking studies suggested that two lysine residues in the second extracellular loop were involved in the covalent binding. In fact, site directed mutagenesis studies confirmed that lysine 153 reacts with the active ester moiety, giving the irreversible adduct [45].
For the rat A3 AR subtype, an irreversible agonist (16) was developed by introducing a benzyl-isothiocyanate group at the N6 position of 5’-(N-methylcarboxamido)adenosine, but, except for binding experiments confirming the irreversible properties of the compound, no significative pharmacological experiments were performed [46].
In addition, several irreversible antagonists for ARs have been reported. We could classify the obtained compounds in two major classes: i) xanthine derivatives (the natural antagonists for ARs), and ii) non-xanthine derivatives.
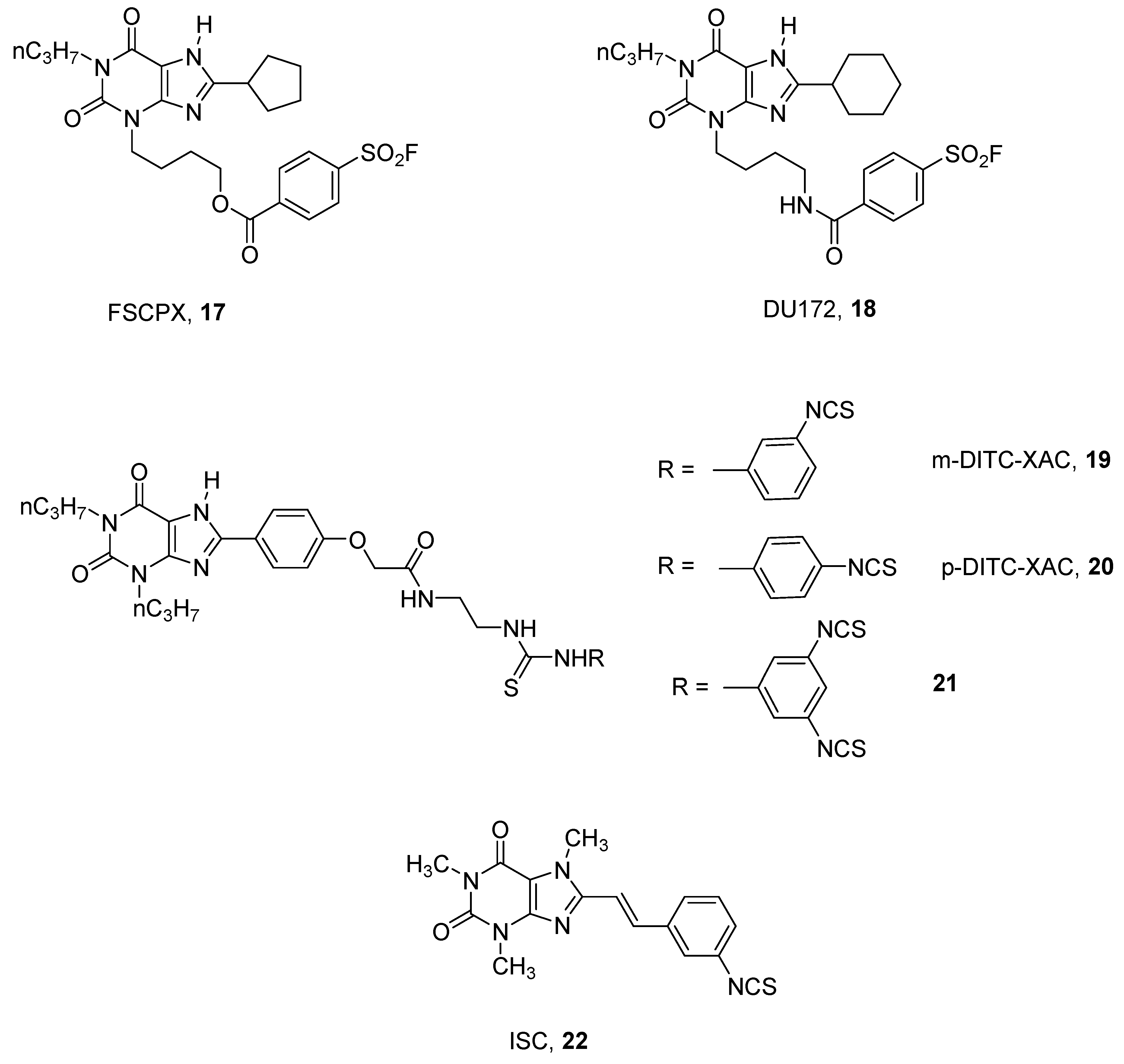
In the xanthine group, several derivatives of the well-known A1 AR antagonist DPCPX (1, 3-dipropyl-8-cyclopentyl xanthine) have been synthesized by introducing chemoreactive groups [47,48,49]. Among this large number of synthesized compounds, the most interesting derivative could be considered to be 8-cyclopentyl-3-N-[3-((3-(4-fluorosulphonyl)benzoyl)-oxy)-propyl]-1-N-propyl-xanthine (FSCPX, 17), which proved to be the best irreversible rat A1 AR antagonist reported [47] (Figure 6).
Several pharmacological studies have been performed using FSCPX (17) as a probe, permitting better understanding of the pathophysiological role of the A1 AR [50,51].
A structurally related derivative to FSCPX (17), bearing a cyclohexyl group at the 8 position and an amido function instead of an ester at the 3 position, named DU172 (18), has been synthesized and utilized to obtain a co-crystalized structure of the human A1 AR. With the help of computational and mutational studies, the structural basis for subtype selectivity versus A2A subtype were identified [15].
A quite similar approach for developing other A1 adenosine covalent antagonists has been utilized using XAC, (N-(2-aminoethyl)-2-[4-(2,3,6,7-tetrahydro-2,6-dioxo-1,3-dipropyl-1H-purin-8-yl)phenoxy]-acetamide), as a template. This approach led to the discovery of several promising covalent agents such as m-DITC-XAC (19), p-DITC-XAC (20) [52], and the trifunctionalized compound 21 [53], which were found to be irreversible ligands for rat A1 ARs in binding studies. In particular, compound 21 was found to be 894-fold selective for A1 vs. A2A ARs (Figure 6) [53].
By modifying the xanthine nucleus, other chemical entities have been developed by Scammells and coworkers. These studies led to some quite interesting covalent antagonists towards hamster receptors but low pharmacological characterization was reported [54].
In this field, some styrylxanthines have been investigated with the aim to obtain novel irreversible A2A AR antagonists. These studies led to the discovery of ISC (8-(3-isothiocyanatostyryl)caffeine, 22), which in binding studies was found to be a good covalent ligand at the rat receptor, with potency in the nanomolar range and good levels of selectivity (Figure 6) [55].
Also in the non-xanthine family, several examples of irreversible AR antagonists have been reported [56,57,58,59].
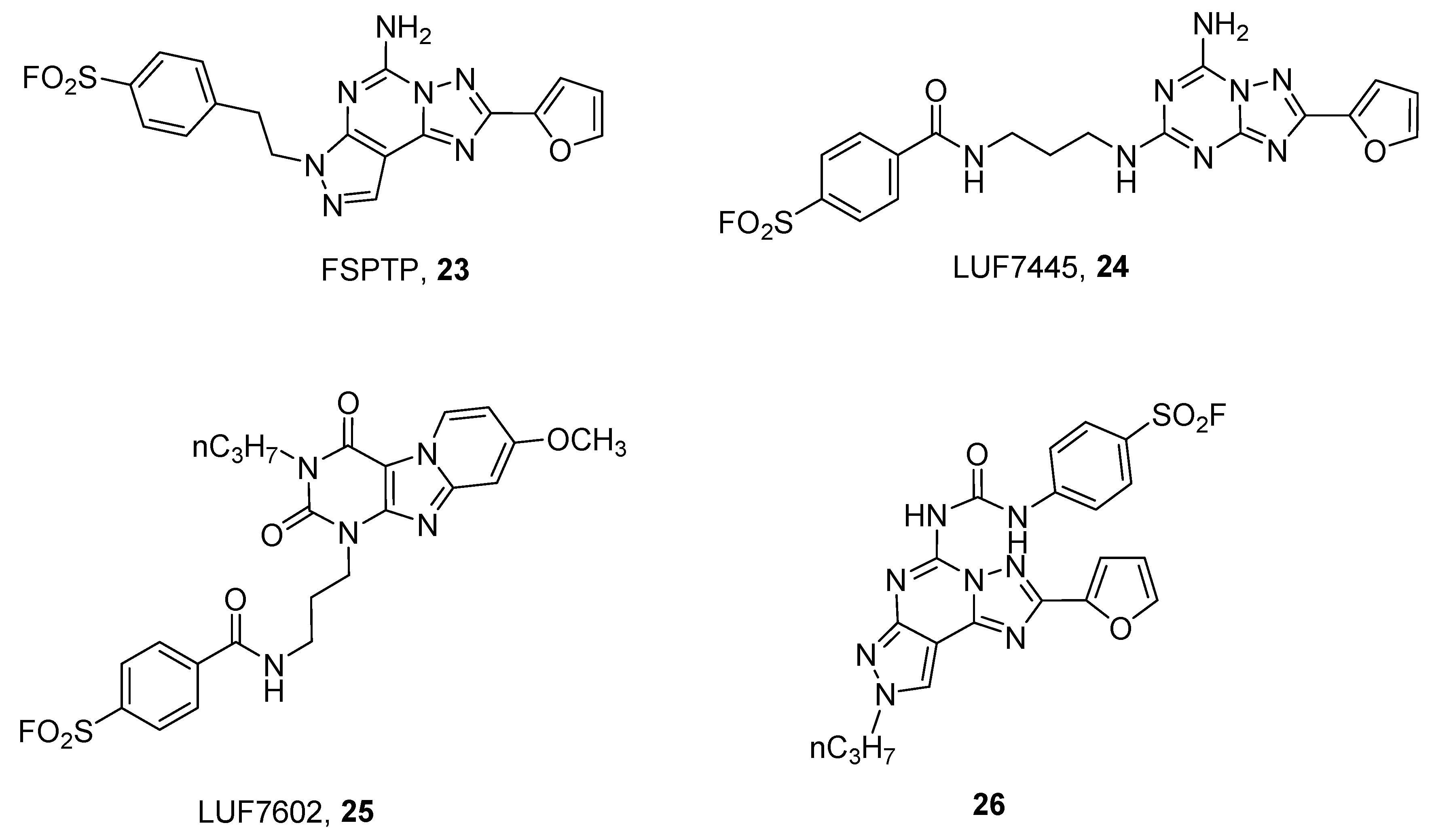
In order to obtain covalent A2A antagonists, insertion of a fluorosulfonyl moiety on the well-known A2A AR antagonists SCH58261 (2-(2-Furanyl)-7-(2-phenylethyl)-7H-pyrazolo [4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-amine) and ZM241385 (4-(2-(7-amino-2-(furan-2-yl)-[1,2,4]triazolo[1,5-a][1,3,5]triazin-5-ylamino)ethyl)phenol) have been developed. This approach led to the discovery of FSPTP (5-amino-7-[2-(4-fluorosulfonyl)phenylethyl]-2-(2-furyl)-pryazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine, 23) and LUF7445 ((4-((3-((7-amino-2-(furan-2-yl)-[1,2,4]triazolo[1,5-a][1,3,5]triazin-5-yl)amino)propyl)carbamoyl)benzene sulfonyl fluoride), 24) as irreversible A2A AR antagonists (Figure 7).
The use of FSPTP (23) in pharmacological studies showed that there is a large receptor reserve for the A2A AR, which mediates the increase in coronary conductance in guinea pigs [56], while LUF7445 (24), through the help of computational and mutational studies permitted identification of lysine 153 as the amino acid involved in covalent binding at the hA2A AR [58].
A quite similar approach has been utilized for developing covalent human A3 AR antagonists, leading to a derivative named LUF7602 (4-((3(8-methoxy-2,4-dioxo-3-propyl-3,4-dihydropyrido[2,1-f]purin-1(2H)yl)propyl)carbamoyl)benzenesulfonyl fluoride, 25) [57] and a pyrazolo-triazolo-pyrimidine compound (26) [59]. The first one showed that tyrosine 265 was involved in the covalent binding, while compound 26, through the help of computational studies, suggested that serine 247 or cysteine 251, both in TM6, could be responsible for covalent binding. Of course, these results clearly indicate that the two compounds have a different binding pose into the receptor binding site.
4. Fluorescent Ligands
The concept of using fluorescent ligands to detect GPCRs in native conditions, including ARs, is not new [60]. However, the urgent need for new fluorescent tools for ARs is given by the possibility of applying new fluorescent techniques to deeply study the receptors in cells, in particular, receptor localization, both on the membrane surface and during their internalization. This would allow for study of desensitization, recycling and homo- or oligo-merization of the receptors [19]. A description of the techniques used to study GPCRs was recently reviewed and comprised fluorescence polarization, confocal microscopy, fluorescence correlation spectroscopy (FCS), flow cytometry, FRET and bioluminescence resonance energy transfer (BRET) [18,19,61,62,63]. Nowadays, thanks to the progress in both synthetic and instrumental techniques, a plethora of fluorophores is available, with a diversity of applicable conjugation strategies that simplify the development of new specific fluorescent ligands [64,65,66]. In fact, the literature reports several examples of fluorescent ligands for all four AR subtypes. A very comprehensive review on fluorescent ligands for ARs was reported by Kozma et al. [67] and extended to some newer derivatives by another two reviews published in 2014 and 2015 [68,69]. Thus, here the most representative fluorescent ligands are reported among with the new fluorescent ligands for the ARs reported in the literature from 2015 to now.
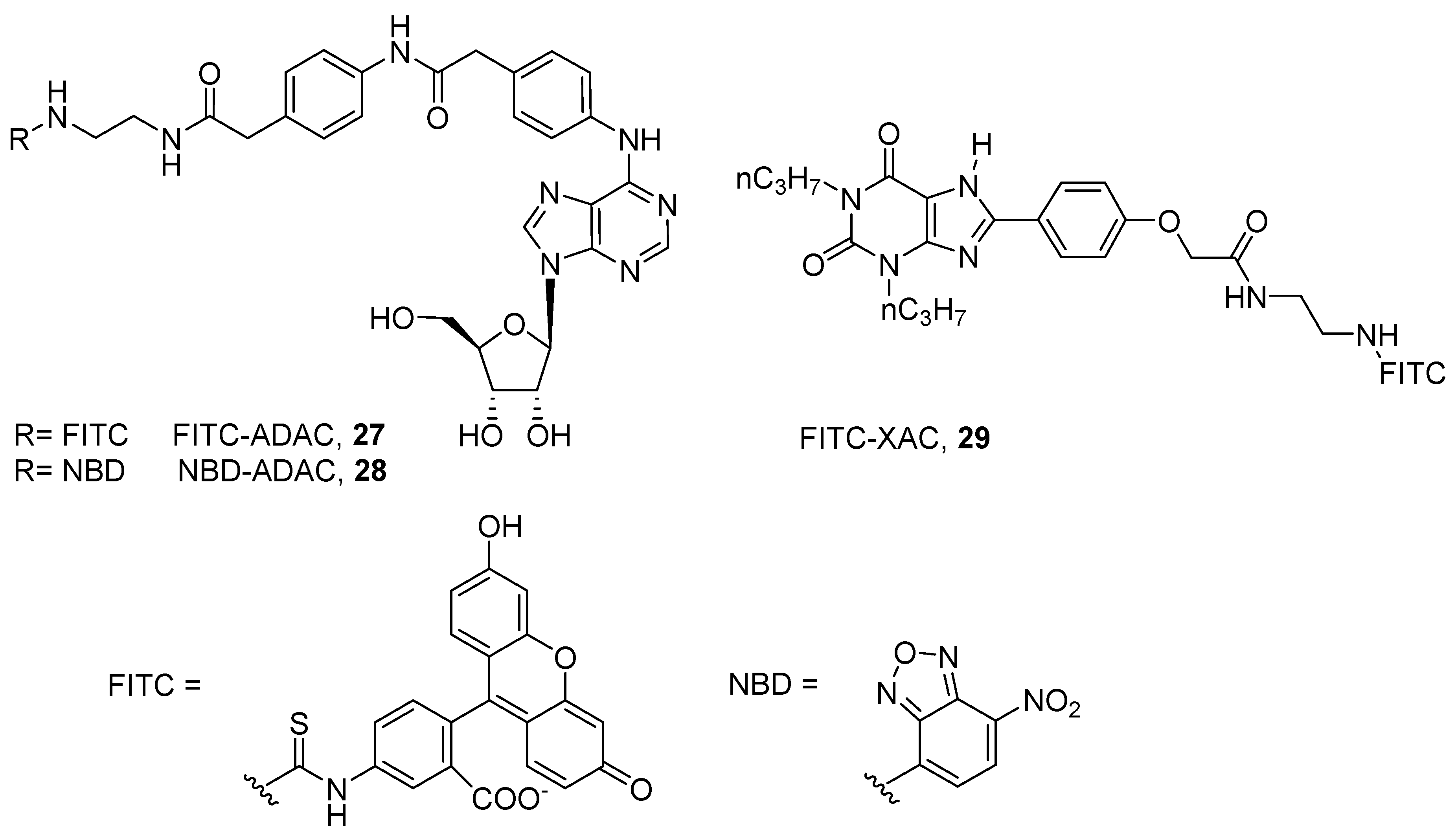
The first fluorescent ligands were developed in 1987 by Jacobson and coworkers to target the rat A1 AR. The authors conjugated the fluorophores fluorescein isothiocyanate (FITC) and nitrobenzoxadiazole (NBD) to the free amino group of the A1 agonist ADAC (FITC-ADAC, 27; NBD-ADAC, 28), maintaining affinity to the receptor (pKi@ratA1 = 8.14, 27; pKi@ratA1 = 8.36, 28). The same approach was used for conjugating FITC to the A1 antagonist XAC, FITC-XAC (29, pKi@ratA1 = 6.90) (Figure 8) [60].
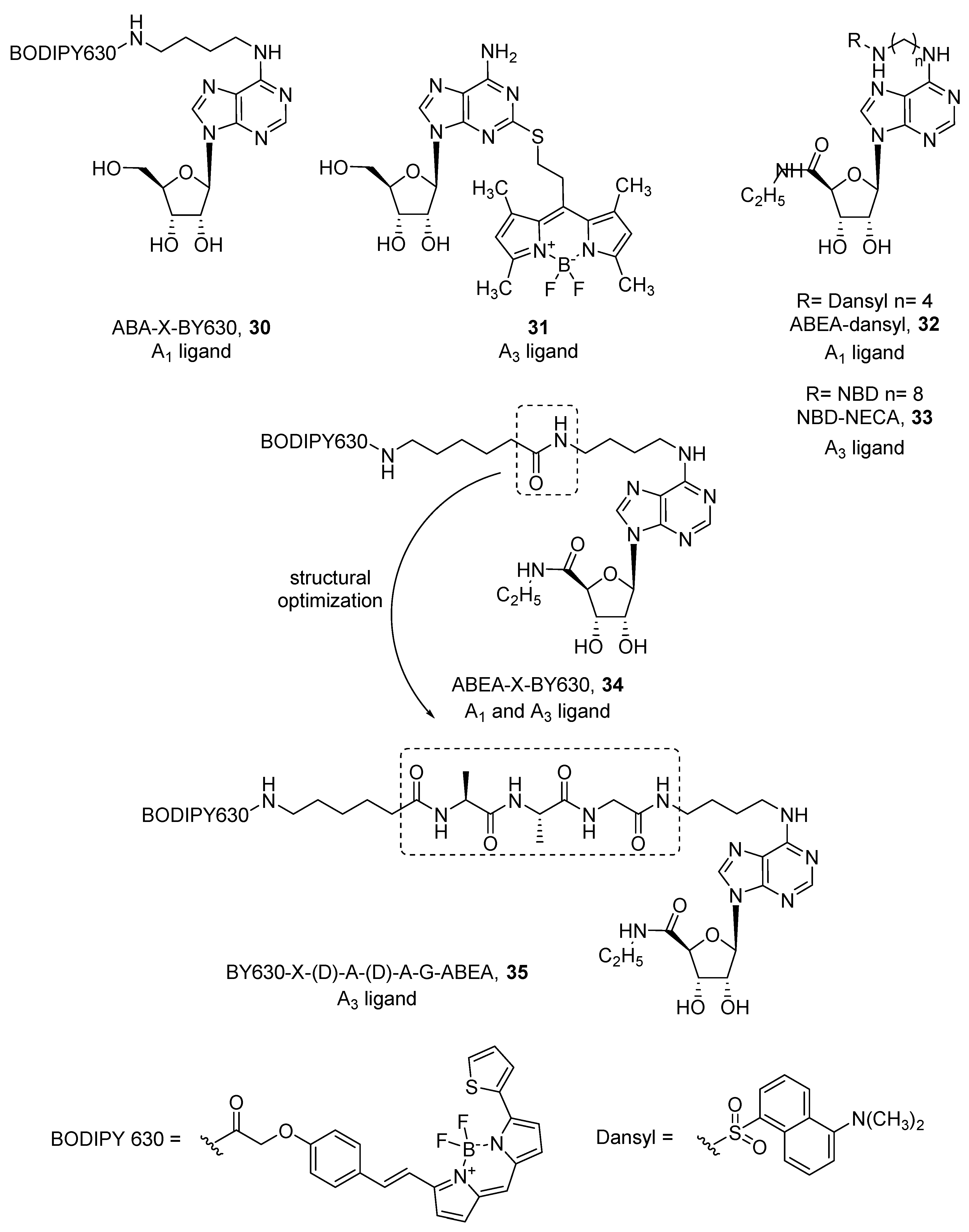
Therefore, fluorescent ligands for human adenosine receptors were developed both from agonists and antagonists. Concerning agonists, only few derivatizations were made from the simple adenosine scaffold. Specifically, in ABA-X-BY630 (30) [70], adenosine was conjugated by a linker at the 6 position, while in compound 31 [71], thioadenosine was used to introduce the BODIPY (boron dipyrromethene) fluorophore at the 2 position of the purine ring (Figure 9). ABA-X-BY630 (30) was developed as an A1 ligand (pKi = 6.65), but compound 31 exhibited a preferential affinity towards the A3 AR subtype (pKi = 6.21), showing more than ten-fold selectivity against A1 and A2A ARs. More efforts were made with the more active, but still not selective, agonist NECA (5’-(N-ethylcarboxamido)adenosine). It has been studied by different groups, where all substitutions were carried out at the 6 position and linkers of different length were used, as were different fluorophores such as dansyl, NBD, BODIPY 630/650, Cy5, Texas Red, and EvoBlue30 [70,72,73,74,75]. In most cases, the introduction of a fluorophore led to A1 AR ligands. Macchia et al. developed a series of dansyl-NECA derivatives that exhibited higher affinity at the A1 AR (i.e., ABEA-dansyl, 32) [75]. Because dansyl excitation wavelength (340 nm) falls in the autofluorescence spectrum of cells and tissues, the authors substituted the dansyl group with a longer excitation wavelength fluorophore such as NBD (λex = 465 nm, λem = 535 nm). Interestingly, NBD led to a detrimental effect on A1 AR, while the affinity towards A3 AR (i.e., NBD-NECA, 33) increased [73] (Figure 9).
In contrast, Middleton et al. developed various NECA-BODIPY 630/650 derivatives as A1 ligands, because red-emitting BODIPY fluorophores become brighter in non-aqueous environments, therefore specific fluorescence on the plasma membrane surface can be more easily visualized. Among them, ABEA-X-BY630 (34) showed high affinity for both A1 and A3 ARs and maintained agonist behavior [70] (Figure 9). This allowed visualization of the receptors at the cell membrane by confocal microscopy and quantification of its binding kinetics. Consequently, it was used as a probe to investigate allosteric interactions given by small molecules and receptor dimerization [76,77]. Unfortunately, ABEA-X-BY630 (34) showed high levels of non-specific cytoplasmic uptake, which prevented its use in long-term studies. In a work aimed to improve these poor imaging properties, an additional tripeptide linker, A-A-G, was introduced between the fluorophore and the agonist to obtain ABEA-A-A-G-X-BY630. All four possible combinations of L and D amino acids were examined, and apart from the (L)A-(L)A-G isomer, all others showed excellent affinity values at the A1 and A3 AR. The ABEA-(D)A-(D)A-G-BY630 (35) agonist was selected for further experiments and because it showed activation of the Gs protein at the A1 AR when tested at high concentrations, this compound was preferentially used as a probe for the A3 AR (Figure 9). As an example, compound 35 was successfully used to investigate the internalization process of A3 ARs and their co-localization with arrestin [78].
As shown in Figure 10, among agonists, the only other compound that was conjugated to a fluorophore in order to obtain, in this case, an agonist for the A2A AR subtype was APEC (2-[2-[4-[2-(2-aminoethylcarbonyl)ethyl]phenyl]ethylamino]-5′-N-ethylcarboxamidoadenosine). It was successfully functionalized with FITC, Alexa Fluor 488 (MRS5206, 36) and Alexa Fluor 532 (MRS5424, 37) [79,80,81]. MRS5206 (36) showed affinity towards both A2A (Ki = 149 nM) and A3 (Ki = 240 nM) ARs and was used to demonstrate that A2A agonist-induced internalization is mediated by a clathrin-dependent mechanism [80]. Instead, the APEC–Alexa Fluor 532 conjugate MRS5424 (37) was developed to be applied in FRET experiments. In fact, the A2A receptor tagged with CFP (cyan fluorescent protein) was able to emit at 480 nm when excited at about 430 nm (blue). Then, emission at 480 nm was able to excite Alexa Fluor 532 in MRS5424 (37), which consequently emitted at 554 nm (yellow). In particular, MRS5424 (37) was used for the investigation of allosterism mediated by A2A/D2 oligomerization [8,81].
APEC was also used to develop quantum dot (QD) conjugates, which are well-known for their emission properties. In particular, a QD was functionalized, through a linker based on (R)-thioctic acid, with a dendron containing multiple molecules of APEC. Dendron was used to increase the solubility of the structure and to ensure a good loading of APEC on the QD. One of the prepared systems, MRS5303, showed affinity towards A2A AR, but further work is needed to obtain a good A2A probe [82].
Finally, Jacobson and coworkers reported different methanocarba derivative-based fluorescent probes for the A3 AR subtype. The functionalized scaffold is that of the A3 agonist MRS3558 ((1S,2R,3S,4R,5S)-4-[2-chloro-6-[(3-chlorophenyl)methylamino]purin-9-yl]-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide) in which the chlorine atom at the 2 position of the purine ring was substituted with an alkyne moiety, and where different fluorophores (e.g., Cy5, squaraine-rotaxane, Alexa Fluor 488, pyrene) were attached directly or through a linker [83,84,85]. Compound MRS5218 (38) is a full agonist of the A3 AR, displaying an IC50 of 1.09 nM in a functional cAMP assay (Figure 10) [83].
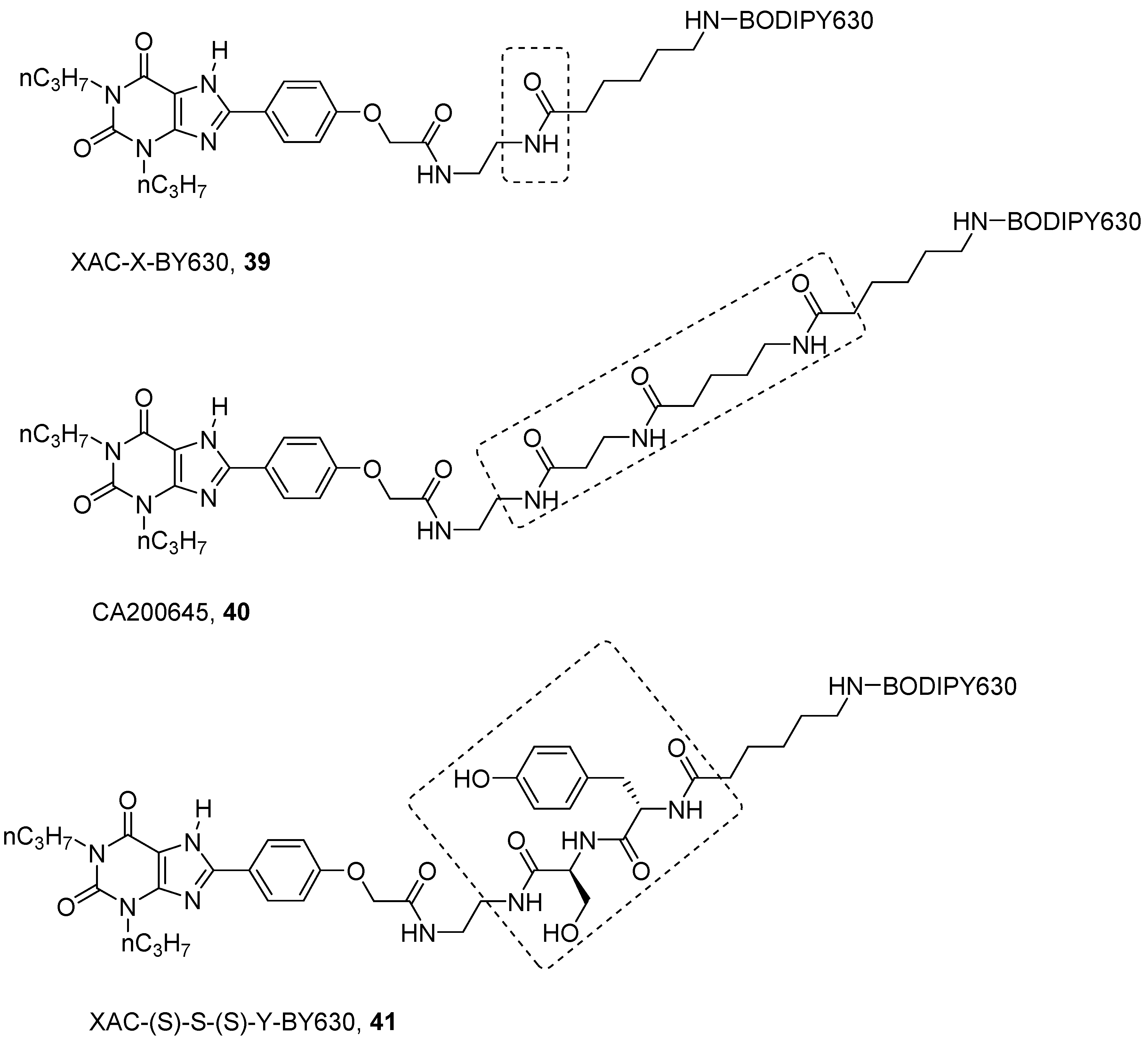
Concerning antagonists, the most studied fluorescent probes were based on the xanthine derivative XAC. As for NECA, several linkers and different fluorophores were introduced on the free amino group in XAC in this case, leading to both A1 and A3 AR fluorescent antagonists [60,72,86,87,88,89]. Among them XAC-X-BY630 (39) and CA200645 (40) are the more representative (Figure 11) [87,90].
The first aim was to develop a probe for the A1 AR, and from SAR analysis, it is known that the introduction of long chains at the 8 position of the xanthine ring are well tolerated by the A1 AR. In fact, XAC-X-BY630 (39) showed affinity in the nanomolar range towards A1 AR and it was used to quantify ligand-receptor binding at a single cell level using FCS [87]. Instead, CA200645 (40), which bears a longer polyamido linker even if it is particularly used as an A3 florescent probe (i.e., for kinetics, allosterism and dimerization studies by FCS and nano-BRET technology [91,92,93]), binds potently both to A1 and A3 ARs, in fact, it was used in a high-content screening assay for both receptors [90].
Further optimization on the linker of XAC/BODIPY 630 derivatives was reported by Vernall et al., where, like NECA derivatives, the authors decided to introduce a peptide linker, instead of a naked polyamido chain like in CA200645 (40) [89]. The aim was to increase selectivity at the A3 AR by gaining specific interactions with the linker. In order to find a less expensive way to assess the SAR of this compound, the BODIPY 630 was replaced by the similarly bulky Fmoc group. The linker was made by two amino acids chosen between alanine, tyrosine, serine and asparagine. The last were chosen for their polarity and, thus, ability to make hydrogen bonds. In addition, tyrosine can also be engaged in π-π interactions. The best results were obtained with XAC-(S)-S-(S)-Y-BY630 (41), which showed a pKD of 9.12 towards the A3 AR, compared to a pKD of 7.26 towards the A1 AR [89]. Compound 41 demonstrated a long residence time at A3 AR (RT = 288 min); thus, when it was used to determine the association and dissociation constants of the ligands, it gave quite different constants with respect to those determined with classical radioligands, such as [3H]PSB-11 ([3H]-(8R)-8-ethyl-1,4,7,8-tetrahydro-4-5H-imidazo[2,1-i]purin-5-one) [92]. This result suggested again that a probe should be properly selected before its use in an experiment.
XAC was also used to prepare GPCR ligand–dendrimer (GLiDe) conjugates. In particular, XAC was functionalized at the free amino group present at the 8 position, with an alkyne-containing chain. Alkyne was then used to perform a copper catalyzed click reaction with an azido-containing G4 (fourth-generation) polyamidoamine (PAMAM) dendrimer to form triazoles. Dendrimers were also conjugated to fluorophores such as Alexa Fluor 488, leading to fluorescent dendrimers MRS5397 and MRS5399. In addition, near infrared dyes were also used by Jacobson and coworkers, leading to MRS5421 (which used NIR dye 800) and MRS5422 (which used NIR dye 700). These compounds showed higher affinity towards the A2A AR, followed by A3 and A1 ARs [88].
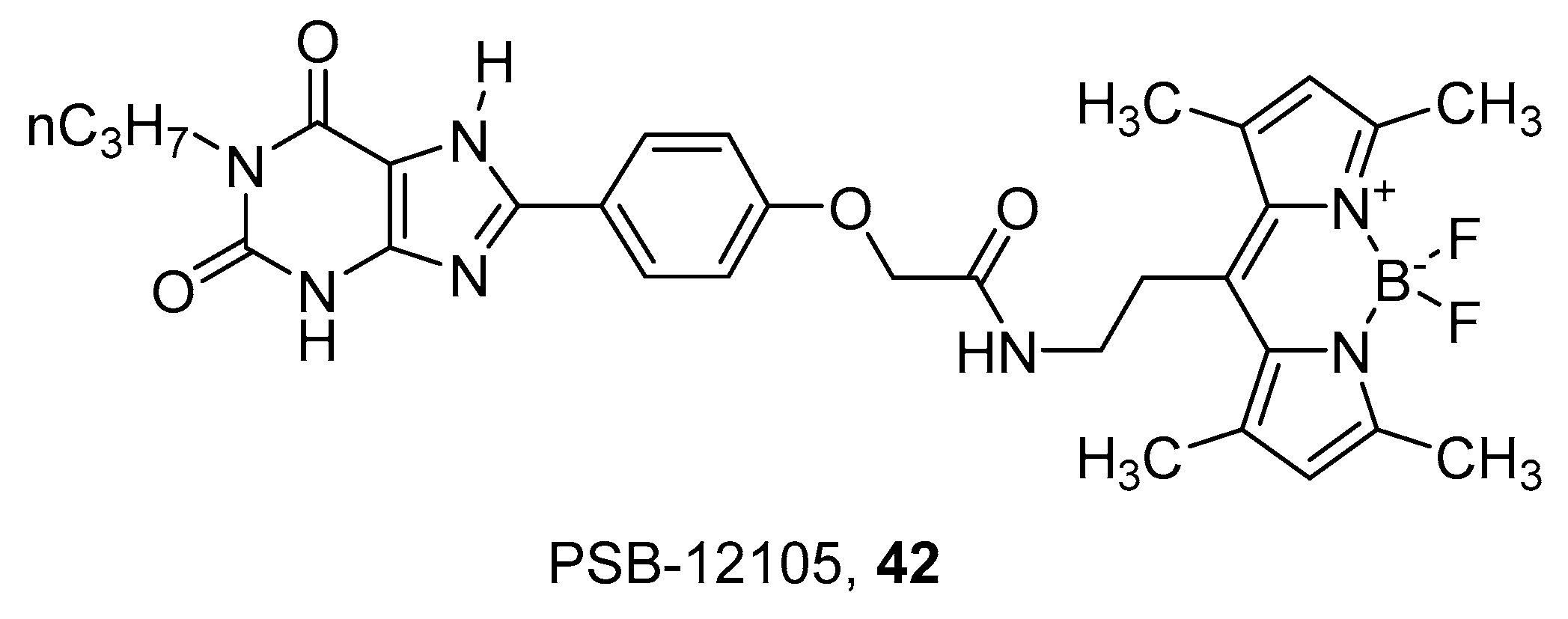
A series of xanthine derivatives was recently developed to obtain fluorescent antagonists towards the A2B AR [94]. The xanthine scaffold was chosen on the basis of known SAR for this receptor, which suggested that 3-unsubstituted 8-phenyl-1-propylxanthine derivatives are generally more potent and selective to A2B AR than the corresponding 1,3-substituted derivatives. Thus, the BODIPY fluorophore was conjugated to the 8-phenyl by means of spacers of different lengths. The best compound of the series was PSB-12105 (42) (Figure 12), which showed nanomolar or subnanomolar affinity towards human, rat and mice receptors and was consequently used to develop a binding assay using flow cytometry [94].
In addition, scaffolds other than xanthines have been used to develop fluorescent antagonists for the adenosine receptors. In particular, tricyclic antagonists that exhibited high affinity and selectivity for the various AR subtypes were used; among them we can find [1,2,4]triazolo[1,5-c]quinazolines [95], [1,2,4]-triazolo[4,3-a]quinoxalin-1-ones [96], pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidines [95,97,98,99,100] and benzo[4,5]imidazo[2,1-c][1,2,4]triazines [101].
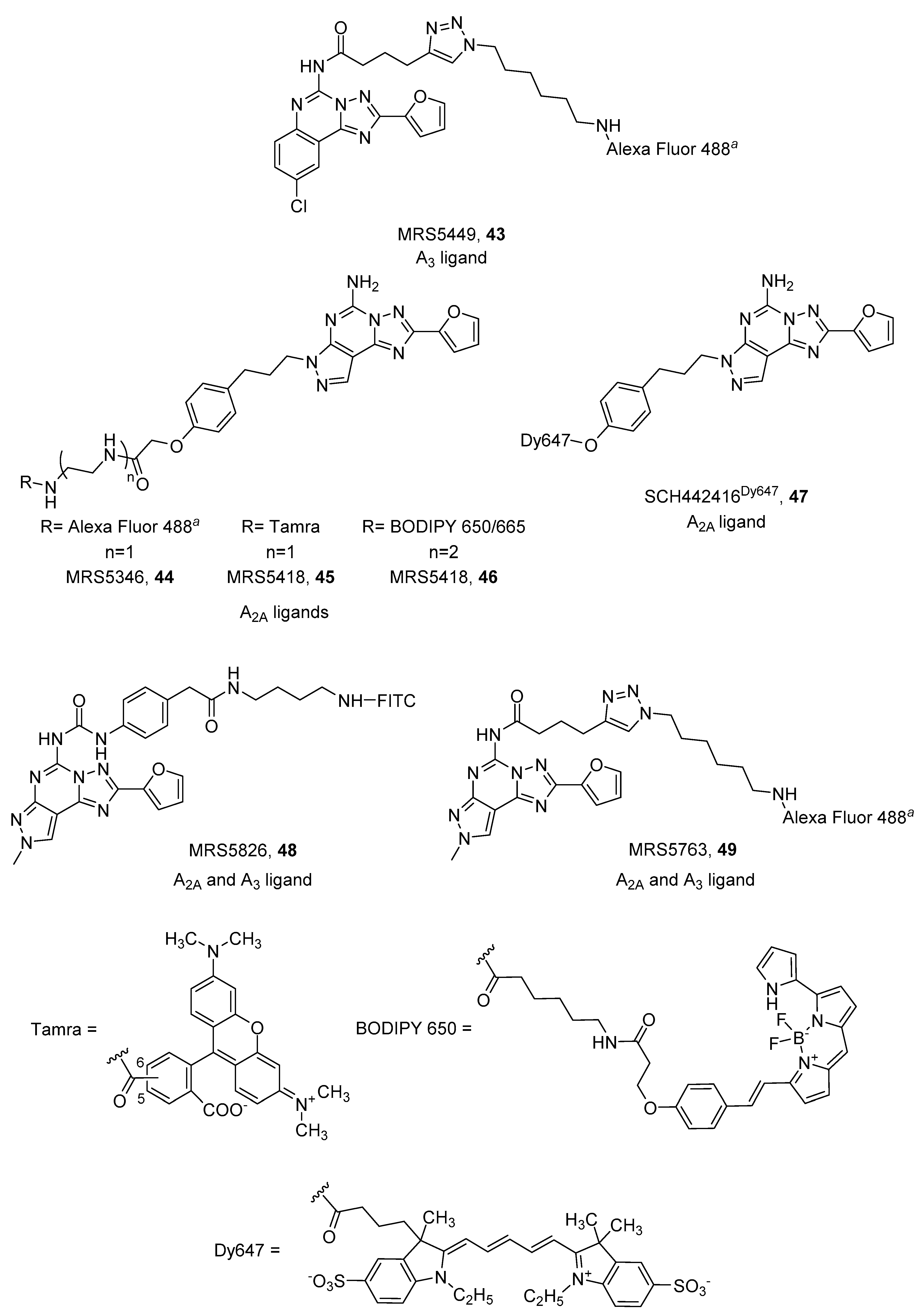
CGS15943 (9-chloro-2-(2-furanyl)-[1,2,4]triazolo[1,5-c]quinazolin-5-amine) is a well-known non-selective AR antagonist that, when conjugated to Alexa Fluor 488 by a triazole-containing linker at the 5 position, led to MRS5449 (43, Figure 13) displaying a radioligand binding Ki value of 6.4 nM at the A3 AR, and was more than 10-fold selective against A1 and A2A ARs. For these characteristics it was chosen for the establishment of an A3 AR biding assay using flow cytometry [95].
In contrast, the analog pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidine derivative SCH442416 (5-Amino-7-[3-(4-methoxy)phenylpropyl]-2-(2-furyl)-pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine), was used to design potent A2A AR fluorescent antagonists. Linkers were introduced on the hydroxy group on the phenylalkyl chain present at the 7 position of the scaffold. Three different fluorophores were introduced: Alexa Fluor 488, Tamra and BODIPY 650/665, leading to MRS5346 (44), MRS5347 (45) and MRS5418 (46), respectively (Figure 13). MRS5418 (46) showed the best affinity (Ki = 15.1 nM) and selectivity for the A2A AR [97,98], but MRS5346 (44) was used in a fluorescence polarization-based receptor binding assay at the A2A AR [99]. Ciruela and coworkers developed another member of this class of A2A antagonists, where they introduced dye 467 directly to the hydroxyl group of SCH442416 (Figure 13). SCH442416Dy647 (47) was employed to undoubtedly demonstrate the presence of A2A/D2 oligomers in native tissues of rat striatum, which is essential information for designing new therapies for Parkinson’s disease [69,100].
The pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidine scaffold was also functionalized at the 5 position, leaving the 7 position unsubstituted and inserting a methyl group at the 8 position: substitutions that should shift selectivity towards the A3 AR [102]. FITC was used as fluorophore and simple diamino alkyl or PEG linkers were used as spacers. These derivatives exhibited equal low micromolar affinity for the A2A and A3 AR subtypes [99]. In an attempt to obtain more potent derivatives, an aromatic ring, which could gain some interactions with the receptor, was introduced. As expected, new compounds (48 and 49) displayed nanomolar affinity at the two receptors (Ki A2A = 60.4 nM, Ki A3 = 73.6 nM, 48; Ki A2A = 90 nM, Ki A3 = 31.8 nM, 49) (Figure 13) [103].
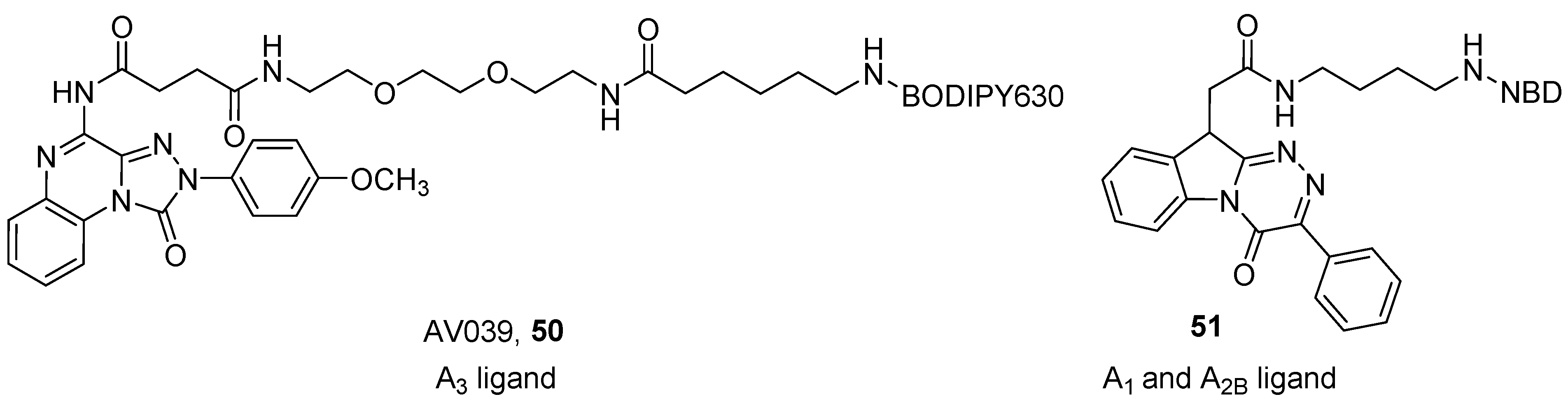
Vernall et al. developed a series of A3 fluorescent derivatives of the 4-amino-2-(4-methoxyphenyl)-[1,2,4]triazolo[4,3-a]quinoxalin-1(2H)-one, by introducing different fluorophores through a polyamido linker at the 4 position of the scaffold [96]. The presence of the p-methoxy moiety on the phenyl group at the 2 position of the triazoloquinoxaline ring and the introduction of an acyl on the amino group at the 4 position, are the characteristics that should confer affinity at the A3 AR. A mini-series of compounds was developed using different linkers (chains from 2 to 12 atoms containing C, N and O atoms) and fluorophores (e.g., BODIPY, Tamra and Cy5). The best compound of the series was AV039 (50), which showed a pKD of about 9 at the human A3 AR and was more than 650-fold selective over other receptor subtypes (Figure 14). This compound was able to specifically label the A3 AR in cells containing a mixed population of ARs, thus it represented an ideal tool for the study of these receptors [92,96].
Recently, Barresi et al. reported a series of benzo[4,5]imidazo[2,1-c][1,2,4]triazines derivatives as fluorescent probes for the A1 and A2B ARs. The authors used NBD as a fluorophore, which was introduced using two linkers of different lengths at the N1 or N10 positions, in order to investigate which position gives the best flexibility to the molecule, to best accommodate the orthosteric binding site [101]. One compound (51) (Figure 14) showed an affinity of about 1 µM at the A1 AR and an IC50 value in the same range towards the A2B AR, and therefore was demonstrated to be a useful tool for detecting these AR subtypes in fluorescence confocal microscopy experiments on cells.
5. Conclusions
From this review, it is clear that even if ARs were discovered more than forty years ago, their complex mechanism of action, which includes, but is not limited to, activation, oligomerization and internalization processes, is not fully elucidated. Over the last years, great progress in the field has been obtained thanks to the advancements made in structural biology. AR probes were continuously applied to new available techniques, which allowed deep investigation of the receptors, but at the same time, obtained information that is useful for designing new, more specific, drugs and probes, which in turn has contributed to the improvement in the AR research field. In particular, a better understanding of the role and the mechanism of action of these receptors could allow design of more specific therapies for several pathological conditions in which these receptors are involved, and for that reason, ARs remain a very interesting and intriguing therapeutic target.
Author Contributions
Conceptualization, S.F. and G.S.; writing—original draft preparation, S.F., L.L., G.S.; writing—review and editing, S.F. and G.S.
Funding
This research was funded by MINISTRO DELL’ISTRUZIONE, DELL’UNIVERSITÀ E DELLA RICERCA, grant number PRIN2017 2017MT3993 and by UNIVERSITÀ DEGLI STUDI DI TRIESTE grant FRA2018.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Iizuka, H.; Adachi, K.; Halprin, K.M.; Levine, V. Adenosine and adenine nucleotides stimulation of skin (epidermal) adenylate cyclase. Biochim. Biophys. Acta Gen. Subj. 1976, 444, 685–693. [Google Scholar] [CrossRef]
- Sawynok, J.; Jhamandas, K.H. Inhibition of acetylcholine release from cholinergic nerves by adenosine, adenine nucleotides and morphine: Antagonism by theophylline. J. Pharmacol. Exp. Ther. 1976, 197, 379–390. [Google Scholar] [PubMed]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of adenosine receptors: The state of the art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef] [PubMed]
- Braun, S.; Levitzki, A. Adenosine receptor permanently coupled to turkey erythrocyte adenylate cyclase. Biochemistry 1979, 18, 2134–2138. [Google Scholar] [CrossRef] [PubMed]
- Bruns, R.F. Adenosine receptor activation by adenine nucleotides requires conversion of the nucleotides to adenosine. Naunyn Schmiedebergs Arch. Pharmacol. 1980, 315, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Klotz, K.N.; Linden, J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 2001, 53, 527–552. [Google Scholar] [PubMed]
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Linden, J.; Müller, C.E. International union of basic and clinical pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—An update. Pharmacol. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Ciruela, F.; Jacobson, K.A.; Fernández-Dueñas, V. Portraying G protein-coupled receptors with fluorescent ligands. ACS Chem. Biol. 2014, 9, 1918–1928. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Milligan, G. Structural and biophysical characterisation of G protein-coupled receptor ligand binding using resonance energy transfer and fluorescent labelling techniques. Biochim. Biophys. Acta Biomembr. 2014, 1838, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Weichert, D.; Gmeiner, P. Covalent Molecular Probes for class A G protein-coupled receptors: Advances and applications. ACS Chem. Biol. 2015, 10, 1376–1386. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Xie, X. Tools for GPCR drug discovery. Acta Pharmacol. Sin. 2012, 33, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Van de Bittner, G.C.; Ricq, E.L.; Hooker, J.M. A Philosophy for CNS radiotracer design. Acc. Chem. Res. 2014, 47, 3127–3134. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.E.; Jacobson, K.A. Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochim. Biophys. Acta Biomembr. 2011, 1808, 1290–1308. [Google Scholar] [CrossRef] [PubMed]
- van Waarde, A.; Dierckx, R.A.J.O.; Zhou, X.; Khanapur, S.; Tsukada, H.; Ishiwata, K.; Luurtsema, G.; de Vries, E.F.J.; Elsinga, P.H. Potential therapeutic applications of adenosine A2A receptor ligands and opportunities for A2A receptor imaging. Med. Res. Rev. 2018, 38, 5–56. [Google Scholar] [CrossRef] [PubMed]
- Glukhova, A.; Thal, D.M.; Nguyen, A.T.; Vecchio, E.A.; Jörg, M.; Scammells, P.J.; May, L.T.; Sexton, P.M.; Christopoulos, A. Structure of the adenosine A1 receptor reveals the basis for subtype selectivity. Cell 2017, 168, 867–877.e13. [Google Scholar] [CrossRef] [PubMed]
- Jörg, M.; Glukhova, A.; Abdul-Ridha, A.; Vecchio, E.A.; Nguyen, A.T.N.; Sexton, P.M.; White, P.J.; May, L.T.; Christopoulos, A.; Scammells, P.J. Novel irreversible agonists acting at the A1 adenosine receptor. J. Med. Chem. 2016, 59, 11182–11194. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Furstenberg, A.; Huber, T. Labeling and single-molecule methods to monitor G proteincoupled receptor dynamics. Chem. Rev. 2017, 117, 186–245. [Google Scholar] [CrossRef] [PubMed]
- Stoddart, L.A.; White, C.W.; Nguyen, K.; Hill, S.J.; Pfleger, K.D.G.G. Fluorescence-and bioluminescence-based approaches to study GPCR ligand binding. Br. J. Pharmacol. 2016, 173, 3028–3037. [Google Scholar] [CrossRef] [PubMed]
- Stoddart, L.A.; Kilpatrick, L.E.; Briddon, S.J.; Hill, S.J. Probing the pharmacology of G protein-coupled receptors with fluorescent ligands. Neuropharmacology 2015, 98, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Kauk, M.; Hoffmann, C. Intramolecular and intermolecular FRET sensors for GPCRs—Monitoring conformational changes and beyond. Trends Pharmacol. Sci. 2018, 39, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Cheong, S.L.; Federico, S.; Venkatesan, G.; Mandel, A.L.; Shao, Y.-M.; Moro, S.; Spalluto, G.; Pastorin, G. The A3 adenosine receptor as multifaceted therapeutic target: Pharmacology, medicinal chemistry, and in silico approaches. Med. Res. Rev. 2013, 33, 235–335. [Google Scholar] [CrossRef] [PubMed]
- Klotz, K. Adenosine receptors and their ligands. Naunyn Schmiedebergs Arch. Pharmacol. 2000, 362, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Moro, S.; Gao, Z.; Jacobson, K.A.; Spalluto, G. Progress in the pursuit of therapeutic adenosine receptor antagonists. Med. Res. Rev. 2006, 26, 131–159. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A.; Pannell, L.K.; Ji, X.; Jarvis, M.F.; Williams, M.; Hutchison, A.J.; Barrington, W.W.; Stiles, G.L. Agonist derived molecular probes for A2 adenosine receptors. J. Mol. Recognit. 1989, 2, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Hinz, S.; Alnouri, W.M.; Pleiss, U.; Müller, C.E. Tritium-labeled agonists as tools for studying adenosine A2B receptors. Purinergic Signal. 2018, 14, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Khanapur, S.; Sijbesma, J.W.; Ishiwata, K.; Elsinga, P.H.; Meerlo, P.; Dierckx, R.A.; van Waarde, A. Use of 11C-MPDX and PET to study adenosine A1 receptor occupancy by nonradioactive agonists and antagonists. J. Nucl. Med. 2014, 55, 315–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, S.; Inaji, M.; Nariai, T.; Oda, K.; Sakata, M.; Toyohara, J.; Ishii, K.; Ishiwata, K.; Maehara, T. Increased Binding Potential of Brain Adenosine A1 Receptor in Chronic Stages of Patients with Diffuse Axonal Injury Measured with [1-methyl-11C] 8-dicyclopropylmethyl-1-methyl-3-propylxanthine Positron Emission Tomography Imaging. J. Neurotrauma 2018, 35, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Mishina, M.; Ishii, K.; Kimura, Y.; Suzuki, M.; Kitamura, S.; Ishibashi, K.; Sakata, M.; Oda, K.; Kobayashi, S.; Kimura, K.; et al. Adenosine A1 receptors measured with 11C-MPDX PET in early Parkinson’s disease. Synapse 2017, 71, e21979. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Gao, Z.; Tyler, R.; Stodden, T.; Li, Y.; Ramsey, J.; Zhao, W.; Wang, G.; Wiers, C.E.; Fowler, J.S.; et al. Preclinical evaluation of the first adenosine A1 receptor partial agonist radioligand for positron emission tomography imaging. J. Med. Chem. 2018, 61, 9966–9975. [Google Scholar] [CrossRef] [PubMed]
- Lahesmaa, M.; Oikonen, V.; Helin, S.; Luoto, P.; Din, U.M.; Pfeifer, A.; Nuutila, P.; Virtanen, K.A. Regulation of human brown adipose tissue by adenosine and A2A receptors—Studies with [15O]H2O and [11C]TMSX PET/CT. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 743–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Khanapur, S.; Huizing, A.P.; Zijlma, R.; Schepers, M.; Dierckx, R.A.J.O.; Van Waarde, A.; De Vries, E.F.J.; Elsinga, P.H. Synthesis and preclinical evaluation of 2-(2-furanyl)-7-[2-[4-[4-(2-[11C]methoxyethoxy)phenyl]-1-piperazinyl]ethyl]7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidine-5-amine ([11C]preladenant) as a PET tracer for the imaging of. J. Med. Chem. 2014, 57, 9204–9210. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Elsinga, P.H.; Khanapur, S.; Dierckx, R.A.J.O.; de Vries, E.F.J.; de Jong, J.R. Radiation dosimetry of a novel adenosine A2A receptor radioligand [11C]preladenant based on PET/CT imaging and ex vivo biodistribution in rats. Mol. Imaging Biol. 2017, 19, 289–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmenhorst, D.; Kroll, T.; Wedekind, F.; Weisshaupt, A.; Beer, S.; Bauer, A. In vivo kinetic and steady-state quantification of 18F-CPFPX binding to rat cerebral A1 Adenosine receptors: Validation by displacement and autoradiographic experiments. J. Nucl. Med. 2013, 54, 1411–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmenhorst, D.; Meyer, P.T.; Matusch, A.; Winz, O.H.; Bauer, A. Caffeine occupancy of human cerebral A1 adenosine receptors: In vivo quantification with 18F-CPFPX and PET. J. Nucl. Med. 2012, 53, 1723–1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabbi-Schroeter, D.; Elmenhorst, D.; Oskamp, A.; Laskowski, S.; Bauer, A.; Kroll, T. Effects of long-term caffeine consumption on the adenosine A1 receptor in the rat brain: An in vivo PET study with [18F]CPFPX. Mol. Imaging Biol. 2018, 20, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Kreft, S.; Bier, D.; Holschbach, M.H.; Schulze, A.; Coenen, H.H. New potent A1 adenosine receptor radioligands for positron emission tomography. Nucl. Med. Biol. 2017, 44, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Wadsak, W.; Mien, L.-K.; Shanab, K.; Ettlinger, D.E.; Haeusler, D.; Sindelar, K.; Lanzenberger, R.R.; Spreitzer, H.; Viernstein, H.; Keppler, B.K.; et al. Preparation and first evaluation of [18F]FE@SUPPY: A new PET tracer for the adenosine A3 receptor. Nucl. Med. Biol. 2008, 35, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Haeusler, D.; Nics, L.; Mien, L.-K.; Ungersboeck, J.; Lanzenberger, R.R.; Shanab, K.; Sindelar, K.M.; Viernstein, H.; Wagner, K.-H.; Dudczak, R.; et al. [18F]FE@SUPPY and [18F]FE@SUPPY:2—Metabolic considerations. Nucl. Med. Biol. 2010, 37, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, M.; Hinz, S.; Deuther-Conrad, W.; Namasivayam, V.; Dukic-Stefanovic, S.; Teodoro, R.; Toussaint, M.; Kranz, M.; Juhl, C.; Steinbach, J.; et al. Radiosynthesis and in vivo evaluation of a fluorine-18 labeled pyrazine based radioligand for PET imaging of the adenosine A2B receptor. Bioorganic Med. Chem. 2018, 26, 4650–4663. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Jacobson, K.A.; Downey, J.M. An irreversible A1-selective adenosine agonist preconditions rabbit heart. Can. J. Cardiol. 1996, 12, 517–521. [Google Scholar] [PubMed]
- Zhang, J.; Belardinelli, L.; Jacobson, K.A.; Otero, D.H.; Baker, S.P. Persistent activation by and receptor reserve for an irreversible A1 -adenosine receptor agonist in DDT 1 MF-2 cells and in guinea pig heart. Mol. Pharmacol. 1997, 52, 491–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klotz, K.N.; Cristalli, G.; Grifantini, M. Photoaffinity labeling of A1-adenosine receptors. J. Biol. Chem. 1985, 260, 14659–14664. [Google Scholar] [PubMed]
- Jacobson, K.A.; Stiles, G.L.; Ji, X. Chemical modification and irreversible inhibition of striatal A2A adenosine receptors. Mol. Pharmacol. 1992, 42, 123–133. [Google Scholar] [PubMed]
- Niiya, K.; Jacobson, K.A.; Silvia, S.K.; Olsson, R.A. Covalent binding of a selective agonist irreversibly activates guinea pig coronary artery A2 adenosine receptors. Naunyn Schmiedebergs Arch. Pharmacol. 1993, 347, 521–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moss, S.M.; Jayasekara, P.S.; Paoletta, S.; Gao, Z.; Jacobson, K.A. Structure-based design of reactive nucleosides for site-specific modification of the A2A adenosine receptor. ACS Med. Chem. Lett. 2014, 5, 1043–1048. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.D.; Gallo-Rodriguez, C.; Jacobson, K.A. A selective agonist affinity label for A3 adenosine receptors. Biochem. Biophys. Res. Commun. 1994, 203, 570–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Muijlwijk-koezen, J.E.; Timmerman, H.; Van Der Sluis, R.P.; Van De Stolpe, A.C.; Menge, W.M.P.B.; Beukers, M.W.; Van Der Graaf, P.H.; Groote, D.; Ijzerman, A.P. Synthesis and use of FSCPX, an irreversible adenosine A1 antagonist, as a receptor knock-down tool. Bioorganic Med. Chem. Lett. 2001, 11, 815–818. [Google Scholar] [CrossRef]
- Beauglehole, A.R.; Baker, P.; Scammells, P.J. New irreversible adenosine A1 antagonists based on FSCPX. Bioorganic Med. Chem. Lett. 2002, 12, 3179–3182. [Google Scholar] [CrossRef]
- Beauglehole, A.R.; Baker, S.P.; Scammells, P.J. Fluorosulfonyl-substituted xanthines as selective irreversible antagonists for the A1—Adenosine receptor. J. Med. Chem. 2000, 43, 4973–4980. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, M.; Shryock, J.C.; Scammells, P.J.; Ruble, J.; Baker, S.P.; Belardinelli, L. A novel irreversible antagonist of the A1-adenosine receptor. Mol. Pharmacol. 1996, 50, 196–205. [Google Scholar] [PubMed]
- Lorenzen, A.; Beukers, M.W.; van der Graaf, P.H.; Lang, H.; van Muijlwijk-koezen, J.; de Groote, M.; Menge, W.; Schwabe, U.; Ijzerman, A.P. Modulation of agonist responses at the A1 adenosine receptor by an irreversible antagonist, receptor-G protein uncoupling and by the G protein activation state. Biochem. Pharmacol. 2002, 64, 1251–1265. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Barone, S.; Kammula, U.; Stiles, G.L. Electrophilic derivatives of purines as irreversible inhibitors of A1 adenosine receptors. J. Med. Chem. 1989, 32, 1043–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boring, D.L.; Ji, X.D.; Zimmet, J.; Taylor, K.E.; Stiles, G.L.; Jacobson, K.A. Trifunctional agents as a design strategy for tailoring ligand properties: Irreversible inhibitors of A1 adenosine receptors. Bioconjugate Chem. 1991, 2, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Scammells, P.J.; Baker, S.P.; Belardinelli, L.; Olsson, R.A. Substituted 1, 3-Dipropylxanthines as irreversible antagonists of A1 adenosine receptors. J. Med. Chem. 1994, 37, 2704–2712. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Callo-rodriguez, C.; Jacobson, K.A. 8-(3-Isothiocyanatostyryl) caffeine is a selective, irreversible inhibitor of striatal A2-Adenosine Receptors. Drug Dev. Res. 1993, 29, 292–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shryock, J.C.; Snowdy, S.; Baraldi, P.G.; Cacciari, B.; Spalluto, G.; Monopoli, A.; Ongini, E.; Baker, S.P.; Belardinelli, L. A2A -Adenosine Receptor reserve for coronary vasodilation. Circulation 1998, 98, 711–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; van Veldhoven, J.P.D.; Offringa, J.; Kuiper, B.J.; Lenselink, E.B.; Heitman, L.H.; van der Es, D.; IJzerman, A.P. Development of covalent ligands for G protein-coupled receptors: A case for the human adenosine A3 receptor. J. Med. Chem. 2019, 62, 3539–3552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Dong, G.; Michiels, T.J.M.; Lenselink, E.B.; Heitman, L.; Louvel, J.; IJzerman, A.P. A covalent antagonist for the human adenosine A2A receptor. Purinergic Signal. 2017, 13, 191–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baraldi, P.G.; Cacciari, B.; Moro, S.; Romagnoli, R.; Ji, X.; Jacobson, K.A.; Gessi, S.; Borea, P.A.; Spalluto, G. Fluorosulfonyl- and bis-(β-chloroethyl)amino-phenylamino functionalized pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine derivatives: Irreversible antagonists at the human A3 adenosine receptor and molecular modeling studies. J. Med. Chem. 2001, 44, 2735–2742. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A.; Ukena, D.; Padgett, W.; Kirk, K.L.; Daly, J.W. Molecular probes for extracellular adenosine receptors. Biochem. Pharmacol. 1987, 36, 1697–1707. [Google Scholar] [CrossRef] [Green Version]
- Cottet, M.; Faklaris, O.; Zwier, J.M.; Trinquet, E.; Pin, J.-P.; Durroux, T. Original fluorescent ligand-based assays open new perspectives in G-protein coupled receptor drug screening. Pharmaceuticals 2011, 4, 202–214. [Google Scholar] [CrossRef] [Green Version]
- Sridharan, R.; Zuber, J.; Connelly, S.M.; Mathew, E.; Dumont, M.E. Fluorescent approaches for understanding interactions of ligands with G protein coupled receptors. Biochim. Biophys. Acta Biomembr. 2014, 1838, 15–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, K.A.; Müller, C.E. Medicinal chemistry of adenosine, P2Y and P2X receptors. Neuropharmacology 2016, 104, 31–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, Y.; Choi, H.-K. Recent conjugation strategies of small organic fluorophores and ligands for cancer-specific bioimaging. Chem. Biol. Interact. 2016, 248, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Ogawa, M.; Alford, R.; Choyke, P.L.; Urano, Y. New strategies for fluorescent probe design in medical diagnostic imaging. Chem. Rev. 2010, 110, 2620–2640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Resch-Genger, U.; Grabolle, M.; Cavaliere-Jaricot, S.; Nitschke, R.; Nann, T. Quantum dots versus organic dyes as fluorescent labels. Nat. Method. 2008, 5, 763–775. [Google Scholar] [CrossRef] [PubMed]
- Kozma, E.; Suresh Jayasekara, P.; Squarcialupi, L.; Paoletta, S.; Moro, S.; Federico, S.; Spalluto, G.; Jacobson, K.A. Fluorescent ligands for adenosine receptors. Bioorganic Med. Chem. Lett. 2013, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernall, A.J.; Hill, S.J.; Kellam, B. The evolving small-molecule fluorescent-conjugate toolbox for Class A GPCRs. Br. J. Pharmacol. 2014, 171, 1073–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciruela, F.; Fernández-Dueñas, V.; Jacobson, K.A. Lighting up G protein-coupled purinergic receptors with engineered fluorescent ligands. Neuropharmacology 2015, 98, 58–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Middleton, R.J.; Briddon, S.J.; Cordeaux, Y.; Yates, A.S.; Dale, C.L.; George, M.W.; Baker, J.G.; Hill, S.J.; Kellam, B. New fluorescent Adenosine A1—Receptor agonists that allow quantification of ligand−receptor interactions in microdomains of single living cells. J. Med. Chem. 2007, 50, 782–793. [Google Scholar] [CrossRef] [PubMed]
- Heisig, F.; Gollos, S.; Freudenthal, S.J.; El-Tayeb, A.; Iqbal, J.; Müller, C.E. Synthesis of BODIPY derivatives substituted with various bioconjugatable linker groups: A construction kit for fluorescent labeling of receptor ligands. J. Fluoresc. 2014, 24, 213–230. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.G.; Middleton, R.; Adams, L.; May, L.T.; Briddon, S.J.; Kellam, B.; Hill, S.J. Influence of fluorophore and linker composition on the pharmacology of fluorescent adenosine A1 receptor ligands: Themed section: Imaging in pharmacology research paper. Br. J. Pharmacol. 2010, 159, 772–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macchia, M.; Salvetti, F.; Bertini, S.; Bussolo, D.; Gattuso, L.; Gesi, M.; Hamdan, M.; Klotz, K.; Laragione, T.; Lucacchini, A.; et al. 7-Nitrobenzofurazan (NBD) derivatives of 5-N-ethylcarboxamidoadenosine (NECA) as new fluorescent probes for human A3 adenosine receptors. Bioorganic Med. Chem. Lett. 2001, 11, 3023–3026. [Google Scholar] [CrossRef]
- Dale, C.L.; Hill, S.J.; Kellam, B. New potent, short-linker BODIPY-630/650TM labelled fluorescent adenosine receptor agonists. Med. Chem. Commun. 2012, 3, 333–338. [Google Scholar] [CrossRef]
- Macchia, M.; Salvetti, F.; Barontini, S.; Calvani, F.; Gesi, M.; Hamdan, M.; Lucacchini, A.; Pellegrini, A.; Soldani, P.; Martini, C. Fluorescent probes for adenosine receptors: Synthesis and biology of N6-dansylaminoalkyl-substituted NECA derivatives. Bioorganic Med. Chem. Lett. 1998, 8, 3223–3228. [Google Scholar] [CrossRef]
- May, L.T.; Self, T.J.; Briddon, S.J.; Hill, S.J. The effect of allosteric modulators on the kinetics of agonist-G protein-coupled receptor interactions in single living cells. Mol. Pharmacol. 2010, 78, 511–523. [Google Scholar] [CrossRef] [PubMed]
- May, L.T.; Bridge, L.J.; Stoddart, L.A.; Briddon, S.J.; Hill, S.J. Allosteric interactions across native adenosine-A3 receptor homodimers: Quantification using single-cell ligand-binding kinetics. FASEB J. 2011, 25, 3465–3476. [Google Scholar] [CrossRef] [PubMed]
- Stoddart, L.A.; Vernall, A.J.; Briddon, S.J.; Kellam, B.; Hill, S.J. Direct visualisation of internalization of the adenosine A3 receptor and localization with arrestin3 using a fluorescent agonist. Neuropharmacology 2015, 98, 68–77. [Google Scholar] [CrossRef] [PubMed]
- McCabe, R.T.; Skolnick, P.; Jacobson, K.A. 2-[2-[4-[2-[2-[1,3-Dihydro-1,1-bis(4-hydroxyphenyl)-3-oxo-5-isobenzofuranthioureidyl]ethylaminocarbonyl]ethyl]phenyl]ethylamino]-5′-N-ethylcarboxamidoadenosine (FITC-APEC): A fluorescent ligand for A2A-Adenosine receptors. J. Fluoresc. 1992, 2, 217–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, F.; Klutz, A.M.; Jacobson, K.A.; Fredholm, B.B.; Schulte, G. Adenosine A2A receptor dynamics studied with the novel fluorescent agonist Alexa488-APEC. Eur. J. Pharmacol. 2008, 590, 36–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Dueñas, V.; Gómez-Soler, M.; Jacobson, K.A.; Kumar, S.T.; Fuxe, K.; Borroto-Escuela, D.O.; Ciruela, F. Molecular determinants of A2AR-D2R allosterism: Role of the intracellular loop 3 of the D2R. J. Neurochem. 2012, 123, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Sanjayan, G.J.; Kecskés, M.; Yoo, L.; Gao, Z.G.; Jacobson, K.A. Nucleoside conjugates of quantum dots for characterization of G protein-coupled receptors: Strategies for immobilizing A2A adenosine receptor agonists. J. Nanobiotechnol. 2010, 8, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tosh, D.K.; Chinn, M.; Ivanov, A.A.; Klutz, A.M.; Gao, Z.; Jacobson, K.A. Functionalized congeners of A3 adenosine receptor-selective nucleosides containing a bicycle [3.1.0]hexane ring system. J. Med. Chem. 2009, 52, 7580–7592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tosh, D.K.; Chinn, M.; Yoo, L.S.; Kang, D.W.; Luecke, H.; Gao, Z.; Jacobson, K.A. 2-Dialkynyl derivatives of (N)-methanocarba nucleosides: ‘Clickable’ A3 adenosine receptor-selective agonists. Bioorganic Med. Chem. 2010, 18, 508–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tosh, D.K.; Deflorian, F.; Phan, K.; Gao, Z.; Wan, T.C.; Gizewski, E.; Auchampach, J.A.; Jacobson, K.A. Structure-guided design of A3 adenosine receptor-selective nucleosides: Combination of 2-arylethynyl and bicyclo[3.1.0]hexane substitutions. J. Med. Chem. 2012, 55, 4847–4860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- May, L.T.; Briddon, S.J.; Hill, S.J. Antagonist selective modulation of adenosine A1 and A3 receptor pharmacology by the food dye brilliant black BN: Evidence for allosteric interactions. Mol. Pharmacol. 2010, 77, 678–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briddon, S.J.; Middleton, R.J.; Cordeaux, Y.; Flavin, F.M.; Weinstein, J.A.; George, M.W.; Kellam, B.; Hill, S.J. Quantitative analysis of the formation and diffusion of A1-adenosine receptor-antagonist complexes in single living cells. Proc. Natl. Acad. Sci. USA 2004, 101, 4673–4678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kecskés, A.; Tosh, D.K.; Wei, Q.; Gao, Z.-G.; Jacobson, K.A. GPCR ligand dendrimer (GLiDe) conjugates: Adenosine receptor interactions of a series of multivalent xanthine antagonists. Bioconjugate Chem. 2011, 22, 1115–1127. [Google Scholar]
- Vernall, A.J.; Stoddart, L.A.; Briddon, S.J.; Ng, H.W.; Laughton, C.A.; Doughty, S.W.; Hill, S.J.; Kellam, B. Conversion of a non-selective adenosine receptor antagonist into A3-selective high affinity fluorescent probes using peptide-based linkers. Org. Biomol. Chem. 2013, 11, 5673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoddart, L.A.; Vernall, A.J.; Denman, J.L.; Briddon, S.J.; Kellam, B.; Hill, S.J. Fragment screening at adenosine-A3 receptors in living cells using a fluorescence-based binding assay. Chem. Biol. 2012, 19, 1105–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corriden, R.; Kilpatrick, L.E.; Kellam, B.; Briddon, S.J.; Hill, S.J. Kinetic analysis of antagonist-occupied adenosine-A3 receptors within membrane microdomains of individual cells provides evidence of receptor dimerization and allosterism. FASEB J. 2014, 28, 4211–4222. [Google Scholar] [CrossRef] [PubMed]
- Bouzo-Lorenzo, M.; Stoddart, L.A.; Xia, L.; IJzerman, A.P.; Heitman, L.H.; Briddon, S.J.; Hill, S.J. A live cell NanoBRET binding assay allows the study of ligand-binding kinetics to the adenosine A3 receptor. Purinergic Signal. 2019, 15, 139–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, S.L.; Soave, M.; Jörg, M.; Scammells, P.J.; Woolard, J.; Hill, S.J. Probe dependence of allosteric enhancers on the binding affinity of adenosine A1—Receptor agonists at rat and human A1—Receptors measured using N ano BRET. Br. J. Pharmacol. 2019, 176, 864–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köse, M.; Gollos, S.; Karcz, T.; Fiene, A.; Heisig, F.; Behrenswerth, A.; Kieć-Kononowicz, K.; Namasivayam, V.; Müller, C.E. Fluorescent-labeled selective adenosine A2B receptor antagonist enables competition binding assay by flow cytometry. J. Med. Chem. 2018, 61, 4301–4316. [Google Scholar] [CrossRef] [PubMed]
- Kozma, E.; Kumar, T.S.; Federico, S.; Phan, K.; Balasubramanian, R.; Gao, Z.-G.; Paoletta, S.; Moro, S.; Spalluto, G.; Jacobson, K.A. Novel fluorescent antagonist as a molecular probe in A3 adenosine receptor binding assays using flow cytometry. Biochem. Pharmacol. 2012, 83, 1552–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernall, A.J.; Stoddart, L.A.; Briddon, S.J.; Hill, S.J.; Kellam, B. Highly potent and selective fluorescent antagonists of the human adenosine A3 receptor based on the 1,2,4-triazolo[4,3-a]quinoxalin-1-one scaffold. J. Med. Chem. 2012, 55, 1771–1782. [Google Scholar] [CrossRef] [PubMed]
- Kecskés, M.; Kumar, T.S.; Yoo, L.; Gao, Z.G.; Jacobson, K.A. Novel Alexa Fluor-488 labeled antagonist of the A2A adenosine receptor: Application to a fluorescence polarization-based receptor binding assay. Biochem. Pharmacol. 2010, 80, 506–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, T.S.; Mishra, S.; Deflorian, F.; Yoo, L.S.; Phan, K.; Kecskés, M.; Szabo, A.; Shinkre, B.; Gao, Z.-G.; Trenkle, W.; et al. Molecular probes for the A2A adenosine receptor based on a pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-amine scaffold. Bioorganic Med. Chem. Lett. 2011, 21, 2740–2745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Federico, S.; Margiotta, E.; Paoletta, S.; Kachler, S.; Klotz, K.-N.; Jacobson, K.A.; Pastorin, G.; Moro, S.; Spalluto, G. Pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidines to develop functionalized ligands to target adenosine receptors: Fluorescent ligands as an example. Medchemcomm 2019, 10, 1094–1108. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Duenas, V.; Taura, J.J.; Cottet, M.; Gomez-Soler, M.; Lopez-Cano, M.; Ledent, C.; Watanabe, M.; Trinquet, E.; Pin, J.-P.; Lujan, R.; et al. Untangling dopamine-adenosine receptor-receptor assembly in experimental parkinsonism in rats. Dis. Model. Mech. 2015, 8, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barresi, E.; Giacomelli, C.; Daniele, S.; Tonazzini, I.; Robello, M.; Salerno, S.; Piano, I.; Cosimelli, B.; Greco, G.; Da, F.; et al. Novel fluorescent triazinobenzimidazole derivatives as probes for labelling human A1 and A2B adenosine receptor subtypes. Bioorganic Med. Chem. 2018, 26, 5885–5895. [Google Scholar] [CrossRef] [PubMed]
- Redenti, S.; Ciancetta, A.; Pastorin, G.; Cacciari, B.; Moro, S.; Spalluto, G.; Federico, S. Pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidines and structurally simplified analogs. Chemistry and SAR profile as adenosine receptor antagonists. Curr. Top. Med. Chem. 2016, 16, 3224–3257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, K.A.; Thatikonda, S.K.; Kozma, E.E.; Spalluto, G.; Moro, S.; Federico, S. Fluorescent Antagonists of the A3 Adenosine Receptor. Patent US9227979B2, 5 January 2016. [Google Scholar]
Figure 1.
Structure of BAY60-6583.

Figure 2.
Structures of 11C labeled adenosine receptor (AR) ligands.

Figure 3.
Structures of 18F-labeled AR ligands.

Figure 4.
Structures of covalent A1 AR agonists.

Figure 5.
Structures of covalent A2A and A3 AR agonists.

Figure 6.
Structures of xanthine derivatives as covalent antagonists for ARs.

Figure 7.
Structures of non-xanthinic derivatives as covalent antagonists for ARs.

Figure 8.
Structures of the first examples of fluorescent ligands for ARs.

Figure 9.
Structures of representative fluorescent agonist ligands for A1 and A3 ARs.

Figure 10.
Structures of representative fluorescent agonist ligands for A2A and A3 ARs.

Figure 11.
Structures of representative fluorescent xanthine antagonist ligands for A1 and A3 ARs.

Figure 12.
Structures of the fluorescent xanthine A2B AR antagonist PSB-12105 (42).

Figure 13.
Structures of tricyclic fluorescent A2A and A3 ARs antagonists. aAlexa Fluor 488 5 isomer.
Figure 13.
Structures of tricyclic fluorescent A2A and A3 ARs antagonists. aAlexa Fluor 488 5 isomer.

Figure 14.
Structures of other tricyclic fluorescent AR antagonists.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Federico, S.; Lassiani, L.; Spalluto, G. Chemical Probes for the Adenosine Receptors. Pharmaceuticals 2019, 12, 168. https://0-doi-org.brum.beds.ac.uk/10.3390/ph12040168
AMA Style
Federico S, Lassiani L, Spalluto G. Chemical Probes for the Adenosine Receptors. Pharmaceuticals. 2019; 12(4):168. https://0-doi-org.brum.beds.ac.uk/10.3390/ph12040168
Chicago/Turabian StyleFederico, Stephanie, Lucia Lassiani, and Giampiero Spalluto. 2019. "Chemical Probes for the Adenosine Receptors" Pharmaceuticals 12, no. 4: 168. https://0-doi-org.brum.beds.ac.uk/10.3390/ph12040168
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.