Development of a High-throughput Agar Colony Formation Assay to Identify Drug Candidates against Medulloblastoma



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
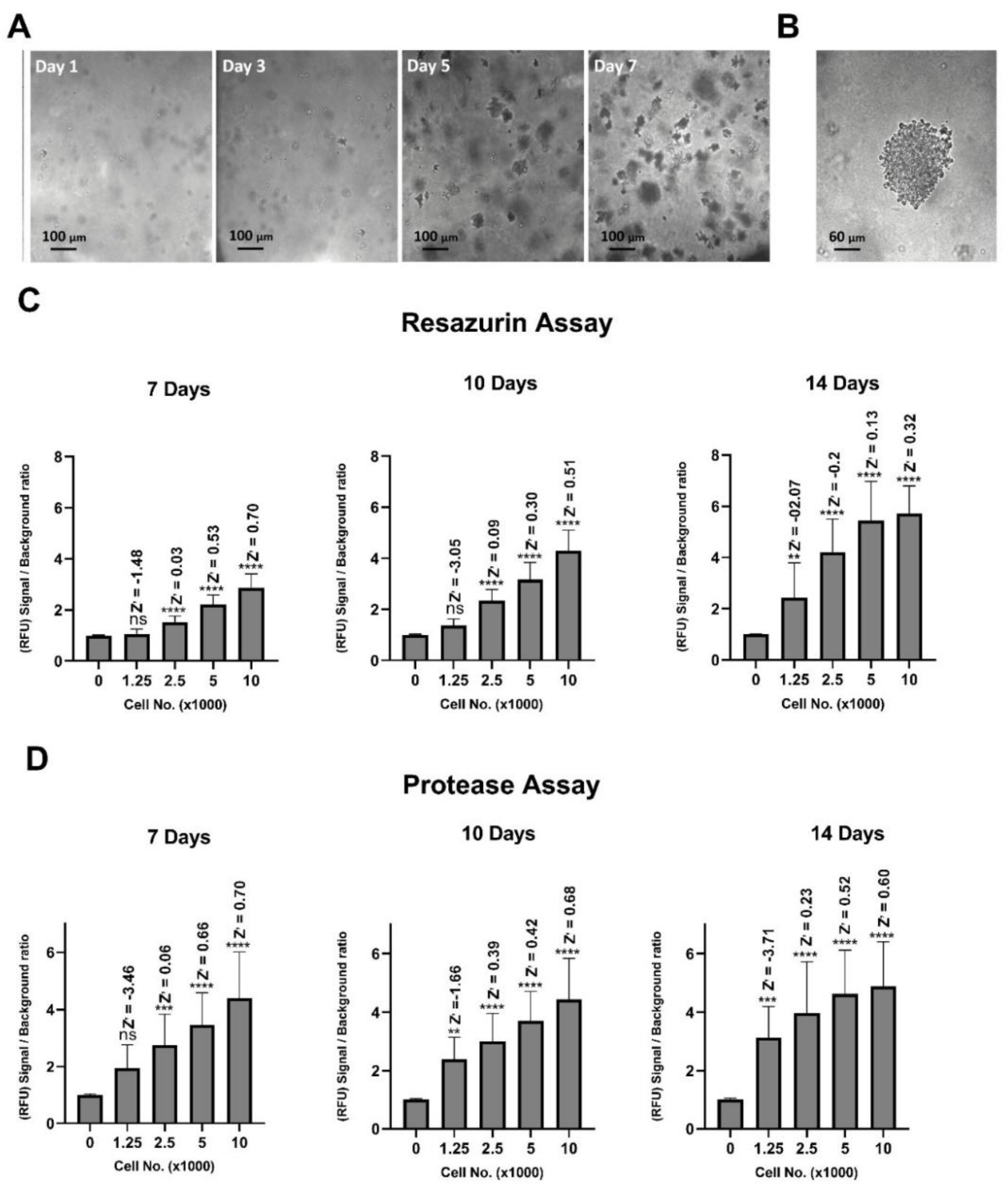
2.1. Optimisation of Cell Seeding Density and Culture Time
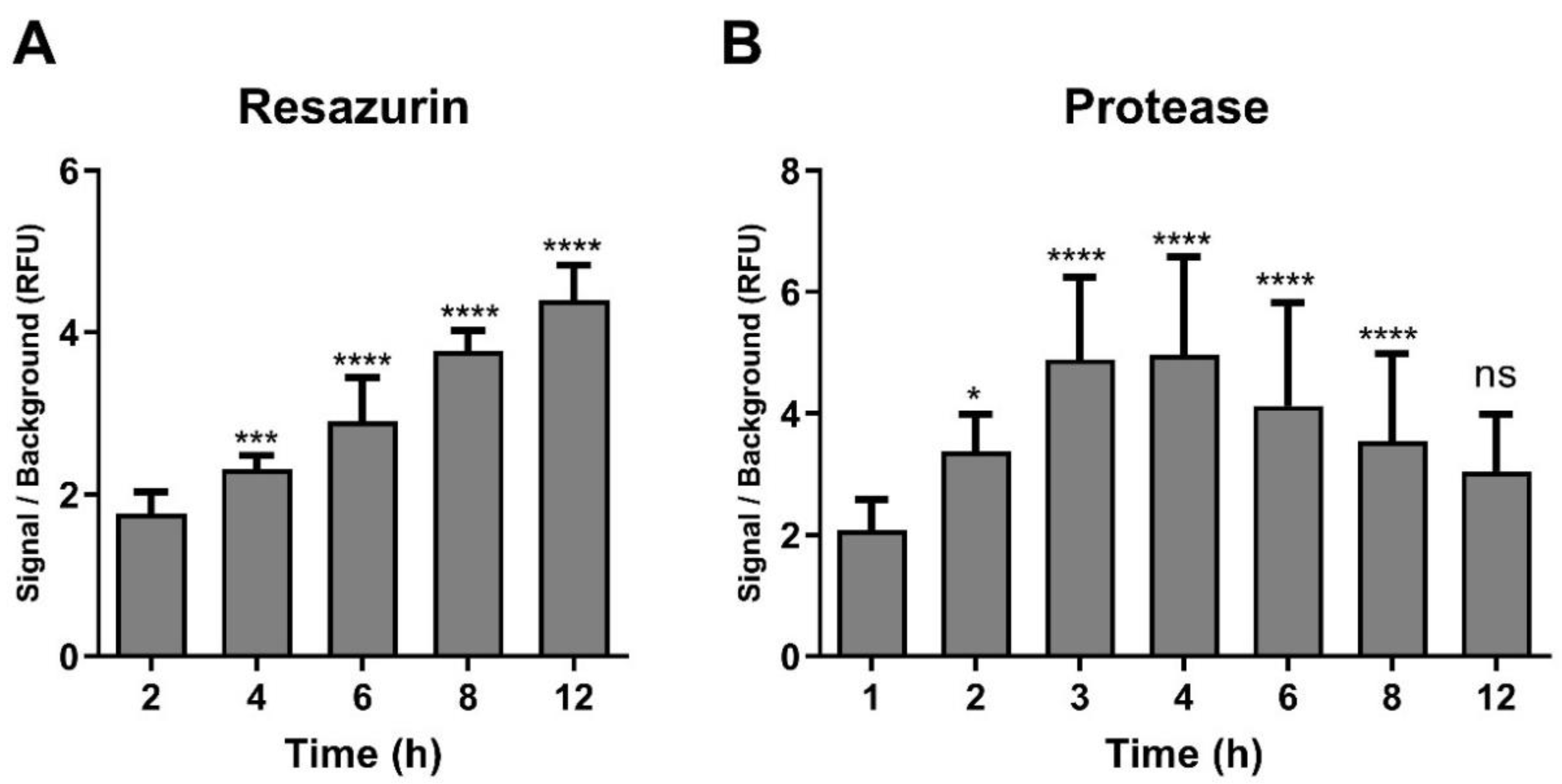
2.2. Optimisation of Incubation Time with Resazurin and GF-AFC Substrates
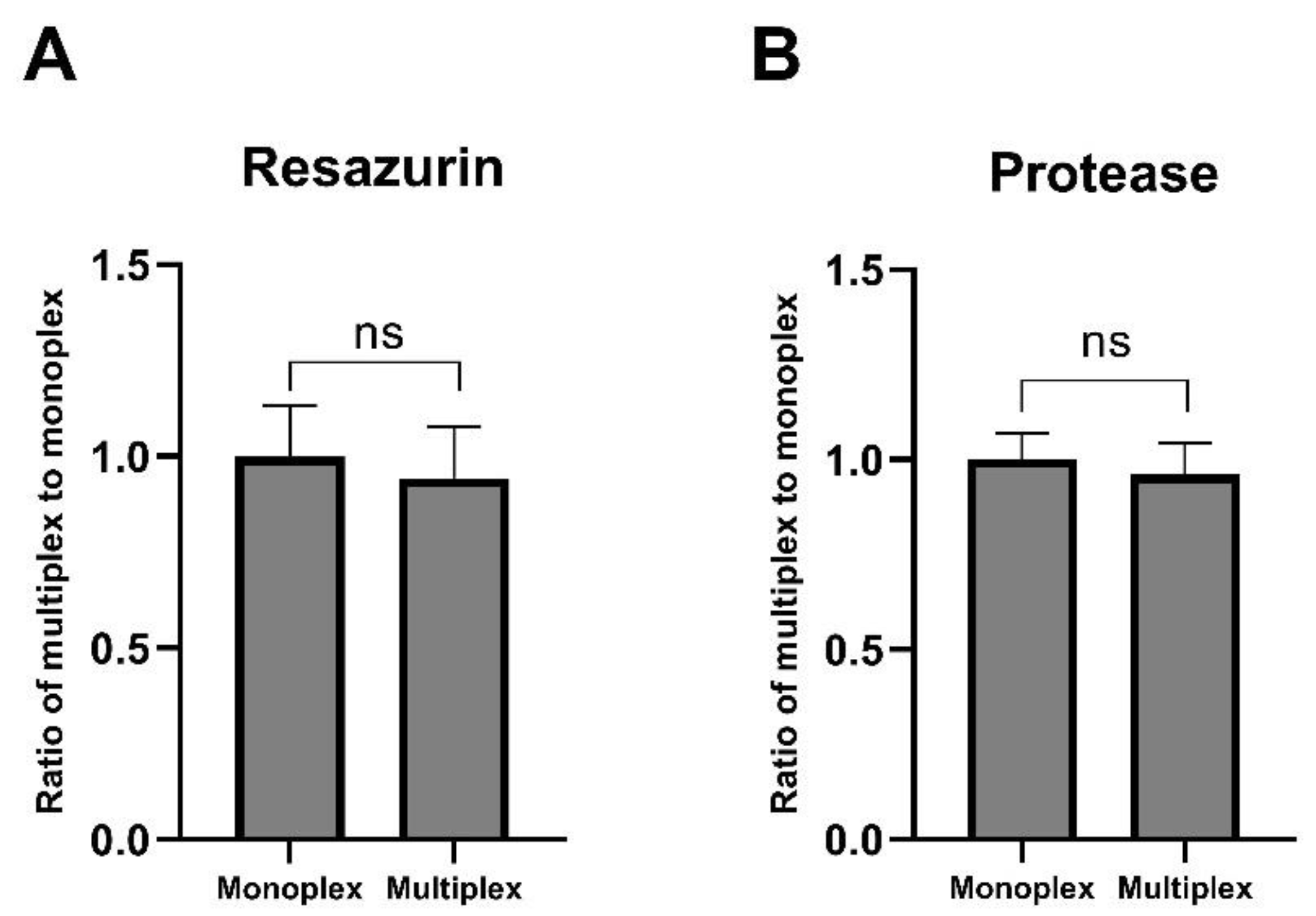
2.3. Multiplex Assay Validation
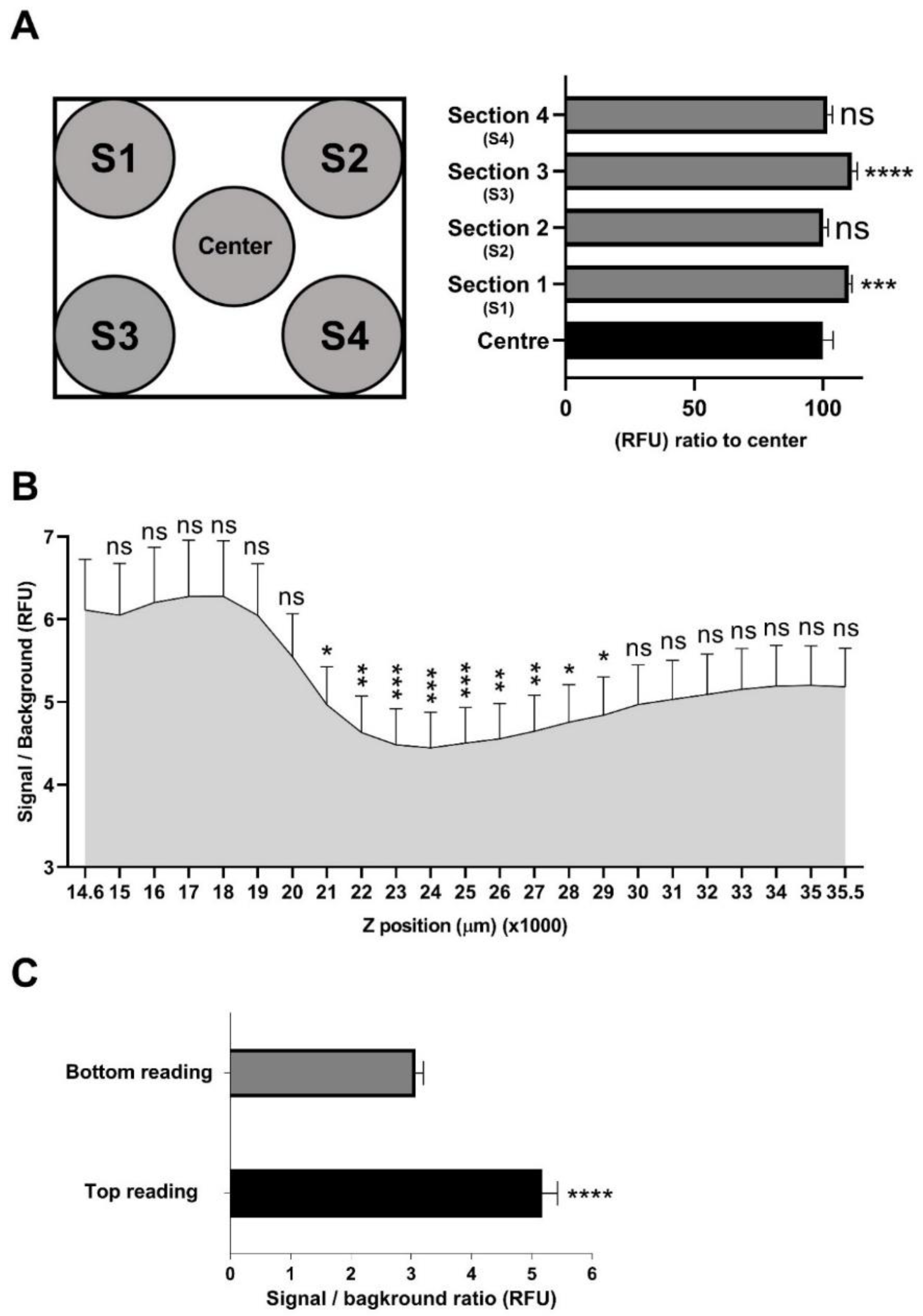
2.4. Assessment of Signal Distribution across a Well
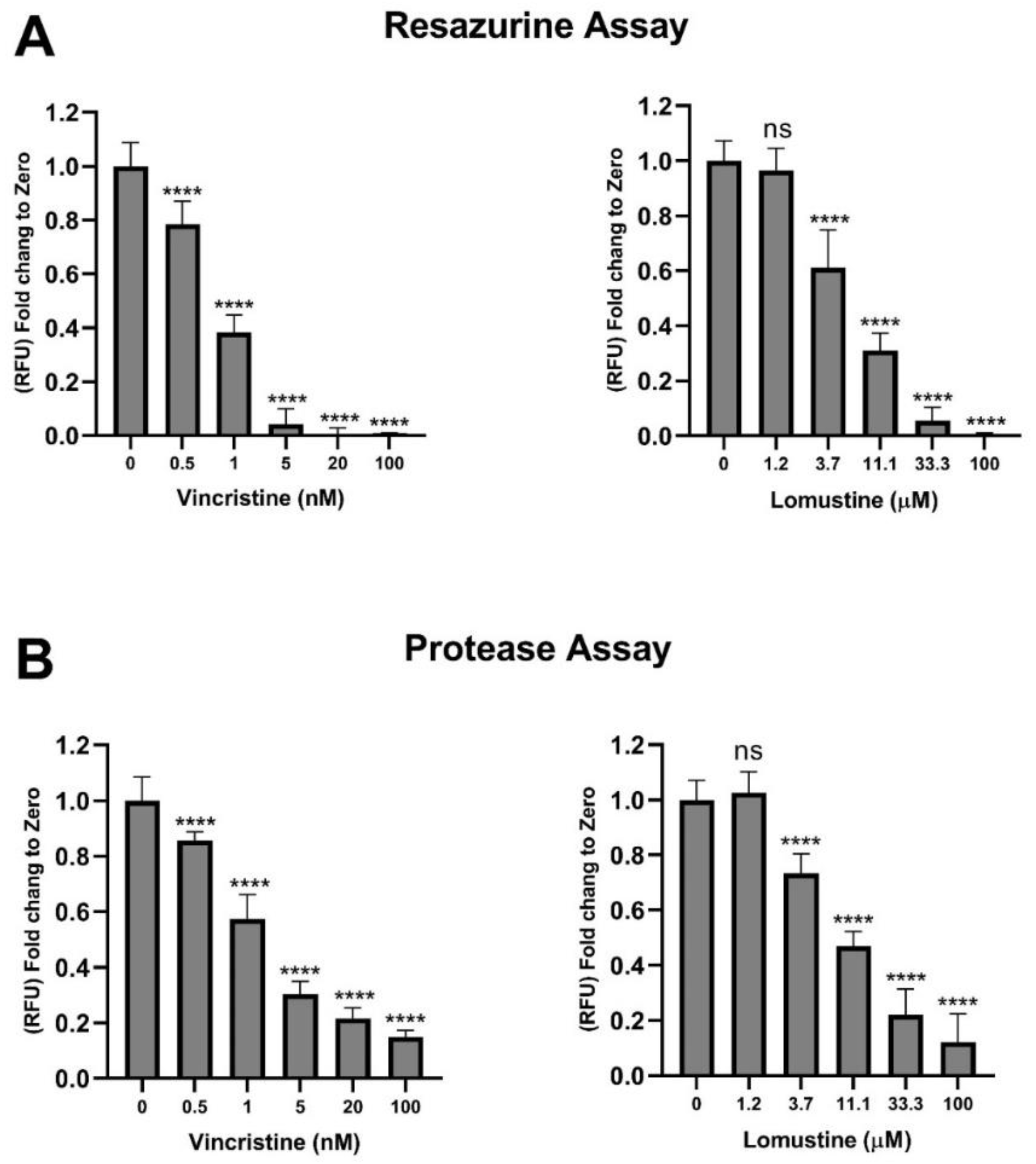
2.5. Assessment of Vincristine and Lomustine Toxicity
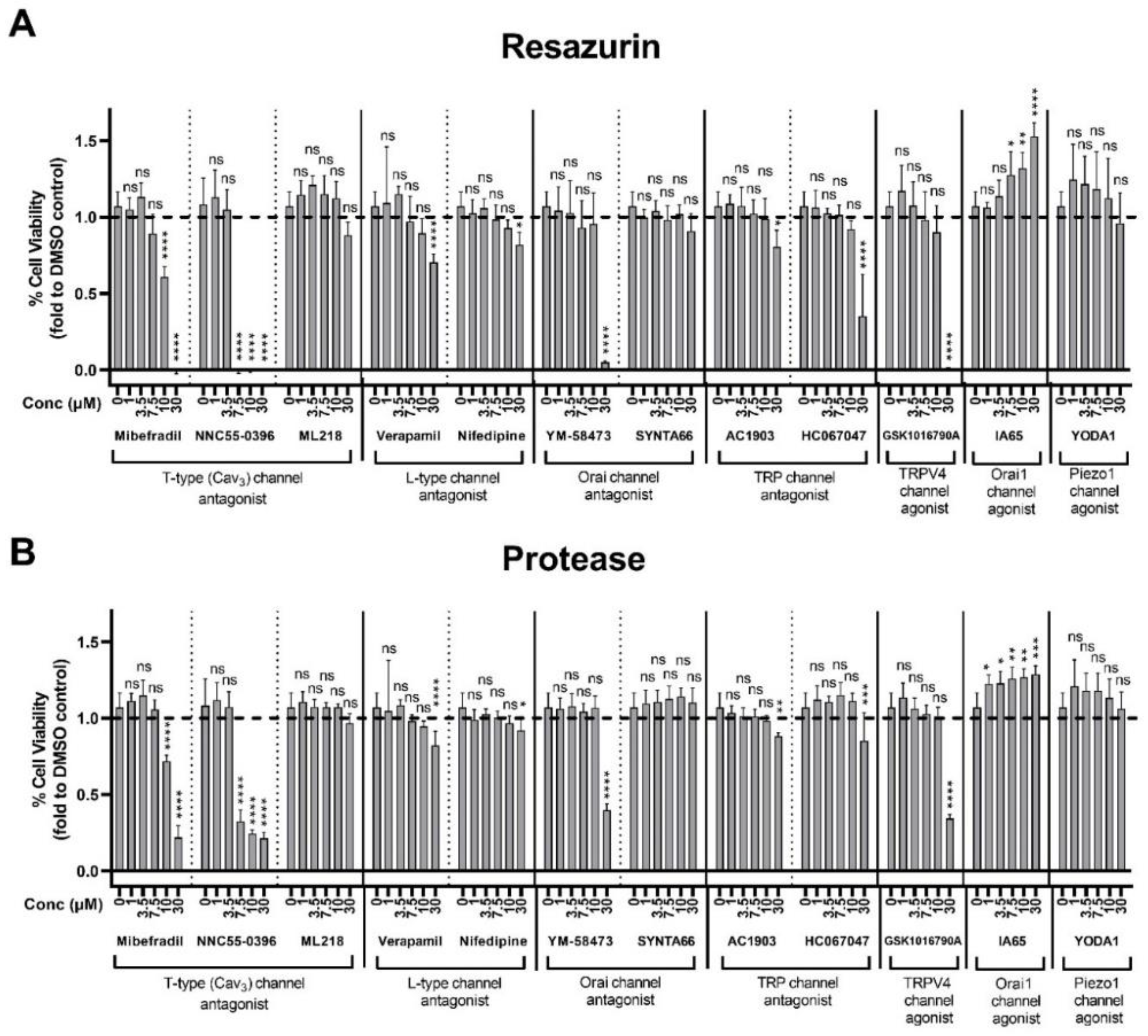
2.6. Assessment of Plasma Membrane Calcium Channel Modulators
3. Discussion
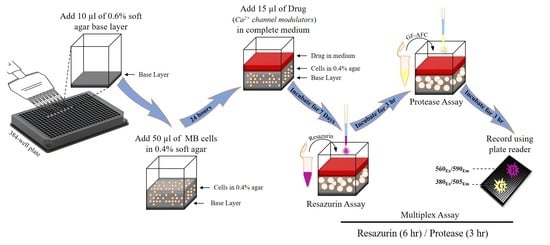
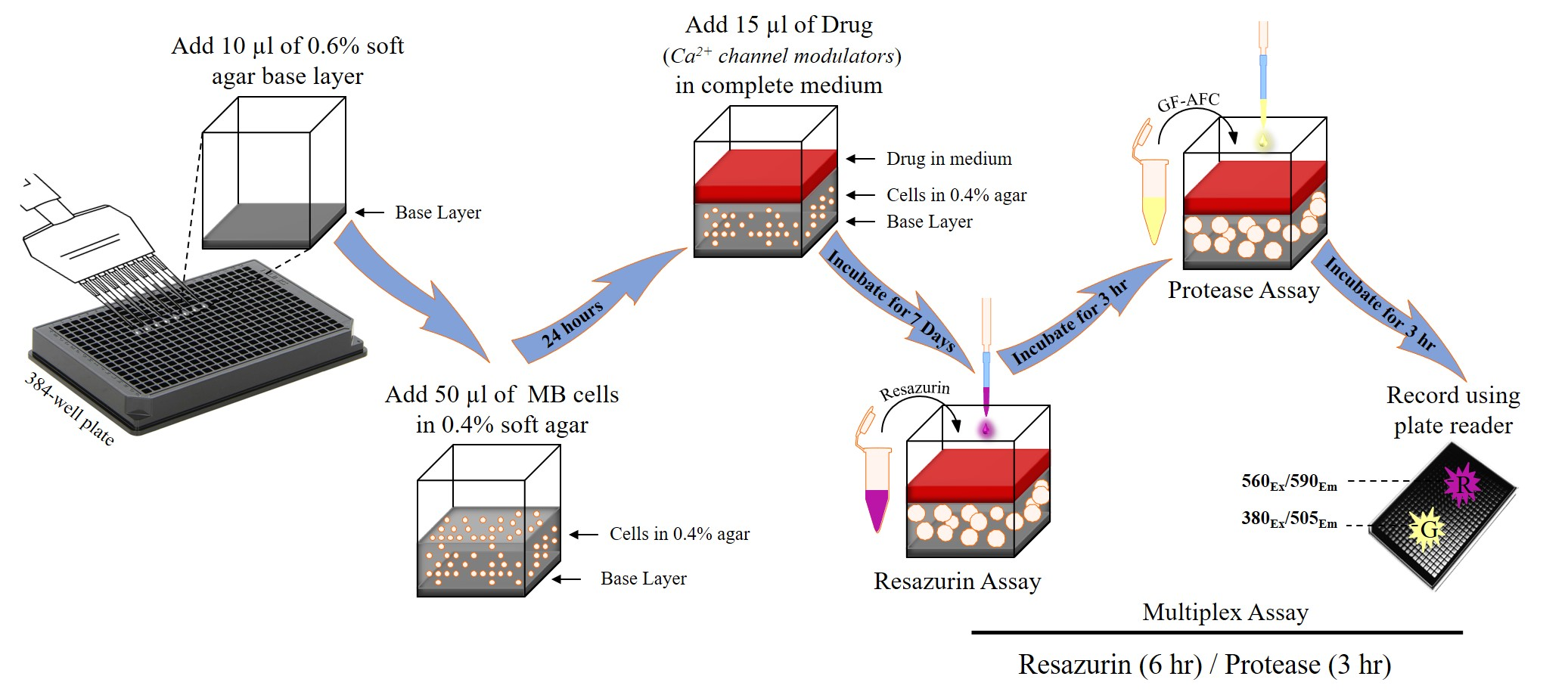
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Culture
4.3. Preparation of Agar Layers
4.4. Cell Viability Measurement
4.5. Addition of Test Compounds
4.6. Calculation of Signal to Background Ratio and Z’-factor
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ramaswamy, V.; Remke, M.; Bouffet, E.; Bailey, S.; Clifford, S.C.; Doz, F.; Kool, M.; Dufour, C.; Vassal, G.; Milde, T. Risk stratification of childhood medulloblastoma in the molecular era: The current consensus. Acta Neuropathol. 2016, 131, 821–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddrey, A.M.; Bergeron, J.A.; Lombardo, E.R.; McDonald, N.K.; Mulne, A.F.; Barenberg, P.D.; Bowers, D.C. Neuropsychological performance and quality of life of 10 year survivors of childhood medulloblastoma. J. Neuro Oncol. 2005, 72, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Robertson, P.L.; Muraszko, K.M.; Holmes, E.J.; Sposto, R.; Packer, R.J.; Gajjar, A.; Dias, M.S.; Allen, J.C. Incidence and severity of postoperative cerebellar mutism syndrome in children with medulloblastoma: A prospective study by the Children’s Oncology Group. J. Neurosurg. Pediatrics 2006, 105, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Noël, G. Medulloblastoma: Optimizing care with a multidisciplinary approach. J. Multidiscip. Healthc. 2019, 12, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, D.P.; Coyle, B.; Walker, D.A.; Grabowska, A.M. In vitro models of medulloblastoma: Choosing the right tool for the job. J. Biotechnol. 2016, 236, 10–25. [Google Scholar] [CrossRef] [PubMed]
- Ward, E.; DeSantis, C.; Robbins, A.; Kohler, B.; Jemal, A. Childhood and adolescent cancer statistics, 2014. CA A Cancer J. Clin. 2014, 64, 83–103. [Google Scholar] [CrossRef]
- Yang, W.Q.; Senger, D.; Muzik, H.; Shi, Z.Q.; Johnson, D.; Brasher, P.M.; Rewcastle, N.B.; Hamilton, M.; Rutka, J.; Wolff, J. Reovirus prolongs survival and reduces the frequency of spinal and leptomeningeal metastases from medulloblastoma. Cancer Res. 2003, 63, 3162–3172. [Google Scholar]
- Fults, D.W.; Taylor, M.D.; Garzia, L. Leptomeningeal dissemination: A sinister pattern of medulloblastoma growth. J. Neurosurg. Pediatrics 2019, 23, 613–621. [Google Scholar] [CrossRef]
- Guadamillas, M.C.; Cerezo, A.; Del Pozo, M.A. Overcoming anoikis–pathways to anchorage-independent growth in cancer. J. Cell Sci. 2011, 124, 3189–3197. [Google Scholar] [CrossRef] [Green Version]
- Hamburger, A.W.; Salmon, S.E. Primary bioassay of human tumor stem cells. Science 1977, 197, 461–463. [Google Scholar] [CrossRef]
- Wang, L.-H. Molecular signaling regulating anchorage-independent growth of cancer cells. Mount Sinai J. Med. N. Y. 2004, 71, 361–367. [Google Scholar]
- Borowicz, S.; Van Scoyk, M.; Avasarala, S.; Rathinam, M.K.K.; Tauler, J.; Bikkavilli, R.K.; Winn, R.A. The soft agar colony formation assay. J. Vis. Exp. 2014, 92, e51998. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.B.; Anzano, M.A.; Wakefield, L.M.; Roche, N.S.; Stern, D.F.; Sporn, M.B. Type beta transforming growth factor: A bifunctional regulator of cellular growth. Proc. Natl. Acad. Sci. USA 1985, 82, 119–123. [Google Scholar] [CrossRef] [Green Version]
- Freedman, V.H.; Shin, S.-I. Cellular tumorigenicity in nude mice: Correlation with cell growth in semi-solid medium. Cell 1974, 3, 355–359. [Google Scholar] [CrossRef]
- Horibata, S.; Vo, T.V.; Subramanian, V.; Thompson, P.R.; Coonrod, S.A. Utilization of the soft agar colony formation assay to identify inhibitors of tumorigenicity in breast cancer cells. J. Vis. Exp. 2015, 99, e52727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, S.N.; Towne, D.L.; Burns, D.J.; Warrior, U. A high-throughput soft agar assay for identification of anticancer compound. J. Biomol. Screen. 2007, 12, 938–945. [Google Scholar] [CrossRef]
- Weaver, V.M.; Lelièvre, S.; Lakins, J.N.; Chrenek, M.A.; Jones, J.C.; Giancotti, F.; Werb, Z.; Bissell, M.J. β4 integrin-dependent formation of polarized three-dimensional architecture confers resistance to apoptosis in normal and malignant mammary epithelium. Cancer Cell 2002, 2, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Bhadriraju, K.; Chen, C.S. Engineering cellular microenvironments to improve cell-based drug testing. Drug Discov. Today 2002, 7, 612–620. [Google Scholar] [CrossRef]
- Fukazawa, H.; Noguchi, K.; Murakami, Y.; Uehara, Y. Mitogen-activated Protein/Extracellular Signal-regulated Kinase Kinase (MEK) Inhibitors Restore Anoikis Sensitivity in Human Breast Cancer Cell Lines with a Constitutively Activated Extracellular-regulated Kinase (ERK) Pathway. Mol. Cancer Ther. 2002, 1, 303–309. [Google Scholar]
- Zhang, J.-H.; Chung, T.D.; Oldenburg, K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef]
- Protocol, C.E.V.A. CellTiter-Fluor™ Cell Viability Assay. Available online: https://www.promega.com.au/resources/protocols/technical-bulletins/101/celltiter-fluor-cell-viability-assay-protocol/ (accessed on 5 November 2020).
- Hall, M.D.; Simeonov, A.; Davis, M.I. Avoiding fluorescence assay interference—The case for diaphorase. Assay Drug Dev. Technol. 2016, 14, 175–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horman, S.R.; To, J.; Orth, A.P. An HTS-compatible 3D colony formation assay to identify tumor-specific chemotherapeutics. J. Biomol. Screen. 2013, 18, 1298–1308. [Google Scholar] [CrossRef] [Green Version]
- Monteith, G.R.; Prevarskaya, N.; Roberts-Thomson, S.J. The calcium–cancer signalling nexus. Nat. Rev. Cancer 2017, 17, 367. [Google Scholar] [CrossRef] [Green Version]
- Maklad, A.; Sharma, A.; Azimi, I. Calcium signaling in brain cancers: Roles and therapeutic targeting. Cancers 2019, 11, 145. [Google Scholar] [CrossRef] [Green Version]
- Prevarskaya, N.; Ouadid-Ahidouch, H.; Skryma, R.; Shuba, Y. Remodelling of Ca2+ transport in cancer: How it contributes to cancer hallmarks? Philos. Trans. R. Soc. 2014, 369, 20130097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteith, G.R.; McAndrew, D.; Faddy, H.M.; Roberts-Thomson, S.J. Calcium and cancer: Targeting Ca2+ transport. Nat. Rev. Cancer 2007, 7, 519–530. [Google Scholar] [CrossRef]
- Azimi, I.; Roberts-Thomson, S.; Monteith, G. Calcium influx pathways in breast cancer: Opportunities for pharmacological intervention. Br. J. Pharmacol. 2014, 171, 945–960. [Google Scholar] [CrossRef] [Green Version]
- Bong, A.H.; Monteith, G.R. Calcium signaling and the therapeutic targeting of cancer cells. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2018, 1865, 1786–1794. [Google Scholar] [CrossRef]
- Roberts-Thomson, S.J.; Chalmers, S.B.; Monteith, G.R. The calcium-signaling toolkit in cancer: Remodeling and targeting. Cold Spring Harb. Perspect. Biol. 2019, 11, a035204. [Google Scholar] [CrossRef] [Green Version]
- Martin, R.L.; Lee, J.-H.; Cribbs, L.L.; Perez-Reyes, E.; Hanck, D.A. Mibefradil block of cloned T-type calcium channels. J. Pharmacol. Exp. Ther. 2000, 295, 302–308. [Google Scholar]
- Huang, L.; Keyser, B.M.; Tagmose, T.M.; Hansen, J.B.; Taylor, J.T.; Zhuang, H.; Zhang, M.; Ragsdale, D.S.; Li, M. NNC 55-0396 [(1S, 2S)-2-(2-(N-[(3-benzimidazol-2-yl) propyl]-N-methylamino) ethyl)-6-fluoro-1, 2, 3, 4-tetrahydro-1-isopropyl-2-naphtyl cyclopropanecarboxylate dihydrochloride]: A new selective inhibitor of T-type calcium channels. J. Pharmacol. Exp. Ther. 2004, 309, 193–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.-W.; Oh, H.A.; Lee, S.H.; Kim, K.C.; Eun, P.H.; Ko, M.J.; Gonzales, E.L.T.; Seung, H.; Kim, S.; Bahn, G.H. T-type calcium channels are required to maintain viability of neural progenitor cells. Biomol. Ther. 2018, 26, 439. [Google Scholar] [CrossRef]
- Xiang, Z.; Thompson, A.D.; Brogan, J.T.; Schulte, M.L.; Melancon, B.J.; Mi, D.; Lewis, L.M.; Zou, B.; Yang, L.; Morrison, R. The discovery and characterization of ML218: A novel, centrally active T-type calcium channel inhibitor with robust effects in STN neurons and in a rodent model of Parkinson’s disease. ACS Chem. Neurosci. 2011, 2, 730–742. [Google Scholar] [CrossRef]
- Zhang, Y.; Cruickshanks, N.; Yuan, F.; Wang, B.; Pahuski, M.; Wulfkuhle, J.; Gallagher, I.; Koeppel, A.F.; Hatef, S.; Papanicolas, C. Targetable T-type calcium channels drive glioblastoma. Cancer Res. 2017, 77, 3479–3490. [Google Scholar] [CrossRef] [Green Version]
- Holdhoff, M.; Ye, X.; Supko, J.G.; Nabors, L.B.; Desai, A.S.; Walbert, T.; Lesser, G.J.; Read, W.L.; Lieberman, F.S.; Lodge, M.A. Timed sequential therapy of the selective T-type calcium channel blocker mibefradil and temozolomide in patients with recurrent high-grade gliomas. Neuro Oncol. 2017, 19, 845–852. [Google Scholar] [CrossRef] [Green Version]
- Quesada, A.; Bui, P.H.; Homanics, G.E.; Hankinson, O.; Handforth, A. Comparison of mibefradil and derivative NNC 55-0396 effects on behavior, cytochrome P450 activity, and tremor in mouse models of essential tremor. Eur. J. Pharmacol. 2011, 659, 30–36. [Google Scholar] [CrossRef] [Green Version]
- Azimi, I.; Stevenson, R.J.; Zhang, X.; Meizoso-Huesca, A.; Xin, P.; Johnson, M.; Flanagan, J.U.; Chalmers, S.B.; Yoast, R.E.; Kapure, J.S. A new selective pharmacological enhancer of the Orai1 Ca2+ channel reveals roles for Orai1 in smooth and skeletal muscle functions. ACS Pharmacol. Transl. Sci. 2020, 3, 135–147. [Google Scholar] [CrossRef] [Green Version]
- Putney, J.W.; Steinckwich-Besançon, N.; Numaga-Tomita, T.; Davis, F.M.; Desai, P.N.; D’Agostin, D.M.; Wu, S.; Bird, G.S. The functions of store-operated calcium channels. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2017, 1864, 900–906. [Google Scholar] [CrossRef]
- Azimi, I.; Milevskiy, M.J.; Chalmers, S.B.; Yapa, K.T.; Robitaille, M.; Henry, C.; Baillie, G.J.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. ORAI1 and ORAI3 in breast cancer molecular subtypes and the identification of ORAI3 as a hypoxia sensitive gene and a regulator of hypoxia responses. Cancers 2019, 11, 208. [Google Scholar] [CrossRef] [Green Version]
- Azimi, I.; Bong, A.H.; Poo, G.X.; Armitage, K.; Lok, D.; Roberts-Thomson, S.J.; Monteith, G.R. Pharmacological inhibition of store-operated calcium entry in MDA-MB-468 basal A breast cancer cells: Consequences on calcium signalling, cell migration and proliferation. Cell. Mol. Life Sci. 2018, 75, 4525–4537. [Google Scholar] [CrossRef]
- Xia, J.; Wang, H.; Huang, H.; Sun, L.; Dong, S.; Huang, N.; Shi, M.; Bin, J.; Liao, Y.; Liao, W. Elevated Orai1 and STIM1 expressions upregulate MACC1 expression to promote tumor cell proliferation, metabolism, migration, and invasion in human gastric cancer. Cancer Lett. 2016, 381, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Motiani, R.K.; Hyzinski-García, M.C.; Zhang, X.; Henkel, M.M.; Abdullaev, I.F.; Kuo, Y.-H.; Matrougui, K.; Mongin, A.A.; Trebak, M. STIM1 and Orai1 mediate CRAC channel activity and are essential for human glioblastoma invasion. Pflügers Arch. Eur. J. Physiol. 2013, 465, 1249–1260. [Google Scholar] [CrossRef] [Green Version]
- Riss, T.L.; Moravec, R.A.; Niles, A.L.; Duellman, S.; Benink, H.A.; Worzella, T.J.; Minor, L. Cell viability assays. In Assay Guidance ManualInternet; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Indianapolis, IN, USA; Bethesda, MD, USA, 2016. [Google Scholar]
- Stoddart, M.J. Cell viability assays: Introduction. In Mammalian Cell Viability; Springer: Berlin, Germany, 2011; pp. 1–6. [Google Scholar]
- Niles, A.L.; Moravec, R.A.; Hesselberth, P.E.; Scurria, M.A.; Daily, W.J.; Riss, T.L. A homogeneous assay to measure live and dead cells in the same sample by detecting different protease markers. Anal. Biochem. 2007, 366, 197–206. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sedeeq, M.; Maklad, A.; Gueven, N.; Azimi, I. Development of a High-throughput Agar Colony Formation Assay to Identify Drug Candidates against Medulloblastoma. Pharmaceuticals 2020, 13, 368. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13110368
Sedeeq M, Maklad A, Gueven N, Azimi I. Development of a High-throughput Agar Colony Formation Assay to Identify Drug Candidates against Medulloblastoma. Pharmaceuticals. 2020; 13(11):368. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13110368
Chicago/Turabian StyleSedeeq, Mohammed, Ahmed Maklad, Nuri Gueven, and Iman Azimi. 2020. "Development of a High-throughput Agar Colony Formation Assay to Identify Drug Candidates against Medulloblastoma" Pharmaceuticals 13, no. 11: 368. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13110368