Enhancing the Low Oral Bioavailability of Sulpiride via Fast Orally Disintegrating Tablets: Formulation, Optimization and In Vivo Characterization



Abstract
:1. Introduction
2. Results and Discussion
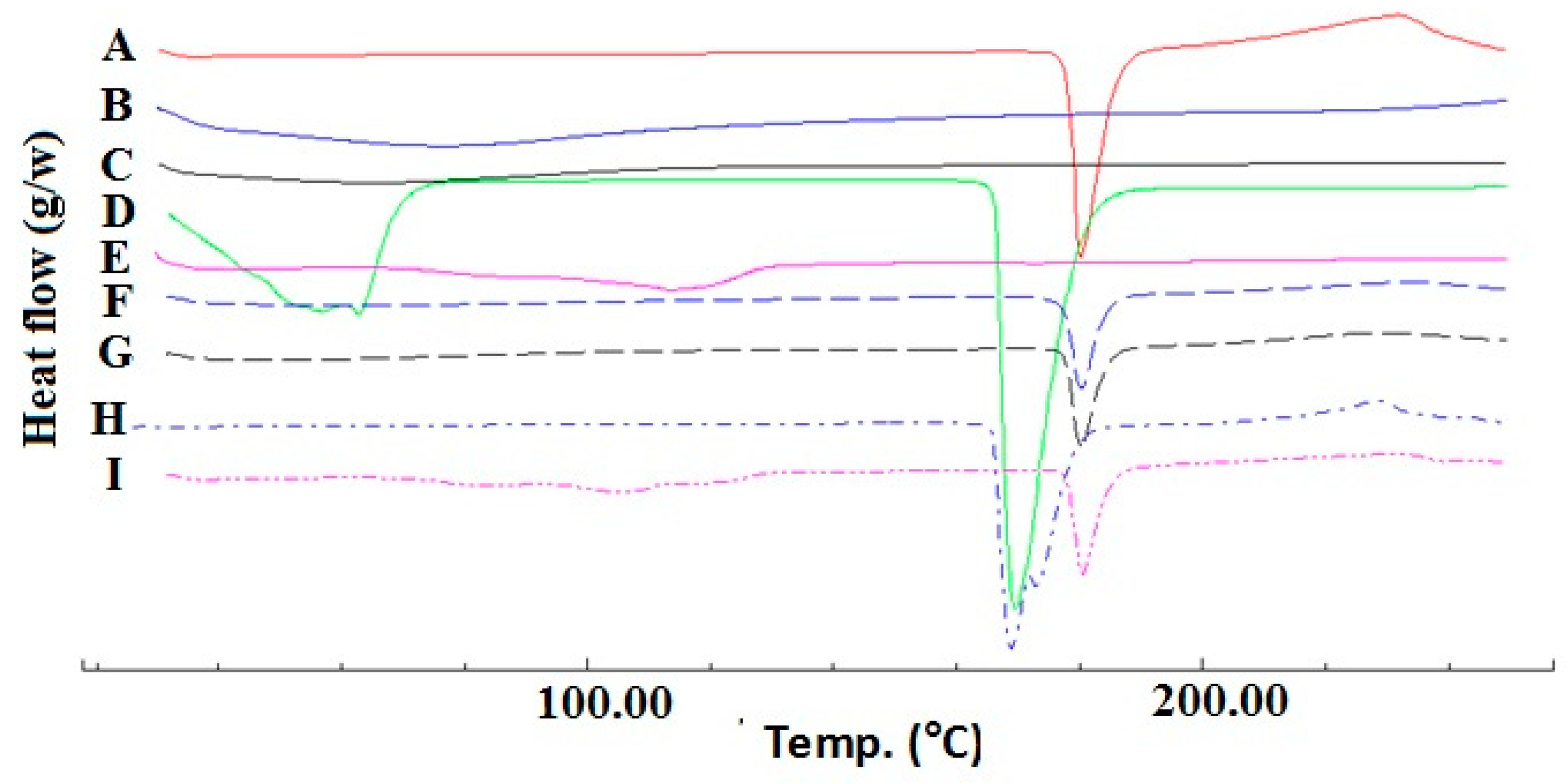
2.1. Compatibility Study
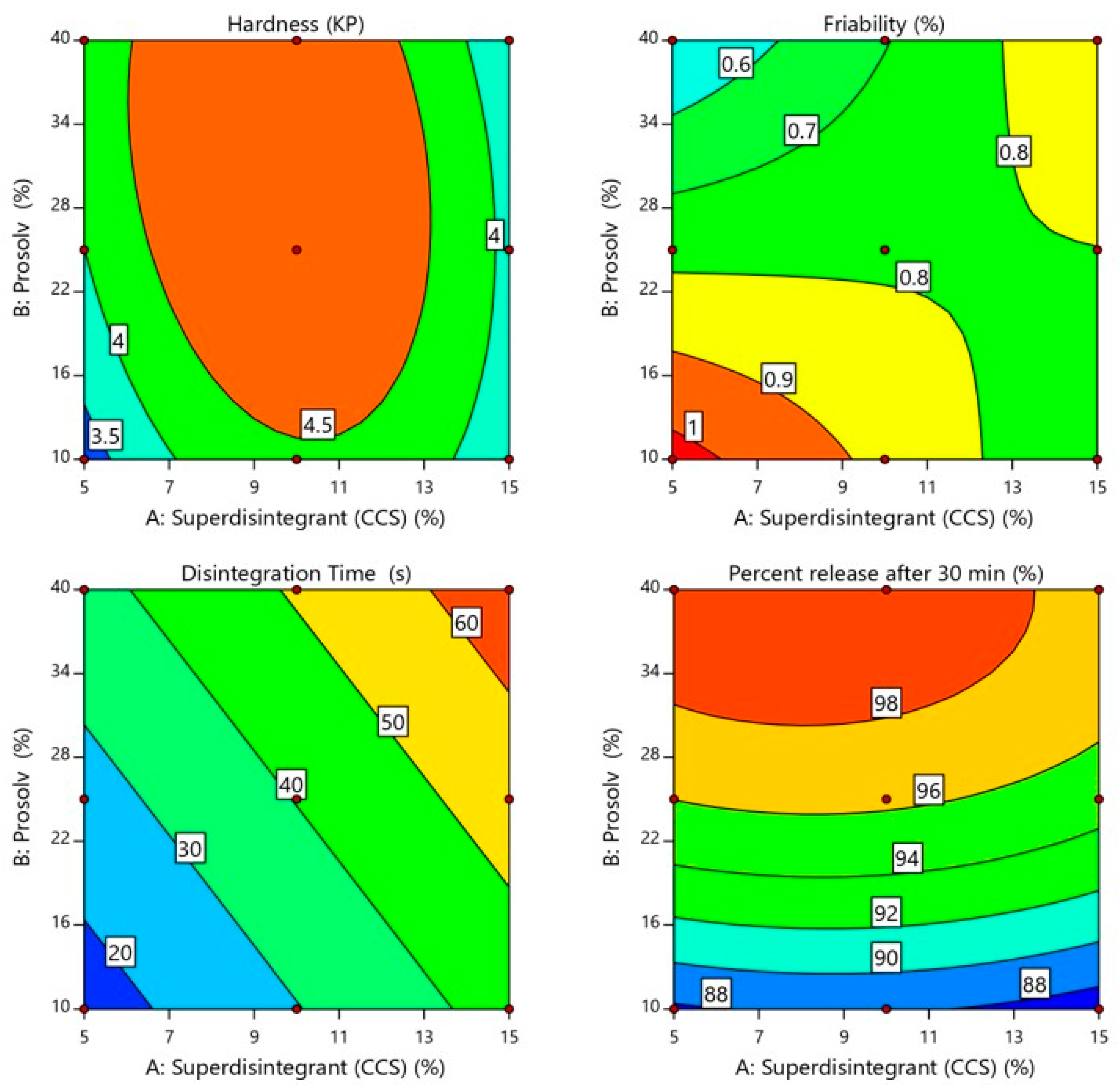
2.2. Effect of Independent Variables on SUL-FDTs Properties
2.2.1. Weight Uniformity and Content Uniformity
2.2.2. Hardness and Friability
2.2.3. In Vitro SUL-FDTs Disintegration
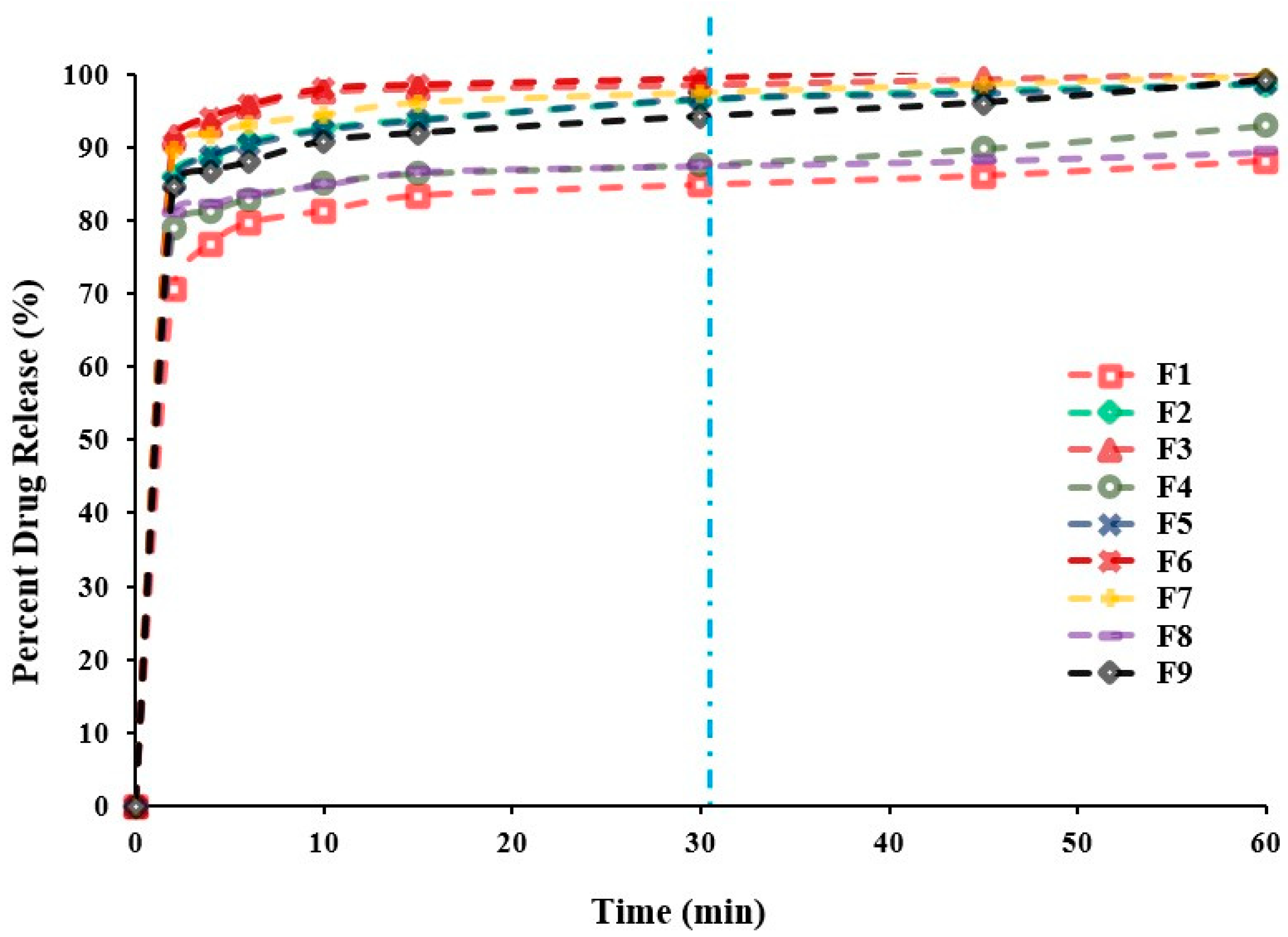
2.2.4. In Vitro Release Study
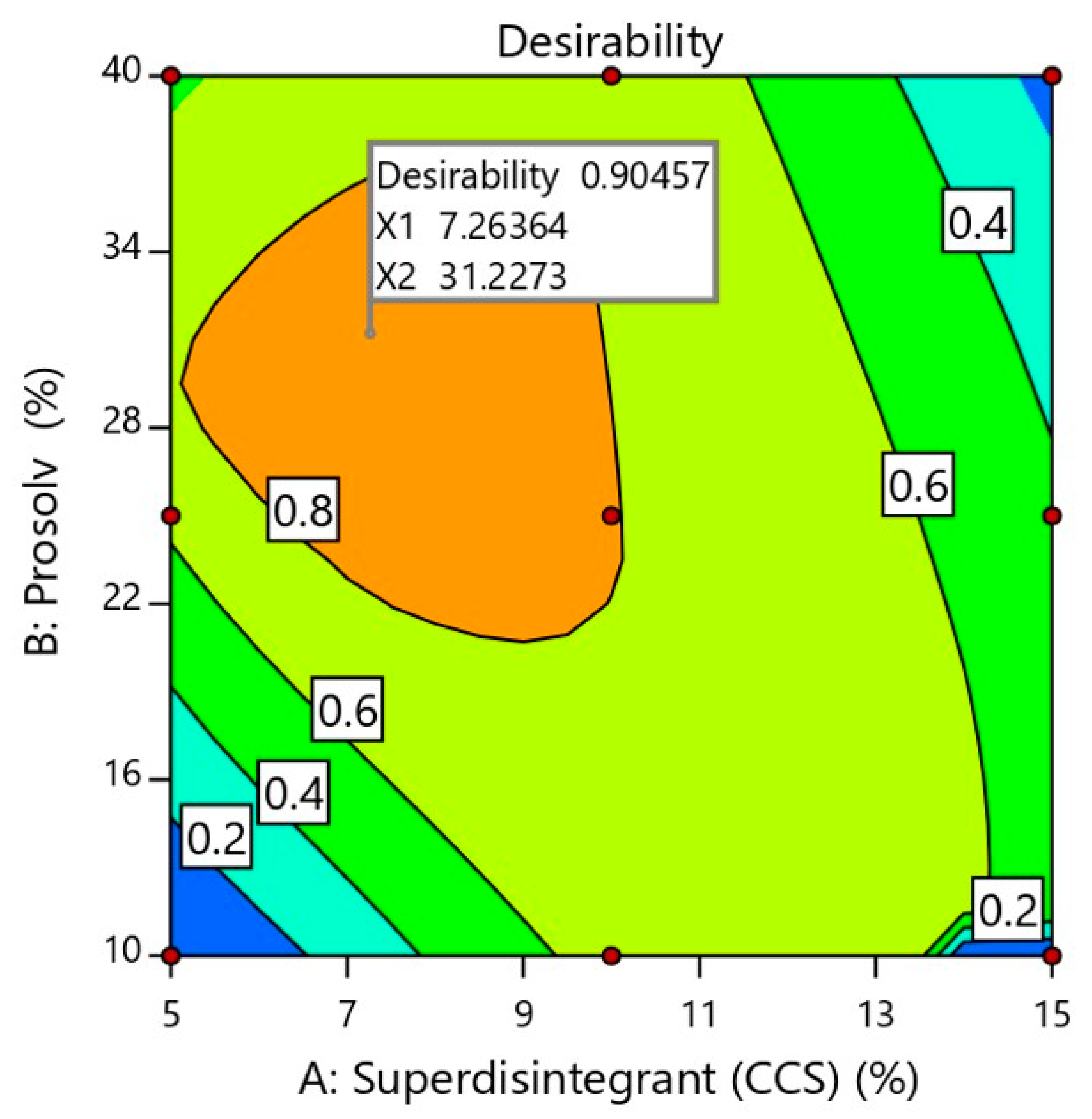
2.2.5. Determination of Optimized Formulation Using Desirability Function
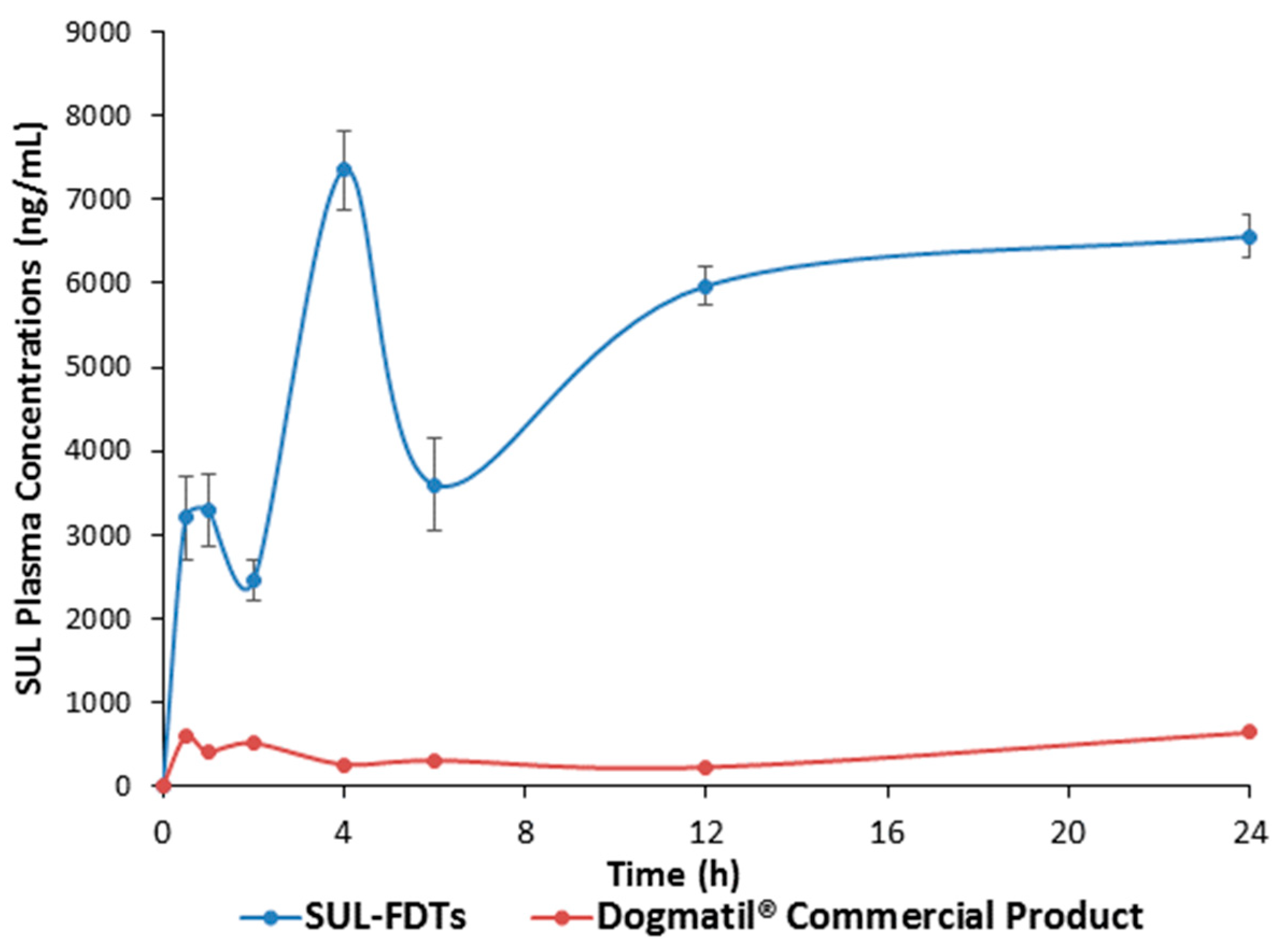
2.2.6. In Vivo Pharmacokinetic Analysis of Optimized Formulation
3. Materials and Methods
3.1. Materials
3.2. Compatibility Study
3.3. Experimental Design and Statistical Analysis
3.4. Preparation of SUL-FDTs
3.5. Characterization of SUL-FDTs
3.5.1. Weight Uniformity
3.5.2. Content Uniformity
3.5.3. Tablet Hardness
3.5.4. Tablet Friability
3.5.5. In Vitro Disintegration
3.5.6. In Vitro Release Study
3.6. In Vivo Study for Optimized Formulation
3.6.1. Study Design and Animals Treatment
3.6.2. Samples Preparation for Analysis
3.6.3. Pharmacokinetic Analysis
3.7. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 4, D1074–D1082. [Google Scholar] [CrossRef]
- Sweetman, S.C. Martindale: The Complete Drug Reference; The Pharmaceutical Press: London, UK, 2007. [Google Scholar]
- Abdallah Mohamed, A.; Mahmoud Mokhtar, I.; Marwa Helmy, A.; Mahmoud, A.M. Intranasal microemulgel as surrogate carrier to enhance low oral bioavialability of sulpiride. Int. J. Pharm. Pharm. Sci. 2016, 8, 188–197. [Google Scholar]
- Watanabe, K.; Sawano, T.; Terada, K.; Endo, T.; Sakata, M.; Sato, J. Studies on Intestinal Absorption of Sulpiride (1): Carrier-Mediated Uptake of Sulpiride in the Human Intestinal Cell Line Caco-2. Biol. Pharm. Bull. 2002, 25, 885–890. [Google Scholar] [CrossRef] [Green Version]
- Baluom, M.; Friedman, M.; Rubinstein, A. Improved intestinal absorption of sulpiride in rats with synchronized oral delivery systems. J. Control. Release 2001, 70, 139–147. [Google Scholar] [CrossRef]
- Kohri, N.; Naasani, I.; Iseki, K.; Miyazaki, K. Improving the oral bioavailability of sulpiride by a gastric-retained form in rabbits. J. Pharm. Pharmacol. 1996, 48, 371–374. [Google Scholar] [CrossRef]
- Akhtar, N.; Ahad, A.; Khar, R.K.; Jaggi, M.; Aqil, M.; Aqil, M.; Iqbal, Z.; Ahmad, F.J.; Talegaonkar, S. The emerging role of P-glycoprotein inhibitors in drug delivery: A patent review. Expert Opin. Ther. Pat. 2011, 21, 561–576. [Google Scholar] [CrossRef] [PubMed]
- Younis, M.A.; El-Zahry, M.R.; Tallat, M.A.; Tawfeek, H.M. Sulpiride gastro-retentive floating microsponges; analytical study, in vitro optimization and in vivo characterization. J. Drug Target. 2020, 28, 386–397. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, W.M.; AlOmrani, A.H.; Yassin, A.E. Novel sulpiride-loaded solid lipid nanoparticles with enhanced intestinal permeability. Int. J. Nanomed. 2013, 9, 129–144. [Google Scholar]
- Tawfeek, H.M.; Faisal, W.A.-O.; Soliman, G.A.-O. Enalapril maleate orally disintegrating tablets: Tableting and in vivo evaluation in hypertensive rats. Pharm. Dev. Technol. 2017, 23, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Alalaiwe, A.; Fayed, M.H.; Alshahrani, S.M.; Alsulays, B.B.; Alshetaili, A.S.; Tawfeek, H.M.; Khafagy, E.-S. Application of design of experiment approach for investigating the effect of partially pre-gelatinized starch on critical quality attributes of rapid orally disintegrating tablets. J. Drug Deliv. Sci. Technol. 2019, 49, 227–234. [Google Scholar] [CrossRef]
- Rezende, R.L.O.; Santoro, M.I.R.M.; Matos, J.R. Stability and compatibility study on enalapril maleate using thermoanalytical techniques. J. Therm. Anal. Calorim. 2008, 93, 881–886. [Google Scholar] [CrossRef]
- Ibrahim, E.H.; El-Faham, T.H.; Mohammed, F.A.; El-Eraky, N.S. Enhancement of solubility and dissolution rate of domperidone by utilizing different techniques. Bull. Pharm. Sci. 2011, 34, 105–120. [Google Scholar] [CrossRef]
- Mora, P.C.; Cirri, M.; Mura, P. Differential scanning calorimetry as a screening technique in compatibility studies of DHEA extended release formulations. J. Pharm. Biomed. 2006, 42, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, B.; Ray, S.; Das, M. Formulation, development and optimization of mouth dissolving tablets of Rizatriptan benzoate. J. Pharm. Investig. 2015, 45, 593–600. [Google Scholar] [CrossRef]
- Khafagy, E.-S.; Fayed, M.H.; Alrabahi, S.H.; Gad, S.; Alshahrani, S.M.; Aldawsari, M. Defining design space for optimization of escitalopram ultra-fast melting tablet using suspension spray-coating technique: In-vitro and in-vivo evaluation. J. Drug Deliv. Sci. Technol. 2020, 57, 101631. [Google Scholar] [CrossRef]
- United States Pharmacopeial Convention. United States Pharmacopeia (USP 38-NF-33); United States Pharmacopeial Convention: Rockville, MD, USA, 2015. [Google Scholar]
- Soroush, H.; Ghorbani-Bidkorbeh, F.; Mortazavi, S.A.; Mehramizi, A. Formulation Optimization and Assessment of Dexamethasone Orally Disintegrating Tablets Using Box-Behnken Design. Iran. J. Pharm. Res. 2018, 17, 1150–1163. [Google Scholar]
- Mostafa, H.F.; Ibrahim, M.A.; Sakr, A. Development and optimization of dextromethorphan hydrobromide oral disintegrating tablets: Effect of formulation and process variables. Pharm. Dev. Technol. 2013, 18, 454–463. [Google Scholar] [CrossRef]
- Solaiman, A.; Suliman, A.S.; Shinde, S.; Naz, S.; Elkordy, A.A. Application of general multilevel factorial design with formulation of fast disintegrating tablets containing croscaremellose sodium and Disintequick MCC-25. Int. J. Pharm. 2016, 501, 87–95. [Google Scholar] [CrossRef]
- Queiroz, A.L.P.; Wood, B.; Faisal, W.; Farag, F.; Garvie-Cook, H.; Glennon, B.; Vucen, S.; Crean, A.M. Application of percolation threshold to disintegration and dissolution of ibuprofen tablets with different microcrystalline cellulose grades. Int. J. Pharm. 2020, 589, 119838. [Google Scholar] [CrossRef]
- Kalný, M.; Grof, Z.; Štěpánek, F. Microstructure based simulation of the disintegration and dissolution of immediate release pharmaceutical tablets. Powder Technol. 2021, 377, 257–268. [Google Scholar] [CrossRef]
- FDA. Guidance for Industry: Orally Disintegrating Tablets; Department of Health and Human Services Food and Drug Administration, Centre for Drug Evaluation and Research (CDER), 2008. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/orally-disintegrating-tablets (accessed on 8 October 2020).
- Desai, P.M.; Liew, C.V.; Heng, P.W.S. Review of Disintegrants and the Disintegration Phenomena. J. Pharm. Sci. 2016, 105, 2545–2555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kachrimanis, K.; Nikolakakis, I.; Malamataris, S. Tensile strength and disintegration of tableted silicified microcrystalline cellulose: Influences of interparticle bonding. J. Pharm. Sci. 2003, 92, 1489–1501. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.R.; Kwon, S.Y.; Choi, D.H.; Park, E.S. Quality by Design (QbD) approach to optimize the formulation of a bilayer combination tablet (Telmiduo®) manufactured via high shear wet granulation. Int. J. Pharm. 2017, 534, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Aodah, H.A.; Fayed, M.H.; Alalaiwe, A.; Alsulays, B.B.; Aldawsari, M.F.; Khafagy, E.-S. Design, Optimization, and Correlation of In Vitro/In Vivo Disintegration of Novel Fast Orally Disintegrating Tablet of High Dose Metformin Hydrochloride Using Moisture Activated Dry Granulation Process and Quality by Design Approach. Pharmaceutics 2020, 12, 598. [Google Scholar] [CrossRef] [PubMed]
- Plusquellec, Y.; Campistron, G.; Staveris, S.; Barre, J.; Jung, L.; Tillement, J.P.; Houin, G. A double-peak phenomenon in the pharmacokinetics of veralipride after oral administration: A double-site model for drug absorption. J. Pharmacokinet. Biopharm. 1987, 15, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Helmy, S.A. Therapeutic drug monitoring and pharmacokinetic compartmental analysis of sulpiride double-peak absorption profile after oral administration to human volunteers. Biopharm. Drug Dispos. 2013, 34, 288–301. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.M.; Takemoto, J.K.; Brocks, D.R.; Yáñez, J.A. Multiple peaking phenomena in pharmacokinetic disposition. Clin. Pharmacokinet. 2010, 49, 351–377. [Google Scholar] [CrossRef]
- Atia, N.N.; Tawfeek, H.M.; Rageh, A.H.; El-Zahry, M.R.; Abdelfattah, A.; Younis, M.A. Novel sublingual tablets of Atorvastatin calcium/Trimetazidine hydrochloride combination; HPTLC quantification, in vitro formulation and characterization. SPJ 2019, 27, 540–549. [Google Scholar] [CrossRef]
- Aboutaleb, A.E.; Abdel-Rahman, S.I.; Ahmed, M.O.; Younis, M.A. Formulation of domperidone in gastro-retentive floating tablets. J. Innov. Pharm. Boil. Sci. 2016, 3, 81–93. [Google Scholar]
- Shazly, G.A.; Tawfeek, H.M.; Ibrahim, M.A.; Auda, S.H.; Elmahdy, M. Formulation and evaluation of fast dissolving tablets containing taste-masked microspheres of diclofenac sodium for sustained release. Dig. J. Nanomater. Biostruct. 2013, 8, 1281–1293. [Google Scholar]
- Gowda, V.; Pabari, R.M.; Kelly, J.G.; Ramtoola, Z. Influence of Prosolv and Prosolv: Mannitol 200 direct compression fillers on the physicomechanical properties of atorvastatin oral dispersible tablets. Pharm. Dev. Technol. 2015, 20, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, U.; Nayak, K. How to Choose Drug Dosage for Human Experiments Based on Drug Dose Used on Animal Experiments: A Review. IJSS Case Rep. Rev. 2014, 1, 31–32. [Google Scholar]
- Shin, J.-W.; Seol, I.-C.; Son, C.-G. Interpretation of animal dose and human equivalent dose for drug development. J Korean Orient. Med. 2010, 31, 1–7. [Google Scholar]
- Tawfeek, H.M.; Roberts, M.; El Hamd, M.A.; Abdellatif, A.A.H.; Younis, M.A. Glibenclamide Mini-tablets with an Enhanced Pharmacokinetic and Pharmacodynamic Performance. AAPS PharmSciTech 2018, 19, 2948–2960. [Google Scholar] [CrossRef] [PubMed]
- Soliman, M.H.; Saad, H.; Gadallah, A.N. Spectrophotometric and chromatographic determination of Sulpiride in pure and dosage forms. J. Environ. Res. Dev. 2010, 4, 125–132. [Google Scholar]
- Abdelbary, A.; Bendas, E.R.; Ramadan, A.A.; Mostafa, D.A. Pharmaceutical and pharmacokinetic evaluation of a novel fast dissolving film formulation of flupentixol dihydrochloride. AAPS PharmSciTech 2014, 15, 1603–1610. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formula | Weight (mg ± SD) | Content Uniformity (% ± SD) (AV) | Breaking Force (KP ± SD) | Friability (% ± SD) | DT (s ± SD) | % Release after 30 min (min ± SD) |
|---|---|---|---|---|---|---|
| 1 | 195 ± 3.20 | 99.8 ± 1.27 (3.048) | 3.25 ± 0.25 | 0.84 ± 0.2 | 15.16 ± 1.47 | 84.79 ± 2.05 |
| 2 | 200.17 ± 1.79 | 100.3 ± 1.74 (4.176) | 4.0 ± 0.5 | 1.01 ± 0.43 | 33.83 ± 5.98 | 96.39 ± 2.03 |
| 3 | 201.8 ± 1.60 | 99.33 ± 1.98 (4.752) | 4.16 ± 0.53 | 0.53 ± 0.07 | 21.16 ± 6.85 | 98.55 ± 1.46 |
| 4 | 197.6 ± 149 | 99.53 ± 1.12 (4.688) | 4.43 ± 0.11 | 0.98 ± 0.24 | 31.66 ± 3.82 | 87.56 ± 0.45 |
| 5 | 199 ± 2.09 | 97.66 ± 1.45 (4.32) | 4.9 ± 0.17 | 0.83 ± 0.3 | 39.83 ± 2.63 | 96.58 ± 0.77 |
| 6 | 198.8 ± 1.66 | 98.3 ± 1.9 (4.76) | 4.9 ± 0.17 | 0.37 ± 0.15 | 66.33 ± 5.24 | 99.39 ± 0.96 |
| 7 | 199 ± 2.28 | 99.03 ± 2.17 (5.208) | 3.58 ± 0.14 | 0.58 ± 0.07 | 40.16 ± 4.35 | 97.35 ± 0.68 |
| 8 | 200 ± 1.26 | 99.1 ± 3.14 (7.536) | 3.9 ± 0.17 | 0.9 ± 0.27 | 51 ± 6.69 | 87.47 ± 0.88 |
| 9 | 200 ± 2.23 | 98 ± 2.3 (6.02) | 3.56 ± 0.11 | 0.9 ± 0.29 | 44 ± 14.31 | 94.15 ± 1.16 |
| Variables | Coefficient Estimate | Sum of Squares | Standard Error | F-Value | p-Value | 95% CI Low | 95% CI High |
| Hardness (Quadratic model) | |||||||
| Model | - | - | - | 496.33 | 0.0001 | - | - |
| Intercept | 4.93 | - | 0.0247 | - | - | 4.86 | 5.01 |
| X1 | −0.0617 | 0.0228 | 0.0135 | 20.83 | 0.0197 | −0.1047 | −0.0187 |
| X2 | 0.2267 | 0.30830 | 0.0135 | 281.43 | 0.0005 | 0.1837 | 0.2697 |
| X1 X2 | −0.2325 | 0.2612 | 0.0165 | 197.40 | 0.0008 | −0.2852 | −0.1798 |
| Friability (2FI model) | |||||||
| Model | - | - | - | 1.180 | 0.4054 | - | - |
| Intercept | 0.7849 | - | 0.0739 | - | - | 0.4959 | 0.9749 |
| X1 | 0.0137 | 0.0011 | 0.0905 | 0.0228 | 0.8859 | −0.2190 | 0.2463 |
| X2 | −0.0900 | 0.0486 | 0.0905 | 0.9887 | 0.3657 | −0.3227 | 0.1427 |
| X1 X2 | 0.1762 | 0.1243 | 0.1109 | 2.53 | 0.1727 | −0.1087 | 0.4612 |
| Disintegration time (linear model) | |||||||
| Model | - | - | - | 9.98 | 0.0124 | - | - |
| Intercept | 40.35 | - | 3.25 | - | - | 32.39 | 48.30 |
| X1 | 14.17 | 1204.45 | 3.98 | 12.66 | 0.0120 | 4.43 | 23.91 |
| X2 | 10.75 | 693.53 | 3.98 | 7.29 | 0.0356 | 1.01 | 20.49 |
| Percent release after 30 min (Quadratic model) | |||||||
| Model | - | - | - | 68.12 | 0.0027 | - | - |
| Intercept | 96.31 | - | 0.5623 | - | - | 94.52 | 98.10 |
| X1 | −0.6183 | 2.29 | 0.3080 | 4.03 | 0.1383 | −1.6 | 0.3617 |
| X2 | 5.40 | 175.18 | 0.3080 | 307.84 | 0.0004 | 4.42 | 6.38 |
| X1 X2 | −0.2075 | 0.1722 | 0.3772 | 0.3027 | 0.6205 | −1.41 | 99.28 |
| Variables | Target | Range | Weight | Importance Co-Efficient |
|---|---|---|---|---|
| In-put | ||||
| Superdisintegrant conc. | In range | 5–15% | 1 | - |
| Prosolv® conc. | In range | 10–40% | 1 | - |
| Out-put | ||||
| Hardness Friability | Maximize 0.7% | 3.25–4.9 KP 0.378–1.01% | 1 | ++++ ++++ |
| Disintegration time | 30 s | 15.16–66.33 s | 1 | ++++ |
| %Release after 30 min | In range | 87.47–99.39% | 1 | - |
| Overall desirability = 0.905 | ||||
| Responses | Predicted Values | Observed Values (mean ± SD) | Relative Error (%) |
|---|---|---|---|
| Weight uniformity (mg) | - | 200.51 ± 2.72 | - |
| Content uniformity (%) (AV) | - | 100.31 ± 1.73 (4.36) | - |
| Hardness (KP) | 4.765 | 4.583 ± 0.52 | 3.819 |
| Friability (%) | 0.70 | 0.731 ± 0.158 | −4.428 |
| Disintegration time (s) | 37.04 | 37.5 ± 1.87 | −1.241 |
| %Release after 30 min | 98.201 | 100.514 ± 1.339 | −2.355 |
| Pharmacokinetic Parameters | Optimized SUL FDT | SUL Commercial Product (Dogmatil®) | p-Value |
|---|---|---|---|
| Cmax1 (ng/mL) | 3299.6 ± 498.8 | 564.5 ± 25.97 | ** <0.01 |
| Tmax1 (h) | 1.0 | 0.5 | |
| Cmax2 (ng/mL) | 7353.5 ± 471.3 | 526.2 ± 12.5 | ** <0.01 |
| Tmax2 (h) | 4.0 | 2.0 | * <0.05 |
| Cmax3 (ng/mL) | 6561.6 ± 247.2 | 673.4 ± 51.5 | ** <0.01 |
| Tmax3 (h) | 24.0 | 24.0 | >0.05 |
| Kabs.1 (h−1) | 1.85 ± 0.06 | 3.02 ± 0.16 | * <0.05 |
| t1/2 abs.1 (h) | 0.37 ± 0.09 | 0.23 ± 0.17 | >0.05 |
| Kabs.2 (h−1) | 0.35 ± 0.08 | 0.66 ± 0.16 | * <0.05 |
| t1/2 abs.2 (h) | 2.02 ± 0.12 | 1.05 ± 0.31 | * <0.05 |
| Kabs.3 (h−1) | 0.058 ± 0.01 | 0.056 ± 0.011 | >0.05 |
| t1/2 abs.3 (h) | 12.01 ± 0.15 | 12.36 ± 0.21 | >0.05 |
| AUC0–24 (ng·h/mL) | 129,948.6 ± 1230 | 9311 ± 482 | ** <0.01 |
| AUC0–∞ (ng·h/mL) | 148,283.8 ± 988.5 | 15,917.45 ± 675 | ** <0.01 |
| AUMC0–24 (ng·h2/mL) | 1,745,461.2 ± 2380 | 193,150.5 ± 1653 | ** <0.01 |
| AUMC0–∞ (ng·h2/mL) | 2,236,763.4 ± 3458 | 416,517.6 ± 1816 | ** <0.01 |
| MRT (h) | 15.08 ± 0.78 | 26.17 ± 0.59 | * <0.05 |
| Kel (h−1) | 0.357 ± 0.081 | 0.318 ± 0.011 | >0.05 |
| t1/2el (h) Relative bioavailability (%) | 1.93 ± 0.052 931.6 ± 36.5 | 2.17 ± 0.33 - | >0.05 - |
| Coded Levels | Superdisintegrant Conc. (%) | Prosolv® Conc. (%) |
|---|---|---|
| −1 | 5 | 10 |
| 0 | 10 | 25 |
| 1 | 15 | 40 |
| Experiment Code | Superdisintegrant Conc. (%) | Prosolv® Conc. (%) |
|---|---|---|
| 1 | 5 | 10 |
| 2 | 5 | 25 |
| 3 | 5 | 40 |
| 4 | 10 | 10 |
| 5 | 10 | 25 |
| 6 | 10 | 40 |
| 7 | 15 | 10 |
| 8 | 15 | 25 |
| 9 | 15 | 40 |
| Ingredients | % w/w |
|---|---|
| Sulpiride | 25 |
| Croscarmellose sodium (CCS) (Vivasol ®) | 5, 10 and 15 |
| Silicified microcrystalline cellulose (Prosolve®) | 10, 25 and 40 |
| Spray dried mannitol | Up to 100 |
| Magnesium stearate | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
M. Tawfeek, H.; Hassan, Y.A.; Aldawsari, M.F.; H. Fayed, M. Enhancing the Low Oral Bioavailability of Sulpiride via Fast Orally Disintegrating Tablets: Formulation, Optimization and In Vivo Characterization. Pharmaceuticals 2020, 13, 446. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13120446
M. Tawfeek H, Hassan YA, Aldawsari MF, H. Fayed M. Enhancing the Low Oral Bioavailability of Sulpiride via Fast Orally Disintegrating Tablets: Formulation, Optimization and In Vivo Characterization. Pharmaceuticals. 2020; 13(12):446. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13120446
Chicago/Turabian StyleM. Tawfeek, Hesham, Yasser A. Hassan, Mohammed F. Aldawsari, and Mohamed H. Fayed. 2020. "Enhancing the Low Oral Bioavailability of Sulpiride via Fast Orally Disintegrating Tablets: Formulation, Optimization and In Vivo Characterization" Pharmaceuticals 13, no. 12: 446. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13120446