Analytical Methods for the Detection and Quantification of ADCs in Biological Matrices

Département Médicaments et Technologies pour la Santé (MTS), CEA, INRAE, SIMoS, Université Paris-Saclay, 91191 Gif-sur-Yvette, France
*
Author to whom correspondence should be addressed.
Pharmaceuticals 2020, 13(12), 462; https://0-doi-org.brum.beds.ac.uk/10.3390/ph13120462
Submission received: 8 October 2020
/
Revised: 1 December 2020
/
Accepted: 11 December 2020
/
Published: 14 December 2020
(This article belongs to the Special Issue Antibody-Drug Conjugates (ADC): 2021)
Abstract
:Understanding pharmacokinetics and biodistribution of antibody–drug conjugates (ADCs) is a one of the critical steps enabling their successful development and optimization. Their complex structure combining large and small molecule characteristics brought out multiple bioanalytical methods to decipher the behavior and fate of both components in vivo. In this respect, these methods must provide insights into different key elements including half-life and blood stability of the construct, premature release of the drug, whole-body biodistribution, and amount of the drug accumulated within the targeted pathological tissues, all of them being directly related to efficacy and safety of the ADC. In this review, we will focus on the main strategies enabling to quantify and characterize ADCs in biological matrices and discuss their associated technical challenges and current limitations.
1. Introduction
Antibody–drug conjugates (ADCs) consist of a small-molecule drug (payload) covalently bound to a monoclonal antibody via a chemical linker. By merging the specific binding properties of antibodies to the potency of small molecules, ADCs are designed to selectively deliver cell-killing agents to targeted pathogenic tissues, while limiting systemic toxicity. Most of the ADCs developed so far are potential anticancer agents and nine of them were approved by the Food and Drug Administration (FDA) for the treatment of various types of cancer (Table 1). Many other ADCs are currently in advanced clinical trials not only for anticancer applications [1,2,3,4,5] but also to treat intracellular Staphylococcus aureus bacterial infection [6], for the targeted delivery of kinase inhibitors [7], as well as ADCs with anti-inflammatory properties [8,9].
In most approved and clinical-stage ADCs, the toxic payloads are microtubule disruptors or DNA-damaging agents [10,11]. In the first and second generations of ADCs, those payloads were conjugated to antibody component in a stochastic manner, leading to heterogeneous mixtures of chemically distinct molecules varying in both drug-to-antibody ratio (DAR) and conjugation sites [1]. This last decade, with the aim to better control the position and the number of drug loads, new conjugation strategies were developed to access a new generation of ADCs with improved homogeneity [1,2,12,13]. Those site-specific reactions rely on the insertion of engineered cysteine residues [14] or unnatural amino acids within the antibodies core [15,16], enzymatic conjugation [17,18,19], cross-link of the reduced interchain disulfides with rebridging chemical reagents [20], and glycan-mediated conjugation [21]. The conjugating linkers between payload and antibody are generally classified into cleavable and noncleavable ones, which can behave quite differently in a biological system [22]. There are several linker types comprising hydrazone [23,24], disulfide [25], dipeptide [26,27], tetrapeptide [28], glucuronide [29], phosphate-ester [30], or noncleavable linker [31], which are sensitive to different triggers such as pH variation, modification of the reducing environment, or local presence of enzymes. Noteworthy, enzymatically cleavable peptide-base linkers have been designed both to be highly stable in circulation and to exhibit a more specific drug release mechanism within the targeted tissues [32]. It was also demonstrated that the in vivo stability of the linker during circulation depends considerably on the location and solvent accessibility to the conjugation site [14].
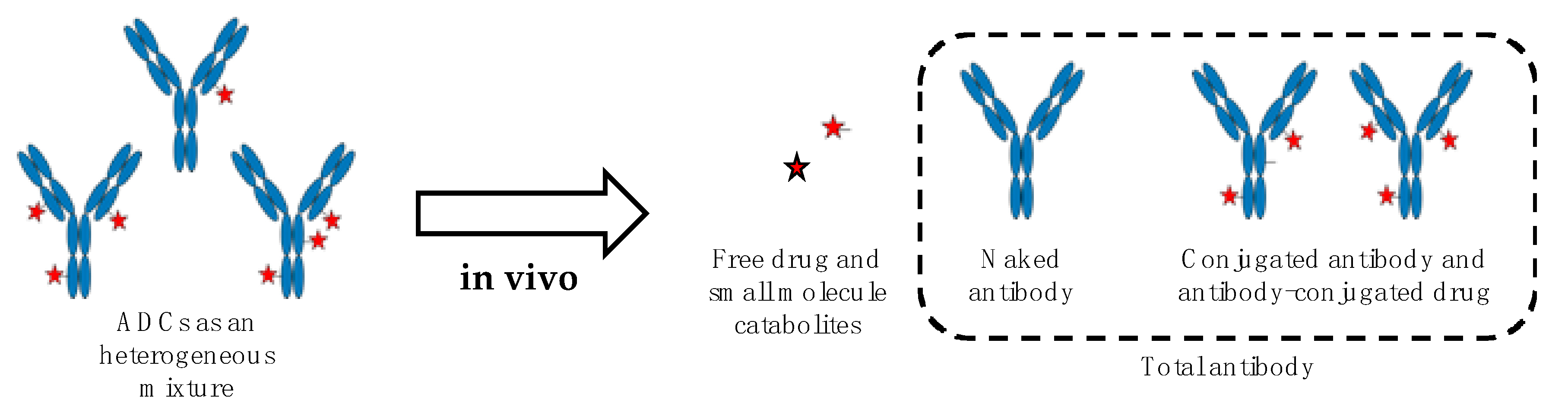
Like for other therapeutics, the understanding of ADCs fate in vivo is critical for their development, optimization, and successful transfer to human. The pharmacology of ADCs is complex and several factors potentially impacting their pharmacokinetics, toxicity, and biodistribution have to be carefully considered. This notably includes their physical characteristics (e.g., composition and size, surface engineering, DAR, conjugation site, and net charge at physiological pH) and their propensity to generate antidrug antibodies (ADAs), the latter correlating with ADCs immunogenicity. All those factors have been recently reviewed [33,34] and are not covered by the present review. In any event and due to their multicomponent nature, ADCs must be considered as a complex and dynamically changing mixture in vivo, driven by undesirable drug release [35], catabolism [36], and variable clearance rate according to DAR [37]. Therefore, in such a scenario, multiple ADCs-derived analytes have to be measured to properly describe the stability and biodistribution of ADCs as well as the amount of drug accumulated within the targeted tissues. The main ADCs-derived analytes comprise the naked antibody, the conjugated antibody, and antibody-conjugated drug, all the three corresponding to total antibody, and the released small molecule drug as well as its metabolites [36,38,39,40] (Figure 1).
To determine systemic (e.g., in plasma) and local (e.g., in tissues) levels of ADCs-derived analytes, two main types of bioanalytical methods can be distinguished. The analytical methods that do not imply any prelabeling of the ADCs components and those requiring to label the antibody part, the payload or both of them. The label-free methods mainly rely on ligand-binding assays (LBAs), liquid chromatography coupled to mass spectrometry (LC–MS/MS), or a combination of both methods. Most of the time in the case of labeled ADCs, a fluorescent reporter or a radioactive isotope are incorporated within the structure of the antibody or/and the drug. In this case, the pharmacokinetics, biodistribution and tissues uptake of ADCs are determined either by noninvasive imaging approaches (e.g., positron emission tomography (PET) or fluorescence molecular tomography (FMT)) or by liquid scintillation counting and ex vivo radio-imaging after animal sacrifice, the latter being obviously restricted to preclinical studies. The present review proposes to outline those different bioanalytical methods and discuss their associated technical challenges aiming at characterizing and accurately quantifying the different ADCs-derived analytes in various biological matrices, with a particular focus on preclinical studies.
2. Label-Free Bioanalytical Methods
2.1. Ligand-Binding Assays (LBAs)
Out of all the LBA formats, ELISA (enzyme-linked immunosorbent assay) relying on an immunoaffinity detection of the protein of interest, is the gold standard of LBA for the analysis of large molecules in complex biological samples. This bioanalytical method offers unique advantages for the quantification of proteins, such as a high sensitivity and a broad range of quantification in a small biological volume without any sample extraction steps. However, the complex multicomponent structure of ADCs and their heterogeneous and dynamically changing nature in vivo present several analytical challenges for LBAs [36,41]. In this respect, the quantification of total antibody requires the use of detection reagents that bind selectively and efficiently to the antibody component. This includes generic anti-human antibody, protein A, G or L, or more specific detection reagent such as anti-idiotype antibody. This approach has been successfully applied to the quantification of several ADCs in various biological matrices, including anti-CD33-Calicheamicin ADC [42], T-DM1 [43], and mAb-MMAE conjugates [14,44,45,46]. LBAs can also utilize antidrug- specific antibody as a capture antibody to quantify conjugated antibody [47,48,49,50] or antidrug antibody as detection reagents [51,52,53,54]. Regarding the choice of primary antibody as capture agent and to avoid any potential interferences from endogenous IgGs, it is essential to evaluate its nonspecific cross-reactivity. For instance, it is recommended to choose antibodies with minimum reactivity to mouse and rat for preclinical studies. Both monoclonal and polyclonal antibodies are suitable as capture and detection agents. Monoclonal antibodies provide mono specificity toward a single epitope, which allows fine detection and quantitation of small differences in antigen. Polyclonal antibodies are used to capture as much antigen as possible, while monoclonal ones are used to detect ADCs with improved specificity. Polyclonal antibodies are much less expensive to produce than monoclonal antibodies. Therefore, an appropriate balance between cost and required specificity has to be systematically considered.
Importantly, conjugated antibody ELISA is very sensitive to the choice of the antidrug antibody, and therefore the key aspect in such assay development is to identify an antidrug antibody that is minimally influenced by the drug load. Indeed, it has been reported that the small molecule drug can indirectly influence the reagents binding to the antibody, resulting in species varying in DAR that are not detected with the same efficiency and accuracy. For instance, purified DAR fractions with high MMAE drug load were under-recovered (around 70% and 55% of recovery for DAR 6 and DAR 8, respectively) in a total antibody ELISA assay using various reagents [55]. As a consequence, the development of such an assay requires the screening of multiple formats and reagents in relevant biological matrices including plasma or serum [47]. Compared to conventional ELISA where reagents are added sequentially, the semihomogeneous assay (SHA), relying on premixing of capture and detection reagents with the sample solution, turns out more effective to accurately detect the whole set of ADCs regardless of their drug loading [55]. Finally, LBAs can also be suitable for the quantification of the drug moiety. Thus, both an anti-MMAE antibody and a HRP-MMAE were used to quantitate free MMAE released from ADC after an in vitro incubation with Cathepsin B [54].
However, LBAs with excellent specificity and accuracy remain difficult to achieve and require the use of efficient and selective capture antibodies. The development of such agents may turn out difficult and time-consuming, making those immunoassays sometimes unpractical in the early phase of ADCs development. Furthermore, their quantitative accuracy, specificity, and reproducibility can be compromised by interferences from endogenous IgGs and degradation of the antibody component. ELISA is very commonly used in quantification of ADCs in plasma but is often matrix- and species-dependent, rendering its transfer across different biological matrices (e.g., from plasma to tissues) sometimes challenging [36,41]. Nevertheless, these limitations can be overcome when the capture and the detection antibodies are finely selected. Thus, by using a total antibody anti-human IgG antibody (Fc-specific), trastuzumab-vc-MMAE can be sensitively and accurately dosed in both plasma and tissue homogenates, with a detection limit down to 1 ng/mL [56].
2.2. Hybrid Ligand-Binding Immuno-Affinity Capture Followed by Liquid Chromatography Coupled to Mass Spectrometry (LB-LC–MS)
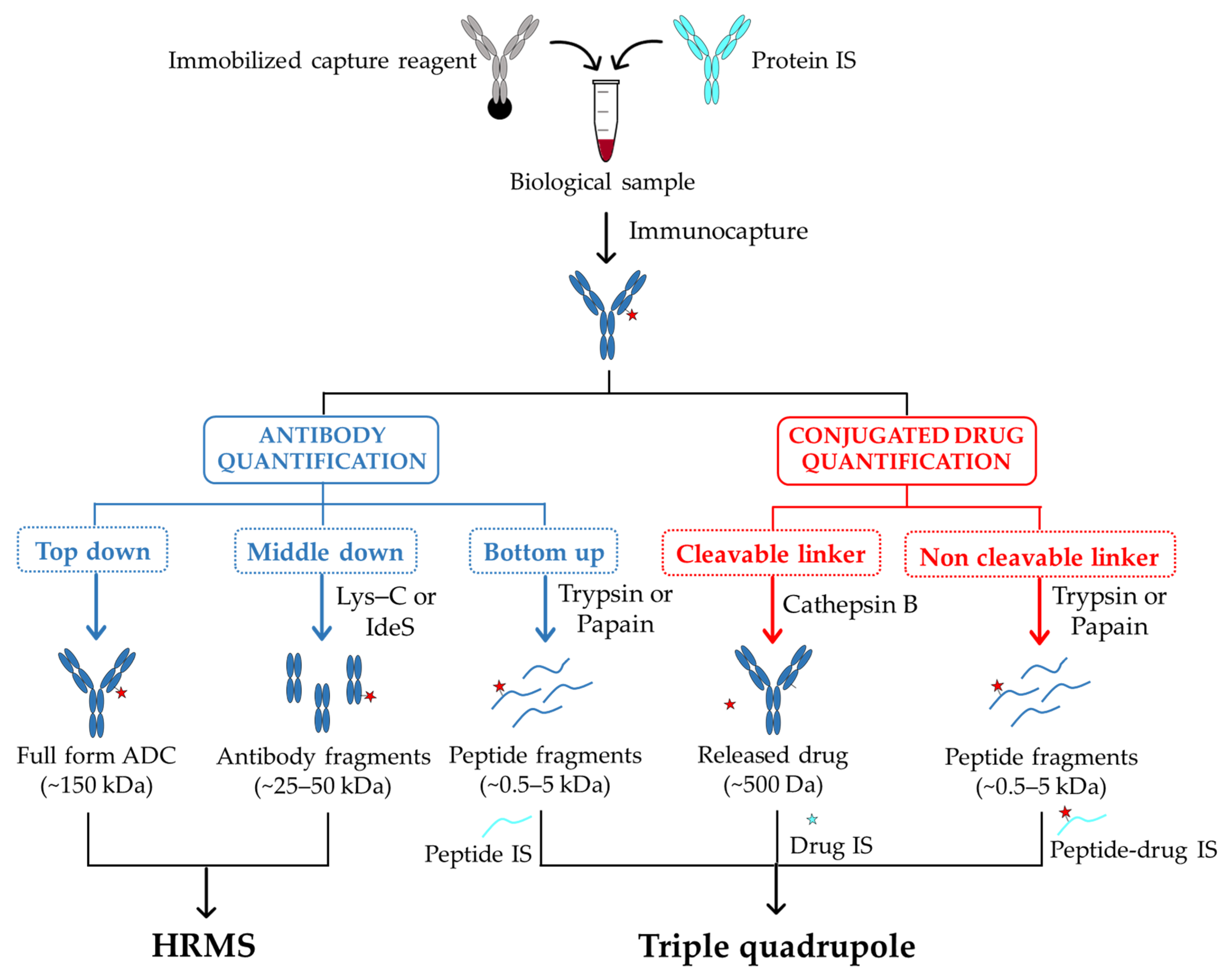
The ligand-binding and LC–MS (LB-LC–MS) hybrid method that combines the strength of both technologies is becoming a gold standard approach in ADC bioanalysis [57,58,59]. In this respect, the ligand-binding affinity enrichment step allows to spare interfering abundant proteins while the LC–MS based method offers quantitative bioanalysis with multiple analytes detection with high specificity within a single sample. In preclinical samples, an immobilized anti-human Fc antibody is commonly used to isolate ADCs from animal plasma whereas a specific anti-idiotype antibody is required in human samples [60]. After washing steps and proteolytic digestion, the target antibodies are unambiguously identified through their MS/MS fragmentation pattern [61].
The quantitative analysis is mostly carried out using the “bottom-up” strategy. In this case, a digestion of the captured ADC is followed by a LC–MS/MS analysis to detect and identify their selected signature peptides (Figure 2). The choice of the signature peptides within the antibody core mainly relies on the biological matrix in which ADCs have to be detected and quantified. In this respect, peptides from the constant region of antibody part will be considered as signature peptides for ADCs characterization in animal models. In human samples and to limit interferences with endogenous human IgGs, signature peptides from antibody variable regions will be preferred. Regarding the quantification, it is performed by comparing signature peptides of target antibody to standard peptides present at defined amounts. Such internal standards must be identical in chemical and physical properties to the peptides of interest, with small differences in mass that enables their discrimination by mass spectrometry. To this purpose, peptides labeled with stable isotopes 13C and 15N are commonly used [62]. In order to compensate potential variations during sample preparation, a stable-isotope-labeled antibody as internal standard can be also incorporated from the beginning of the enrichment process [63]. Although this workflow offers a very good sensitivity of quantification, the signature peptides only provide limited information about potential structural changes of the entire ADC and may therefore lead to a misestimation of their concentration in sample solutions.
To overcome these limitations, the “top-down” strategy that allows assessing qualitative and quantitative changes simultaneously, has come out as a complementary analysis to the “bottom-up” approach. In this case, the whole target protein is analyzed using high-resolution mass spectrometry (HRMS) with no further digestion step required [64] (Figure 2). This approach has been used to efficiently quantify intact antibodies in biological samples [65,66], and variations resulting from proteolytic digestion step remain limited. Nevertheless, compared to peptide level quantification, LC–MS quantification of intact ADCs presents many other challenges. Indeed, due to their larger size and heterogeneity, their mass signal can be dispersed among multiple charge states, resulting in a loss of detection sensitivity [67]. Further, antibody internal standards are almost systematically required for the absolute quantification of ADCs [65]. In some cases, this turns out problematic when the internal standard coelutes with the target specie, thus contributing in complicating the spectra deconvolution [67]. However, by applying a narrow-window of extracted-ion chromatogram (EIC) and a deconvolution method with no internal standard, an accurate quantification with an acceptable detection threshold (5 µg/mL) can be achieved [67]. When a high-resolution accurate-mass (HR/AM) mass spectrometry is used, this “top-down” strategy enables to characterize the in vivo biotransformations of trastuzumab emtansine (T-DM1 or Kadcyla) in circulation in tumor-bearing mice [68]. In this case, by combining an affinity capture with a biotinylated HER2 extracellular domain (EDC) and mass analyses on total antibody using a HR/AM orbitrap, the authors provided new data on T-DM1 catabolism, including maleimide exchange, loss of payload, and linker-drug hydrolysis.
A good compromise between high sensitivity and the access to dynamic changes of ADCs in biological matrixes can be obtained with a “middle-down” approach implying ADCs digestion into smaller subunits (subfragments) before LC–MS analysis (Figure 2). In this case, a partial digestion with Lys-C of a protein A bound IgG allows the direct characterization of the released Fab fragment (50 kDa), and its accurate quantification [69]. An additional step of disulfide bridges reduction after digestion using the endopeptidase IdeS has also been reported to generate three approximately 25 kDa fragments (Lc, Fc/2, Fd) that can be directly analyzed by LC–MS [70]. In all cases, the reduced size of analytes results in an improved sensitivity and resolution of mass spectra compared to the intact ADC.
In a comparable manner to LBAs, one of the main limitations of affinity capture combined with LC−MS is that it requires specific capture reagents capable of selectively bind the targeted ADCs, as well as its derived analytes. In addition, the majority of LB-LC–MS focus on Fab-conjugated ADCs and are not readily applicable to Fc-conjugated ones. In such a context, a generic and universal affinity capture LC−MS assay has been recently developed to evaluate biotransformation of ADCs in preclinical studies, independently of the antibody type, conjugation site (Fab, Fc), or conjugation technologies [71]. In this case, the ADCs are captured from mouse serum with generic antihuman F(ab′)2 coated beads and, depending on the site of conjugation, either directly eluted or digested “on-bead” with IdeS enzyme before elution. The resulting ADC sub fragments are finally analyzed by LC−HRMS. The advantages of this method are that it can be performed using commercially available generic reagents and requires sample preparation time of less than 7 h.
The hybrid immuno-affinity capture LC–MS is also suitable for the quantification of the conjugated payload [72]. The characteristic of the linker moiety mainly determines the sample processing strategy for the conjugated payload analysis. In the case of ADC incorporating an enzyme- sensitive dipeptide Val-Cit linker (e.g., brentuximab vedotin), the release of payload is performed by digestion with proteases such as cathepsin B or papain [54,72,73,74,75]. For noncleavable linkers, the whole antibody is first digested by a protease and the resulting drug-conjugated peptide is the surrogate analyte to monitor by LC–MS [76].
Interestingly, this LB-LC–MS/MS method has also been reported for the quantitative assessment of albumin linker-payload adduct. This mainly encompasses thiol-maleimide based ADCs that can undergo deconjugation through a thiol exchange with the Cys34 from the serum albumin in circulation [52]. In this case, the capture reagents are antipayload [77] or an antialbumin antibodies [78].
2.3. Nano-Surface and Molecular-Orientation Limited (nSMOL) Proteolysis
Shotgun proteomics enables quantification of fully digested peptides by trypsin using liquid chromatography MS (LC/MS). With the aim to apply a comparable technology to monoclonal antibodies and ADCs quantitation in biological matrices, Shimada and coworkers recently developed an elegant approach consisting in limiting protease access to a specific region of antibody substrate, thus enabling the generation of a restricted array of peptides fragments (signature peptides) subsequently analyzed by mass spectrometry [79,80]. This method, named nano-surface and molecular orientation limited (nSMOL) proteolysis, allows the selective and orientational proteolysis of the antibodies Fab region and relies on three distinct critical steps: (i) the miniaturization of the reaction field while increasing the surface area of the protease reaction through the use of nanoparticles, (ii) the antibody immobilization onto resin via protein A/G affinity binding, and (iii) the Fab region-selective proteolysis by limiting protease access to the substrate by tuning the particle size (protease) and the pore diameter of the resin immobilizing the antibodies. This approach has notably permitted to quantify Trastuzumab emtansine (T-DM1) in human plasma [80]. In the same study, by using nSMOL for the antibody and polarity-selective liquid–liquid partition for the cytotoxic drugs, it was possible to obtain an overall picture of antibody–drug conjugate T-DM1 pharmacokinetics and released payloads in samples from patients with breast cancer. Noteworthy, nSMOL proteolysis also enables to accurately quantify antibody drug in coexistence with antidrug antibodies (ADAs) [81].
2.4. MS Methods to Assess Pharmacokinetics and Biodistribution of Payload
Conventional small-molecule LC–MS/MS assay is designed to measure drug that is no longer bound to the antibody. This includes the portion released during circulation phase as well as that liberated during ADC catabolism. A step of protein precipitation or solid phase extraction (SPE) is systematically performed to remove all the plasma proteins prior to LC analysis. Molecular and fragments masses are used to determine the structure of the major metabolites and UV absorption at characteristic wavelength enables the quantification of different entities. The sensitivity of the assay is one of the major challenges associated with the free drug quantification since the unconjugated drug can be present at a very low concentration in biological samples. For instance, it has been reported that the quantification of the free DM1 payload in clinical samples of patients treated every 3 weeks was not possible as the signal was below the lower limit of quantification (LLOQ) of the method (0.20 ng/mL) [82]. In another study, the quantification of the unconjugated DM4 has only been performed during the first 24 h of a 3-week study using a LC–MS/MS method with a LLOQ of 0.50 ng/mL [83]. However, the method sensitivity remains difficult to predict since it depends both on the payload considered and the performances of mass spectrometry device used.
Matrix assisted laser desorption/ionization imaging mass spectrometry (MALDI–IMS) can be also used to directly detect the distribution of payload within tumor tissues [84]. In this study, the authors conducted quantitative and semiquantitative analyses of MMAE in subcutaneous tumors by using LC–MS/MS and MALDI–IMS, respectively. Interestingly, by exploiting MALDI–IMS conditions dedicated to low-molecular weight substances, they visualized only free MMAE without interference from conjugated MMAE. Further, by relying on a specific MS pattern of the MMAE payload, the latter can be observed with high reliability and accuracy in tumor tissues.
A label-free method has also been described for the quantification of the Cys-Mc-MMAF metabolite in homogenates of tumor, liver, lung, kidney, and heart (Figure 3). This has been achieved by a LC–MS/MS analysis using the amino acid linker-drug Cys-Mc-MMAD as an internal standard [85]. The direct quantification of the released payload Lys-MCC-DM1 on tumor and liver sections has also been achieved by liquid extraction surface analysis micro-liquid chromatography–tandem mass spectrometry (LESA-µLC–MS/MS, Figure 3) [86].
3. Analytical Methods Requiring a Prelabeling of ADCs
3.1. Noninvasive Molecular Imaging Approaches
Imaging of radiolabeled ADCs by positron emission tomography (PET), or immuno-PET, has been employed for the noninvasive quantification of ADCs uptake in normal and tumor tissues (Figure 3) [87]. For immuno-PET agents, different radioactive isotopes can be used, including 64Cu, 86Y, 89Zr, and 124I. This approach has been notably exploited to challenge the stability of an original bifunctional platinum (II) linker incorporated within the core of 89Zr-labeled Trastuzumab, and conjugated to two different payloads (Desferal or auristatin) in a preclinical model [88] or to assess the targeting capacity of novel mAb806-antibody–drug conjugates in malignant mesothelioma [89]. Noninvasive in vivo imaging technologies such as single-photon emission computed tomography (SPECT) have been also used to determine the whole-body biodistribution of ADCs using a 89Zr or a 111In radiolabeling on the antibody component [90,91,92,93]. An alternative method based on fluorescence molecular tomography (FMT) has been also reported to assess the whole-body distribution of an antibody labeled with a near-infrared fluorophore [94] (Figure 3).
3.2. Ex Vivo “Cut and Count” Techniques
The evaluation of ADCs pharmacokinetics and biodistribution in preclinical models can also be performed by either labeling the antibodies component with 125I [48,95,96,97], 123I [98], or 111In [91,97,99,100] or the drug with 3H [48,86,101]. In this case, the pharmacokinetics can be obtained by collecting blood samples, within which the content in radioactivity is assessed by liquid scintillation counting (Figure 3). The biodistribution is examined after animal sacrifice at different time points, tissues harvesting and ex vivo “cut and count” techniques enabling to quantify the radioactivity levels in different organs using beta [101,102] or gamma counters [91,92,99,103]. Of note, two examples of quantitative whole-body autoradiography (QWBA) have been recently reported to study the tissue distribution in depth of 125I labeled antibody [96], and a 3H-labeled DM1 payload [86]. Regarding the latter and beyond the possibility to accurately quantify the amount of payload accumulated within the tumor, the use of liquid extraction surface analysis coupled to micro-liquid chromatography–tandem mass spectrometry allowed to identify a unique catabolite distributed in the tumor and liver tissue.
3.3. Dual Radiolabeling
Surprisingly, though the dual radiolabeling of both ADCs components should provide the most comprehensive understanding of ADCs in vivo performances, only a very few examples have been reported to date [102,103,104,105]. The biodistribution of the antibody component and the payload-related species in different organs has been thus described in a case of a 3H antibody and 14C-labeled MMAF [102], and a 89Zr antibody and 131I-labeled tubulysin analogue [103]. Digital autoradiography systems can acquire data from multiple radioisotopes and accurately discriminate them based on their difference in emission energies. Thus, the use of cryo-imaging quantitative autoradiography (CIQA) has been described to study the distribution to the tumor of a 111In antibody and 3H-labeled MMAE, where the 3H signal has been acquired after a delay of more than 10 111In half-lives (2.8 days) [104]. More recently, a dual labeling strategy has been used to challenge the stability of the Pt(II)-based linker mentioned above [105]. Of note, dual imaging is not achievable by noninvasive PET imaging since the use of two radioelements at the same time remains challenging mainly due to energy interference.
3.4. Limitations of Analytical Methods Involving Radiolabeling
Although allowing a high sensitivity of detection, quantitative radio imaging studies provide no information on the identity of ADC-related species and drug metabolites present in the tissues. To this purpose, these imaging methods must be associated to qualitative bioanalytical methods. In this respect, tissues extraction followed by a SEC-HPLC or standard HPLC analyses coupled to a radioactivity detector can partly circumvent these issues [86,101]. Further, a single labeling on antibody component or payload does not allow to monitor the whole set of ADCs-derived analytes. To overcome these limitations, different and complementary technologies (e.g., liquid scintillation counting, liquid chromatography/mass spectrometry, enzyme-linked immunosorbent assay, and size exclusion chromatography) have to be used to access the pharmacokinetics of the total antibody, the released small molecule drug as well as its metabolites.
Whether it refers to single or dual labeling, these methods all require expensive radioisotopes and dedicated facilities that complicate their clinical transfer. In addition, the radioactive tags are often incorporated randomly within the ADCs core, with potential consequences on their in vivo behavior. In this respect, the incorporated tag may cause immunoreactivity or alter the characteristics of ADCs binding and distribution.
4. Conclusions
ADME characterization of ADCs is a complex process due to the multicomponent and heterogeneous nature of these conjugates. While conventional large- and small-molecule methods can be used for ADC bioanalysis, it is however important to understand their limitations to develop ADCs-dedicated bioanalytical strategies.
These recent years, the hybrid LB-LC–MS method has become the most versatile assay in ADCs bioanalysis since they can provide information not only on the entire molecule but also on ADC-derived analytes. When combined with a highly specific LC–MS method, LBA has overcome its intrinsic limitations, mainly related to interferences with highly abundant proteins, to enable unambiguous identification of ADC-derived analytes in many different biological samples. In addition, recent emerging technologies such as an enrichment strategy via pH regulation and ionic solvent strengths [106] are completing the array of LC–MS quantification of biotherapeutics in biological matrices and should be soon applicable to ADCs.
The use of radiolabeled ADCs to support pharmacokinetic and biodistribution studies is undoubtedly a way to reach high detection threshold. More specifically, designing a dual radiolabeled ADC, harboring distinct radioactive tags on the protein component and the small molecule drug, could appear as a particularly valuable strategy to monitor the in vivo fate of both entities simultaneously and to accurately and sensitively measure their relative amount during the circulation and distribution phases. Combined with whole-body imaging, such an approach can provide useful information on overall stability of the conjugate as well as its biodistribution within target-expressing and nontarget-expressing tissues, thus assessing the ADCs efficacy and potential toxicity. However, imaging and accurate quantification by radioactive counting must be associated to qualitative analyses such as HPLC or SEC-HPLC to provide information about the identity of the radio emitting species. Furthermore, the dual radiolabeling must neither interfere with the physicochemical properties of both ADCs components, nor add further heterogeneity that could alter ADCs binding properties and in vivo behavior. Accordingly, the conception of dual labeled ADCs must imperatively rely on efficient radiolabeling reactions that do not affect the drug properties, namely traceless reactions, and on site-specific conjugation reactions aiming at controlling the modification made within the antibody core. Once these limitations are overcome, the dual labeling strategy could become a central methodology to rapidly challenge and optimize various ADCs in preclinical models, thus accelerating their transfer to clinical studies.
Author Contributions
Writing—review and editing, H.C. and L.D.; supervision, L.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by CEA program “amont-aval”.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ADA | Antidrug antibody |
| ADC | Antibody–drug conjugate |
| ADME | Absorption, distribution, metabolism and excretion |
| CIQA | Cryo-imaging quantitative autoradiography |
| DAR | Drug-to-antibody ratio |
| DM1 | Mertansine |
| DM4 | Ravtansine |
| DOTA | Tetraxetan |
| DTPA | Pentetic acid |
| EIC | Extracted-ion chromatogram |
| ELISA | Enzyme-linked immunosorbent assay |
| EPR | Enhanced permeability and retention |
| Fab | Fragment antigen binding |
| Fc | Fragment crystallizable |
| FcRn | Neonatal Fc receptor |
| FDA | Food and Drug Administration |
| FMT | Fluorescence molecular tomography |
| HER-2 | Human epidermal growth factor receptor 2 |
| HR/AM | High resolution accurate-mass |
| HRMS | High resolution mass spectrometry |
| HRP | Horseradish peroxidase |
| IgG | Immunoglobulin G |
| IS | Internal standard |
| LBA | Ligand binding assay |
| LB-LC–MS | Ligand-binding immuno-affinity capture followed by LC–MS analysis |
| LC–MS | Liquid chromatography tandem mass spectrometry |
| LESA–µLC–MS | Liquid extraction surface analysis coupled to µLC–MS |
| LLOQ | Lower limit of quantification |
| MMAE | Monomethyl auristatin E |
| MMAF | Monomethyl auristatin F |
| nSMOL | Nano-surface and molecular-orientation limited proteolysis |
| PET | Positron emission tomography |
| QWBA | Quantitative whole-body autoradiography |
| SEC | Size exclusion chromatography |
| SHA | Semihomogeneous assay |
| SPE | Solid phase extraction |
| SPECT | Single-photon emission computed tomography |
| T-DM1 | Trastuzumab emtansine/Kadcyla® |
References
- Joubert, N.; Beck, A.; Dumontet, C.; Denevault-Sabourin, C. Antibody–Drug Conjugates: The Last Decade. Pharmaceuticals 2020, 13, 245. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Liu, D. Antibody-drug conjugates in clinical trials for lymphoid malignancies and multiple myeloma. J. Hematol. Oncol. 2019, 12, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Coats, S.; Williams, M.; Kebble, B.; Dixit, R.; Tseng, L.; Yao, N.; Tice, D.A.; Soria, J. Antibody–Drug Conjugates: Future Directions in Clinical and Translational Strategies to Improve the Therapeutic Index. Clin. Cancer Res. 2019, 25, 5441–5449. [Google Scholar] [CrossRef] [Green Version]
- Chau, C.H.; Steeg, P.S.; Figg, W.D. Therapeutics Antibody–Drug conjugates for cancer. Therapeutics 2019, 394, 793–804. [Google Scholar] [CrossRef]
- Lehar, S.M.; Pillow, T.; Xu, M.; Staben, L.; Kajihara, K.K.; Vandlen, R.; DePalatis, L.; Raab, H.; Hazenbos, W.L.; Hiroshi Morisaki, J.; et al. Novel antibody-antibiotic conjugate eliminates intracellular S. aureus. Nature 2015, 527, 323–328. [Google Scholar] [CrossRef]
- Wang, R.E.; Liu, T.; Wang, Y.; Cao, Y.; Du, J.; Luo, X.; Deshmukh, V.; Kim, C.H.; Lawson, B.R.; Tremblay, M.S.; et al. An immunosuppressive antibody-drug conjugate. J. Am. Chem. Soc. 2015, 137, 3229–3232. [Google Scholar] [CrossRef] [Green Version]
- Lim, R.K.V.; Yu, S.; Cheng, B.; Li, S.; Kim, N.J.; Cao, Y.; Chi, V.; Kim, J.Y.; Chatterjee, A.K.; Schultz, P.G.; et al. Targeted Delivery of LXR Agonist Using a Site-Specific Antibody-Drug Conjugate. Bioconjug. Chem. 2015, 26, 2216–2222. [Google Scholar] [CrossRef]
- Svendsen, P.; Graversen, J.H.; Etzerodt, A.; Hager, H.; Røge, R.; Grønbæk, H.; Christensen, E.I.; Møller, H.J.; Vilstrup, H.; Moestrup, S.K. Antibody-Directed Glucocorticoid Targeting to CD163 in M2-type Macrophages Attenuates Fructose-Induced Liver Inflammatory Changes. Mol. Ther. Methods Clin. Dev. 2017, 4, 50–61. [Google Scholar] [CrossRef] [Green Version]
- Pahl, A.; Lutz, C.; Hechler, T. Amanitins and their development as a payload for antibody-drug conjugates. Drug Discov. Today Technol. 2018, 30, 85–89. [Google Scholar] [CrossRef]
- Masters, J.C.; Nickens, D.J.; Xuan, D.; Shazer, R.L.; Amantea, M. Clinical toxicity of antibody drug conjugates: A meta-analysis of payloads. Invest. New Drugs 2017, 36, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q. Site-specific antibody conjugation for ADC and beyond. Biomedicines 2017, 5, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, R.M.; Coumbe, B.G.T.; Josephs, D.H.; Mele, S.; Ilieva, K.M.; Cheung, A. Antibody structure and engineering considerations for the design and function of Antibody Drug Conjugates (ADCs). Oncoimmunology 2018, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932. [Google Scholar] [CrossRef]
- Hallam, T.J.; Smider, V.V. Unnatural amino acids in novel antibody conjugates. Future Med. Chem. 2014, 6, 1309–1324. [Google Scholar] [CrossRef]
- Hofer, T.; Skeffington, L.R.; Chapman, C.M.; Rader, C. Molecularly defined antibody conjugation through a selenocysteine interface. Biochemistry 2009, 48, 12047–12057. [Google Scholar] [CrossRef] [Green Version]
- Dennler, P.; Chiotellis, A.; Fischer, E.; Brégeon, D.; Belmant, C.; Gauthier, L.; Lhospice, F.; Romagne, F.; Schibli, R. Transglutaminase-based chemo-enzymatic conjugation approach yields homogeneous antibody-drug conjugates. Bioconjug. Chem. 2014, 25, 569–578. [Google Scholar] [CrossRef]
- Pishesha, N.; Ingram, J.R.; Ploegh, H.L. Sortase A: A Model for Transpeptidation and Its Biological Applications. Annu. Rev. Cell Dev. Biol. 2018, 34, 163–188. [Google Scholar] [CrossRef]
- Rabuka, D.; Rush, J.S.; Dehart, G.W.; Wu, P.; Bertozzi, C.R. Site-specific chemical protein conjugation using genetically encoded aldehyde tags. Nat. Protoc. 2012, 7, 1052–1067. [Google Scholar] [CrossRef] [Green Version]
- Badescu, G.; Bryant, P.; Bird, M.; Henseleit, K.; Swierkosz, J.; Parekh, V.; Tommasi, R.; Pawlisz, E.; Jurlewicz, K.; Farys, M.; et al. Bridging disulfides for stable and defined antibody drug conjugates. Bioconjug. Chem. 2014, 25, 1124–1136. [Google Scholar] [CrossRef]
- Van Geel, R.; Wijdeven, M.A.; Heesbeen, R.; Verkade, J.M.M.; Wasiel, A.A.; Van Berkel, S.S.; Van Delft, F.L. Chemoenzymatic Conjugation of Toxic Payloads to the Globally Conserved N-Glycan of Native mAbs Provides Homogeneous and Highly Efficacious Antibody-Drug Conjugates. Bioconjug. Chem. 2015, 26, 2233–2242. [Google Scholar] [CrossRef]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable linkers in antibody-drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef] [PubMed]
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G.; et al. Approval summary: Gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res. 2001, 7, 1490–1496. [Google Scholar] [PubMed]
- Lamb, Y.N. Inotuzumab Ozogamicin: First Global Approval. Drugs 2017, 77, 1603–1610. [Google Scholar] [CrossRef] [PubMed]
- Pillow, T.H.; Schutten, M.; Yu, S.F.; Ohri, R.; Sadowsky, J.; Poon, K.A.; Solis, W.; Zhong, F.; Del Rosario, G.; Go, M.A.T.; et al. Modulating therapeutic activity and toxicity of pyrrolobenzodiazepine antibody-drug conjugates with self-immolative disulfide linkers. Mol. Cancer Ther. 2017, 16, 871–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Claro, R.A.; McGinn, K.; Kwitkowski, V.; Bullock, J.; Khandelwal, A.; Habtemariam, B.; Ouyang, Y.; Saber, H.; Lee, K.; Koti, K.; et al. U.S. Food and Drug Administration approval summary: Brentuximab vedotin for the treatment of relapsed Hodgkin lymphoma or relapsed systemic anaplastic large-cell lymphoma. Clin. Cancer Res. 2012, 18, 5845–5849. [Google Scholar] [CrossRef] [Green Version]
- Deeks, E.D. Polatuzumab Vedotin: First Global Approval. Drugs 2019, 79, 1467–1475. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Y.J.; Shao, L.H.; Li, Y. Cathepsin B-cleavable doxorubicin prodrugs for targeted cancer therapy (Review). Int. J. Oncol. 2013, 42, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Jeffrey, S.C.; Andreyka, J.B.; Bernhardt, S.X.; Kissler, K.M.; Kline, T.; Lenox, J.S.; Moser, R.F.; Nguyen, M.T.; Okeley, N.M.; Stone, I.J.; et al. Development and properties of β-glucuronide linkers for monoclonal antibody-drug conjugates. Bioconjug. Chem. 2006, 17, 831–840. [Google Scholar] [CrossRef]
- Kern, J.C.; Cancilla, M.; Dooney, D.; Kwasnjuk, K.; Zhang, R.; Beaumont, M.; Figueroa, I.; Hsieh, S.C.; Liang, L.; Tomazela, D.; et al. Discovery of Pyrophosphate Diesters as Tunable, Soluble, and Bioorthogonal Linkers for Site-Specific Antibody-Drug Conjugates. J. Am. Chem. Soc. 2016, 138, 1430–1445. [Google Scholar] [CrossRef]
- Erickson, H.K.; Widdison, W.C.; Mayo, M.F.; Whiteman, K.; Audette, C.; Wilhelm, S.D.; Singh, R. Tumor delivery and in vivo processing of disulfide-linked and thioether-linked antibody-maytansinoid conjugates. Bioconjug. Chem. 2010, 21, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Doronina, S.O.; Mendelsohn, B.A.; Bovee, T.D.; Cerveny, C.G.; Alley, S.C.; Meyer, D.L.; Oflazoglu, E.; Toki, B.E.; Sanderson, R.J.; Zabinski, R.F.; et al. Enhanced activity of monomethylauristatin F through monoclonal antibody delivery: Effects of linker technology on efficacy and toxicity. Bioconjug. Chem. 2006, 17, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Lucas, A.; Price, L.; Schorzman, A.; Storrie, M.; Piscitelli, J.; Razo, J.; Zamboni, W. Factors Affecting the Pharmacology of Antibody–Drug Conjugates. Antibodies 2018, 7, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, A.T.; Robinson, R.; Schorzman, A.N.; Piscitelli, J.A.; Razo, J.F.; Zamboni, W.C. Pharmacologic Considerations in the Disposition of Antibodies and Antibody-Drug Conjugates in Preclinical Models and in Patients. Antibodies 2019, 8, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, B.Q.; Xu, K.; Liu, L.; Raab, H.; Bhakta, S.; Kenrick, M.; Parsons-Reponte, K.L.; Tien, J.; Yu, S.F.; Mai, E.; et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat. Biotechnol. 2012, 30, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Gorovits, B.; Alley, S.C.; Bilic, S.; Booth, B.; Kaur, S.; Oldfield, P.; Purushothama, S.; Rao, C.; Shord, S.; Siguenza, P. Bioanalysis of antibody-drug conjugates: American Association of Pharmaceutical Scientists Antibody-Drug Conjugate Working Group position paper. Bioanalysis 2013, 5, 997–1006. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Ponte, J.F.; Yoder, N.C.; Laleau, R.; Coccia, J.; Lanieri, L.; Qiu, Q.; Wu, R.; Hong, E.; Bogalhas, M.; et al. Effects of Drug-Antibody Ratio on Pharmacokinetics, Biodistribution, Efficacy, and Tolerability of Antibody-Maytansinoid Conjugates. Bioconjug. Chem. 2017, 28, 1371–1381. [Google Scholar] [CrossRef]
- Kamath, A.V.; Iyer, S. Preclinical Pharmacokinetic Considerations for the Development of Antibody Drug Conjugates. Pharm. Res. 2015, 32, 3470–3479. [Google Scholar] [CrossRef] [Green Version]
- Sukumaran, S.; Zhang, C.; Leipold, D.D.; Saad, O.M.; Xu, K.; Gadkar, K.; Samineni, D.; Wang, B.; Milojic-blair, M.; Rubinfeld, B.; et al. Development and Translational Application of an Integrated, Mechanistic Model of Antibody-Drug Conjugate Pharmacokinetics. AAPS J. 2016. [Google Scholar] [CrossRef]
- Lin, K.; Tibbitts, J. Pharmacokinetic considerations for antibody drug conjugates. Pharm. Res. 2012, 29, 2354–2366. [Google Scholar] [CrossRef]
- Kumar, S.; King, L.E.; Clark, T.H.; Gorovits, B. Antibody-drug conjugates nonclinical support: From early to late nonclinical bioanalysis using ligand-binding assays. Bioanalysis 2015, 7, 1605–1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowell, J.A.; Korth-Bradley, J. Pharmacokinetics of Gemtuzumab Ozogamicin, an Antibody-Targeted Chemotherapy Agent for the Treatment of Patients with Acute Myeloid Leukemia in First Relapse. J. Clin. Pharmacol. 2001, 41, 1206–1214. [Google Scholar] [CrossRef]
- Lewis Phillips, G.D.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blättler, W.A.; Lambert, J.M.; Chari, R.V.J.; Lutz, R.J.; et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollack, V.A.; Alvarez, E.; Tse, K.F.; Torgov, M.Y.; Xie, S.; Shenoy, S.G.; MacDougall, J.R.; Arrol, S.; Zhong, H.; Gerwien, R.W.; et al. Treatment parameters modulating regression of human melanoma xenografts by an antibody–Drug conjugate (CR011-vcMMAE) targeting GPNMB. Cancer Chemother. Pharmacol. 2007, 60, 423–435. [Google Scholar] [CrossRef]
- Singh, A.P.; Shah, D.K. Measurement and Mathematical Characterization of Cell-Level Pharmacokinetics of Antibody-Drug Conjugates: A Case Study with Trastuzumab-vc-MMAE s. Drug Metab. Dispos. 2017, 45, 1120–1132. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.P.; Guo, L.; Verma, A.; Wong, G.G.; Shah, D.K. A Cell-Level Systems PK-PD Model to Characterize In Vivo Efficacy of ADCs. Pharmaceutics 2019, 11, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephan, J.P.; Chan, P.; Lee, C.; Nelson, C.; Elliott, J.M.; Bechtel, C.; Raab, H.; Xie, D.; Akutagawa, J.; Baudys, J.; et al. Anti-CD22-MCC-DM1 and MC-MMAF conjugates: Impact of assay format on pharmacokinetic parameters determination. Bioconjug. Chem. 2008, 19, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Audette, C.; Hoffee, M.; Lambert, J.M.; Blättler, W. Erratum: Pharmacokinetics and biodistribution of the antitumor immunoconjugate, cantuzumab mertansine (huC242-DM1), and its two components in mice (Journal of Pharmacology and Experimental Therapeutics (2004) 308 (1073-1082)). J. Pharmacol. Exp. Ther. 2004, 310, 844. [Google Scholar] [CrossRef]
- Kovtun, Y.V.; Audette, C.A.; Ye, Y.; Kovtun, Y.V.; Audette, C.A.; Ye, Y.; Xie, H.; Ruberti, M.F.; Phinney, S.J.; Leece, B.A.; et al. Antibody-Drug Conjugates Designed to Eradicate Tumors with Homogeneous and Heterogeneous Expression of the Target Antigen with Homogeneous and Heterogeneous Expression of the Target Antigen. Cancer Res. 2006, 66, 3214–3221. [Google Scholar] [CrossRef] [Green Version]
- Advani, A.; Coiffier, B.; Czuczman, M.S.; Dreyling, M.; Foran, J.; Gine, E.; Gisselbrecht, C.; Ketterer, N.; Nasta, S.; Rohatiner, A.; et al. Safety, Pharmacokinetics, and Preliminary Clinical Activity of Inotuzumab Ozogamicin, a Novel Immunoconjugate for the Treatment of B-Cell Non-Hodgkin’ s Lymphoma: Results of a Phase I Study. J. Clin. Oncol. 2010, 28, 2085–2093. [Google Scholar] [CrossRef]
- DiJoseph, J.F.; Armellino, D.C.; Boghaert, E.R.; Khandke, K.; Dougher, M.M.; Sridharan, L.; Kunz, A.; Hamann, P.R.; Gorovits, B.; Udata, C.; et al. Antibody-targeted chemotherapy with CMC-544: A CD22-targeted immunoconjugate of calicheamicin for the treatment of B-lymphoid malignancies. Blood 2004, 103, 1807–1814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alley, S.C.; Benjamin, D.R.; Jeffrey, S.C.; Okeley, N.M.; Meyer, D.L.; Sanderson, R.J.; Senter, P.D. Contribution of linker stability to the activities of anticancer immunoconjugates. Bioconjug. Chem. 2008, 19, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, B.A.W.; Ochoa, L.; Hammond, L.A.; Patnaik, A.; Edwards, T.; Takimoto, C.; Smith, L.; De Bono, J.; Schwartz, G.; Mays, T.; et al. Directed to the CanAg Antigen: A Phase I, Pharmacokinetic, and Biologic Correlative Study. J. Clin. Oncol. 2003, 21, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, R.J.; Hering, M.A.; James, S.F.; Sun, M.M.C.; Doronina, S.O.; Siadak, A.W.; Senter, P.D.; Wahl, A.F. In vivo drug-linker stability of an anti-CD30 dipeptide-linked auristatin immunoconjugate. Clin. Cancer Res. 2005, 11, 843–852. [Google Scholar]
- Kozak, K.R.; Tsai, S.P.; Fourie-O’Donohue, A.; Dela Cruz Chuh, J.; Roth, L.; Cook, R.; Chan, E.; Chan, P.; Darwish, M.; Ohri, R.; et al. Total antibody quantification for MMAE-conjugated antibody-drug conjugates: Impact of assay format and reagents. Bioconjug. Chem. 2013, 24, 772–779. [Google Scholar] [CrossRef]
- Chang, H.-P.; Shah, D.K. Determination of ADC concentration by Ligand-Binding Assays. In Methods in Molecular Biology 2078; Springer: Berlin, Germany, 2020; pp. 361–369. ISBN 9781493999286. [Google Scholar]
- Ramagiri, S.; Moore, I. Hybridizing LBA with LC-MS/MS: The new norm for biologics quantification. Bioanalysis 2016, 8, 483–486. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Huo, S.; Xue, C.; An, B.; Qu, J. Current LC-MS-based strategies for characterization and quanti fi cation of antibody-drug conjugates. J. Pharm. Anal. 2020, 10, 209–220. [Google Scholar] [CrossRef]
- Botzanowski, T.; Wagner-Rousset, E.; Rabuka, D.; Beck, A.; Drake, P.M.; Cianf, S. Insights from native mass spectrometry approaches for top- and middle- level characterization of site-specific antibody-drug conjugates. MAbs 2017, 9, 801–811. [Google Scholar] [CrossRef]
- Fung, E.N.; Bryan, P.; Kozhich, A. Techniques for quantitative LC-MS/MS analysis of protein therapeutics: Advances in enzyme digestion and immunocapture. Bioanalysis 2016, 8, 847–856. [Google Scholar] [CrossRef]
- Van Den Broek, I.; Van Dongen, W.D. LC-MS-based quantification of intact proteins: Perspective for clinical and bioanalytical applications. Bioanalysis 2015, 7, 1943–1958. [Google Scholar] [CrossRef]
- Wang, J.; Gu, H.; Liu, A.; Kozhich, A.; Rangan, V.; Myler, H.; Luo, L.; Wong, R.; Sun, H.; Wang, B.; et al. Antibody-drug conjugate bioanalysis using LB-LC-MS/MS hybrid assays: Strategies, methodology and correlation to ligand-binding assays. Bioanalysis 2016, 8, 1383–1401. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ortiz, R.; Tran, L.; Hall, M.; Spahr, C.; Walker, K.; Laudemann, J.; Miller, S.; Salimi-Moosavi, H.; Lee, J.W. General LC-MS/MS method approach to quantify therapeutic monoclonal antibodies using a common whole antibody internal standard with application to preclinical studies. Anal. Chem. 2012, 84, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Su, D.; Ng, C.; Liu, L.; Yu, S.F.; Pillow, T.H.; Del Rosario, G.; Darwish, M.; Lee, B.C.; Ohri, R.; et al. High-Resolution Accurate-Mass Mass Spectrometry Enabling In-Depth Characterization of in Vivo Biotransformations for Intact Antibody-Drug Conjugates. Anal. Chem. 2017, 89, 5476–5483. [Google Scholar] [CrossRef]
- Jian, W.; Kang, L.; Burton, L.; Weng, N. A workflow for absolute quantitation of large therapeutic proteins in biological samples at intact level using LC-HRMS. Bioanalysis 2016, 8, 1679–1691. [Google Scholar] [CrossRef] [PubMed]
- Lanshoeft, C.; Cianférani, S.; Heudi, O. Generic Hybrid Ligand Binding Assay Liquid Chromatography High-Resolution Mass Spectrometry-Based Workflow for Multiplexed Human Immunoglobulin G1 Quantification at the Intact Protein Level: Application to Preclinical Pharmacokinetic Studies. Anal. Chem. 2017, 89, 2628–2635. [Google Scholar] [CrossRef]
- Jin, W.; Burton, L.; Moore, I. LC–HRMS quantitation of intact antibody drug conjugate trastuzumab emtansine from rat plasma. Bioanalysis 2018, 10, 851–862. [Google Scholar] [CrossRef]
- He, J.; Yu, S.F.; Yee, S.; Kaur, S.; Xu, K. Characterization of in vivo biotransformations for trastuzumab emtansine by high-resolution accurate-mass mass spectrometry. MAbs 2018, 10, 960–967. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Manuilov, A.V.; Chumsae, C.; Babineau, M.L.; Tarcsa, E. Quantitation of a recombinant monoclonal antibody in monkey serum by liquid chromatography-mass spectrometry. Anal. Biochem. 2011, 414, 147–153. [Google Scholar] [CrossRef]
- Wagner-Rousset, E.; Janin-Bussat, M.C.; Colas, O.; Excoffier, M.; Ayoub, D.; Haeuw, J.F.; Rilatt, I.; Perez, M.; Corvaïa, N.; Beck, A. Antibody-drug conjugate model fast characterization by LC-MS following IdeS proteolytic digestion. MAbs 2014, 6, 173–184. [Google Scholar] [CrossRef] [Green Version]
- Kotapati, S.; Passmore, D.; Yamazoe, S.; Kishore, R.; Sanku, K.; Cardarelli, P.M.; Deshpande, S.; Strop, P.; Dollinger, G.; Rajpal, A. Article A Universal Affinity Capture LC-MS Assay for Evaluation of Biotransformation of Site-Specific Antibody Drug Conjugates in Preclinical Studies A Universal Affinity Capture LC-MS Assay for Evaluation of Biotransformation of Site-Specific Antibody Dr. Anal. Chem. 2019, 92, 2065–2073. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Kozhich, A.; Passmore, D.; Gu, H.; Wong, R.; Zambito, F.; Rangan, V.S.; Myler, H.; Aubry, A.F.; Arnold, M.E.; et al. Quantitative bioanalysis of antibody-conjugated payload in monkey plasma using a hybrid immuno-capture LC-MS/MS approach: Assay development, validation, and a case study. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2015, 1002, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Rago, B.; Tumey, L.N.; Wei, C.; Barletta, F.; Clark, T.; Hansel, S.; Han, X. Quantitative Conjugated Payload Measurement Using Enzymatic Release of Antibody−Drug Conjugate with Cleavable Linker. Bioconjug. Chem. 2017, 28, 620–626. [Google Scholar] [CrossRef]
- Sanderson, R.J.; Nicholas, N.D.; Baker Lee, C.; Hengel, S.M.; Lyon, R.P.; Benjamin, D.R.; Alley, S.C. Antibody-conjugated drug assay for protease-cleavable antibody-drug conjugates. Bioanalysis 2016, 8, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gu, C.; Gruenhagen, J.; Yehl, P.; Chetwyn, N.P.; Medley, C.D. An enzymatic deconjugation method for the analysis of small molecule active drugs on antibody-drug conjugates. MAbs 2016, 8, 698–705. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.; Goldberg, S.; Lin, T.; Dudkin, V.; Widdison, W.; Harris, L.; Wilhelm, S.; Jmeian, Y.; Davis, D.; O’Neil, K.; et al. LC/MS/MS Bioanalysis of Protein-Drug Conjugates—The Importance of Incorporating Succinimide Hydrolysis Products. Anal. Chem. 2018, 90, 5314–5321. [Google Scholar] [CrossRef]
- Wei, C.; Zhang, G.; Clark, T.; Barletta, F.; Tumey, L.N.; Rago, B.; Hansel, S.; Han, X. Where Did the Linker-Payload Go? A Quantitative Investigation on the Destination of the Released Linker-Payload from an Antibody-Drug Conjugate with a Maleimide Linker in Plasma. Anal. Chem. 2016, 88, 4979–4986. [Google Scholar] [CrossRef]
- Dong, L.; Li, C.; Locuson, C.; Chen, S.; Qian, M.G. A Two-Step Immunocapture LC/MS/MS Assay for Plasma Stability and Payload Migration Assessment of Cysteine-Maleimide-Based Antibody Drug Conjugates. Anal. Chem. 2018, 90, 5989–5994. [Google Scholar] [CrossRef]
- Iwamoto, N.; Shimada, T.; Umino, Y.; Aoki, C.; Aoki, Y.; Sato, T.-A.; Hamada, A.; Nakagama, H. Selective detection of complementarity-determining regions of monoclonal antibody by limiting protease access to the substrate: Nano-. Analyst 2014, 139, 576–580. [Google Scholar] [CrossRef]
- Iwamoto, N.; Shimomura, A.; Tamura, K.; Hamada, A. LC–MS bioanalysis of Trastuzumab and released emtansine using nano-surface and molecular-orientation limited (nSMOL) proteolysis and liquid–liquid partition in plasma of Trastuzumab emtansine-treated breast cancer patients. J. Pharm. Biomed. Anal. 2017, 145, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, N.; Hamada, A.; Shimada, T. Antibody drug quantitation in coexistence with anti-drug antibodies on nSMOL bioanalysis. Anal. Biochem. 2018, 540–541, 30–37. [Google Scholar] [CrossRef]
- Heudi, O.; Barteau, S.; Picard, F.; Kretz, O. Quantitative analysis of maytansinoid (DM1) in human serum by on-line solid phase extraction coupled with liquid chromatography tandem mass spectrometry—Method validation and its application to clinical samples. J. Pharm. Biomed. Anal. 2016, 120, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Sullivan, M.; Espinosa, O.; Yang, L. A sensitive LC-MS/MS method for the determination of free maytansinoid DM4 concentrations—Method development, validation, and application to the nonclinical studies of antitumor agent DM4 conjugated hu-anti-Cripto MAb B3F6 (B3F6-DM4) in rats and monkeys. Int. J. Mass Spectrom. 2012, 312, 53–60. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Furuta, M.; Manabe, S.; Koga, Y.; Yasunaga, M.; Matsumura, Y. Imaging mass spectrometry for the precise design of antibody-drug conjugates. Sci. Rep. 2016, 6, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Leal, M.; Wentland, J.A.; Han, X.; Zhang, Y.; Rago, B.; Duriga, N.; Spriggs, F.; Kadar, E.; Song, W.; McNally, J.; et al. Preclinical Development of an anti-5T4 Antibody-Drug Conjugate: Pharmacokinetics in Mice, Rats, and NHP and Tumor/Tissue Distribution in Mice. Bioconjug. Chem. 2015, 26, 2223–2232. [Google Scholar] [CrossRef] [PubMed]
- Walles, M.; Rudolph, B.; Wolf, T.; Bourgailh, J.; Suetterlin, M.; Moenius, T.; Peraus, G.; Heudi, O.; Elbast, W.; Lanshoeft, C.; et al. New insights in tissue distribution, metabolism, and excretion of [3H]-labeled antibody maytansinoid conjugates in female tumor-bearing nude rats. Drug Metab. Dispos. 2016, 44, 897–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmon, K.S.; Azhdarinia, A. Application of Immuno-PET in Antibody—Drug Conjugate Development. Mol. Imaging 2018, 17, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Sijbrandi, N.J.; Merkul, E.; Muns, J.A.; Waalboer, D.C.J.; Adamzek, K.; Bolijn, M.; Somsen, G.W.; Haselberg, R.; Steverink, P.J.G.M.; Houthoff, H.; et al. A Novel Platinum (II)—Based Bifunctional ADC Linker Benchmarked Using 89 Zr-Desferal and Auristatin F—Conjugated Trastuzumab. Cancer Res. 2017, 77, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Chia, P.; Parakh, S.; Tsao, M.; Pham, N.; Gan, H.K.; Cao, D.; Burvenich, I.J.G.; Rigopoulos, A.; Reilly, E.B.; John, T.; et al. Targeting and Efficacy of Novel mAb806-Antibody-Drug Conjugates in Malignant Mesothelioma. Pharmaceuticals 2020, 13, 289. [Google Scholar] [CrossRef]
- Lütje, S.; Gerrits, D.; Molkenboer-kuenen, J.D.; Herrmann, K.; Fracasso, G.; Colombatti, M.; Boerman, O.C.; Heskamp, S. Characterization of Site-Specifically Conjugated Monomethyl Auristatin E–And Duocarmycin-Based Anti-PSMA Antibody–Drug Conjugates for Treatment of PSMA-Expressing Tumors. J. Nucl. Med. 2018, 59, 494–502. [Google Scholar] [CrossRef] [Green Version]
- Boswell, C.A.; Yadav, D.B.; Mundo, E.E.; Yu, S.F.; Lacap, J.A.; Fourie-O’Donohue, A.; Kozak, K.R.; Ferl, G.Z.; Zhang, C.; Ho, J.; et al. Biodistribution and efficacy of an anti-TENB2 antibody-drug conjugate in a patient-derived model of prostate cancer. Oncotarget 2019, 10, 6234–6244. [Google Scholar] [CrossRef] [Green Version]
- Boswell, C.A.; Mundo, E.E.; Zhang, C.; Stainton, S.L.; Yu, S.F.; Lacap, J.A.; Mao, W.; Kozak, K.R.; Fourie, A.; Polakis, P.; et al. Differential effects of predosing on tumor and tissue uptake of an 111in-labeled anti-TENB2 antibody-drug conjugate. J. Nucl. Med. 2012, 53, 1454–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ter Weele, E.J.; van Scheltinga, A.G.T.T.; Kosterink, J.G.W.; Pot, L.; Vedelaar, S.R.; Lamberts, L.E.; Williams, S.P.; Lub-de Hooge, M.N.; de Vries, E.G.E. Imaging the distribution of an antibody-drug conjugate constituent targeting mesothelin with 89Zr and IRDye 800CW in mice bearing human pancreatic tumor xenografts. Oncotarget 2015, 6, 42081–42090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giddabasappa, A.; Gupta, V.R.; Norberg, R.; Gupta, P.; Spilker, M.E.; Wentland, J.; Rago, B.; Eswaraka, J.; Leal, M.; Sapra, P. Biodistribution and targeting of anti-5t4 antibody-drug conjugate using fluorescence molecular tomography. Mol. Cancer Ther. 2016, 15, 2530–2540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Zhao, X.; Sun, X.; Li, J.; Wang, W.; Zhang, L.; Gou, S. Preclinical pharmacokinetics of a novel anti-c-Met antibody–drug conjugate, SHR-A1403, in rodents and non-human primates. Xenobiotica 2019, 49, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Saad, O.M.; Shen, B.Q.; Xu, K.; Khojasteh, S.C.; Girish, S.; Kaur, S. Bioanalytical approaches for characterizing catabolism of antibody-drug conjugates. Bioanalysis 2015, 7, 1583–1604. [Google Scholar] [CrossRef] [PubMed]
- Mandler, R.; Kobayashi, H.; Hinson, E.R.; Brechbiel, M.W.; Waldmann, T.A. Herceptin-Geldanamycin Immunoconjugates: Pharmacokinetics, Biodistribution, and Enhanced Antitumor Activity. Cancer Res. 2004, 64, 1460–1467. [Google Scholar] [CrossRef] [Green Version]
- Journal, A.I.; Gong, J.; Guo, F.; Cheng, W.; Fan, H.; Miao, Q. Preliminary biological evaluation of 123I-labelled anti-CD30-LDM in CD30-positive lymphomas murine models. Artif. Cells Nanomed. Biotechnol. 2020, 48, 408–414. [Google Scholar] [CrossRef]
- Boswell, C.A.; Mundo, E.E.; Zhang, C.; Bumbaca, D.; Valle, N.R.; Kozak, K.R.; Fourie, A.; Chuh, J.; Koppada, N.; Saad, O.; et al. Impact of drug conjugation on pharmacokinetics and tissue distribution of Anti-STEAP1 antibody-drug conjugates in rats. Bioconjug. Chem. 2011, 22, 1994–2004. [Google Scholar] [CrossRef]
- Herbertson, R.A.; Tebbutt, N.C.; Lee, F.T.; MacFarlane, D.J.; Chappell, B.; Micallef, N.; Lee, S.T.; Saunder, T.; Hopkins, W.; Smyth, F.E.; et al. Phase I biodistribution and pharmacokinetic study of Lewis Y-targeting immunoconjugate CMD-193 in patients with advanced epithelial cancers. Clin. Cancer Res. 2009, 15, 6709–6715. [Google Scholar] [CrossRef] [Green Version]
- Shen, B.-Q.; Bumbaca, D.; Saad, O.; Yue, Q.; V Pastuskovas, C.; Cyrus Khojasteh, S.; Tibbitts, J.; Kaur, S.; Wang, B.; Chu, Y.-W.; et al. Catabolic Fate and Pharmacokinetic Characterization of Trastuzumab Emtansine (T-DM1): An Emphasis on Preclinical and Clinical Catabolism. Curr. Drug Metab. 2012, 13, 901–910. [Google Scholar] [CrossRef]
- Alley, S.C.; Zhang, X.; Okeley, N.M.; Anderson, M.; Law, C.L.; Senter, P.D.; Benjamin, D.R. The pharmacologic basis for antibody-auristatin conjugate activity. J. Pharmacol. Exp. Ther. 2009, 330, 932–938. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.; Vugts, D.J.; Visser, G.W.M.; Stigter-Van Walsum, M.; Bolijn, M.; Spiga, M.; Lazzari, P.; Shankar, E.; Sani, M.; Zanda, M.; et al. Development of novel ADCs: Conjugation of tubulysin analogues to trastuzumab monitored by dual radiolabeling. Cancer Res. 2014, 74, 5700–5710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilovich, O.; Qutaish, M.; Hesterman, J.Y.; Orcutt, K.; Hoppin, J.; Polyak, I.; Seaman, M.; Abu-Yousif, A.O.; Cvet, D.; Bradley, D.P. Dual-isotope cryoimaging quantitative autoradiography: Investigating antibody-drug conjugate distribution and payload delivery through imaging. J. Nucl. Med. 2018, 59, 1461–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muns, J.A.; Montserrat, V.; Houthoff, H.; Codée-van der Schilden, K.; Zwaagstra, O.; Sijbrandi, N.J.; Merkul, E.; Van Dogen, G.A.M.S. In vivo Characterization of Platinum(II)-base linker Technology for the Development of Antibody—Drug Conjugates: Taking Advantage of Dual Labeling with. J. Nucl. Med. 2018, 59, 1146–1152. [Google Scholar] [CrossRef] [Green Version]
- An, B.; Zhang, M.; Pu, J.; Shen, S.; Qu, Y.; Chen, Y.; Huo, S.; Wang, X.; Polli, J.R.; Balthasar, J.P.; et al. High-throughput, sensitive LC-MS quantification of biotherapeutics and biomarkers using an antibody-free, peptide-level multiple-mechanism enrichment via strategically regulated pH, ionic and solvent strengths The Department of Pharmaceutical Sciences. Anal. Chem. 2019, 91, 3475–3483. [Google Scholar] [CrossRef]
Figure 1.
The catabolic fate of ADCs increases the complexity of the mixture in vivo giving rise to multiple analytes to be measured. The antibody is colored in blue, the free drug and its metabolites are represented as red stars.
Figure 1.
The catabolic fate of ADCs increases the complexity of the mixture in vivo giving rise to multiple analytes to be measured. The antibody is colored in blue, the free drug and its metabolites are represented as red stars.

Figure 2.
A typical workflow for the bioanalysis of ADCs in biological sample using LB-LC–MS/MS methods. The antibody and the peptide fragments are colored in blue, the free drug and its metabolites are represented as red stars. The internal standards, Peptide IS and Peptide-drug IS, are colored in cyan. The Drug IS is represented as a cyan star. IS: internal standard
Figure 2.
A typical workflow for the bioanalysis of ADCs in biological sample using LB-LC–MS/MS methods. The antibody and the peptide fragments are colored in blue, the free drug and its metabolites are represented as red stars. The internal standards, Peptide IS and Peptide-drug IS, are colored in cyan. The Drug IS is represented as a cyan star. IS: internal standard

Figure 3.
Overview of the different in vivo and ex vivo methods used to study the biodistribution of ADCs in preclinical model. PET: positron emission tomography. SPECT: single-photon emission computed tomography. FMT: fluorescence molecular tomography. QWBA: quantitative whole-body autoradiography. LC–MS/MS: liquid chromatography tandem mass spectrometry. LESA–µLC–MS/MS: liquid extraction surface analysis coupled to liquid chromatography tandem mass spectrometry. PET/SPECT, FMT, and QWBA images were extracted from doi:10.18632/oncotarget.27263; doi:10.1158/1535-7163.MCT-15-1012 and doi:10.1124/dmd.115.069021, respectively.
Figure 3.
Overview of the different in vivo and ex vivo methods used to study the biodistribution of ADCs in preclinical model. PET: positron emission tomography. SPECT: single-photon emission computed tomography. FMT: fluorescence molecular tomography. QWBA: quantitative whole-body autoradiography. LC–MS/MS: liquid chromatography tandem mass spectrometry. LESA–µLC–MS/MS: liquid extraction surface analysis coupled to liquid chromatography tandem mass spectrometry. PET/SPECT, FMT, and QWBA images were extracted from doi:10.18632/oncotarget.27263; doi:10.1158/1535-7163.MCT-15-1012 and doi:10.1124/dmd.115.069021, respectively.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Antibody–drug conjugates (ADCs) approved by the Food and Drug Administration (FDA).
| ADC | FDA Approval | Indication | Target Antigen | Antibody | Linker | Payload | Conjugation | Average DAR |
|---|---|---|---|---|---|---|---|---|
| Mylotarg® | 2000, withdrawn in 2010 and reapproved in 2017 | Relapsed or refractory acute myeloid leukemia | CD33 | IgG4 humanized | Hydrazone (cleavable) | Calicheamicine | Lysine | 2.5 |
| Adcetris® | 2011 | Cutaneous anaplastic large cell lymphoma | CD30 | IgG1 chimeric | Val-Cit (cleavable) | MMAE | Cysteine | 4 |
| Kadcyla® | 2013 | HER2-positive metastatic breast cancer | HER2 | IgG1 humanized | SMCC * (noncleavable) | DM1 | Lysine | 3.5 |
| Besponsa® | 2017 | Acute lymphoblastic leukemia | CD22 | IgG4 humanized | Hydrazone (cleavable) | Calicheamicine | Lysine | 6 |
| Polivy® | 2019 | Diffuse large B-cell lymphoma | CD79b | IgG1 humanized | Val-Cit (cleavable) | MMAE | Cysteine | 4 |
| Enhertu® | 2019 | HER2-positive metastatic breast cancer | HER2 | IgG1 humanized | Gly-Gly-Phe-Gly (cleavable) | DXd | Cysteine | 7.5 |
| Padcev® | 2019 | Locally advanced or metastatic urothelial cancer | Nectin-4 | IgG1 fully human | Val-Cit (cleavable) | MMAE | Cysteine | 3.8 |
| Trodelvy® | 2020 | Triple negative breast cancer | Trop-2 | IgG1 humanized | Carbonate (cleavable) | SN-38 | Cysteine | 7.6 |
| Blenrep® | 2020 | Relapsed or refractory multiple myeloma | BCMA | IgG1 humanized | MC * (noncleavable) | MMAF | Cysteine | 4 |
* SMCC: Succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate; MC: Maleimido-caproyl.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Cahuzac, H.; Devel, L. Analytical Methods for the Detection and Quantification of ADCs in Biological Matrices. Pharmaceuticals 2020, 13, 462. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13120462
AMA Style
Cahuzac H, Devel L. Analytical Methods for the Detection and Quantification of ADCs in Biological Matrices. Pharmaceuticals. 2020; 13(12):462. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13120462
Chicago/Turabian StyleCahuzac, Héloïse, and Laurent Devel. 2020. "Analytical Methods for the Detection and Quantification of ADCs in Biological Matrices" Pharmaceuticals 13, no. 12: 462. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13120462
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.