4.2. Chemistry
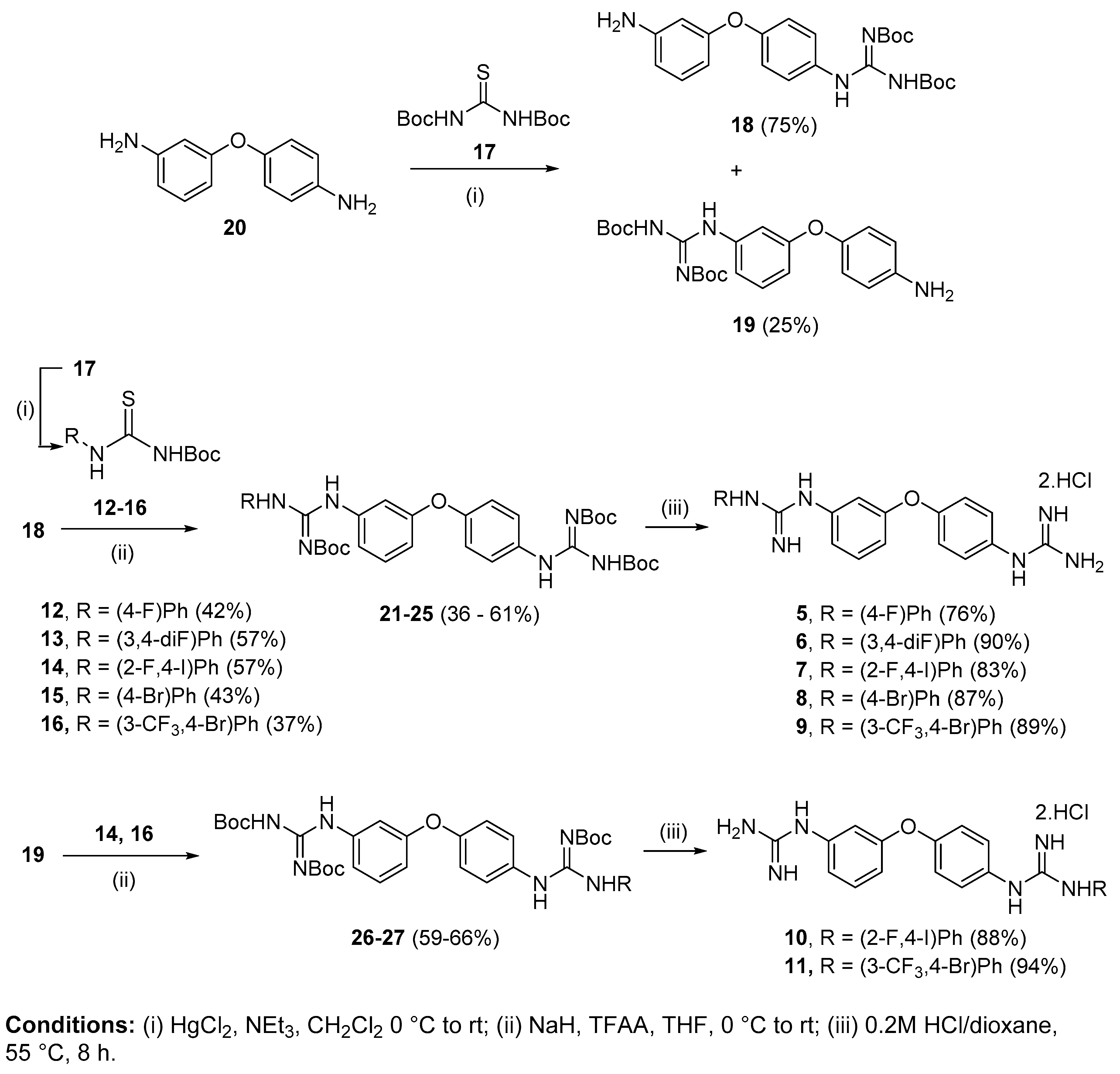
4.2.1. 1-(4-Fluorophenyl)-2-(3-(4-guanidinophenoxy)phenyl)guanidine dihydrochloride (5)
Following Method A (see ESI), 21 (80 mg, 0.12 mmol) was dissolved in 4 M HCl in dioxane (0.54 mL, 2.16 mmol) and in additional dioxane (0.07 mL) until a final concentration of 0.2 M was reached. After 8 h stirring at 55 °C, the reaction was adjudged complete (TLC), solvents were evaporated and the residue was purified by silica gel (CH3Cl:MeOH) chromatography to afford the pure hydrochloride salt as a white-yellow solid (35 mg, 76%). Mp: decomp. > 180 °C. δH (400 MHz, CD3OD): 7.00 (dd, J = 8.3, 2.4, 1H, H-4), 7.04 (t, J = 2.1 Hz, 1H, H-2), 7.13–7.17 (m, 3H, H-8 and H-8′ and H-6), 7.19–7.24 (m, 2H, H-13 and H-13′), 7.31 (d, J = 8.9 Hz, 2H, H-9 and H-9′), 7.36–7.39 (m, 2H, H-12 and H-12′), 7.47 (t, J = 8.1 Hz, 1H, H-5). δC (100 MHz, CD3OD): 116.5 (CH Ar, C-2), 117.8 (d, J = 23.3 Hz, 2 CH Ar, C-13 and C-13′), 118.6 (CH Ar, C-4), 121.1 (CH Ar, C-6), 121.5 (2 CH Ar, C-8 and C-8′), 129.95 (d, J = 8.8 Hz, 2 CH Ar, C-12 and C-12′), 128.96 (2 CH Ar, C-9 and C-9′), 131.6 (qC), 132.3 (d, J = 3.1 Hz, qC, C-11), 132.4 (CH Ar, C-5), 138.0 (qC), 156.6 (qC), 157.4 (qC), 158.4 (qC), 159.5 (qC), 163.2 (d, J = 246.1 Hz, qC, C-14). δF (376 MHz, CD3OD): −115.93 (m). νmax(ATR)/cm−1: 3110 (NH), 3052 (NH), 2922, 2330, 2134, 1655 (C=N), 1582 (C=N), 1505 (C-N), 1486, 1404, 1212 (C-O), 1066 (C-F), 834, 792, 552. HRMS (m/z ESI+): found: 379.1687 (M+ + H), C20H20N6OF requires: 379.1683. HPLC: 99.7% (tR: 22.9 min).
4.2.2. 1-(3:4-Di-fluorophenyl)-2-(3-(4-guanidinophenoxy)phenyl)guanidine dihydrochloride (6)
Following Method A (see ESI), 22 (53 mg, 0.08 mmol) was dissolved in 4 M HCl in dioxane (0.36 mL, 1.37 mmol) and in additional dioxane (0.06 mL) until a final concentration of 0.2 M was reached. After 8 h stirring at 55 °C, the reaction was adjudged complete (TLC), solvents were evaporated and the residue was purified by silica gel chromatography (CHCl3:MeOH) to afford the pure hydrochloride salt as a white solid (34 mg, 90%). Mp: 158–160 °C. δH (400 MHz, CD3OD): 7.00 (dd, J = 8.3, 2.4 Hz, 1H, H-4), 7.05 (t, J = 2.2 Hz, 1H, H-2), 7.13–7.20 (m, 4H, H-8 and H-8′, H-6 and H-16), 7.32 (d, J = 8.9 Hz, 2H, H-9 and H-9′), 7.34–7.41 (m, 2H, H-12 and H-15), 7.47 (t, J = 8.1 Hz, 1H, H-5). δC (100 MHz, CD3OD): 116.3 (d, J = 19.7 Hz, C-12 or C-15), 116.5 (CH Ar, C-2), 118.6 (CH Ar, C-4), 119.5 (d, J = 18.8 Hz, C-12 or C-15), 121.1 (CH Ar, C-6), 121.5 (2 CH Ar, C-8 and C-8′), 123.4 (dd, J = 6.7, 3.7 Hz, C-16), 129.0 (2 CH Ar, C-9 and C-9′), 131.6 (qC), 132.4 (CH Ar, C-5), 133.0 (dd, J = 8.3, 3.6 Hz, qC, C-11), 137.9 (qC), 150.8 (dd, J = 247.8, 12.6 Hz, qC, C-13 or C-14), 151.8 (dd, J = 248.7, 13.7 Hz, qC, C-13 or C-14), 156.5 (qC), 157.4 (qC), 158.3 (qC), 159.5 (qC). δF (376 MHz, CD3OD): −137.23 (m), −141.12 (m). νmax(ATR)/cm−1: 3228 (NH), 3040 (NH), 2923, 2853, 1655 (C=N), 1579 (C=N), 1505, 1485, 1401, 1259, 1211 (C-F), 1149, 972, 825, 771, 694, 649, 609, 587. HRMS (m/z ESI+): found: 397.1598 (M+ + H), C20H19N6OF2 requires: 397.1588. HPLC: 96.8% (tR: 23.2 min).
4.2.3. 1-(2-Fluoro-4-iodophenyl)-2-(3-(4-guanidinophenoxy)phenyl)guanidine dihydrochloride (7)
Following Method A (see ESI), 23 (357 mg, 0.44 mmol) was dissolved in 4 M HCl in dioxane (2 mL, 7.92 mmol) and in additional dioxane (0.2 mL) until a final concentration of 0.2 M was reached. After 8 h stirring at 55 °C, the reaction was adjudged complete (TLC), solvents were evaporated and the residue was purified by silica gel chromatography (CH3Cl:MeOH) to afford the pure hydrochloride salt as a white solid (210 mg, 83%). Mp: decomp. > 180 °C. δH (400 MHz, CD3OD): 6.99–7.01 (m, 2H, H-2 and H-4), 7.11–7.14 (m, 1H, H-6), 7.15 (d, J = 8.9 Hz, 2H, H-8 and H-8′), 7.20 (t, J = 8.2 Hz, 1H, H-5), 7.33 (d, J = 8.9 Hz, 2H, H-9 and H-9′), 7.44–7.49 (m, 1H, H-15), 7.64–7.66 (m, 1H, H-16), 7.69 (dd, J = 9.6, 1.8 Hz, 1H, H-13). δC (100 MHz, CD3OD): 93.4 (d, J = 7.5 Hz, qC, C-14), 116.4 (CH Ar, C-2), 118.6 (CH Ar, C-4), 121.0 (CH Ar, C-6), 121.5 (2 CH Ar, C-8 and C-8′), 124.1 (d, J = 12.4 Hz, qC, C-11), 127.3 (d, J = 22.2 Hz, CH Ar, C-13), 128.9 (2 CH Ar, C-9 and C-9′), 130.9 (CH Ar, C-5), 131.5 (qC), 132.4 (CH Ar, C-15), 136.0 (d, J = 3.9 Hz, CH Ar, C-16), 137.9 (qC), 156.4 (qC), 158.0 (d, J = 254.4 Hz, qC, C-12), 157.3 (qC), 158.3 (qC), 159.5 (qC). δF (376, CD3OD):—121.10 (t, J = 8.9 Hz). νmax(ATR)/cm−1: 3318 (NH), 3098 (NH), 2958, 1661 (C=N), 1620, 1579, 1485, 1214 (C-F), 1149, 625, 609, 576 (C-I), 566. HRMS (m/z ESI+): found 505.0646 (M+ + H), C20H19N6OFI requires: 505.0649. HPLC: 99.2% (tR: 25.3 min).
4.2.4. 1-(4-Bromophenyl)-2-(3-(4-guanidinophenoxy)phenyl)guanidine dihydrochloride (8)
Following Method A (see ESI), 24 (212 mg, 0.29 mmol) was dissolved in 4 M HCl in dioxane (1.30 mL, 5.22 mmol) and in additional dioxane (0.12 mL) until a final concentration of 0.2 M was reached. After 8 h stirring at 55 °C, the reaction was adjudged complete (TLC), solvents were evaporated and the residue was purified by flash chromatography to afford the pure hydrochloride salt as a light-yellow solid (128 mg, 87%). Mp: decomp. > 110 °C. δH (400 MHz, CD3OD): 6.99 (dd, J = 8.2, 2.3 Hz, 1H, H-4), 7.03 (t, J = 2.2 Hz, 1H, H-2), 7.12–7.16 (m, 3H, H-6 and H-8 and H-8′), 7.27 (d, J = 8.7 Hz, 2H, H-12 and H-12′ or H-13 and H-13′), 7.31 (d, J = 8.8 Hz, 2H, H-9 and H-9′), 7.46 (t, J = 8.1 Hz, 1H, H-5), 7.61 (d, J = 8.7 Hz, 2H, H-12 and H-12′ or H-13 and H-13′). δC (100 MHz, CD3OD): 116.3 (CH Ar, C-2), 118.5 (CH Ar, C-4), 120.9 (CH Ar, C-6), 121.5 (2 CH Ar, C-8 and C-8′), 121.7 (qC, C-14), 127.9 (2 CH Ar, C-12 and C-12′ or C-13 and C-13′), 128.9 (2 CH Ar, C-9 and C-9′), 131.5 (qC), 132.4 (CH Ar, C-5), 134.1 (CH Ar, C-12 and C-12′ or C-13 and C-13′), 135.8 (qC), 138.0 (qC), 156.2 (qC), 157.4 (qC), 158.3 (qC), 159.5 (qC). νmax(ATR)/cm−1: 3124 (NH), 3044 (NH), 1655, 1571 (C=N), 1504 (C=N), 1484, 1405, 1213 (C-O), 1070 (C-Br), 1010, 617-567. HRMS (m/z APCI+): found: 439.0857 (M+ + H), C20H20BrN6O requires: 439.0876. HPLC: 99.8% (tR: 24.8 min).
4.2.5. 1-(4-Bromo-3-(trifluoromethyl)phenyl)-2-(3-(4-guanidinophenoxy)phenyl)guanidine dihydrochloride (9)
Following Method A (see ESI), 25 (566 mg, 0.70 mmol) was dissolved in 4 M HCl in dioxane (3.15 mL, 12.6 mmol) and in additional dioxane (0.35 mL) until a final concentration of 0.2 M was reached. After 8 h stirring at 55 °C, the reaction was adjudged complete (TLC), solvents were evaporated and the residue was purified by silica gel chromatography (CHCl3:MeOH) to afford the pure hydrochloride salt as a white solid (361 mg, 89%). Mp: decomp. >136 °C. δH (400 MHz, CD3OD): 6.99 (dd, J = 8.0, 1.9 Hz, 1H, H-4), 7.06 (t, J = 2.1 Hz, 1H, H-2), 7.14–7.16 (m, 3H, H-8 and H-8′ and H-6), 7.32 (d, J = 8.9 Hz, 2H, H-9 and H-9′), 7.46 (t, J = 8.1 Hz, 1H, H-5), 7.51 (dd, J = 8.6, 2.4 Hz, 1H, H-16), 7.74 (d, J = 2.4 Hz, 1H, H-12), 7.88 (d, J = 8.6 Hz, 1H, H-15). δC (100 MHz, CD3OD): 116.2 (CH Ar, C-2), 118.3 (qC, C-14), 118.5 (CH Ar, C-4), 120.8 (CH Ar, C-6), 121.5 (2 CH Ar, C-8 and C-8′), 123.9 (d, J = 260.3 Hz, qCF3), 125.3 (m, CH Ar, C-12), 128.9 (2 CH Ar, C-9 and C-9′), 130.5 (CH Ar, C-16), 131.6 (qC), 132.3 (d, J = 31.7 Hz, qC, C-13), 132.4 (CH Ar, C-5), 136.9 (qC), 137.8 (CH Ar, C-15), 138.0 (qC), 156.1 (qC), 157.3 (qC), 158.3 (qC), 159.6 (qC). δF (376 MHz, CD3OD):—64.73 (s). νmax(ATR)/cm−1: 3119 (NH), 3053 (NH), 1663 (C=O), 1584 (C=N), 1478, 1412, 1320 (C-F), 1238, 1214, 1174, 1129 (CF3), 1099 (C-Br), 1023, 828, 581—558. HRMS (m/z ESI+): found 507.0766 (M+ + H), C21H19N6OF3Br requires: 507.0756. HPLC: 99.9% (tR: 26.3 min).
4.2.6. 1-(2-Fluoro-4-iodophenyl)-2-(4-(3-guanidinophenoxy)phenyl)guanidine dihydrochloride (10)
Following Method A (see ESI), 26 (310 mg, 0.39 mmol) was dissolved in 4 M HCl in dioxane (1.73 mL, 6.93 mmol) and in additional dioxane (0.17 mL) until a final concentration of 0.2 M was reached. After 8 h stirring at 55 °C, the reaction was adjudged complete (TLC), solvents were evaporated and the residue was purified by flash chromatography to afford the pure hydrochloride salt as a white solid (198 mg, 88%). Mp: decomp. >150 °C. δH (400 MHz, CD3OD): 6.97 (t, J = 2.1 Hz, 1H, H-2), 7.00 (dd, J = 8.2, 2.3 Hz, 1H, H-4), 7.08 (dd, J = 7.6, 1.5 Hz, 1H, H-6), 7.16 (d, J = 8.8 Hz, 2H, H-8 and H-8′), 7.22 (t, J = 8.2 Hz, 1H, H-5 or H-15), 7.36 (d, J = 8.8 Hz, 2H, H-9 and H-9′), 7.47 (t, J = 8.1 Hz, 1H, H-5 or H-15), 7.66 (d, J = 9.1 Hz, 1H, H-16), 7.70 (dd, J = 9.6, 1.7 Hz, 1H, H-13). δC (100 MHz, CD3OD): 93.5 (d, J = 7.5 Hz, qC, C-14), 116.6 (CH Ar, C-2), 118.6 (CH Ar, C-4), 121.3 (CH Ar, C-6), 121.6 (2 CH Ar, C-8 and C-8′), 124.0 (d, J = 12.5 Hz, qC, C-11), 127.3 (d, J = 22.2 Hz, C-13), 128.7 (2 CH Ar, C-9 and C-9′), 131.1 (CH Ar, C-5 or C-15), 131.6 (qC), 132.4 (CH Ar, C-5 or C-15), 136.0 (d, J = 3.9 Hz, C-16), 137.7 (qC), 156.8 (qC), 157.3 (qC), 157.9 (qC), 158.2 (d, J = 254.5 Hz, qC, C-12), 159.5 (qC). δF (376 MHz, CD3OD): −121.04 (t, J = 8.8 Hz). νmax(ATR)/cm−1: 3335 (NH), 3265 (NH), 3180 (NH), 3052, 2868, 2325, 1616 (C=N), 1560 (C=N), 1504, 1400, 1226 (C-F), 1162, 1109, 971, 875, 789, 684—573 (C-I). HRMS (m/z ESI+): found: 505.0645 (M+ + H), C20H19N6OFI requires: 505.0649. HPLC: 99.9% (tR: 25.9 min).
4.2.7. 1-(4-Bromo-3-(trifluoromethyl)phenyl)-2-(4-(3-guanidinophenoxy)phenyl)guanidine dihydrochloride (11)
Following Method A (see ESI), 27 (148 mg, 0.18 mmol) was dissolved in 4 M HCl in dioxane (0.81 mL, 3.24 mmol) and in additional dioxane (0.10 mL) until a final concentration of 0.2 M was reached. After 8 h stirring at 55 °C, the reaction was adjudged complete (TLC), solvents were evaporated and the residue was purified by flash chromatography to afford the pure hydrochloride salt as a white solid (100 mg, 94%). Mp: decomp. > 95 °C. δH (400 MHz, CD3OD): 6.98–7.02 (m, 2H, H-2 and H-4), 7.09 (ddd, 1H, J = 8.0, 1.9, 0.9 Hz, H-6), 7.17 (d, J = 8.9 Hz, 2H, H-8 and H-8′), 7.41 (d, J = 9.0 Hz, 2H, H-9 and H-9′), 7.49 (t, J = 8.0 Hz, 1H, H-5), 7.55 (dd, J = 8.5, 2.6 Hz, 1H, H-16), 7.77 (d, J = 2.6 Hz, 1H, H-12), 7.91 (d, J = 8.6 Hz, 1H, H-15). δC (100 MHz, CD3OD): 116.6 (CH Ar, C-2 or C-4), 118.5 (qC, C-14), 118.6 (CH Ar, C-2 or C-4), 121.3 (CH Ar, C-6), 121.6 (2 CH Ar, C-8 and C-8′), 123.9 (d, J = 272.8 Hz, qCF3), 125.5 (q, J = 5.6 Hz, CH Ar, C-12), 128.5 (2 CH Ar, C-9 and C-9′), 130.8 (CH Ar, C-16), 131.7 (qC), 132.3 (q, J = 31.6 Hz, qC, C-13), 132.4 (CH Ar, C-5), 136.7 (qC), 137.7 (qC), 137.8 (CH Ar, C-15), 156.5 (qC), 157.3 (qC), 157.9 (qC), 159.6 (qC). δF (376 MHz, CD3OD):—64.30 (s). νmax(ATR)/cm−1: 3309 (NH), 3116 (NH), 3053 (NH), 2837, 2280, 1663 (C=N), 1577 (C=N), 1505, 1486, 1405, 1320, 1258 (C-O), 1214 (CF3), 1129, 1023 (C-Br), 829, 595—575. HRMS (m/z ESI+): found: 507.0750 (M+ + H), C21H19N6OBrF3 requires: 507.0756. HPLC: 99.9% (tR: 26.6 min).
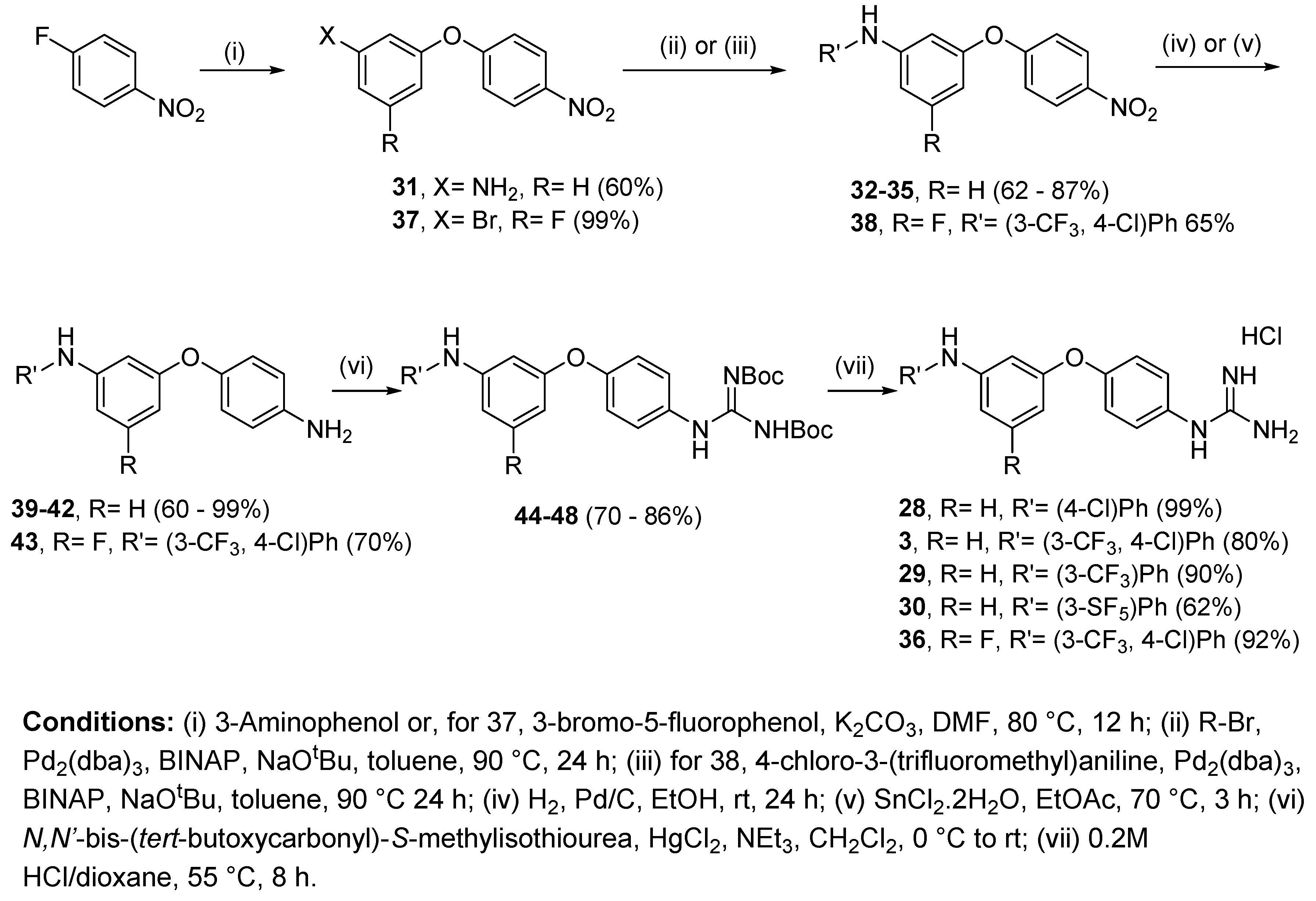
4.2.8. 1-(4-(3-((4-Chlorophenyl)amino)phenoxy)phenyl)guanidine hydrochloride (28)
Following Method A (see ESI), 44 (97 mg, 0.18 mmol) was dissolved in 4 M HCl in dioxane (0.53 mL, 2.10 mmol) and in additional dioxane (0.9 mL) until a final concentration of 0.2 M was reached. After 8 h stirring at 55 °C, the reaction was adjudged complete (TLC), solvents were evaporated and the residue was purified by silica gel chromatography (CHCl3:MeOH) to afford the pure hydrochloride salt as a purple solid (69 mg, 99%). Mp: 50–52 °C. δH (400 MHz, CD3OD): 6.51 (dd, J = 8.3, 1.9 Hz, 1H, H-4), 6.73 (t, J = 2.2 Hz, 1H, H-2), 6.85 (dd, J = 7.8, 1.8 Hz, 1H, H-6), 7.05 (d, J = 8.9 Hz, 2H, H-12 and H-12′), 7.08 (d, J = 8.9 Hz, 2H, H-8 and H-8′), 7.18 (d, J = 8.9 Hz, 2H, H-13 and H-13′), 7.22 (t, J = 8.2 Hz, 1H, H-5), 7.27 (d, J = 8.9 Hz, 2H, H-9 and H-9′). δC (100 MHz, CD3OD): 108.6 (CH Ar, C-2), 111.8 (CH Ar, C-4), 113.7 (CH Ar, C-6), 120.0 (2 CH Ar, C-12 and C-12′), 120.7 (2 CH Ar, C-8 and C-8′), 126.1 (qC, C-14), 128.8 (2 CH Ar, C-9 and C-9′), 130.1 (2 CH Ar, C-13 and C-13′), 130.6 (qC), 131.5 (CH Ar, C-5), 143.4 (qC), 146.7 (qC), 158.4 (qC), 158.5 (qC), 158.9 (qC). νmax(ATR)/cm−1: 3297 (N-H), 3126 (N-H), 1688, 1586 (C=N), 1502, 1485 (C-N), 1325, 1216 (C-O), 1142 (C-Cl), 997, 972, 823, 770, 689, 604, 588, 570. HRMS (m/z ESI+): found 353.1177 (M+ + H. C19H18N4OCl requires: 353.1169). HPLC: 98.0% (tR: 32.3 min).
4.2.9. 1-(4-(3-((4-Chloro-3-(trifluoromethyl)phenyl)amino)phenoxy)phenyl)guanidine hydrochloride (3)
Following Method A (see ESI), 45 (112 mg, 0.18 mmol) was dissolved in 4 M HCl in dioxane (0.54 mL, 2.16 mmol) and in additional dioxane (0.36 mL) until a final concentration of 0.2 M was reached. After 8 h stirring at 55 °C, the reaction was adjudged complete (TLC), solvents were evaporated and the residue was purified by flash chromatography to afford the pure hydrochloride salt as a light brown solid (59 mg, 80%). Mp: 58–60 °C. δH (400 MHz, CD3OD): 6.62 (dd, J = 8.7, 2.3 Hz, 1H, H-4), 6.78 (t, J = 2.2 Hz, 1H, H-2), 6.90 (dd, J = 8.1, 2.1 Hz, 1H, H-6), 7.11 (d, J = 8.9 Hz, 2H, H-8 and H-8’), 7.24 (dd, J = 8.8, 2.8 Hz, 1H, H-16), 7.27–7.31 (m, 3H, H-9, H-9’ and H-5) 7.36–7.39 (m, 2H, H-12, H-15). δC (100 MHz, CD3OD): 109.8 (CH Ar, C-2), 113.1 (CH Ar, C-4), 114.8 (CH Ar, C-6), 116.1 (q, J = 5.6 Hz, CH Ar, C-12), 121.0 (2 CH Ar, C-8 and C-8’), 121.5 (CH Ar, C-16), 122.1 (qC, C-14), 124.4 (d, J = 272.5 Hz, qCF3), 129.0 (2 CH Ar, C-9 and C-9’), 129.6 (q, J = 31.0, qC, C-13), 131.0 (qC), 131.8 (CH Ar, C-5), 133.4 (CH Ar, C-15), 144.4 (qC), 145.3 (qC), 158.2 (qC), 158.4 (qC), 159.2 (qC). δF (376 MHz, CD3OD):—64.18 (s). νmax(ATR)/cm−1: 3295 (NH), 3163, 2923, 2853, 2400, 1664 (C=O), 1595 (C=N), 1504, 1482, 1441, 1333, 1258, 1217 (CF3), 1127, 1112 (C-Cl), 1027, 999, 977, 825. HRMS (m/z ESI+): found 421.1044 (M+ + H. C20H17ClF3N4O requires: 421.1043). HPLC: 97.8% (tR: 32.9 min).
4.2.10. 1-(4-(3-((3-(Trifluoromethyl)phenyl)amino)phenoxy)phenyl)guanidine hydrochloride (29)
Following Method A (see ESI), 46 (371 mg, 0.63 mmol) was dissolved in 4 M HCl in dioxane (1.90 mL, 7.56 mmol) and in additional dioxane (1.25 mL) until a final concentration of 0.2 M was reached. After 6 h stirring at 55 °C, the reaction was adjudged complete (TLC), solvents were evaporated and the residue was purified by flash chromatography to afford the pure hydrochloride salt as a white solid (242 mg, 90%). Mp: 93–95 °C. δH (400 MHz, CD3OD): 6.59 (dd, J = 8.1, 2.3 Hz, 1H, H-4), 6.78 (t, J = 2.2 Hz, 1H, H-2), 6.90 (dd, J = 7.9, 1.9 Hz, 1H, H-6), 7.07–7.13 (m, 3H, H-8 and 8′ and H-12 or H-14), 7.26–7.30 (m, 5H, H-9 and 9′, H-12 or H-14, H-16 and H-5 or H-15), 7.37 (t, J = 8.3 Hz, 1H, H-5 or H-15). δC (100 MHz, CD3OD): 109.4 (CH Ar, C-2), 112.6 (CH Ar, C-4), 113.9 (q, J = 4.0 Hz, CH Ar, C-12 or C-14), 114.6 (CH Ar, C-6), 117.3 (q, J = 4.0 Hz, CH Ar, C-12 or C-14), 119.9 (d, J = 280.7 Hz, qCF3), 120.9 (2 CH Ar, C-8 and C-8′), 121.2 (CH Ar, C-16), 128.9 (2 CH Ar, C-9 and C-9′), 130.8 (qC), 131.1 (CH Ar, C-5 or C-15), 131.7 (CH Ar, C-5 or C-15), 132.6 (d, J = 31.9 Hz, qC, C-13), 145.7 (qC), 145.9 (qC), 158.3 (qC), 158.4 (qC), 159.1 (qC). δF (376 MHz, CD3OD): −64.42 (s). νmax(ATR)/cm−1: 3301 (NH), 3135 (NH), 1665, 1587 (C=N), 1490, 1486, 1335 (C-N), 1216 (C-O), 1161, 1116 (CF3), 1067 (C-Cl), 976, 836, 785, 689. HRMS (m/z ESI+): found 387.1438 (M+ + H. C20H18N4OF3 requires: 387.1433). HPLC: 97.5% (tR: 31.7 min).
4.2.11. 1-(4-(3-((3-(Pentafluorosulfanyl)phenyl)amino)phenoxy)phenyl)guanidine hydrochloride (30)
Following Method A (see ESI), 47 (272 mg, 0.42 mmol) was dissolved in 4 M HCl in dioxane (1.27 mL, 5.06 mmol) and in additional dioxane (0.83 mL) until a final concentration of 0.2 M was reached. After 6 h stirring at 55 °C, the reaction was adjudged complete (TLC), solvents were evaporated and the residue was purified by silica gel chromatography (CHCl3:MeOH) to afford the pure hydrochloride salt as an orange solid (127 mg, 62%). Mp: 104–106 °C. δH (400 MHz, CD3OD): 6.61 (dd, J = 8.2, 2.3 Hz, 1H, H-4), 6.78 (t, J = 2.2. Hz, 1H, H-2), 6.90 (dd, J = 8.1, 2.1 Hz, 1H, H-6), 7.11 (d, J = 8.9 Hz, H-8 and H-8′), 7.22–7.31 (m, 5H, H-9 and H-9′, H-5 or H-15, H-14 and H-16), 7.34–7.38 (m, 1H, H-5 or H-15), 7.44 (t, J = 2.2 Hz, 1H, H-12). δC (100 MHz, CD3OD): 109.6 (CH Ar, C-2), 112.9 (CH Ar, C-4), 114.6 (CH Ar, C-6), 114.9 (p, J = 4.6 Hz, CH Ar, C-12), 118.0 (p, J = 4.7 Hz, CH Ar, C-14), 120.8 (CH Ar, C-16), 120.9 (2 CH Ar, C-8 and C-8′), 128.8 (2 CH Ar, C-9 and C-9′), 130.6 (CH Ar, C-5 or C-15), 130.8 (qC), 131.8 (CH Ar, C-5 or C-15), 145.6 (qC), 145.7 (qC), 155.9 (p, J = 16.4 Hz, qC, C-13), 158.2 (qC), 158.3 (qC), 159.2 (qC). δF (376 MHz, CD3OD): −64.34 (s). νmax(ATR)/cm−1: 3273 (NH), 3150 (NH), 1669, 1593 (C=N), 1487 (C-N), 1218 (C-O), 834 (SF5), 567. HRMS (m/z ESI+): found 445.1124 (M+ + H. C19H18N4OSF5 requires: 445.1121). HPLC: 95.4% (tR: 32.3 min).
4.2.12. 1-(4-(3-((4-Chloro-3-(trifluormethyl)phenyl)amino)-5-fluorophenoxy)phenyl)guanidine hydrochloride (36)
Following Method A (see ESI), 48 (166 mg, 0.26 mmol) was dissolved in 4 M HCl in dioxane (0.78 mL, 3.12 mmol) and in additional dioxane (0.51 mL) until a final concentration of 0.2 M was reached. After 8 h stirring at 55 °C, the reaction was adjudged complete (TLC), solvents were evaporated and the residue was purified by silica gel chromatography (CHCl3:MeOH) to afford the pure hydrochloride salt as a light brown solid (114 mg, 92%). Mp: 92–94 °C. δH (600 MHz, CD3OD): 6.31 (dt, J = 9.9, 2.1 Hz, 1H, H-4), 6.55 (s, 1H, H-2), 6.58 (dt, J = 10.7, 2.0 Hz, 1H, H-6), 7.16 (d, J = 8.8 Hz, 2H, H-8 and H-8′), 7.29 (dd, J = 8.8, 2.7 Hz, 1H, H-16), 7.32 (d, J = 8.8 Hz, 2H, H-9 and H-9′), 7.40 (d, J = 2.6 Hz, 1H, H-12), 7.43 (d, J = 8.7 Hz, 1H, H-15). δC (150 MHz, CD3OD): 99.4 (d, J = 25.7 Hz, CH Ar, C-4), 100.3 (d, J = 25.5 Hz, CH Ar, C-6), 103.8 (d, J = 2.6 Hz, CH Ar, C-2), 117.4 (q, J = 5.5 Hz, CH Ar, C-12), 121.8 (2 CH Ar, C-8 and C-8′), 122.7 (CH Ar, C-16), 123.4 (qC, C-14), 124.3 (d, J = 272.2 Hz, qCF3), 128.9 (2 CH Ar, C-9 and C-9′), 129.8 (q, J = 31.0, qC, C-13), 131.7 (qC), 133.5 (CH Ar, C-15), 143.4 (qC), 146.7 (d, J = 13.2 Hz, qC, C-1 or C-3), 157.1 (qC), 158.4 (qC), 160.7 (d, J = 13.7 Hz, qC, C-1 or C-3), 165.8 (d, J = 243.4 Hz, qC, C-5). δF (376 MHz, CD3OD): −64.67 (s), −112.53 (s). νmax(ATR)/cm−1: 3285 (NH), 3139 (NH), 1666, 1601 (C=N), 1504, 1476, 1323 (CF3), 1216 (C-O), 1112 (C-F), 1020 (C-Cl), 994, 823, 660. HRMS (m/z ESI+): found 439.0945 (M+ + H. C20H16N4OF4Cl requires: 439.0943). HPLC: 95.7% (tR: 33.1 min).
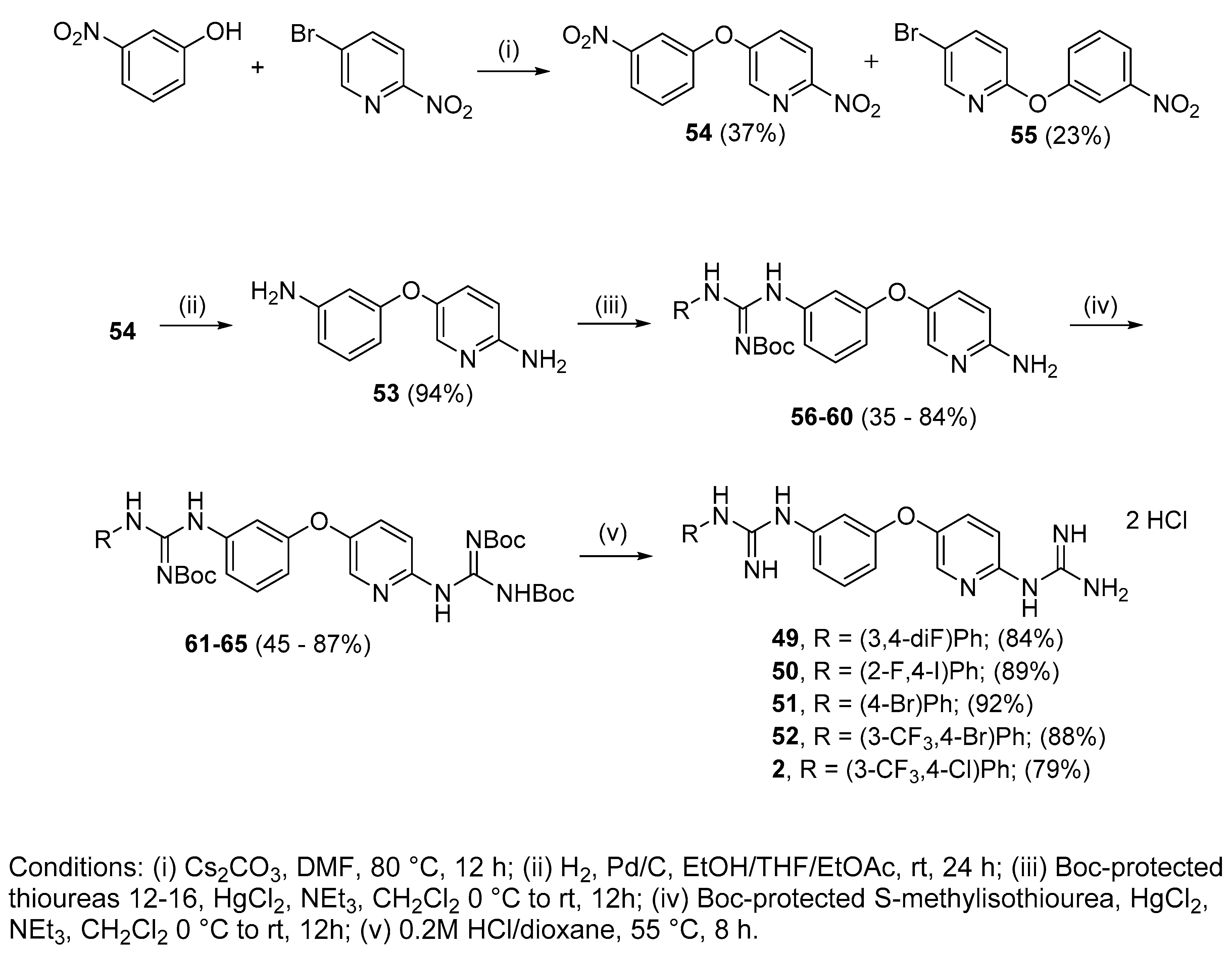
4.2.13. 1-(3,4-Di-fluorophenyl)-3-(3-((6-guanidinopyridin-3-yl)oxy)phenyl)guanidine dihydrochloride (49)
Following Method A (see ESI), 61 (176 mg, 0.25 mmol) was dissolved in 4 M HCl in dioxane (1.14 mL, 4.54 mmol) and in additional dioxane (0.11 mL) until a final concentration of 0.2 M was reached. After 8 h stirring at 55 °C, the reaction was adjudged complete (TLC), solvents were evaporated and the residue was purified by silica gel chromatography (CHCl3:MeOH) to afford the pure hydrochloride salt as a white solid (101 mg, 84%). Mp: decomp. above 110 °C. δH (400 MHz, CD3OD): 6.62 (dd, J = 7.5, 2.4 Hz, 1H, H-4), 6.77–6.81 (m, 1H, H-17), 6.93 (dd, J = 8.0, 1.8 Hz, 1H, H-6), 6.99 (d, J = 8.9 Hz, 1H, H-9), 7.00–7.13 (m, 3H, H-2, H-13 and H-16), 7.25 (t, J = 8.1 Hz, 1H, H-5), 7.49 (dd, J = 8.9, 2.9 Hz, 1H, H-8), 8.05 (d, J = 2.9 Hz, 1H, H-11). δC (100 MHz, CD3OD): 112.0 (d, J = 18.8 Hz, CH Ar, C-13 or C-16), 112.4 (CH Ar, C-2), 113.0 (CH Ar, C-4), 116.4 (CH Ar, C-9), 118.0 (CH Ar, C-6), 118.1 (d, J = 17.9 Hz, CH Ar, C-13 or C-16), 119.1 (dd, J = 5.6, 3.1 Hz, CH Ar, C-17), 131.3 (CH Ar, C-5), 131.4 (CH Ar, C-8), 138.5 (CH Ar, C-11), 143.9 (dd, J = 7.4, 2.2 Hz, qC, C-12), 146.6 (qC), 147.2 (dd, J = 240.4, 12.9 Hz, qC, C-14 or C-15), 150.1 (qC), 151.1 (qC), 151.4 (dd, J = 245.1, 13.4 Hz, qC, C-14 or C-15), 152.5 (qC), 157.4 (qC), 159.0 (qC). δF (376 MHz, CD3OD): −139.72 (m), −149.19 (m). νmax(ATR)/cm−1: 3313 (NH), 3154 (NH), 2922, 2861, 1682, 1625 (C=N), 1507, 1473 (C-F), 1375, 1228 (C-F), 1166, 1146 (C-O), 866, 830, 770, 570, 557. HRMS (m/z ESI+): found 398.1548 (M+ + H. C19H18N7OF2 requires: 398.1541). HPLC: 99.8% (tR: 23.6 min).
4.2.14. 1-(2-Fluoro-4-iodophenyl)-3-(3-((6-guanidinopyridin-3-yl)oxy)phenyl)guanidine dihydrochloride (50)
Following Method A (see ESI), 62 (104 mg, 0.13 mmol) was dissolved in 4 M HCl in dioxane (0.58 mL, 2.32 mmol) and in additional dioxane (0.10 mL) until a final concentration of 0.2 M was reached. After 8 h stirring at 55 °C, the reaction was adjudged complete (TLC), solvents were evaporated and the residue was purified by silica gel chromatography (CHCl3:MeOH) to afford the pure hydrochloride salt as a white solid (67 mg, 89%). Mp: decomp. above 120 °C. δH (400 MHz, CD3OD): 6.98 (ddd, J = 8.3, 2.4, 0.7 Hz, 1H, H-4), 7.02 (t, J = 2.2 Hz, 1H, H-2), 7.11–7.18 (m, 3H, H-5 or H-16, H-6 and H-9), 7.46 (t, J = 8.1 Hz, 1H, H-5 or H-16), 7.61–7.64 (m, 2H, H-17 and H-8), 7.67 (dd, J = 9.7, 1.8 Hz, 1H, H-14), 8.16 (d, J = 2.9 Hz, 1H, H-11). δC (100 MHz, CD3OD): 92.7 (qC, C-15), 115.5 (CH Ar, C-2), 115.6 (CH Ar, C-9), 117.6 (CH Ar, C-4), 120.9 (CH Ar, C-6), 127.2 (d, J = 22.2 Hz, CH Ar, C-14), 127.3 (d, J = 14.1 Hz, qC, C-12), 130.6 (CH Ar, C-5 or C-16), 132.1 (CH Ar, C-8), 132.5 (CH Ar, C-5 or C-16), 136.0 (CH Ar, J = 3.9 Hz, C-17), 138.7 (qC), 139.1 (CH Ar, C-11), 149.2 (qC), 151.2 (qC), 156.2 (qC), 156.9 (qC), 157.9 (qAr, J = 253.9 Hz, C-13), 159.4 (qC). δF (376 MHz CD3OD):—121.55 (t, J = 8.3 Hz). νmax(ATR)/cm−1: 3277 (NH), 3122 (NH), 2923, 2849, 1680, 1660, 1623–1570 (C=N), 1474 (C-F), 1227 (C-O), 1160, 1026, 945, 600 (C-I). HRMS (m/z ESI+): found 506.0609 (M+ + H. C19H18N7OFI requires: 506.0602). HPLC: 98.1% (tR: 25.7 min).
4.2.15. 1-(4-Bromophenyl)-3-(3-((6-guanidinopyridin-3-yl)oxy)phenyl)guanidine dihydrochloride (51)
Following Method A (see ESI), 63 (184 mg, 0.24 mmol) was dissolved in 4 M HCl in dioxane (1.08 mL, 4.4 mmol) and in additional dioxane (0.12 mL) until a final concentration of 0.2 M was reached. After 8 h stirring at 55 °C, the reaction was adjudged complete (TLC), solvents were evaporated and the residue was purified by silica gel chromatography (CHCl3:MeOH) to afford the pure hydrochloride salt as a white solid (113 mg, 92%). Mp: decomp. above 124 °C. δH (400 MHz, CD3OD): 6.99–7.01 (m, 1H, H-4), 7.04 (bs, 1H, H-2), 7.14–7.16 (m, 2H, H-6 and H-9), 7.28 (d, J = 8.5 Hz, 2H, H-13 and H-13′ or H-14 and H-14′), 7.48 (t, J = 8.1 Hz, 1H, H-5), 7.60–7.63 (m, 3H, H-13 and H-13′ or H-14 and H-14′and H-8), 8.16 (d, J = 2.6 Hz, 1H, H-11). δC (100 MHz, CD3OD): 115.6 (CH Ar, C-6 or C-9), 115.8 (CH Ar, C-2), 118.0 (CH Ar, C-4), 121.1 (CH Ar, C-6 or C-9), 121.7 (qC, C-15), 127.9 (CH Ar, C-13 and C-13′ or C-14 and C-14′), 132.1 (CH Ar, C-8), 132.5 (CH Ar, C-5), 134.1 (CH Ar, C-13 and C-13′ or C-14 and C-14′), 135.7 (qC), 138.2 (qC), 139.0 (CH Ar, C-11), 149.2 (qC), 151.2 (qC), 156.1 (qC), 156.8 (qC), 159.4 (qC). νmax(ATR)/cm−1: 3256 (NH), 3114 (NH), 2971, 1680, 1660, 1619 (C=N), 1566 (C=N), 1474, 1376 (C-N), 1226 (C-O), 1069 (C-Br), 1010, 832, 637—584. HRMS (m/z ESI+): found 440.0837 (M+ + H. C19H19N7OBr requires: 440.0834). HPLC: 97.5% (tR: 25.2 min).
4.2.16. 1-(4-Bromo-3-(trifluoromethyl)phenyl)-3-(3-((6-guanidinopyridin-3-yl)oxy)phenyl) guanidine dihydrochloride (52)
Following Method A (see ESI), 64 (358 mg, 0.44 mmol) was dissolved in 4 M HCl in dioxane (2 mL, 7.97 mmol) and in additional dioxane (0.2 mL) until a final concentration of 0.2 M was reached. After 8 h stirring at 55 °C, the reaction was adjudged complete (TLC), solvents were evaporated and the residue was purified by silica gel chromatography (CHCl3:MeOH) to afford the pure hydrochloride salt as a white solid (225 mg, 88%). Mp: decomp. above 170 °C. δH (400 MHz, CD3OD): 6.66 (dd, J = 8.1, 2.4 Hz, 1H, H-4), 6.96 (dd, J = 8.0, 1.9 Hz, 1H, H-6), 7.04 (d, J = 8.9 Hz, 1H, H-9), 7.08 (t, J = 2.1 Hz, 1H, H-2), 7.20 (dd, J = 8.6, 2.5 Hz, 1H, H-17), 7.28 (t, J = 8.1 Hz, 1H, H-5), 7.48 (d, J = 2.5, 1H, H-13), 7.56 (dd, J = 8.9, 2.9 Hz, 1H, H-8), 7.60 (d, J = 8.6 Hz, 1H, H-16), 8.10 (d, J = 2.7 Hz, 1H, H-11). δC (100 MHz, CD3OD): 111.8 (d, J = 1.8 Hz, qC, C-15), 112.4 (CH Ar, C-2), 113.3 (CH Ar, C-4), 115.5 (CH Ar, C-9), 117.8 (CH Ar, C-6), 122.4 (q, J = 5.5 Hz, CH Ar, C-13), 124.4 (d, J = 272.6 Hz, qCF3), 127.7 (CH Ar, C-17), 130.9 (d, J = 30.8 Hz, qC, C-14), 131.4 (CH Ar, C-5), 131.6 (CH Ar, C-8), 136.6 (CH Ar, C-16), 138.6 (CH Ar, C-11), 146.1 (qC), 147.2 (qC), 148.9 (qC), 151.8 (qC), 152.4 (qC), 156.9 (qC), 159.0 (qC). δF (376 MHz, CD3OD):—63.96 (s). νmax(ATR)/cm−1: 3281 (NH), 3142 (NH), 2922, 2849, 1680, 1625 (C=N), 1566 (C=N), 1473, 1319, 1227 (CF3), 1129 (C-Br), 1023, 832, 592-583. HRMS (m/z ESI+): found 508.0710 (M+ + H. C20H18N7OF3Br requires: 508.0708). HPLC: 99.8% (tR: 27.5 min).
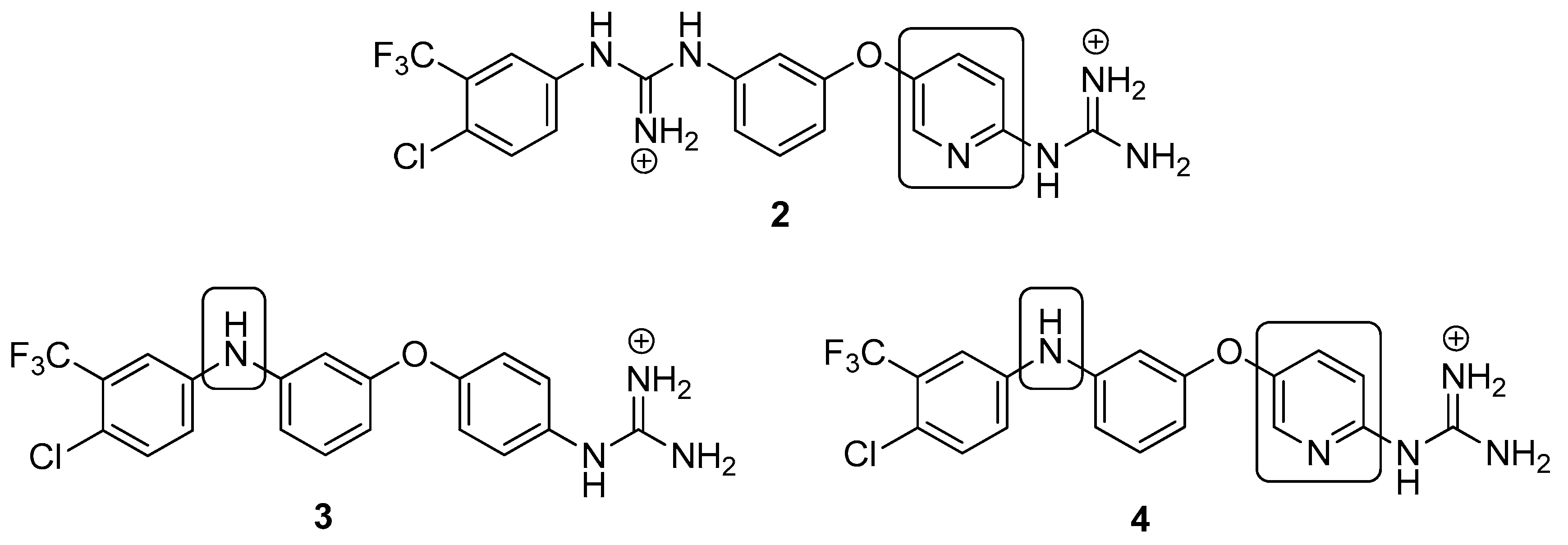
4.2.17. 1-(4-Chloro-3-(trifluoromethyl)phenyl)-3-(3-((6-guanidinopyridin-3-yl)oxy)phenyl) guanidine dihydrochloride (2)
Following Method A (see ESI), 65 (113 mg, 0.15 mmol) was dissolved in 4M HCl in dioxane (0.67 mL, 2.70 mmol) and in additional dioxane (0.10 mL) until a final concentration of 0.2M was reached. After 6 h stirring at 55 °C, the reaction was adjudged complete (TLC), solvents were evaporated and the residue was purified by flash chromatography to afford the pure hydrochloride salt as a white solid (63 mg, 79%). Mp: 169–171 °C. δH (400 MHz, CD3OD): 6.93 (dd, J = 8.3, 2.3 Hz, 1H, H-4), 7.06 (t, J = 2.1 Hz, 1H, H-2), 7.11–7.13 (m, 2H, H-6 and H-9), 7.44 (t, J = 8.2 Hz, 1H, H-5), 7.53 (dd, J = 8.6, 2.5 Hz, 1H, H-17), 7.60–7.65 (m, 2H, H-16 and H-8), 7.69 (d, J = 2.4 Hz, 1H, H-13), 8.15 (d, J = 2.9 Hz, 1H, H-11). δC (100 MHz, CD3OD): 115.1 (CH Ar, C-2), 115.6 (CH Ar, C-9), 117.1 (CH Ar, C-4), 120.5 (CH Ar, C-6), 123.9 (d, J = 272.5 Hz, qCF3) 124.5 (q, J = 5.5 Hz, CH Ar, C-13), 129.9 (qC, C-15), 130.1 (CH Ar, C-17), 130.2 (d, J = 34.7 Hz, qC, C-14), 132.0 (CH Ar, C-8 or C-16), 132.3 (CH Ar, C-5), 134.0 (CH Ar, C-8 or C-16), 138.3 (qC), 139.0 (CH Ar, C-11), 139.7 (qC), 149.1 (qC), 151.3 (qC), 155.5 (qC), 156.9 (qC), 159.3 (qC). δF (376 MHz, CD3OD):—64.24 (s). νmax(ATR)/cm−1: 3297 (NH), 3121 (NH), 2923, 2854, 1625 (C=N), 1581 (C=N), 1474 (CF3), 1320, 1227 (C-O), 1130 (C-Cl), 1032, 832, 589, 557. HRMS (m/z ESI+): found: 464.1222 (M+ + H. C20H18N7OF3Cl requires: 464.1213). HPLC: 96.9% (tR: 26.8 min).
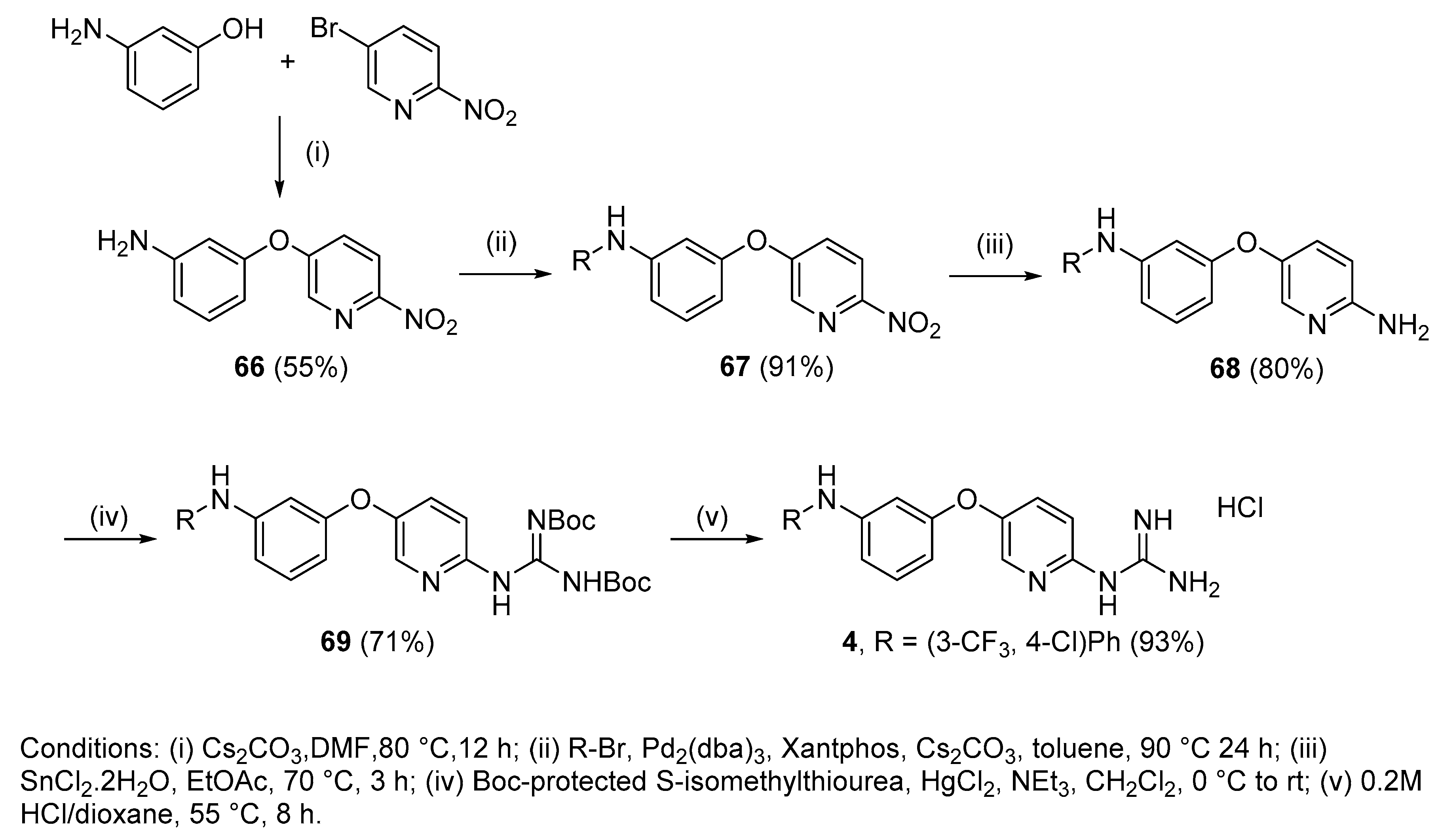
4.2.18. 1-(5-(3-((4-Chloro-3-(trifluoromethyl)phenyl)amino)phenoxy)pyridin-2-yl)guanidine hydrochloride (4)
Following Method A (see ESI), 69 (200 mg, 0.32 mmol) was dissolved in 4 M HCl in dioxane (0.96 mL, 3.86 mmol) and in additional dioxane (0.65 mL) until a final concentration of 0.2 M was reached. After 8 h stirring at 55 °C, the reaction was adjudged complete (TLC), solvents were evaporated and the residue was purified by silica gel chromatography (CHCl3:MeOH) to afford the pure hydrochloride salt as a white solid (136 mg, 93%). Mp: 89–91 °C. δH (400 MHz, CD3OD): 6.60 (dd, J = 8.2, 2.3 Hz, 1H, H-4), 6.75 (t, J = 2.2 Hz,1H, H-2), 6.90 (dd, J = 8.1, 2.0 Hz, 1H, H-6), 7.08 (d, J = 8.9 Hz, 1H, H-9), 7.24 (dd, J = 8.7, 2.7 Hz, 1H, H-17), 7.29 (t, J = 8.1 Hz, 1H, H-5), 7.37–7.39 (m, 2H, H-13 and H-16), 7.59 (dd, J = 8.9, 2.9 Hz, 1H, H-8), 8.13 (d, J = 2.9 Hz, 1H, H-11). δC (100 MHz, CD3OD): 108.7 (CH Ar, C-2), 112.2 (CH Ar, C-4), 114.7 (CH Ar, C-6), 115.5 (CH Ar, C-9), 116.2 (q, J = 5.5 Hz, CH Ar, C-13), 121.8 (CH Ar, C-17), 122.4 (qC, C-15), 124.3 (d, J = 272.4 Hz, qCF3), 129.6 (d, J = 31.0 Hz, qC, C-14), 131.8 (CH Ar, C-8), 132.0 (CH Ar, C-5), 133.4 (CH Ar, C-16), 138.7 (CH Ar, C-11), 144.2 (qC), 145.6 (qC), 148.8 (qC), 151.8 (qC), 156.9 (qC), 159.4 (qC). δF (376 MHz, CD3OD):—64.15 (s). νmax(ATR)/cm−1: 3264 (NH), 2923, 2863, 1684, 1629, 1595, 1474 (C=N), 1400, 1332, 1229 (C-O), 1129 (CF3), 1115 (C-Cl), 977, 998. HRMS (m/z ESI+): found 422.0987 (M+ + H. C19H16N5OClF3 requires: 422.0995). HPLC: 98.6% (tR: 33.3 min).
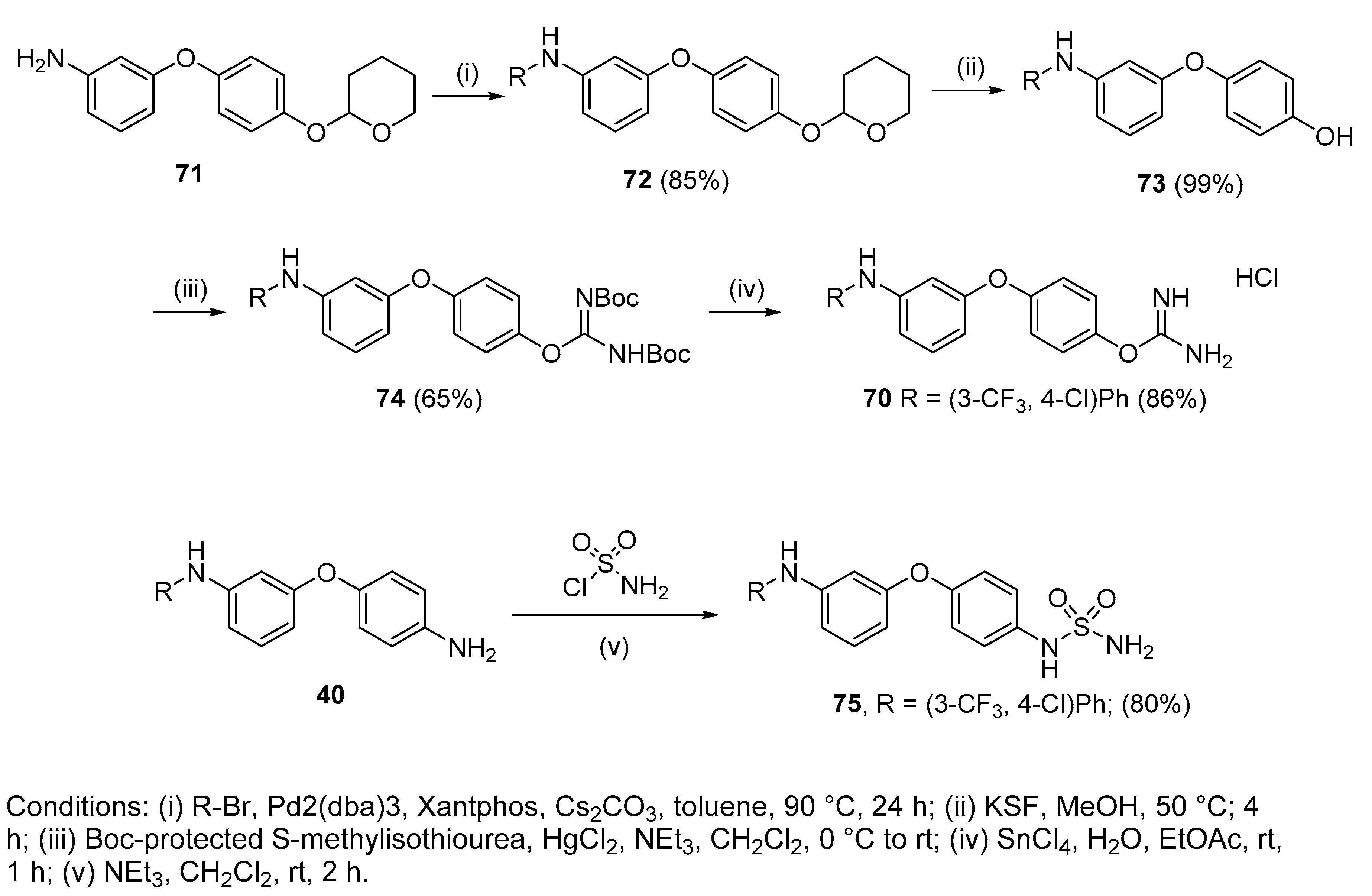
4.2.19. 4-(3-((4-Chloro-3-(trifluoromethyl)phenyl)amino)phenoxy)phenyl carbamimidate hydrochloride (70)
To a stirred solution of 74 (205 mg, 0.33 mmol, 1eq) in EtOAc was added SnCl4 (0.15 mL, 1.32 mmol, 4 eq). After 2 h of stirring at room temperature, the solvent and the excess of SnCl4 were evaporated in vacuo. The remaining liquid was purified by silica gel chromatography(CHCl3:Acetone) to afford the pure hydrochloride salt (130 mg, 86%) as a colourless gum. δH (400 MHz, DMSO-d6): 6.61 (dd, J = 7.6, 2.3 Hz, 1H, H-4), 6.76 (t, J = 2.2 Hz, 1H, H-2), 6.92 (dd, J = 7.8, 1.7 Hz, 1H, H-6), 7.16 (d, J = 9.1 Hz, 2H, H-8 and H-8′ or H-9 and H-9′), 7.30–7.34 (m, 2H, H-5 and H-16), 7.37 (d, J = 9.0 Hz, 2H, H-8 and H-8′ or H-9 and H-9′), 7.40 (d, J = 2.7 Hz, 1H, H-12), 7.50 (d, J = 8.8 Hz, 1H, H-15), 8.61 (bs, 4H, NH), 8.88 (bs, NH). δC (100 MHz, DMSO-d6): 107.8 (CH Ar, C-2), 111.2 (CH Ar, C-4), 113.1 (CH Ar, C-6), 114.7 (q, J = 5.5 Hz, CH Ar, C-12), 119.4 (qC, C-14), 120.4 (CH Ar, C-5 or C-16), 120.5 (2 CH Ar, C-8 and C-8′ or C-9 and C-9′), 122.0 (d, J = 227.5 Hz, qCF3), 123.1 (2 CH Ar, C-8 and C-8′ or C-9 and C-9′), 127.2 (d, J = 30.7 Hz, qC, C-13), 130.9 (CH Ar, C-5 or C-16), 132.7 (CH Ar, C-15), 142.8 (qC), 143.5 (qC), 145.2 (qC), 155.2 (qC), 157.6 (qC), 161.1 (qC). δF (376 MHz, CD3OD):—61.59 (s). νmax(ATR)/cm−1: 3285 (NH), 2924, 2854, 1693, 1655, 1593 (C=N), 1481 (C-N), 1400, 1258, 1231, 1195, 1175 (C-O), 1128 (CF3), 1111 (C-Cl), 1027, 824, 681, 665. HRMS (m/z ESI+): found: 422.0883 (M+ + H. C20H16N3O2ClF3 requires: 422.0883). HPLC: 96.2% (tR: 32.2 min).
4.2.20. 4’-Sulfonamide-3-[4-chloro-3 trifluoromethylphenylamino]diphenylether (75)
Compound 40 (100 mg, 0.26 mmol, 1 eq.), sulfamoyl chloride (30 mg, 0.26 mmol, 1 eq.) and NEt3 (0.05 mL, 0.29 mmol, 1.1 eq.) were dissolved in CH2Cl2 (2 mL) and stirred at overnight at room temperature. The mixture was then washed with water and the organic layer extracted with EtOAc, washed with brine, dried over MgSO4, concentrated under vacuum and purified by silica gel chromatography (hexanes:EtOAc) to get 75 as a light brown solid (95 mg, 80%). Mp: 124–126 °C. δH (400 MHz, CD3OD): 6.54 (dd, J = 8.2, 2.3 Hz, 1H, H-4), 6.69 (t, J = 2.2 Hz, 1H, H-2), 6.84 (dd, J = 8.1, 2.1 Hz, 1H, H-6), 6.99 (d, J = 9.0 Hz, 2H, H-8 and H-8′), 7.21–7.26 (m, 4H, H-9 and H-9′, H-16, H-5), 7.34–7.38 (m, 2H, H-12, H-15). δC (100 MHz, CD3OD): 109.1 (CH Ar, C-2), 112.2 (CH Ar, C-4), 113.8 (CH Ar, C-6), 116.1 (q, J = 5.5 Hz, C-12, CH Ar), 121.1 (2 CH Ar, C-8 and C-8′), 121.2 (CH Ar, C-16), 121.9 (qC, C-14), 123.2 (2 CH Ar, C-9 and C-9′), 124.4 (d, J = 272.5 Hz, qCF3), 129.7 (d, J = 30.9 Hz, qC, C-13), 131.5 (CH Ar, C-5), 133.3 (CH Ar, C-15), 136.1 (qC), 144.6 (qC), 145.1 (qC), 154.4 (qC), 160.5 (qC). δF (376 MHz, CD3OD):—64.19 (s). νmax (ATR)/cm−1: 3404 (NH), 3279 (NH), 1596 (S=O), 1489 (S=O), 1143 (C-O), 1153 (CF3), 1125 (C-N), 830 (C-Cl), 821. HRMS (m/z ESI−): found: 456.0397 (M−—H. C19H14N3O3SClF3 requires: 456.0397). HPLC: 99.3% (tR: 35.4 min).

 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}