Effects of Carbamazepine, Lacosamide and Zonisamide on Gliotransmitter Release Associated with Activated Astroglial Hemichannels


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
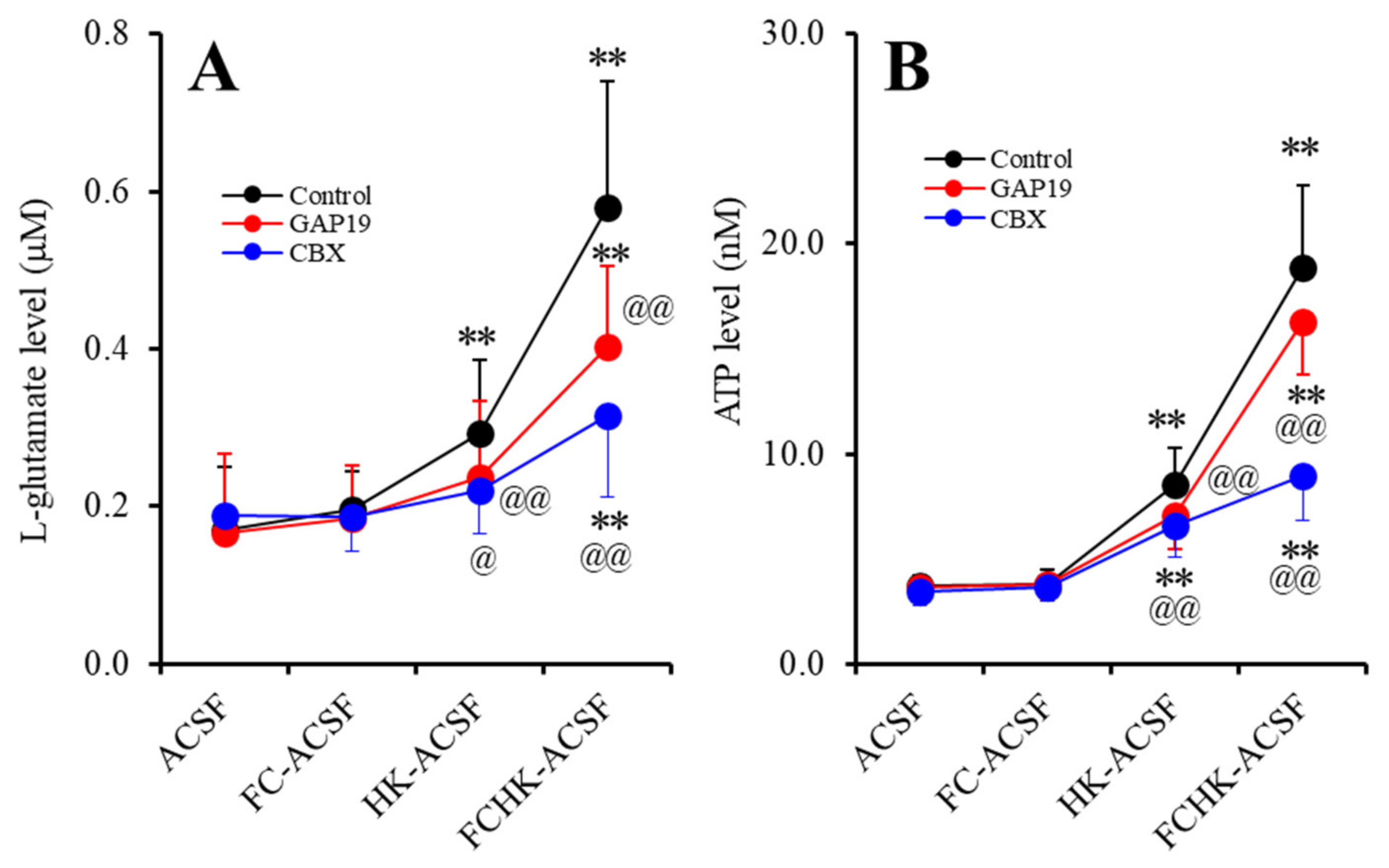
2.1. Effects of the Levels of Extracellular Ca2+ and K+ on the Astroglial Release of l-Glutamate and ATP (Study_1)
2.2. Effects of the Hemichannel Inhibitors on the Repetitive FCHK-ASCF-Evoked Astroglial Release of l-Glutamate and ATP
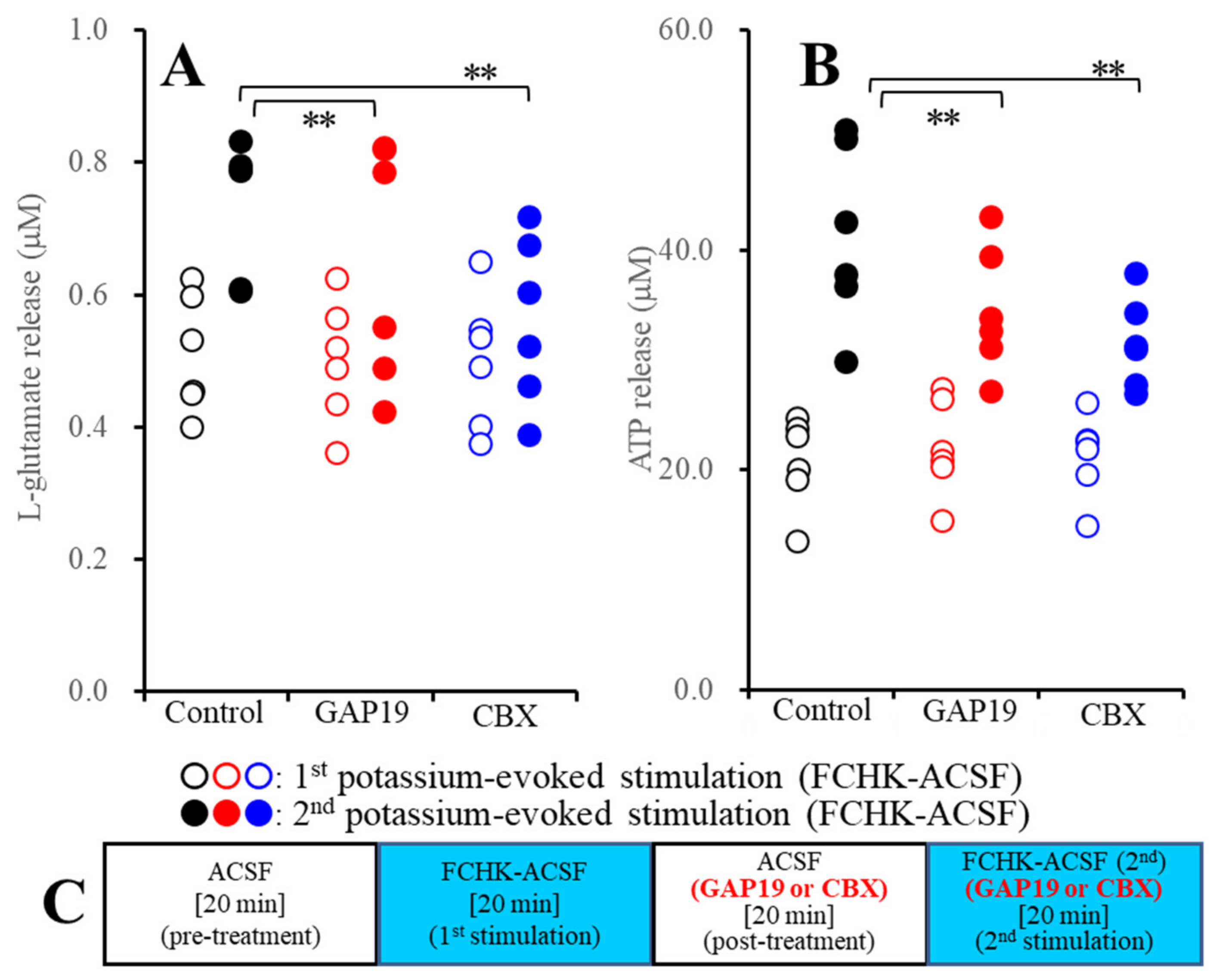
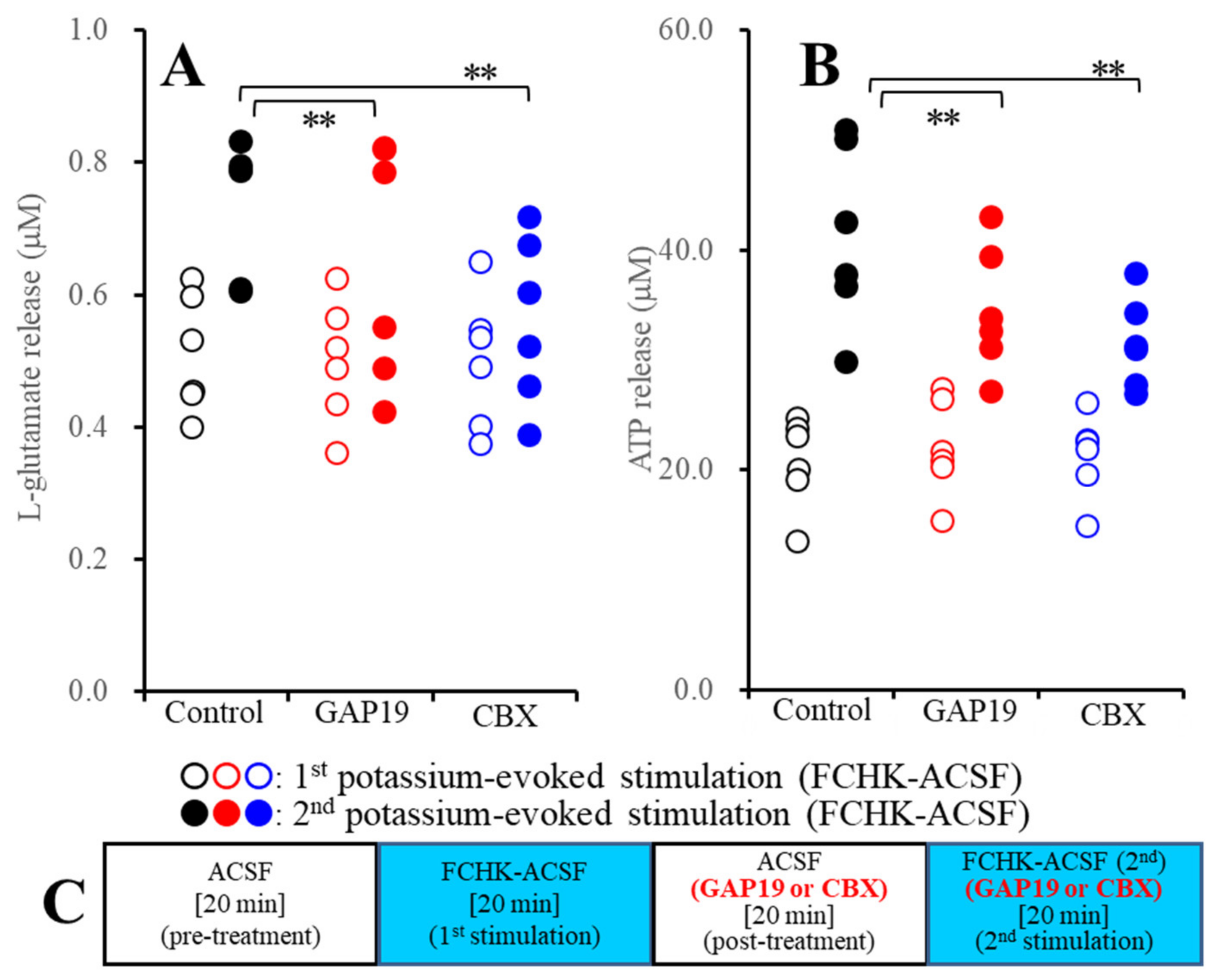
2.2.1. Effects of GAP19 and Carbenoxolone (CBX) on the FCHK-ACSF-Evoked Astroglial Release of l-Glutamate and ATP through Activated Hemichannels (Study_2)
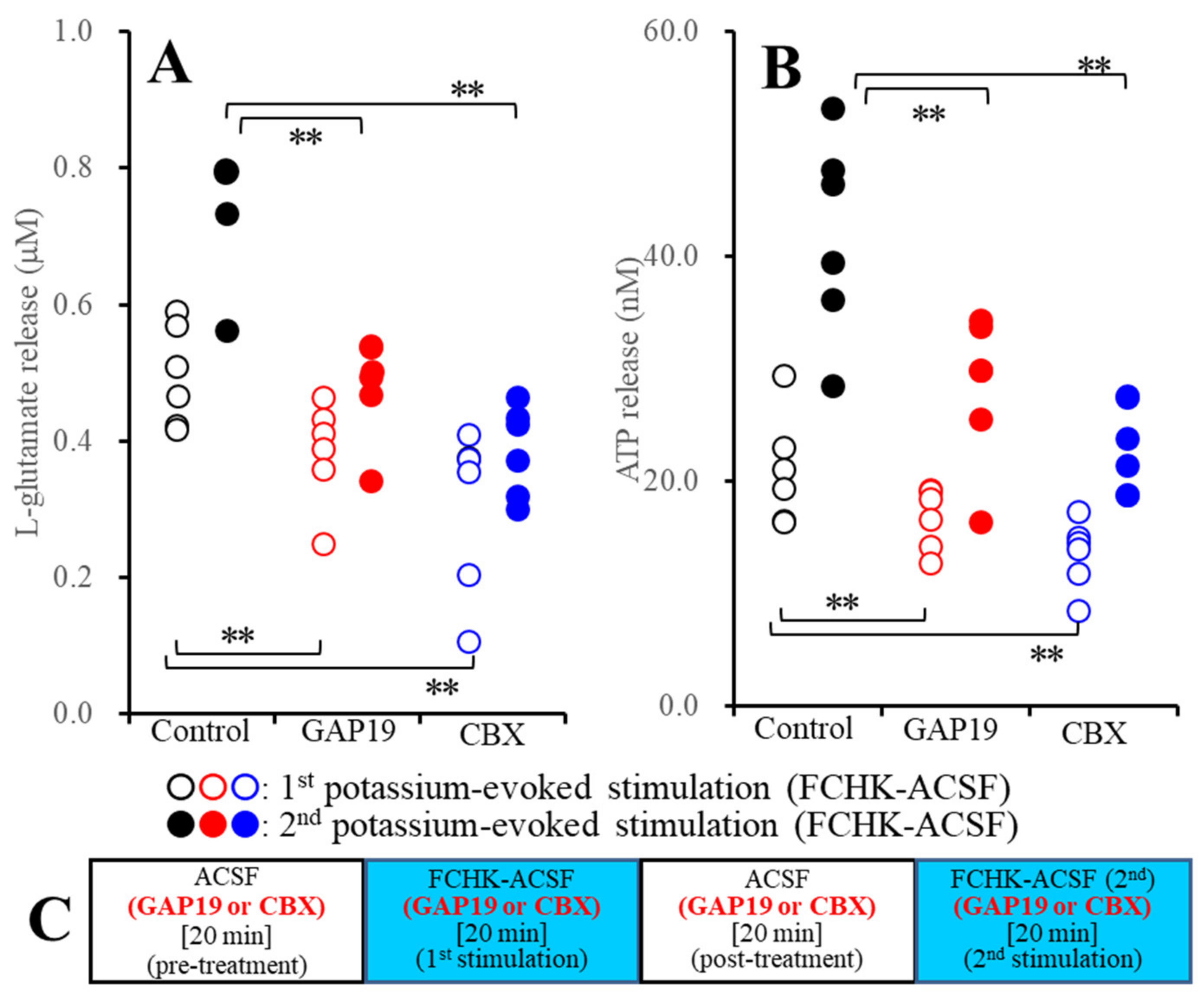
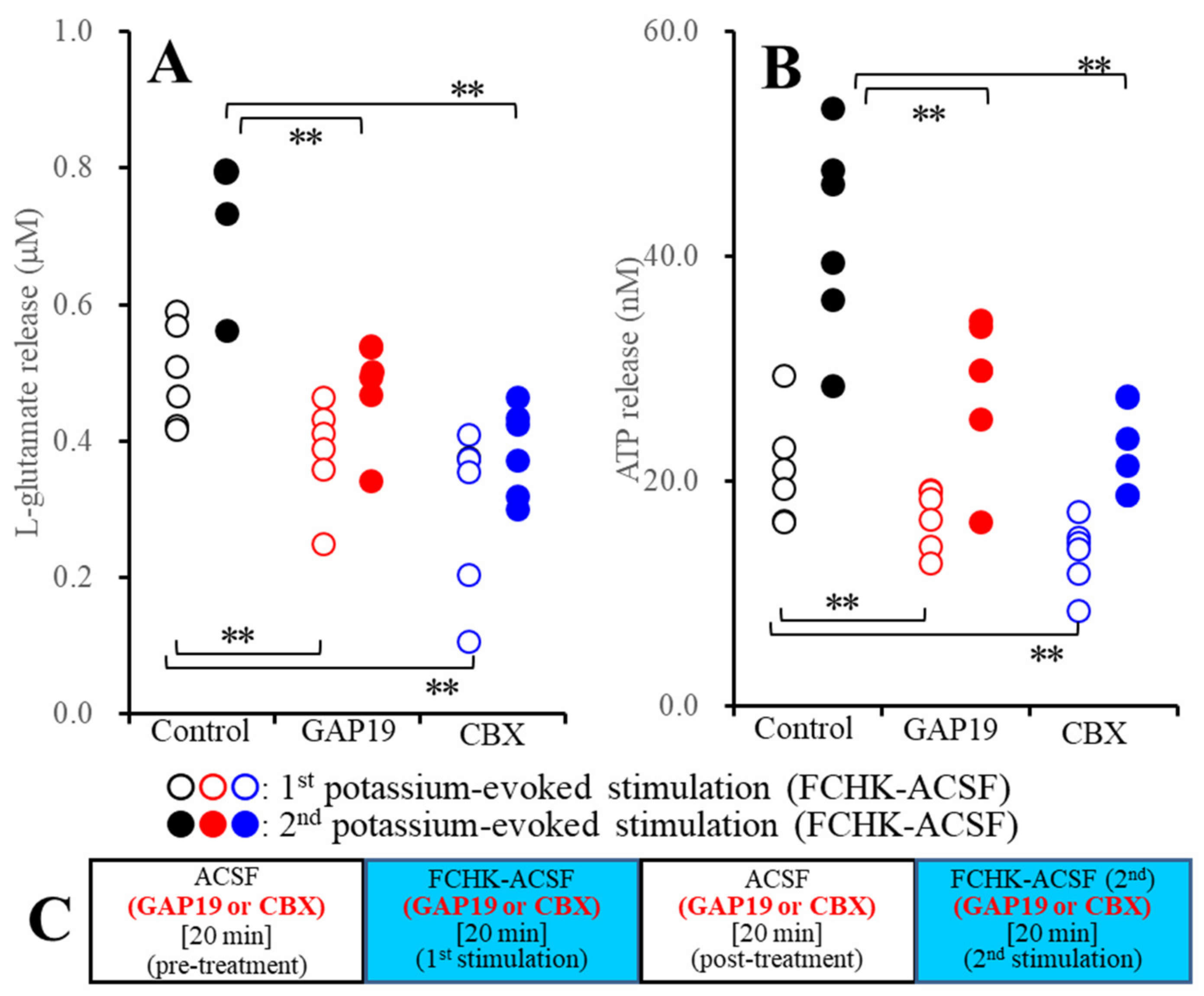
2.2.2. Effects of GAP19 and CBX on the Repetitive FCHK-ACSF-Evoked Astroglial Release of l-Glutamate and ATP through Unactivated and Activated Hemichannels (Study_3)
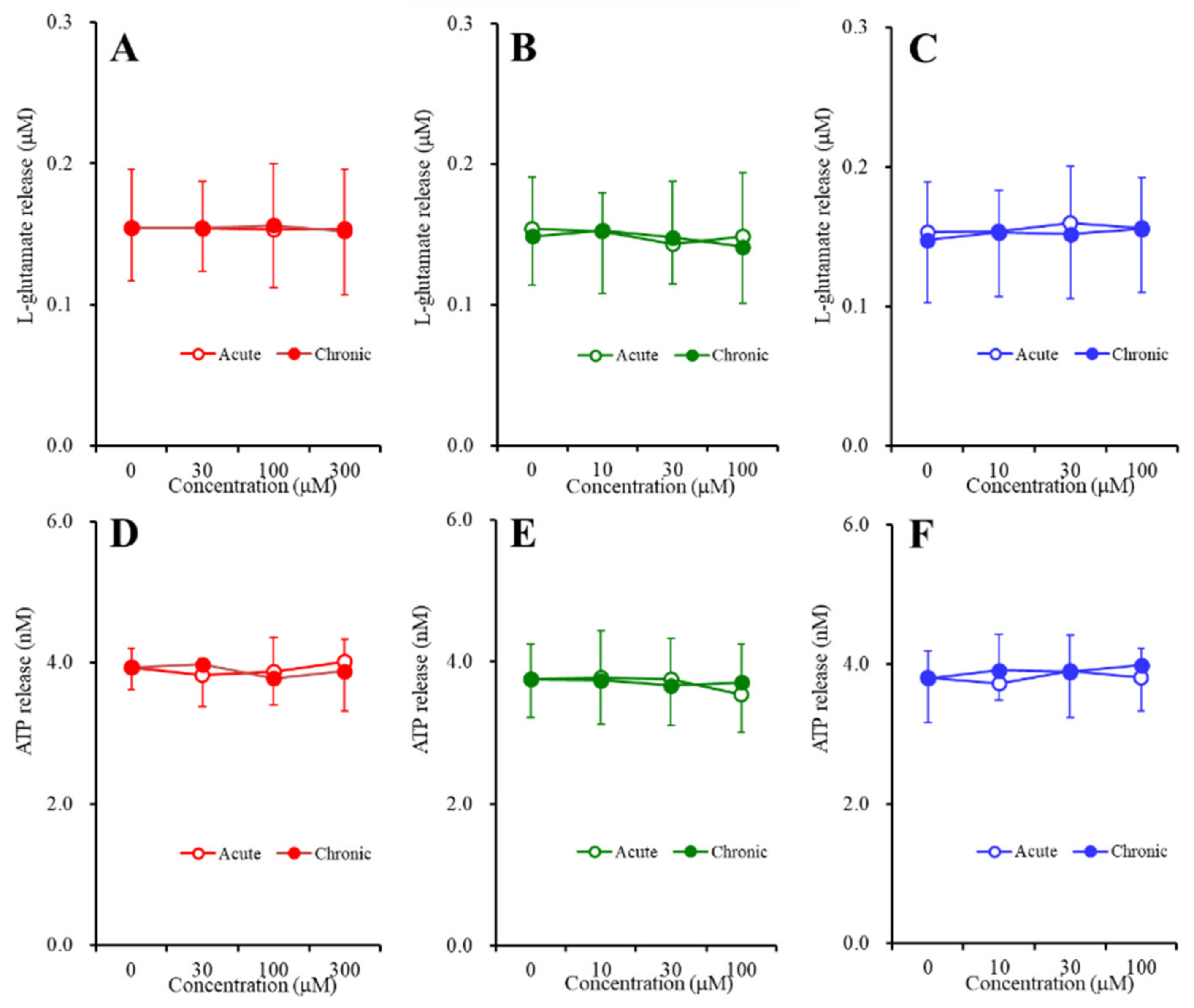
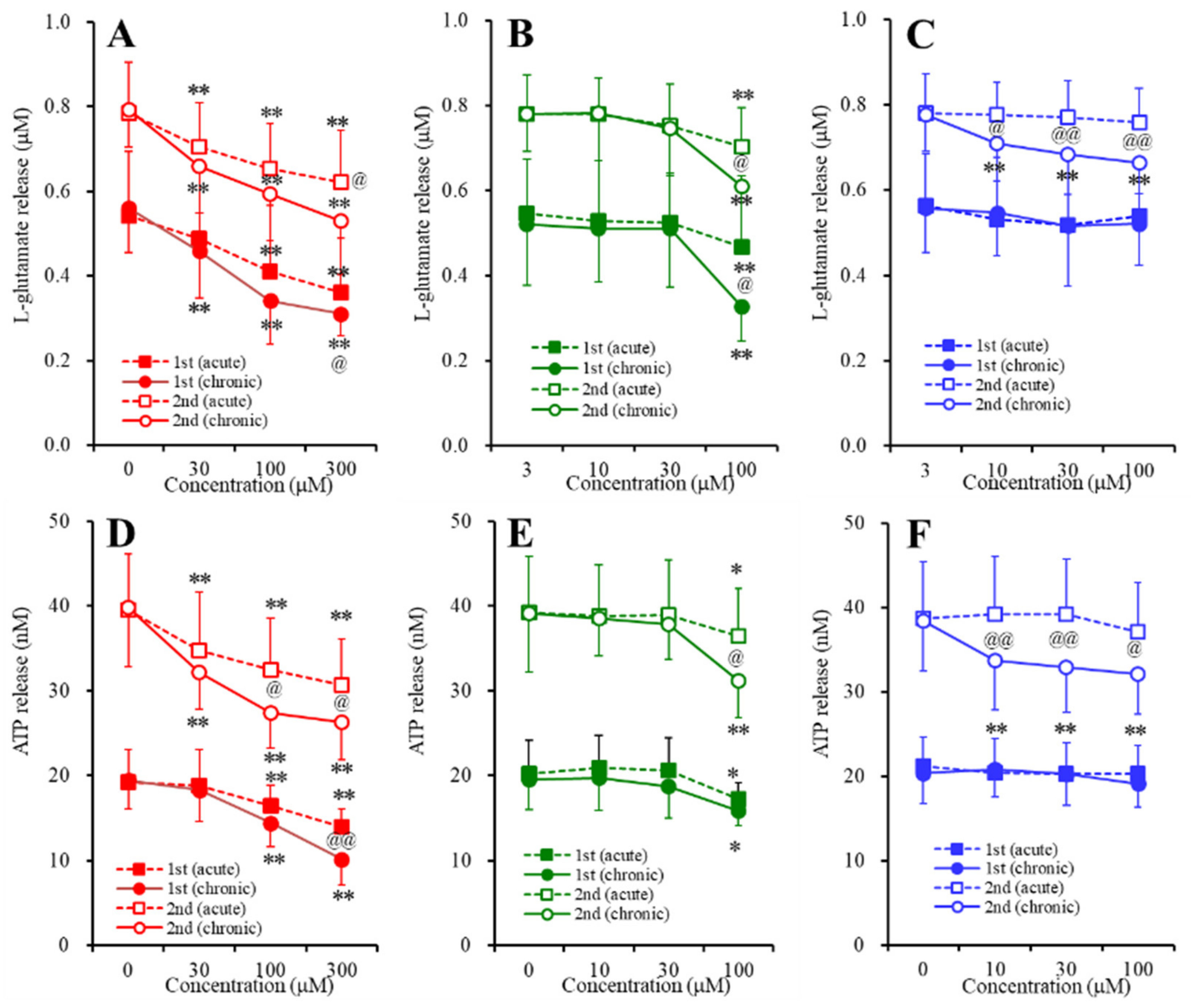
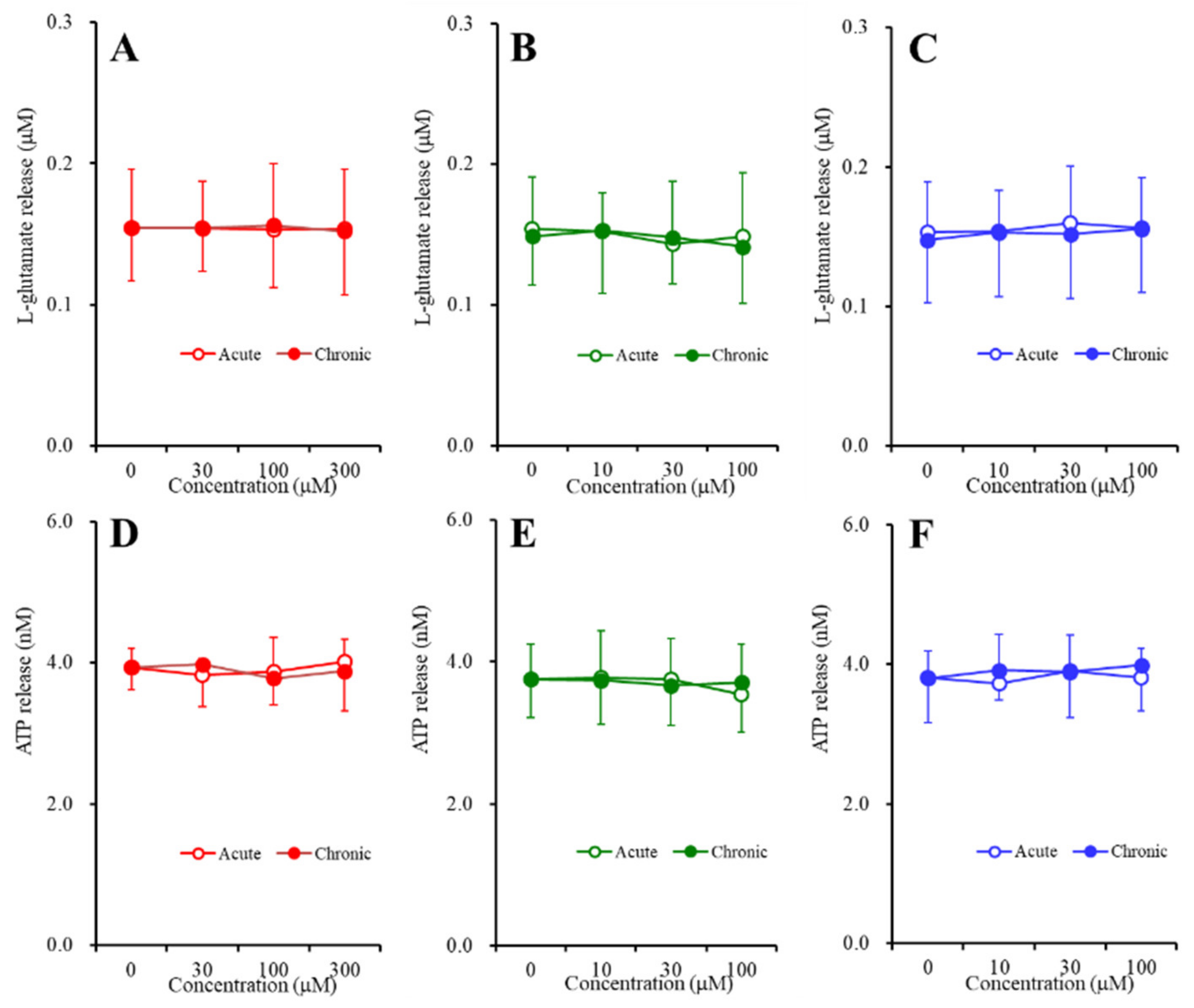
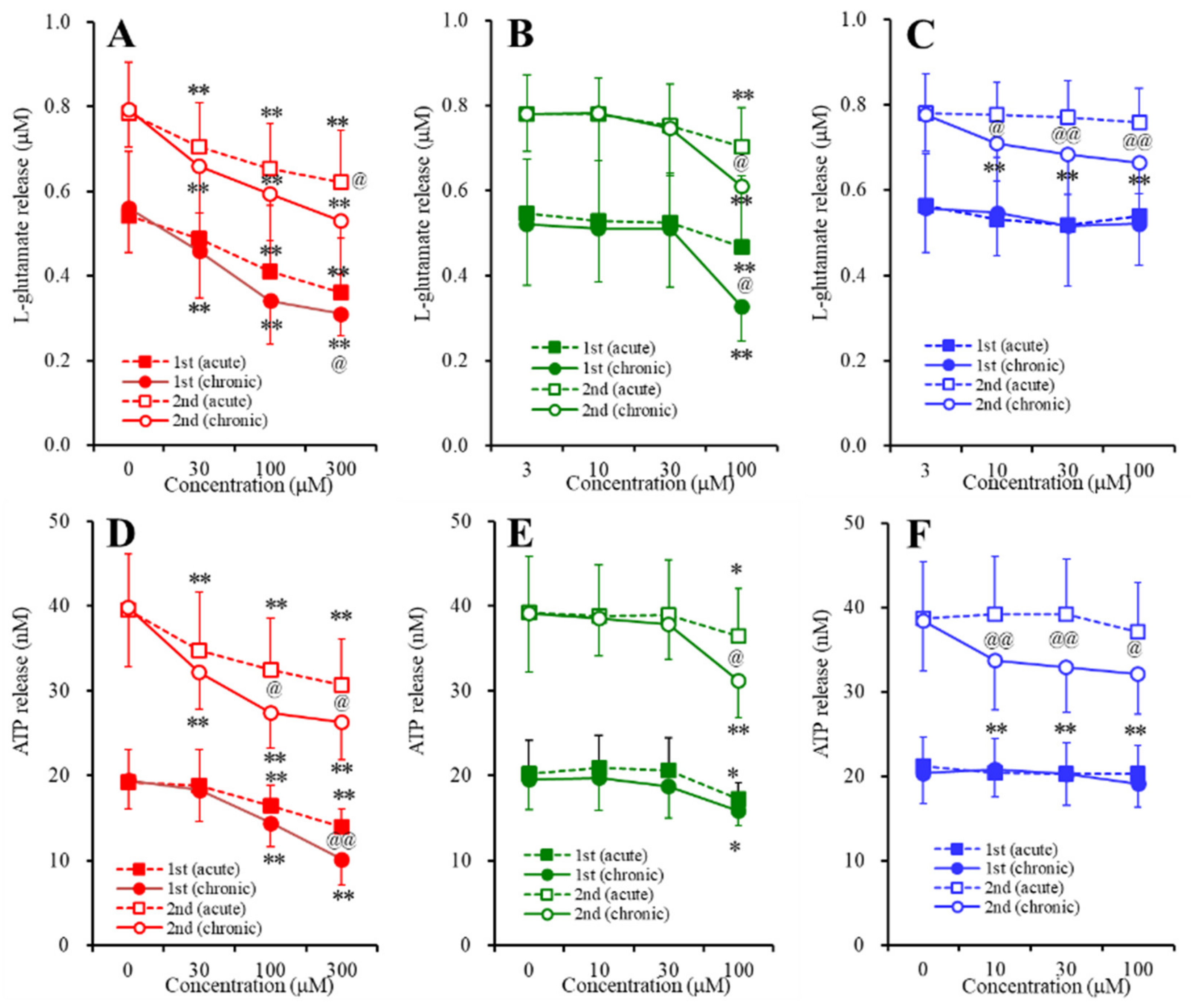
2.3. Concentration-Dependent Effects of Carbamazepine (CBZ), Lacosamide (LCM), and Zonisamide (ZNS) on the Astroglial Release of l-Glutamate and ATP (Study_4)
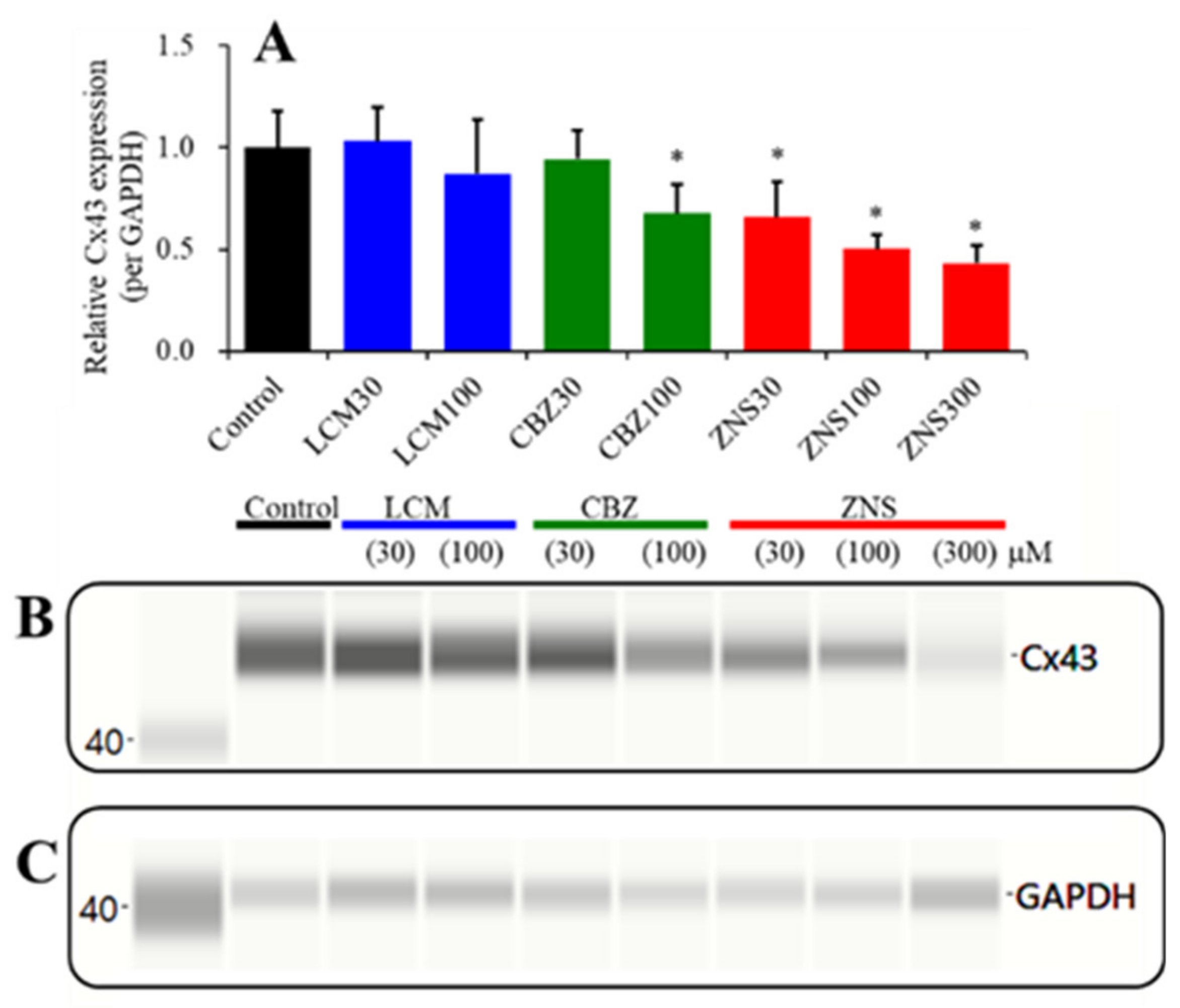
2.4. Concentration-Dependent Effects of the Subchronic Administration of LCM, CBZ, and ZNS on Cx43 Expression in the Plasma Membrane Fraction of Primary Cultured Astrocytes (Study_4)
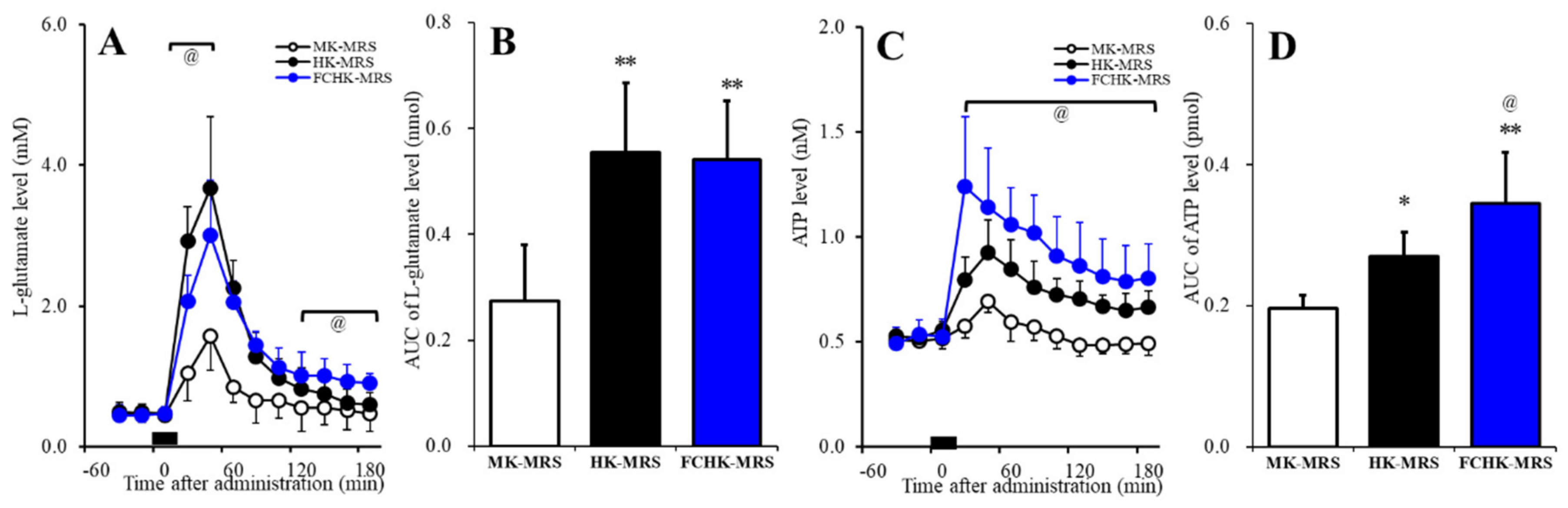
2.5. Effects of the Extracellular K+ and Ca2+ Levels on the Release of l-Glutamate and ATP in the Orbitofrontal Cortex (OFC)
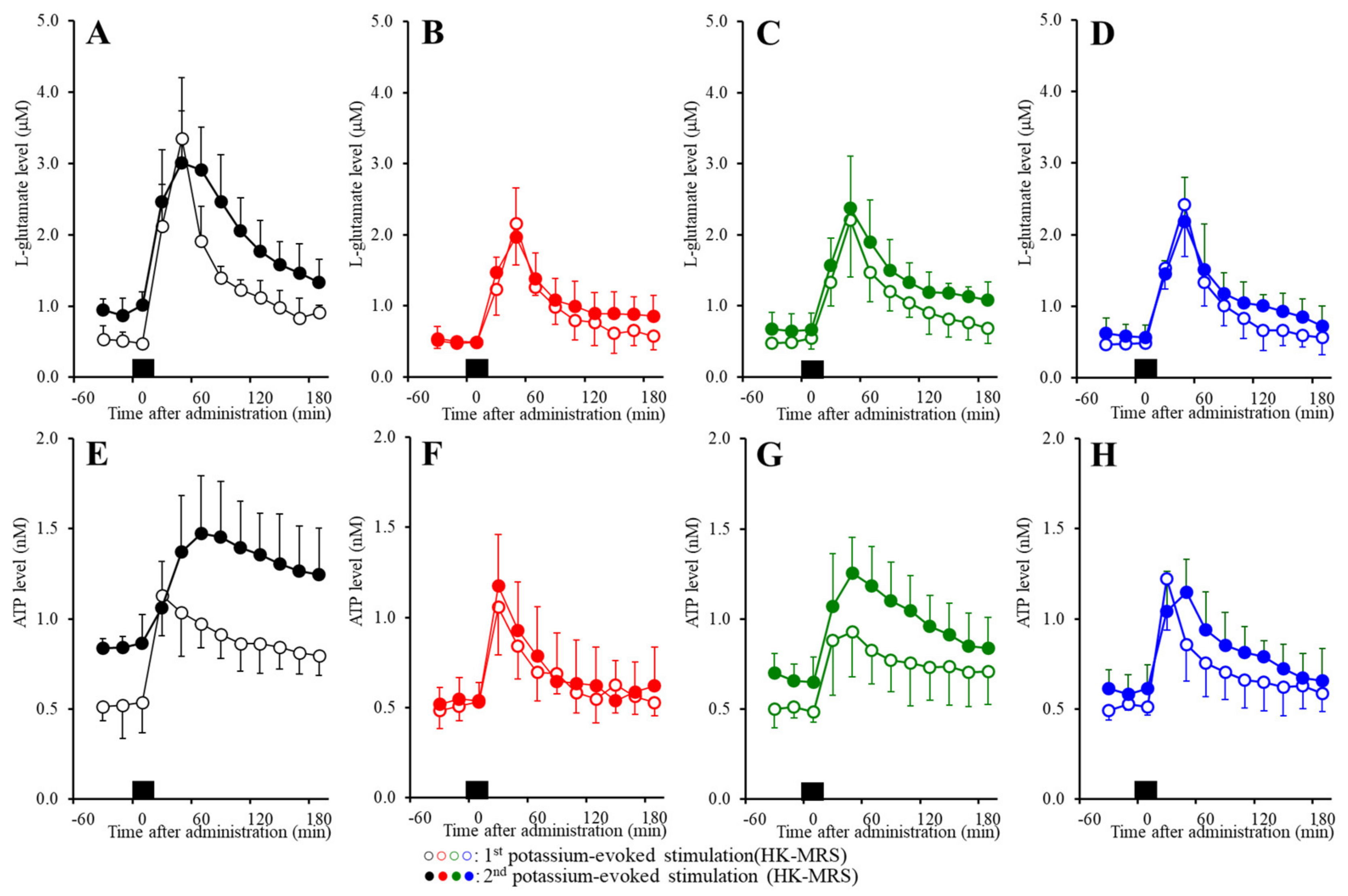
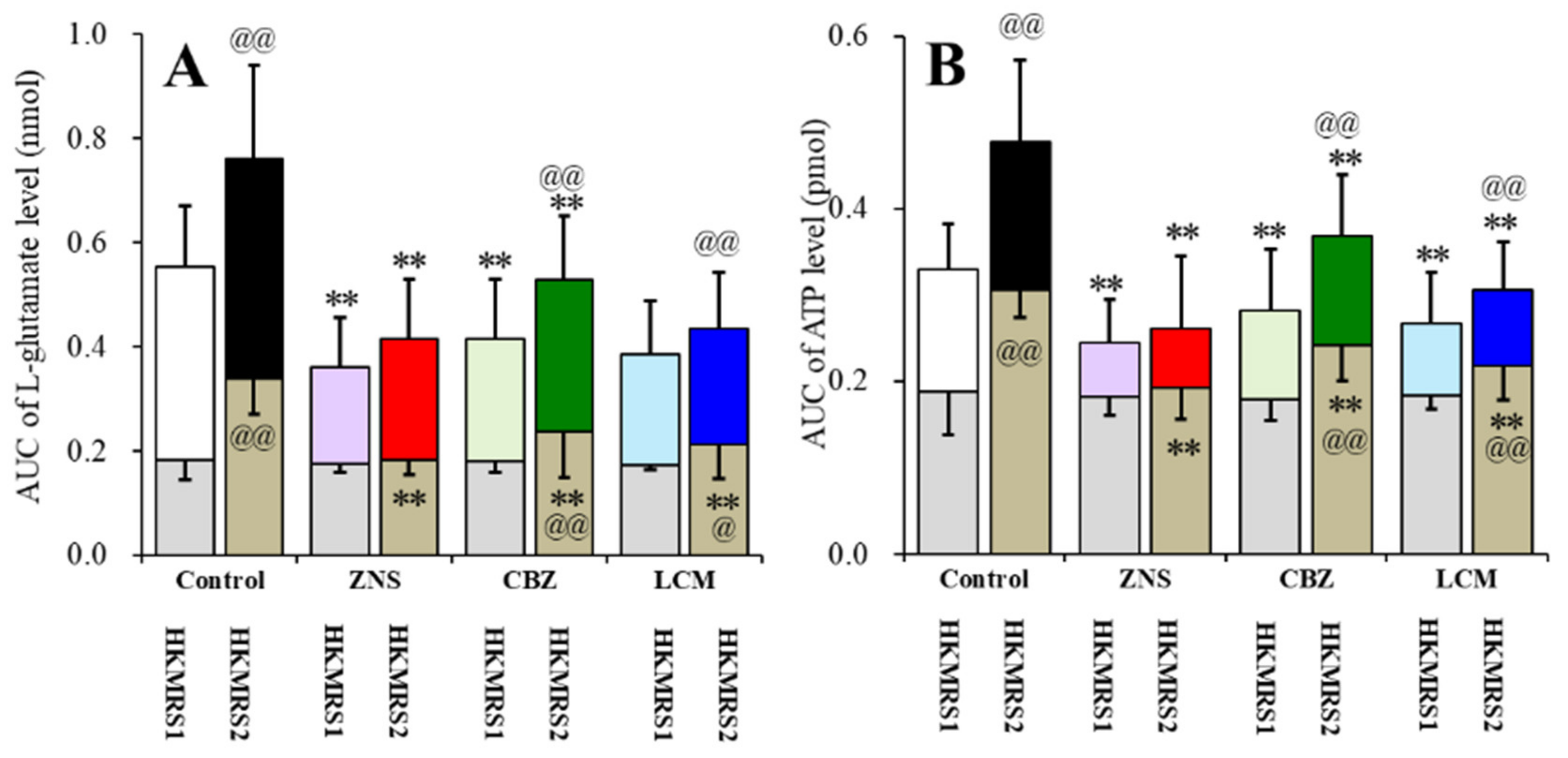
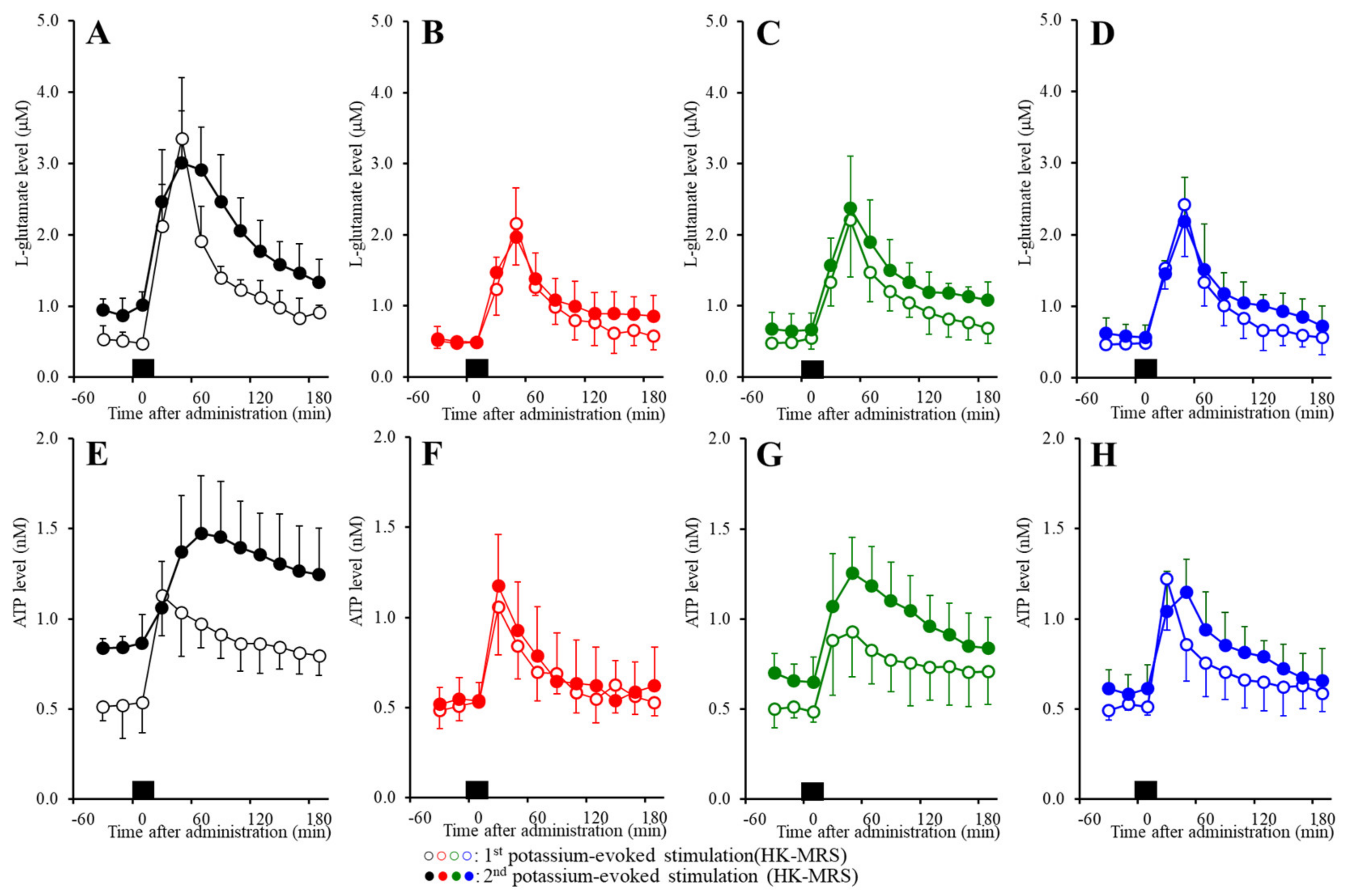
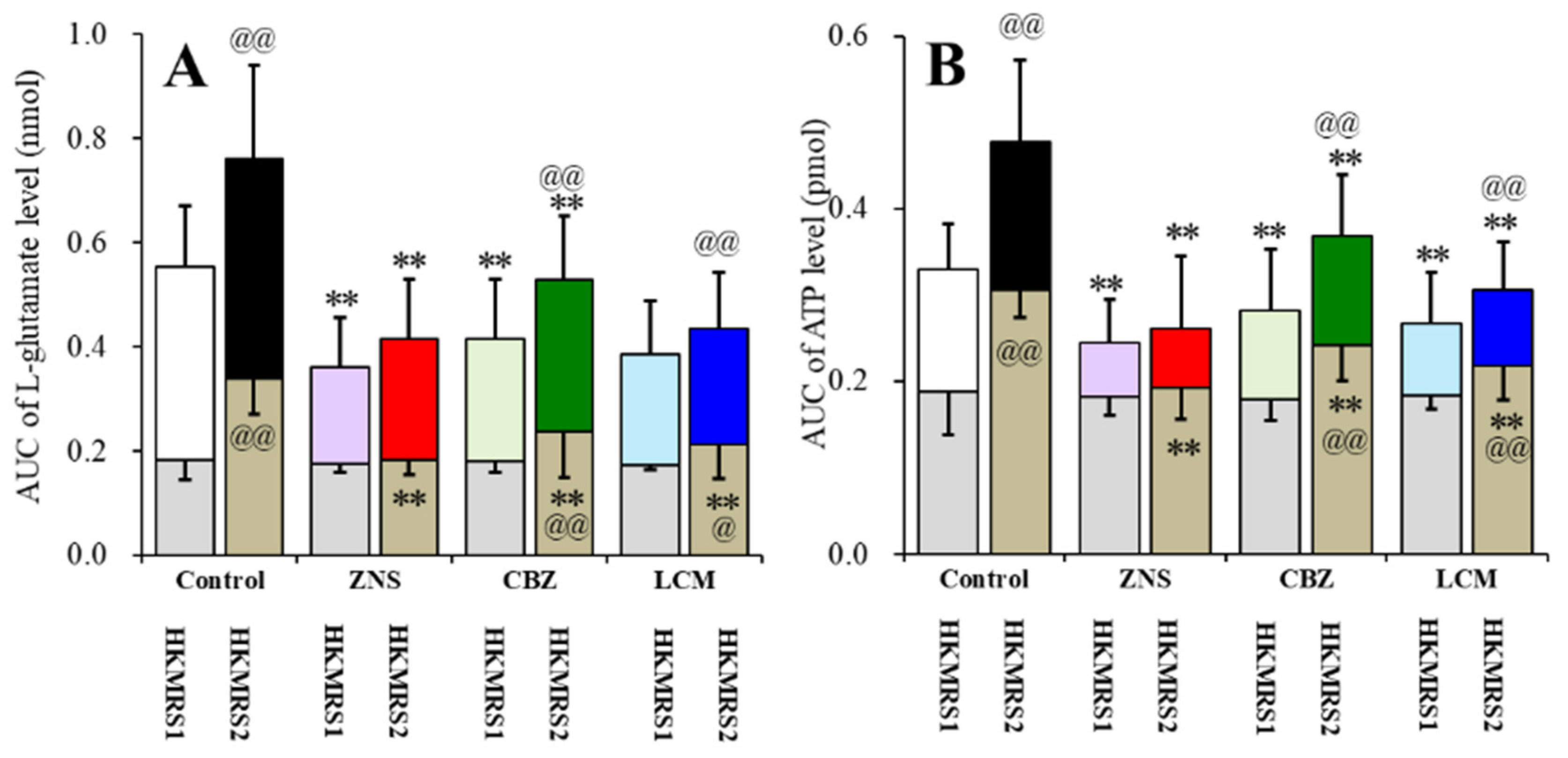
2.6. Effects of the Subchronic Administration of a Therapeutic-Relevant Dose of CBZ, LCM, and ZNS on the Repetitive FCHK-MRS-Evoked Release of l-Glutamate and ATP in the OFC
3. Discussion
3.1. Regulation Mechanisms of the Astroglial Gliotransmitter Release Associated with Cx43-Containing Hemichannels
3.2. Inhibitory Effects of the VDSC-Inhibiting Acticonvulsants CBZ, LCM, and ZNS on Activated Cx43 Hemichannels
4. Materials and Methods
4.1. Experimental Animals
4.2. Preparation of the Primary Cultured Astrocytes
4.3. Simple Western Analysis
4.4. Preparation of the Microdialysis System
4.5. Ultra-High-Performance Liquid Chromatography (UHPLC)
4.6. Ultra-High-Performance Liquid Chromatography with Mass Spectrometry (LCMS)
4.7. Chemical Agents
4.8. Data Analysis
4.9. Nomenclature of Targets and Ligands
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sills, G.J.; Rogawski, M.A. Mechanisms of action of currently used antiseizure drugs. Neuropharmacology 2020, 168, 107966. [Google Scholar] [CrossRef]
- Meador, K.J.; Loring, D.W.; Boyd, A.; Echauz, J.; LaRoche, S.; Velez-Ruiz, N.; Korb, P.; Byrnes, W.; Dilley, D.; Borghs, S.; et al. Randomized double-blind comparison of cognitive and EEG effects of lacosamide and carbamazepine. Epilepsy Behav. 2016, 62, 267–275. [Google Scholar] [CrossRef]
- Arif, H.; Buchsbaum, R.; Weintraub, D.; Pierro, J.; Resor, S.R., Jr.; Hirsch, L.J. Patient-reported cognitive side effects of antiepileptic drugs: Predictors and comparison of all commonly used antiepileptic drugs. Epilepsy Behav. 2009, 14, 202–209. [Google Scholar] [CrossRef]
- Rogawski, M.A.; Tofighy, A.; White, H.S.; Matagne, A.; Wolff, C. Current understanding of the mechanism of action of the antiepileptic drug lacosamide. Epilepsy Res. 2015, 110, 189–205. [Google Scholar] [CrossRef]
- Ijff, D.M.; Aldenkamp, A.P. Cognitive side-effects of antiepileptic drugs in children. Handb. Clin. Neurol. 2013, 111, 707–718. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Zhu, G.; Yoshida, S.; Kanai, K.; Hirose, S.; Kaneko, S. Exocytosis mechanism as a new targeting site for mechanisms of action of antiepileptic drugs. Life Sci. 2002, 72, 465–473. [Google Scholar] [CrossRef]
- Fukuyama, K.; Fukuzawa, M.; Shiroyama, T.; Okada, M. Pathogenesis and pathophysiology of autosomal dominant sleep-related hypermotor epilepsy with S284L-mutant alpha4 subunit of nicotinic ACh receptor. Br. J. Pharm. 2020, 177, 2143–2162. [Google Scholar] [CrossRef]
- Fukuyama, K.; Fukuzawa, M.; Ruri, O.; Okada, M. Upregulated connexin 43 induced by loss-of-functional S284L-mutant alpha4 subunit of nicotinic ACh receptor contributes to pathomechanisms of autosomal dominant sleep-related hypermotor epilepsy. Pharmaceuticals 2020, 13, 58. [Google Scholar] [CrossRef] [Green Version]
- Fukuyama, K.; Fukuzawa, M.; Okada, M. Upregulated and hyperactivated thalamic connexin 43 plays important roles in pathomechanisms of cognitive impairment and seizure of autosomal dominant sleep-related hypermotor epilepsy with S284L-mutant α4 subunit of nicotinic ACh receptor. Pharmaceuticals 2020, 13, 99. [Google Scholar] [CrossRef]
- Fukuyama, K.; Fukuzawa, M.; Shiroyama, T.; Okada, M. Pathomechanism of nocturnal paroxysmal dystonia in autosomal dominant sleep-related hypermotor epilepsy with S284L-mutant α4 subunit of nicotinic ACh receptor. Biomed Pharm. 2020, 126, 110070. [Google Scholar] [CrossRef]
- Shiba, Y.; Mori, F.; Yamada, J.; Migita, K.; Nikaido, Y.; Wakabayashi, K.; Kaneko, S.; Okada, M.; Hirose, S.; Ueno, S. Spontaneous epileptic seizures in transgenic rats harboring a human ADNFLE missense mutation in the beta2-subunit of the nicotinic acetylcholine receptor. Neurosci. Res. 2015, 100, 46–54. [Google Scholar] [CrossRef] [Green Version]
- Yamada, J.; Zhu, G.; Okada, M.; Hirose, S.; Yoshida, S.; Shiba, Y.; Migita, K.; Mori, F.; Sugawara, T.; Chen, L.; et al. A novel prophylactic effect of furosemide treatment on autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE). Epilepsy Res. 2013, 107, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Okada, M.; Yoshida, S.; Ueno, S.; Mori, F.; Takahara, T.; Saito, R.; Miura, Y.; Kishi, A.; Tomiyama, M.; et al. Rats harboring S284L Chrna4 mutation show attenuation of synaptic and extrasynaptic GABAergic transmission and exhibit the nocturnal frontal lobe epilepsy phenotype. J. Neurosci. 2008, 28, 12465–12476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tinuper, P.; Bisulli, F.; Cross, J.H.; Hesdorffer, D.; Kahane, P.; Nobili, L.; Provini, F.; Scheffer, I.E.; Tassi, L.; Vignatelli, L.; et al. Definition and diagnostic criteria of sleep-related hypermotor epilepsy. Neurology 2016, 86, 1834–1842. [Google Scholar] [CrossRef]
- Scheffer, I.E.; Bhatia, K.P.; Lopes-Cendes, I.; Fish, D.R.; Marsden, C.D.; Andermann, F.; Andermann, E.; Desbiens, R.; Cendes, F.; Manson, J.I.; et al. Autosomal dominant frontal epilepsy misdiagnosed as sleep disorder. Lancet 1994, 343, 515–517. [Google Scholar] [CrossRef]
- Provini, F.; Plazzi, G.; Tinuper, P.; Vandi, S.; Lugaresi, E.; Montagna, P. Nocturnal frontal lobe epilepsy. A clinical and polygraphic overview of 100 consecutive cases. Brain 1999, 122, 1017–1031. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Zhu, G.; Yoshida, S.; Kaneko, S. Validation criteria for genetic animal models of epilepsy. Epilepsy Seizure 2010, 3, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Asioli, G.M.; Rossi, S.; Bisulli, F.; Licchetta, L.; Tinuper, P.; Provini, F. Therapy in sleep-related hypermotor epilepsy (SHE). Curr. Treat. Options Neurol. 2020, 22, 1. [Google Scholar] [CrossRef]
- Miyajima, T.; Kumada, T.; Saito, K.; Fujii, T. Autism in siblings with autosomal dominant nocturnal frontal lobe epilepsy. Brain Dev. 2013, 35, 155–157. [Google Scholar] [CrossRef]
- Fukuyama, K.; Tanahashi, S.; Hoshikawa, M.; Shinagawa, R.; Okada, M. Zonisamide regulates basal ganglia transmission via astroglial kynurenine pathway. Neuropharmacology 2014, 76, 137–145. [Google Scholar] [CrossRef]
- Medina-Ceja, L.; Salazar-Sanchez, J.C.; Ortega-Ibarra, J.; Morales-Villagran, A. Connexins-based hemichannels/channels and their relationship with inflammation, seizures and epilepsy. Int. J. Mol. Sci. 2019, 20, 5976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kar, R.; Batra, N.; Riquelme, M.A.; Jiang, J.X. Biological role of connexin intercellular channels and hemichannels. Arch. Biochem. Biophys. 2012, 524, 2–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fasciani, I.; Temperan, A.; Perez-Atencio, L.F.; Escudero, A.; Martinez-Montero, P.; Molano, J.; Gomez-Hernandez, J.M.; Paino, C.L.; Gonzalez-Nieto, D.; Barrio, L.C. Regulation of connexin hemichannel activity by membrane potential and the extracellular calcium in health and disease. Neuropharmacology 2013, 75, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, K.; Okada, M. Effects of levetiracetam on astroglial release of kynurenine-pathway metabolites. Br. J. Pharm. 2018, 175, 4253–4265. [Google Scholar] [CrossRef] [Green Version]
- He, J.T.; Li, X.Y.; Yang, L.; Zhao, X. Astroglial connexins and cognition: Memory formation or deterioration? Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef]
- Hussein, A.M.; Ghalwash, M.; Magdy, K.; Abulseoud, O.A. Beta lactams antibiotic ceftriaxone modulates seizures, oxidative stress and connexin 43 expression in hippocampus of pentylenetetrazole kindled rats. J. Epilepsy Res. 2016, 6, 8–15. [Google Scholar] [CrossRef]
- Das, A.; Wallace, G.C.T.; Holmes, C.; McDowell, M.L.; Smith, J.A.; Marshall, J.D.; Bonilha, L.; Edwards, J.C.; Glazier, S.S.; Ray, S.K.; et al. Hippocampal tissue of patients with refractory temporal lobe epilepsy is associated with astrocyte activation, inflammation, and altered expression of channels and receptors. Neuroscience 2012, 220, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Garbelli, R.; Frassoni, C.; Condorelli, D.F.; Trovato-Salinaro, A.; Musso, N.; Medici, V.; Tassi, L.; Bentivoglio, M.; Spreafico, R. Expression of connexin 43 in the human epileptic and drug-resistant cerebral cortex. Neurology 2011, 76, 895–902. [Google Scholar] [CrossRef]
- Li, Q.; Li, Q.Q.; Jia, J.N.; Liu, Z.Q.; Zhou, H.H.; Mao, X.Y. Targeting gap junction in epilepsy: Perspectives and challenges. Biomed. Pharm. 2019, 109, 57–65. [Google Scholar] [CrossRef]
- Fukuyama, K.; Okubo, R.; Murata, M.; Shiroyama, T.; Okada, M. Activation of astroglial connexin is involved in concentration-dependent double-edged sword clinical action of clozapine. Cells 2020, 9, 414. [Google Scholar] [CrossRef] [Green Version]
- Rusakov, D.A. Depletion of extracellular Ca2+ prompts astroglia to moderate synaptic network activity. Sci. Signal 2012, 5, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; De Bock, M.; Decrock, E.; Bol, M.; Gadicherla, A.; Bultynck, G.; Leybaert, L. Connexin targeting peptides as inhibitors of voltage-and intracellular Ca2+-triggered Cx43 hemichannel opening. Neuropharmacology 2013, 75, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Yanguas, S.C.; Willebrords, J.; Vinken, M. Models and methods for in vitro testing of hepatic gap junctional communication. Toxicol. Vitro 2015, 30, 569–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, C.L.; Lai, Y.J.; Chi, P.C.; Chen, L.C.; Tseng, Y.M.; Kuo, J.Y.; Lin, C.I.; Chen, Y.C.; Lin, S.J.; Yeh, H.I. Dose-related ethanol intake, Cx43 and Nav1.5 remodeling: Exploring insights of altered ventricular conduction and QRS fragmentation in excessive alcohol users. J. Mol. Cell Cardiol. 2018, 114, 150–160. [Google Scholar] [CrossRef]
- Yamamura, S.; Hoshikawa, M.; Dai, K.; Saito, H.; Suzuki, N.; Niwa, O.; Okada, M. ONO-2506 inhibits spike-wave discharges in a genetic animal model without affecting traditional convulsive tests via gliotransmission regulation. Br. J. Pharm. 2013, 168, 1088–1100. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Okada, M.; Zhu, G.; Kaneko, S. Effects of zonisamide on neurotransmitter exocytosis associated with ryanodine receptors. Epilepsy Res. 2005, 67, 153–162. [Google Scholar] [CrossRef]
- Kawata, Y.; Okada, M.; Murakami, T.; Mizuno, K.; Wada, K.; Kondo, T.; Kaneko, S. Effects of zonisamide on K+ and Ca2+ evoked release of monoamine as well as K+ evoked intracellular Ca2+ mobilization in rat hippocampus. Epilepsy Res. 1999, 35, 173–182. [Google Scholar] [CrossRef]
- Qiao, X.; Werkman, T.R.; Gorter, J.A.; Wadman, W.J.; van Vliet, E.A. Expression of sodium channel alpha subunits 1.1, 1.2 and 1.6 in rat hippocampus after kainic acid-induced epilepsy. Epilepsy Res. 2013, 106, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Carmignoto, G.; Haydon, P.G. Astrocyte calcium signaling and epilepsy. Glia 2012, 60, 1227–1233. [Google Scholar] [CrossRef] [Green Version]
- Rusakov, D.; Fine, A. Extracellular Ca2+ depletion contributes to fast activity-dependent modulation of synaptic transmission in the brain. Neuron 2003, 37, 287. [Google Scholar] [CrossRef] [Green Version]
- Omura, T.; Kaneko, M.; Okuma, Y.; Matsubara, K.; Nomura, Y. Endoplasmic reticulum stress and Parkinson’s disease: The role of HRD1 in averting apoptosis in neurodegenerative disease. Oxid. Med. Cell Longev. 2013, 2013, 239854. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, S.; Hamaguchi, T.; Ohoyama, K.; Sugiura, Y.; Suzuki, D.; Kanehara, S.; Nakagawa, M.; Motomura, E.; Matsumoto, T.; Tanii, H.; et al. Topiramate and zonisamide prevent paradoxical intoxication induced by carbamazepine and phenytoin. Epilepsy Res. 2009, 84, 172–186. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.M.; Khanna, R. Specific binding of lacosamide to collapsin response mediator protein 2 (CRMP2) and direct impairment of its canonical function: Implications for the therapeutic potential of lacosamide. Mol. Neurobiol. 2015, 51, 599–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGrath, J.C.; Lilley, E. Implementing guidelines on reporting research using animals (ARRIVE etc.): New requirements for publication in BJP. Br. J. Pharm. 2015, 172, 3189–3193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, M.; Fukuyama, K.; Shiroyama, T.; Ueda, Y. Carbamazepine attenuates astroglial L-glutamate release induced by pro-inflammatory cytokines via chronically activation of adenosine A2A receptor. Int. J. Mol. Sci. 2019, 20, 3727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, M.; Fukuyama, K.; Shiroyama, T.; Ueda, Y. Lurasidone inhibits NMDA antagonist-induced functional abnormality of thalamocortical glutamatergic transmission via 5-HT7 receptor blockade. Br. J. Pharm. 2019, 176, 4002–4018. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K.; Okubo, R.; Shiroyama, T.; Ueda, Y. Lurasidone sub-chronically activates serotonergic transmission via desensitization of 5-HT1A and 5-HT7 receptors in dorsal raphe nucleus. Pharmaceuticals 2019, 12, 149. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Okubo, R.; Fukuyama, K. Vortioxetine subchronically activates serotonergic transmission via desensitization of serotonin 5-HT1A receptor with 5-HT3 receptor inhibition in rats. Int. J. Mol. Sci. 2019, 20, 6235. [Google Scholar] [CrossRef] [Green Version]
- Paxinos, G.; Watson, C. The Rat Brain: In Stereotoxic Coordinates, 6th ed.; Academic Press: San Diego, CA, USA, 2007. [Google Scholar]
- Okada, M.; Fukuyama, K.; Kawano, Y.; Shiroyama, T.; Suzuki, D.; Ueda, Y. Effects of acute and sub-chronic administrations of guanfacine on catecholaminergic transmissions in the orbitofrontal cortex. Neuropharmacology 2019, 156, 107547. [Google Scholar] [CrossRef]
- Kawata, Y.; Okada, M.; Murakami, T.; Kamata, A.; Zhu, G.; Kaneko, S. Pharmacological discrimination between effects of carbamazepine on hippocampal basal, Ca2+- and K+-evoked serotonin release. Br. J. Pharm. 2001, 133, 557–567. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Wada, K.; Kiryu, K.; Kawata, Y.; Mizuno, K.; Kondo, T.; Tasaki, H.; Kaneko, S. Effects of Ca2+ channel antagonists on striatal dopamine and DOPA release, studied by in vivo microdialysis. Br. J. Pharm. 1998, 123, 805–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, M.; Kawata, Y.; Mizuno, K.; Wada, K.; Kondo, T.; Kaneko, S. Interaction between Ca2+, K+, carbamazepine and zonisamide on hippocampal extracellular glutamate monitored with a microdialysis electrode. Br. J. Pharm. 1998, 124, 1277–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mtui, E.; Gruener, G.; Dockery, P. Fitzgerald’s Clinical Neuroanatomy and Neuroscience, 7th ed.; Elsevier: Philadelphia, PA, USA, 2015. [Google Scholar]
- Karlsen, A.S.; Korbo, S.; Uylings, H.B.; Pakkenberg, B. A stereological study of the mediodorsal thalamic nucleus in Down syndrome. Neuroscience 2014, 279, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Hasegawa, T.; Suzuki, D.; Motomura, E.; Okada, M. Amantadine combines astroglial system Xc− activation with glutamate/NMDA receptor inhibition. Biomolecules 2019, 9, 191. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Hirano, T.; Mizuno, K.; Chiba, T.; Kawata, Y.; Kiryu, K.; Wada, K.; Tasaki, H.; Kaneko, S. Biphasic effects of carbamazepine on the dopaminergic system in rat striatum and hippocampus. Epilepsy Res. 1997, 28, 143–153. [Google Scholar] [CrossRef]
- Okada, M.; Hirano, T.; Mizuno, K.; Kawata, Y.; Wada, K.; Murakami, T.; Tasaki, H.; Kaneko, S. Effects of carbamazepine on hippocampal serotonergic system. Epilepsy Res. 1998, 31, 187–198. [Google Scholar] [CrossRef]
- Tanahashi, S.; Yamamura, S.; Nakagawa, M.; Motomura, E.; Okada, M. Effect of lamotrigine and carbamazepine on corticotropin-releasing factor-associated serotonergic transmission in rat dorsal raphe nucleus. Psychopharmacology 2012, 220, 599–610. [Google Scholar] [CrossRef]
- Yamamura, S.; Ohoyama, K.; Nagase, H.; Okada, M. Zonisamide enhances delta receptor-associated neurotransmitter release in striato-pallidal pathway. Neuropharmacology 2009, 57, 322–331. [Google Scholar] [CrossRef]
- Masuda, Y.; Utsui, Y.; Shiraishi, Y.; Karasawa, T.; Yoshida, K.; Shimizu, M. Relationships between plasma concentrations of diphenylhydantoin, phenobarbital, carbamazepine, and 3-sulfamoylmethyl-1,2-benzisoxazole (AD-810), a new anticonvulsant agent, and their anticonvulsant or neurotoxic effects in experimental animals. Epilepsia 1979, 20, 623–633. [Google Scholar] [CrossRef]
- Greenaway, C.; Ratnaraj, N.; Sander, J.W.; Patsalos, P.N. Saliva and serum lacosamide concentrations in patients with epilepsy. Epilepsia 2011, 52, 258–263. [Google Scholar] [CrossRef]
- Wasterlain, C.G.; Stöhr, T.; Matagne, A. The acute and chronic effects of the novel anticonvulsant lacosamide in an experimental model of status epilepticus. Epilepsy Res. 2011, 94, 10–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, M.; Kaneko, S.; Hirano, T.; Mizuno, K.; Kondo, T.; Otani, K.; Fukushima, Y. Effects of zonisamide on dopaminergic system. Epilepsy Res. 1995, 22, 193–205. [Google Scholar] [CrossRef]
- Okada, M.; Hirano, T.; Kawata, Y.; Murakami, T.; Wada, K.; Mizuno, K.; Kondo, T.; Kaneko, S. Biphasic effects of zonisamide on serotonergic system in rat hippocampus. Epilepsy Res. 1999, 34, 187–197. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K.; Kawano, Y.; Shiroyama, T.; Ueda, Y. Memantine protects thalamocortical hyper-glutamatergic transmission induced by NMDA receptor antagonism via activation of system xc−. Pharm. Res. Perspect. 2019, 7, e00457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duerrschmidt, N.; Hagen, A.; Gaertner, C.; Wermke, A.; Nowicki, M.; Spanel-Borowski, K.; Stepan, H.; Mohr, F.W.; Dhein, S. Nicotine effects on human endothelial intercellular communication via alpha4beta2 and alpha3beta2 nicotinic acetylcholine receptor subtypes. Naunyn Schmiedebergs Arch. Pharm. 2012, 385, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Curtis, M.J.; Alexander, S.; Cirino, G.; Docherty, J.R.; George, C.H.; Giembycz, M.A.; Hoyer, D.; Insel, P.A.; Izzo, A.A.; Ji, Y.; et al. Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. Br. J. Pharm. 2018, 175, 987–993. [Google Scholar] [CrossRef] [Green Version]
- Harding, S.D.; Sharman, J.L.; Faccenda, E.; Southan, C.; Pawson, A.J.; Ireland, S.; Gray, A.J.G.; Bruce, L.; Alexander, S.P.H.; Anderton, S.; et al. The IUPHAR/BPS Guide to pharmacology in 2018: Updates and expansion to encompass the new guide to immunopharmacology. Nucleic. Acids Res. 2018, 46, D1091–D1106. [Google Scholar] [CrossRef] [Green Version]
- Alexander, S.P.H.; Christopoulos, A.; Davenport, A.P.; Kelly, E.; Mathie, A.; Peters, J.A.; Veale, E.L.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; et al. The concise guide to pharmacology 2019/20: G protein-coupled receptors. Br. J. Pharm. 2019, 176, S21–S141. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fukuyama, K.; Ueda, Y.; Okada, M. Effects of Carbamazepine, Lacosamide and Zonisamide on Gliotransmitter Release Associated with Activated Astroglial Hemichannels. Pharmaceuticals 2020, 13, 117. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13060117
Fukuyama K, Ueda Y, Okada M. Effects of Carbamazepine, Lacosamide and Zonisamide on Gliotransmitter Release Associated with Activated Astroglial Hemichannels. Pharmaceuticals. 2020; 13(6):117. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13060117
Chicago/Turabian StyleFukuyama, Kouji, Yuto Ueda, and Motohiro Okada. 2020. "Effects of Carbamazepine, Lacosamide and Zonisamide on Gliotransmitter Release Associated with Activated Astroglial Hemichannels" Pharmaceuticals 13, no. 6: 117. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13060117