Reprogramming of the Antibacterial Drug Vancomycin Results in Potent Antiviral Agents Devoid of Antibacterial Activity

 , ,
, ,
Abstract
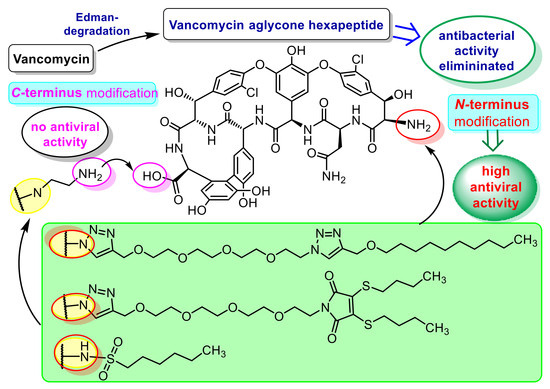
:
1. Introduction
2. Results and Discussion
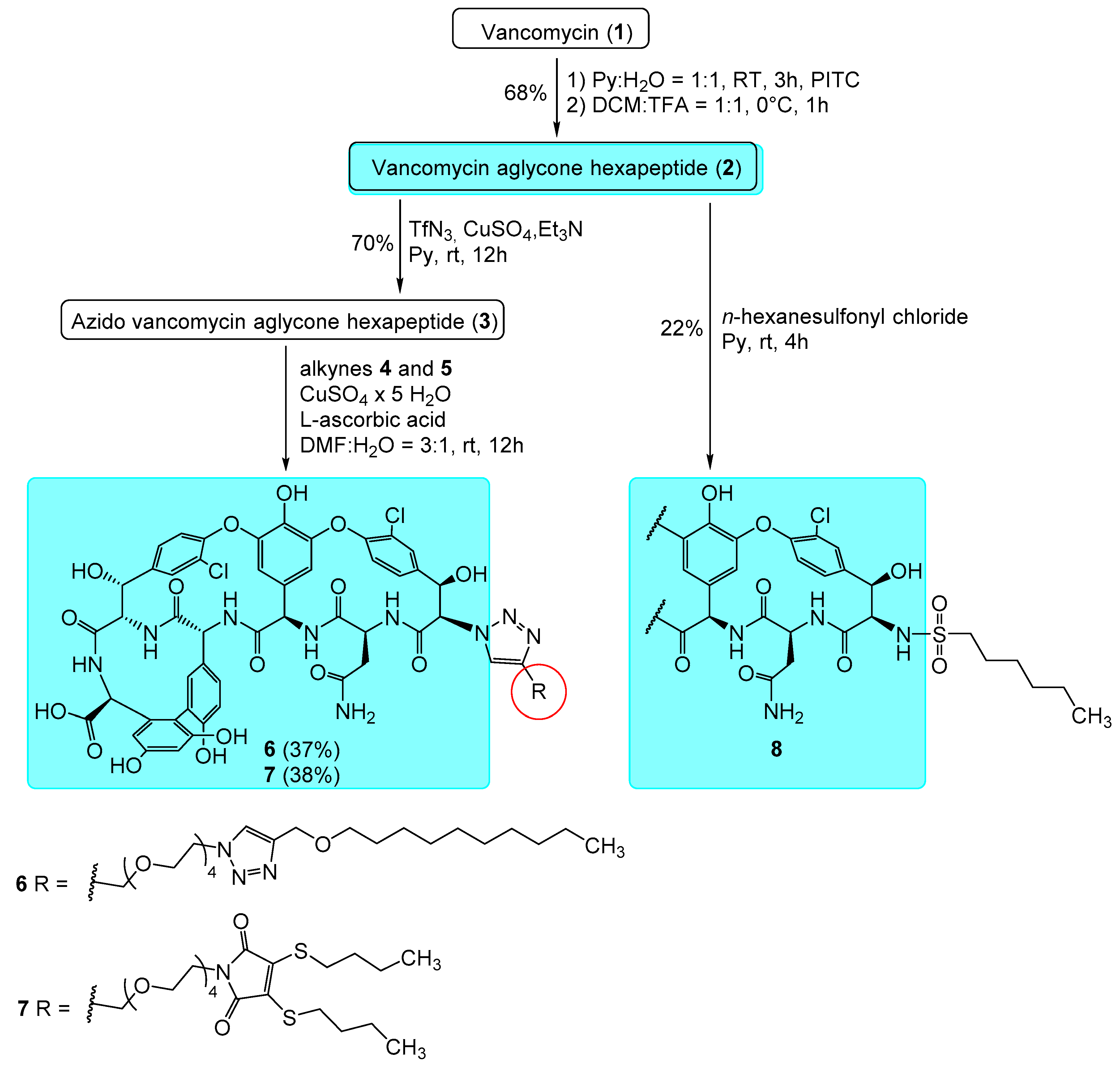
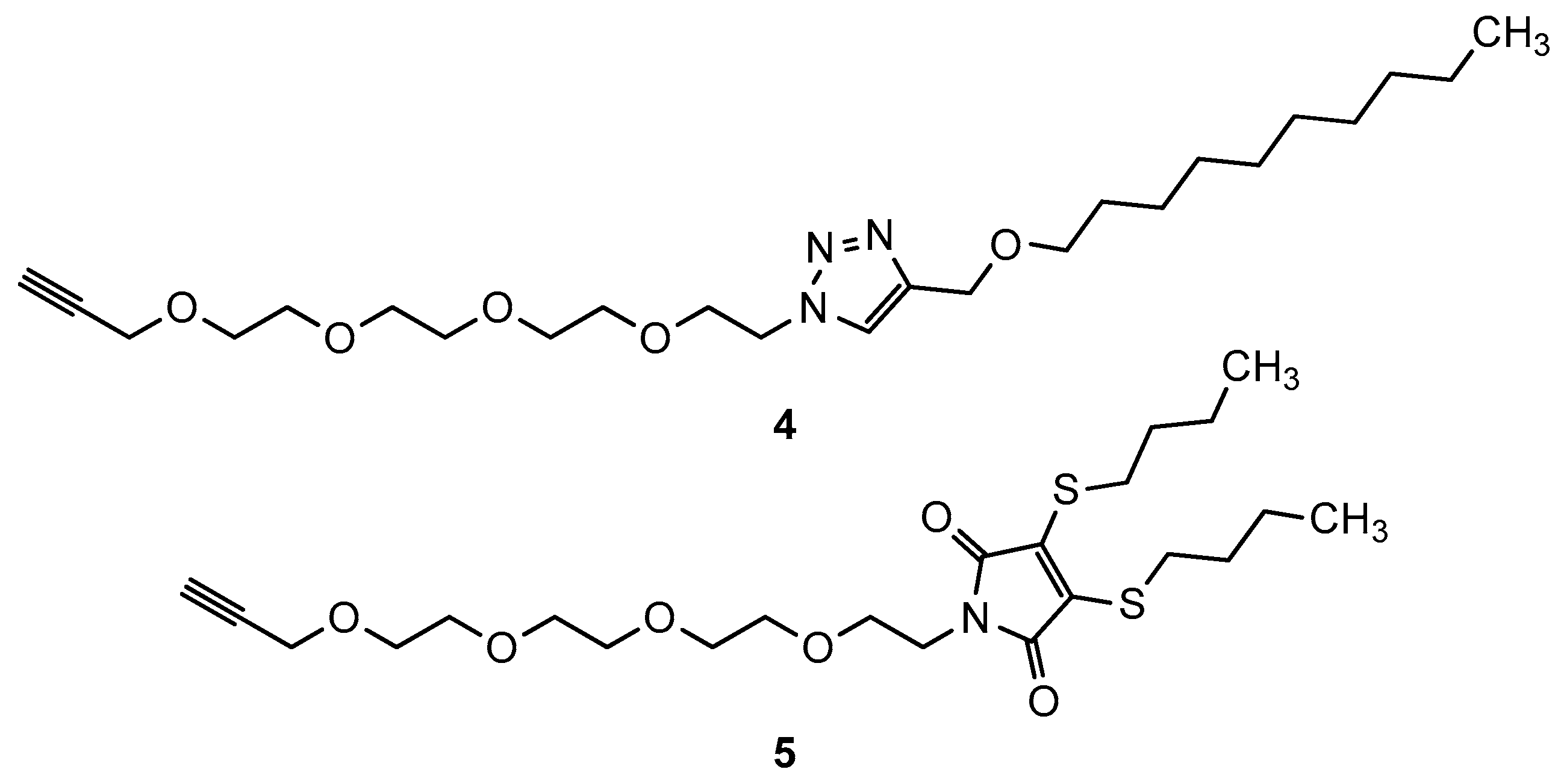
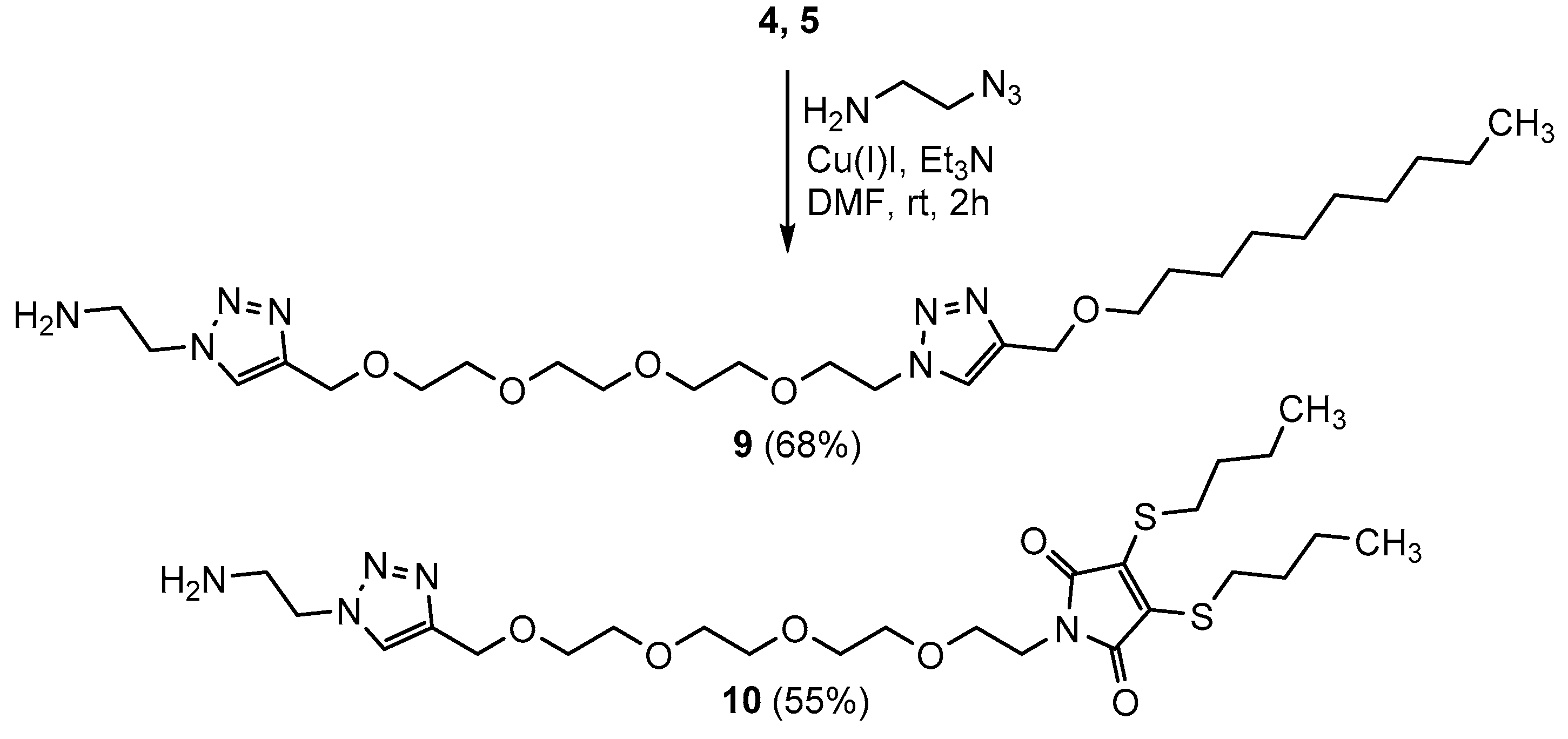
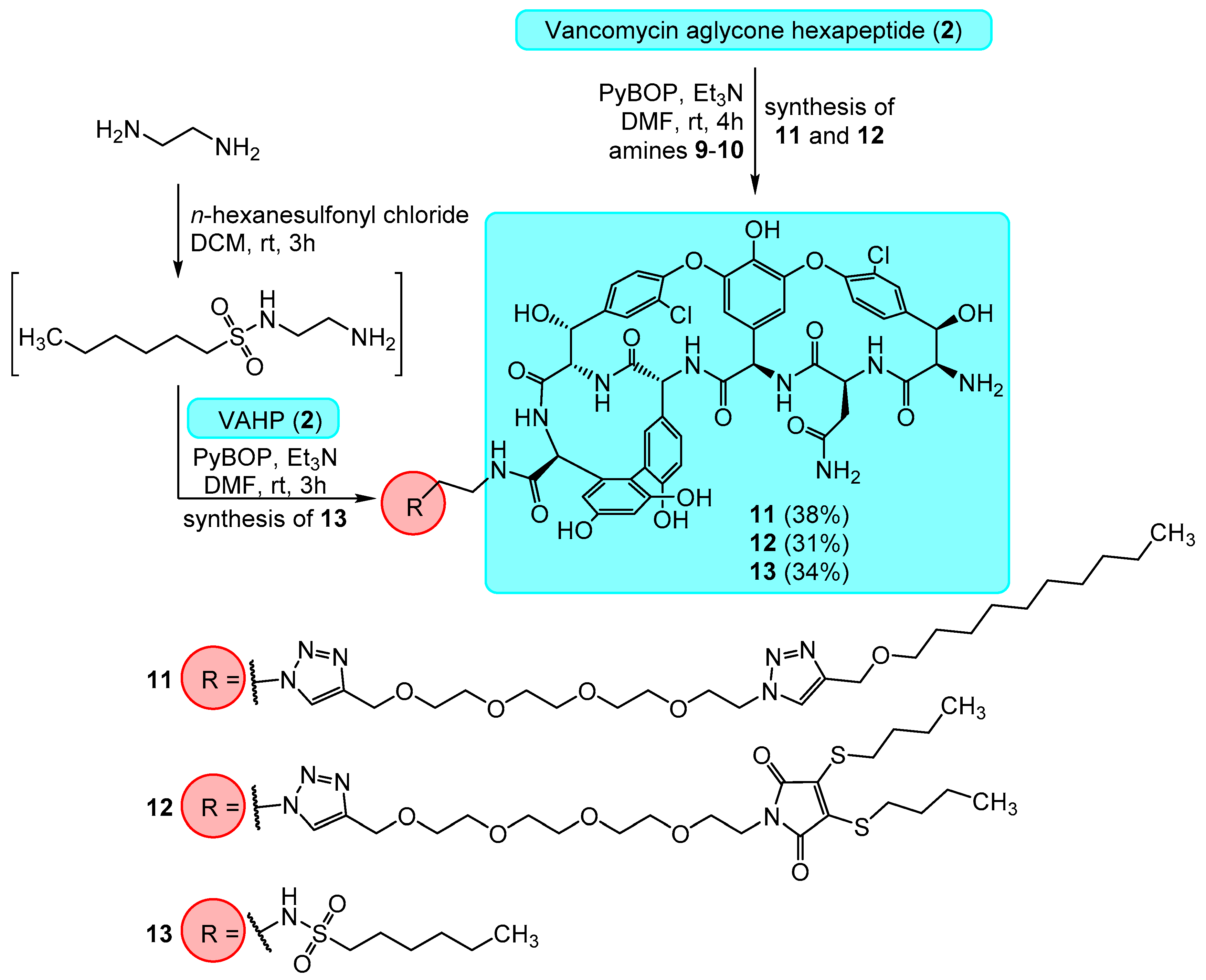
2.1. Chemistry
2.2. Biology
2.2.1. Antibacterial Evaluation
2.2.2. Antiviral Evaluation
3. Materials and Methods
3.1. Chemistry
Synthesis
3.2. Antiviral Procedures
3.2.1. Anti-Influenza Virus Activity
3.2.2. Other Antiviral Procedures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Coleman, B.L.; Fadel, S.A.; Fitzpatrick, T.; Thomas, S.M. Risk factors for serious outcomes associated with influenza illness in high- versus low- and middle-income countries: Systematic literature review and meta-analysis. Influenza Other Respir. Viruses 2018, 12, 22–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, M.; Fouchier, R.A.M. Influenza A virus transmission via respiratory aerosols or droplets as it relates to pandemic potential. FEMS Microbiol. Rev. 2016, 40, 68–85. [Google Scholar] [CrossRef] [PubMed]
- Von Itzstein, M. The war against influenza: Discovery and development of sialidase inhibitors. Nat. Rev. Drug Discov. 2007, 6, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Noshi, T.; Kitano, M.; Taniguchi, K.; Yamamoto, A.; Omoto, S.; Baba, K.; Hashimoto, T.; Ishida, K.; Kushima, Y.; Hattori, K.; et al. In vitro characterization of baloxavir acid, a first-in-class cap-dependent endonuclease inhibitor of the influenza virus polymerase PA subunit. Antivir. Res. 2018, 160, 109–117. [Google Scholar] [CrossRef]
- Furuta, Y.; Takahashi, K.; Shiraki, K.; Sakamoto, K.; Smee, D.F.; Barnard, D.L.; Gowen, B.B.; Julander, J.G.; Morrey, J.D. T-705 (favipiravir) and related compounds: Novel broad-spectrum inhibitors of RNA viral infections. Antivir. Res. 2009, 82, 95–102. [Google Scholar] [CrossRef]
- Dong, G.; Peng, C.; Luo, J.; Wang, C.; Han, L.; Wu, B.; Ji, G.; He, H. Adamantane-resistant influenza A viruses in the world (1902–2013): Frequency and distribution of M2 gene mutations. PLoS ONE 2015, 10, e0119115. [Google Scholar] [CrossRef] [Green Version]
- Samson, M.; Pizzorno, A.; Abed, Y.; Boivin, G. Influenza virus resistance to neuraminidase inhibitors. Antivir. Res. 2013, 98, 174–185. [Google Scholar] [CrossRef]
- Moscona, A. Global transmission of oseltamivir-resistant influenza. N. Engl. J. Med. 2009, 360, 953–956. [Google Scholar] [CrossRef] [Green Version]
- McKimm-Breschkin, J.L. Influenza neuraminidase inhibitors: Antiviral action and mechanisms of resistance. Influenza Other Respir. Viruses 2013, 7, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Vanderlinden, E.; Naesens, L. Emerging antiviral strategies to interfere with influenza virus entry. Med. Res. Rev. 2014, 34, 301–339. [Google Scholar] [CrossRef] [Green Version]
- Naesens, L.; Stevaert, A.; Vanderlinden, E. Antiviral therapies on the horizon for influenza. Curr. Opin. Pharmacol. 2016, 30, 106–115. [Google Scholar] [CrossRef] [Green Version]
- Yip, T.-F.; Selim, A.S.M.; Lian, I.; Lee, S.M.-Y. Advancements in host-based interventions for influenza treatment. Front. Immunol. 2018, 9, 1547. [Google Scholar] [CrossRef]
- Colson, P.; Raoult, D. Fighting viruses with antibiotics: An overlooked path. Int. J. Antimicrob. Agents 2016, 48, 349–352. [Google Scholar] [CrossRef]
- Pizzorno, A.; Padey, B.; Terrier, O.; Rosa-Calatrava, M. Drug repurposing approaches for the treatment of influenza viral infection: Reviving old drugs to fight against a long-lived enemy. Front. Immunol. 2019, 10, 531. [Google Scholar] [CrossRef]
- Wang, Y.; Cui, R.; Li, G.; Gao, Q.; Yuan, S.; Altmeyer, R.; Zou, G. Teicoplanin inhibits Ebola pseudovirus infection in cell culture. Antivir. Res. 2016, 125, 1–7. [Google Scholar] [CrossRef]
- Zhou, N.; Pan, T.; Zhang, J.; Li, Q.; Zhang, X.; Bai, C.; Huang, F.; Peng, T.; Zhang, J.; Liu, C.; et al. Glycopeptide antibiotics potently inhibit cathepsin L in the late endosome/lysosome and block the entry of Ebola virus, Middle East respiratory syndrome coronavirus (MERS-CoV), and severe acute respiratory syndrome coronavirus (SARS-CoV). J. Biol. Chem. 2016, 291, 9218–9232. [Google Scholar] [CrossRef] [Green Version]
- Balzarini, J.; Pannecouque, C.; De Clercq, E.; Pavlov, A.Y.; Printsevskaya, S.S.; Miroshnikova, O.V.; Reznikova, M.I.; Preobrazhenskaya, M.N. Antiretroviral activity of semisynthetic derivatives of glycopeptide antibiotics. J. Med. Chem. 2003, 46, 2755–2764. [Google Scholar] [CrossRef]
- Preobrazhenskaya, M.N.; Olsufyeva, E.N. Polycyclic peptide and glycopeptide antibiotics and their derivatives as inhibitors of HIV entry. Antivir. Res. 2006, 71, 227–236. [Google Scholar] [CrossRef]
- Balzarini, J.; Keyaerts, E.; Vijgen, L.; Egberink, H.; De Clercq, E.; Van Ranst, M.; Printsevskaya, S.S.; Olsufyeva, E.N.; Solovieva, S.E.; Preobrazhenskaya, M.N. Inhibition of feline (FIPV) and human (SARS) coronavirus by semisynthetic derivatives of glycopeptide antibiotics. Antivir. Res. 2006, 72, 20–33. [Google Scholar] [CrossRef]
- Obeid, S.; Printsevskaya, S.S.; Olsufyeva, E.N.; Dallmeier, K.; Durantel, D.; Zoulim, F.; Preobrazhenskaya, M.N.; Neyts, J.; Paeshuyse, J. Inhibition of hepatitis C virus replication by semi-synthetic derivatives of glycopeptide antibiotics. J. Antimicrob. Chemother. 2011, 66, 1287–1294. [Google Scholar] [CrossRef] [Green Version]
- De Burghgraeve, T.; Kaptein, S.J.; Ayala-Nunez, N.V.; Mondotte, J.A.; Pastorino, B.; Printsevskaya, S.S.; de Lamballerie, X.; Jacobs, M.; Preobrazhenskaya, M.; Gamarnik, A.V.; et al. An analogue of the antibiotic teicoplanin prevents Flavivirus entry in vitro. PLoS ONE 2012, 7, e37244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naesens, L.; Vanderlinden, E.; Rőth, E.; Jekő, J.; Andrei, G.; Snoeck, R.; Pannecouque, C.; Illyés, E.; Batta, G.; Herczegh, P.; et al. Anti-influenza virus activity and structure–activity relationship of aglycoristocetin derivatives with cyclobutenedione carrying hydrophobic chains. Antivir. Res. 2009, 82, 89–94. [Google Scholar] [CrossRef]
- Vanderlinden, E.; Vanstreels, E.; Boons, E.; ter Veer, W.; Huckriede, A.; Daelemans, D.; Van Lommel, A.; Roth, E.; Sztaricskai, F.; Herczegh, P.; et al. Intracytoplasmic trapping of influenza virus by a lipophilic derivative of aglycoristocetin. J. Virol. 2012, 86, 9416–9431. [Google Scholar] [CrossRef] [Green Version]
- Pintér, G.; Batta, G.; Kéki, S.; Mándi, A.; Komáromi, I.; Takács-Novák, K.; Sztaricskai, F.; Rőth, E.; Ostorházi, E.; Rozgonyi, F.; et al. Diazo transfer-click reaction route to new, lipophilic teicoplanin and ristocetin aglycon derivatives with high antibacterial and anti-influenza virus activity: An aggregation and receptor binding study. J. Med. Chem. 2009, 52, 6053–6061. [Google Scholar] [CrossRef]
- Sipos, A.; Máté, G.; Rőth, E.; Borbás, A.; Batta, G.; Bereczki, I.; Kéki, S.; Jóna, I.; Ostorházi, E.; Rozgonyi, F.; et al. Synthesis of fluorescent ristocetin aglycone derivatives with remarkable antibacterial and antiviral activities. Eur. J. Med. Chem. 2012, 58, 361–367. [Google Scholar] [CrossRef]
- Sipos, A.; Török, Z.; Rőth, E.; Kiss-Szikszai, A.; Batta, G.; Bereczki, I.; Fejes, Z.; Borbás, A.; Ostorházi, E.; Rozgonyi, F.; et al. Synthesis of isoindole and benzoisoindole derivatives of teicoplanin pseudoaglycone with remarkable antibacterial and antiviral activities. Bioorg. Med. Chem. Lett. 2012, 22, 7092–7096. [Google Scholar] [CrossRef]
- Bereczki, I.; Mándi, A.; Rőth, E.; Borbás, A.; Fizil, Á.; Komáromi, I.; Sipos, A.; Kurtán, T.; Batta, G.; Ostorházi, E.; et al. A few atoms make the difference: Synthetic, CD, NMR and computational studies on antiviral and antibacterial activities of glycopeptide antibiotic aglycone derivatives. Eur. J. Med. Chem. 2015, 94, 73–86. [Google Scholar] [CrossRef] [Green Version]
- Bereczki, I.; Kicsák, M.; Dobray, L.; Borbás, A.; Batta, G.; Kéki, S.; Nikodém, É.N.; Ostorházi, E.; Rozgonyi, F.; Vanderlinden, E.; et al. Semisynthetic teicoplanin derivatives as new influenza virus binding inhibitors: Synthesis and antiviral studies. Bioorg. Med. Chem. Lett. 2014, 24, 3251–3254. [Google Scholar] [CrossRef]
- Szűcs, Z.; Csávás, M.; Rőth, E.; Borbás, A.; Batta, G.; Perret, F.; Ostorházi, E.; Szatmári, R.; Vanderlinden, E.; Naesens, L.; et al. Synthesis and biological evaluation of lipophilic teicoplanin pseudoaglycone derivatives containing a substituted triazole function. J. Antibiot. 2017, 70, 152–157. [Google Scholar] [CrossRef]
- Szűcs, Z.; Kelemen, V.; Thai, S.L.; Csávás, M.; Rőth, E.; Batta, G.; Stevaert, A.; Vanderlinden, E.; Naesens, L.; Herczegh, P.; et al. Structure-activity relationship studies of lipophilic teicoplanin pseudoaglycon derivatives as new anti-influenza virus agents. Eur. J. Med. Chem. 2018, 157, 1017–1030. [Google Scholar] [CrossRef]
- Printsevskaya, S.S.; Solovieva, S.E.; Olsufyeva, E.N.; Mirchink, E.P.; Isakova, E.B.; De Clercq, E.; Balzarini, J.; Preobrazhenskaya, M.N. Structure-activity relationship studies of a series of antiviral and antibacterial aglycon derivatives of the glycopeptide antibiotics vancomycin, eremomycin, and dechloroeremomycin. J. Med. Chem. 2005, 48, 3885–3890. [Google Scholar] [CrossRef]
- Malabarba, A.; Ciabatti, R.; Kettenring, J.; Ferrari, P.; Vékey, K.; Bellasio, E.; Denaro, M. Structural modifications of the active site in teicoplanin and related glycopeptides. 1. Reductive hydrolysis of the 1,2- and 2,3-peptide bonds. J. Org. Chem. 1996, 61, 2137–2150. [Google Scholar] [CrossRef]
- Cavalleri, B.; Ferrari, P.; Malabarba, A.; Magni, A.; Pallanza, R.; Gallo, G.G. Teicoplanin, antibiotics from Actinoplanes teichomyceticus nov. sp. VIII. Opening of the polypeptide chain of teicoplanin aglycone under hydrolytic conditions. J. Antibiot. 1987, 40, 49–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malabarba, A.; Ciabatti, R.; Maggini, M.; Ferrari, P.; Colombo, L.; Denaro, M. Structural modifications of the active site in teicoplanin and related glycopeptides. 2. Deglucoteicoplanin-derived tetrapeptide. J. Org. Chem. 1996, 61, 2151–2157. [Google Scholar] [CrossRef]
- Booth, P.M.; Stone, D.J.M.; Williams, D.H. The Edman degradation of vancomycin - Preparation of vancomycin hexapeptide. J. Chem. Soc Chem Commun. 1987, 1987, 1694–1695. [Google Scholar] [CrossRef]
- Crane, C.M.; Boger, D.L. Synthesis and evaluation of vancomycin aglycon analogues that bear modifications in the N-terminal d-leucyl amino acid. J. Med. Chem. 2009, 52, 1471–1476. [Google Scholar] [CrossRef] [Green Version]
- Agalave, S.G.; Maujan, S.R.; Pore, V.S. Click chemistry: 1,2,3-triazoles as pharmacophores. Chem. Asian J. 2011, 6, 2696–2718. [Google Scholar] [CrossRef]
- Cihan-Üstündag, G.; Zopun, M.; Vanderlinden, E.; Ozkirimli, E.; Persoons, L.; Capan, G.; Naesens, L. Superior inhibition of influenza virus hemagglutinin-mediated fusion by indole-substituted spirothiazolidinones. Bioorg. Med. Chem. 2020, 28, 115130. [Google Scholar] [CrossRef]
- Vrijens, P.; Noppen, S.; Boogaerts, T.; Vanstreels, E.; Ronca, R.; Chiodelli, P.; Laporte, M.; Vanderlinden, E.; Liekens, S.; Stevaert, A.; et al. Influenza virus entry via the GM3 ganglioside-mediated platelet-derived growth factor receptor beta signalling pathway. J. Gen. Virol. 2019, 100, 583–601. [Google Scholar] [CrossRef]
- Stevaert, A.; Dallocchio, R.; Dessi, A.; Pala, N.; Rogolino, D.; Sechi, M.; Naesens, L. Mutational analysis of the binding pockets of the diketo acid inhibitor L-742,001 in the influenza virus PA endonuclease. J. Virol. 2013, 87, 10524–10538. [Google Scholar] [CrossRef] [Green Version]
- Apaydin, Ç.B.; Cesur, N.; Stevaert, A.; Naesens, L.; Cesur, Z. Synthesis and anti-coronavirus activity of a series of 1-thia-4-azaspiro [4.5]decan-3-one derivatives. Arch. Pharm. 2019, 352, e1800330. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacteria | In Vitro MIC in μg/mL | |||||||
|---|---|---|---|---|---|---|---|---|
| TEI | VAN | 6 | 7 | 8 | 11 | 12 | 13 | |
| B. subtilis ATCC 6633 | 0.5 | 0.5 | 32 | 32 | 32 | 32 | 256 | 256 |
| S. aureus MSSA ATCC 29213 | 0.5 | 0.5 | 128 | 256 | 256 | 256 | 256 | 256 |
| S. aureus MRSA ATCC 33591 | 0.5 | 0.5 | 128 | 256 | 256 | 256 | 256 | 256 |
| S. epidermidis ATCC 35984 | 4 | 2 | 32 | 32 | 32 | 256 | 128 | 128 |
| S. epidermidis mecA | 16 | 4 | 32 | 32 | 64 | 256 | 256 | 128 |
| E. faecalis ATCC 29212 | 1 | 1 | 32 | 32 | 32 | 128 | 128 | 64 |
| E. faecalis 15,376 VanA | 256 | 256 | 128 | 256 | 256 | 256 | 256 | 256 |
| E. faecalis ATCC 51,299 VanB | 0.5 | 128 | 128 | 256 | 128 | 256 | 256 | 128 |
| Compound | CC50 2 (µM) | Antiviral EC50 3 (μM) | ||
|---|---|---|---|---|
| Influenza A/H1N1 | Influenza A/H3N2 | Influenza B | ||
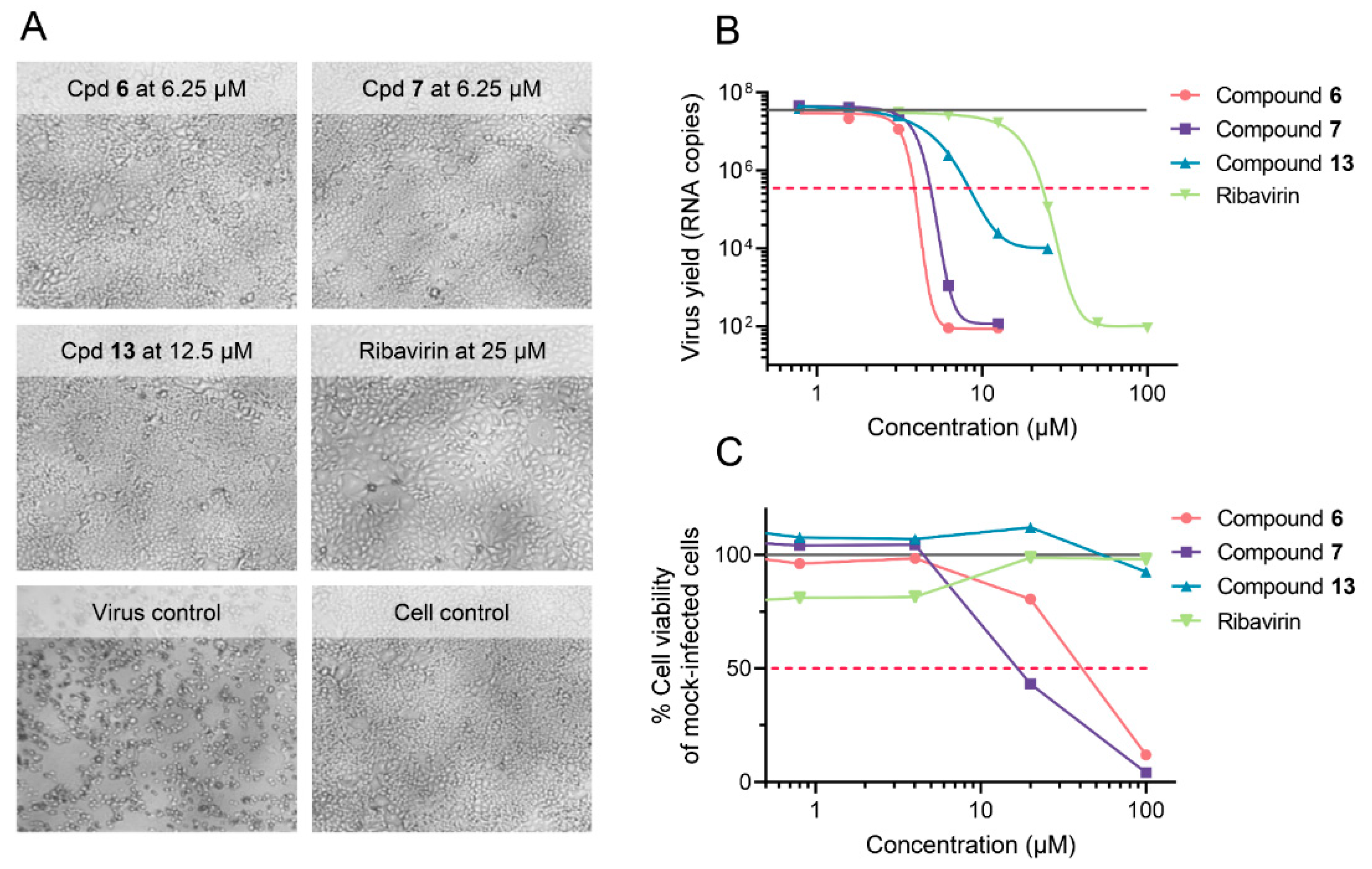
| 6 | 41 | 4.1 | 1.4 | 3.2 |
| 7 | 18 | 3.6 | 2.0 | 3.2 |
| 8 | 100 | >100 | >100 | >100 |
| 11 | ≥20 | >100 | >100 | >100 |
| 12 | 100 | 12 | >100 | >100 |
| 13 | >100 | 34 | 14 | >100 |
| Ribavirin | >100 | 7.0 | 6.4 | 7.2 |
| Zanamivir | >100 | 0.4 | 9.0 | 4.5 |
| Compound | CC50 2 (µM) | Antiviral EC50 3 (µM) - cell line 4 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| HEL | HeLa | Vero | HEL | HeLa | Vero | ||||||
| HSV-1 | HSV-2 | HSV-1/TK- | Vaccinia Virus | Human Coronavirus 229E | RSV | Yellow Fever Virus | Zika Virus | ||||
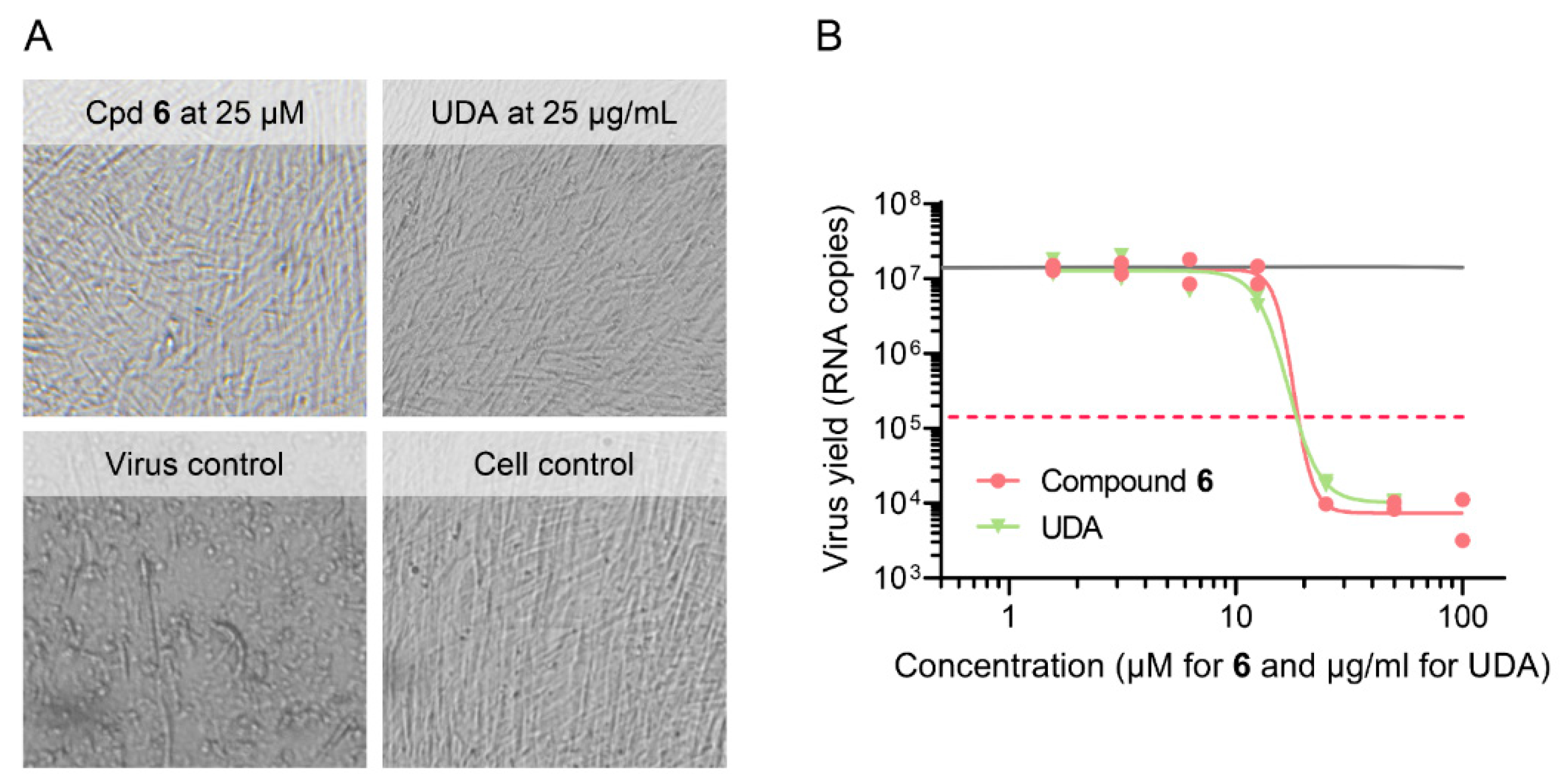
| 6 | >100 | >100 | >100 | 20 | 7.8 | 7.4 | 7.2 | 11 | 7.7 | >100 | >100 |
| 7 | >100 | >100 | >100 | 43 | 6.5 | 11 | 32 | 32 | 60 | 55 | 14 |
| Cidofovir | >250 | >250 | >250 | 2.4 | 1.0 | 5.8 | 37 | - | - | - | - |
| Aciclovir | >250 | >250 | >250 | 2.4 | 0.05 | 146 | >250 | - | - | - | - |
| Ganciclovir | >100 | >100 | >100 | 0.1 | 0.03 | 8.9 | >100 | - | - | - | - |
| UDA 5 | >100 | >100 | >100 | - | - | - | - | 1.8 | - | - | - |
| Ribavirin | >250 | >250 | >250 | - | - | - | - | - | 5.0 | 119 | - |
| Mycophenolic acid | >100 | >100 | >100 | - | - | - | - | - | - | 0.7 | 0.8 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szűcs, Z.; Naesens, L.; Stevaert, A.; Ostorházi, E.; Batta, G.; Herczegh, P.; Borbás, A. Reprogramming of the Antibacterial Drug Vancomycin Results in Potent Antiviral Agents Devoid of Antibacterial Activity. Pharmaceuticals 2020, 13, 139. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13070139
Szűcs Z, Naesens L, Stevaert A, Ostorházi E, Batta G, Herczegh P, Borbás A. Reprogramming of the Antibacterial Drug Vancomycin Results in Potent Antiviral Agents Devoid of Antibacterial Activity. Pharmaceuticals. 2020; 13(7):139. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13070139
Chicago/Turabian StyleSzűcs, Zsolt, Lieve Naesens, Annelies Stevaert, Eszter Ostorházi, Gyula Batta, Pál Herczegh, and Anikó Borbás. 2020. "Reprogramming of the Antibacterial Drug Vancomycin Results in Potent Antiviral Agents Devoid of Antibacterial Activity" Pharmaceuticals 13, no. 7: 139. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13070139