Autophagy as a Potential Therapy for Malignant Glioma

 , , ,
, , ,
Abstract
:
1. Introduction
2. Pathways and Molecular Mechanisms of Autophagy
2.1. The Autophagic Process
2.1.1. Induction
2.1.2. Nucleation
2.1.3. Elongation and Completion
2.1.4. Fusion with Lysosomes and Degradation
2.2. AMPK and mTOR in Autophagy Regulation
2.3. Regulation of Autophagy at a Nuclear Level by Transcription Factors
2.3.1. P53
2.3.2. TFEB
2.3.3. FoxO
2.3.4. HIF
2.3.5. PTEN
2.3.6. E2F1 and NF-κB
3. Autophagy in Glioma
3.1. Autophagy as a Tumor Suppressor in Glioma
3.2. Autophagy as a Tumoral Promotor in Glioma
4. Treatment Options for Glioblastoma
4.1. The Standard Care: Temozolomide
4.2. Arsenic Trioxide
4.3. Chloroquine
4.4. Metformin
4.5. Small-Molecule Inhibitors
4.5.1. Erlotinib
4.5.2. Gefitinib
4.5.3. Dacomitinib
4.5.4. Imatinib
4.5.5. Sunitinib
4.5.6. Sorafenib
4.6. Targeting Downstream Intracellular Effector Molecules
4.6.1. The RAS/RAF/MAPK Pathway
4.6.2. Lonafarnib
4.6.3. Vemurafenib
4.7. Inhibition of the PI3K/AKT/mTOR Pathway
4.7.1. Temsirolimus
4.7.2. Everolimus
4.8. Novel and Promising Strategies in Pre-Clinical Stages
4.8.1. MicroRNAs
4.8.2. BH3 Mimetics
4.8.3. Cannabinoids
4.8.4. Histone Deacetylase Inhibitors
4.8.5. Proteasome Inhibitors
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 3-MA | 3-methyladenine |
| AMPK | 5′-AMP-activated protein kinase |
| ATF4 | Activating transcription factor 4 |
| STAT3 | Activator of transcription-3 |
| AMBRA-1 | Activating Molecule in Beclin 1-Regulated Autophagy |
| AP-1 | Activator protein 1 |
| AMP | Adenosine monophosphate |
| ATP | Adenosine triphosphate |
| AMPK | AMP-activated kinase |
| AA | Anaplastic astrocytoma |
| ARP | Autophagosome recognition particle |
| ATG | Autophagy-related genes |
| Bif-1 | Bax-interacting factor 1 |
| Bcl-2 | B-cell lymphoma 2 |
| Bcl-xL | B-cell lymphoma-extra-large |
| BCL2L11 | Bcl-2-like protein 11 |
| BNIP3 | BH3-type proteins in the Bcl-2 family |
| CHOP | Binding Protein Homology Protein |
| CaMKKβ | Ca2+/calmodulin-dependent kinase kinase |
| Cav-1 | Caveolin-1 |
| CNS | Central nervous system |
| CMA | Chaperone-mediated autophagy |
| JNK | c-Jun N-terminal kinase |
| CSF1R | Colony-stimulating factor-1 |
| C-VPS | C vacuolar protein |
| cyt c | Cytochrome c |
| DAPK2 | Death-associated protein kinase 2 |
| DRAM | DNA damage-regulated autophagy modulator |
| DHA | Docosahexaenoic acid |
| ESCRT-III | Endosomal sorting complexes required for transport III |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial–mesenchymal transition |
| ERK | Extracellular-signal-regulated kinase |
| FTIs | Farnesyltransferase inhibitors |
| FADD | Fas-associated death domain |
| FKBP-12 | FK-binding protein-12 |
| FLT3 | Fms-like tyrosine kinase-3 |
| FAK | Focal adhesion kinase |
| FDA | Food and Drug Administration |
| FOXO | Forkhead box class O |
| GBM | Glioblastoma multiforme |
| GSC | Glioma Stem Cells |
| GSK3β | Glycogen Synthase Kinase 3 Beta |
| GEF | Guanine nucleotide exchange factor |
| GBP | Guanylate binding proteins-3 |
| GTP | Guanylyl triphosphate |
| HSP | Heat shock protein |
| BH | Homologue domains |
| HIF-1 | Hypoxia-inducible factor 1-alpha |
| HIV | Human Immunodeficiency Virus |
| IRGM | Immunity-related GTPase M |
| IAP | Inhibitor of apoptosis |
| KEAP1 | Kelch-like ECH-associated protein 1 |
| LIR | LC3-interacting regions |
| LKB1 | Liver kinase B1 |
| LAMP2A | Lysosomal-associate membrane protein 2A receptor |
| MMP | Matrix metallopeptidases |
| MAGE | Melanoma antigen |
| MGMT | Methylguanine-O6-methyltransferase |
| LC3-PE | Microtubule-associated protein 1A/1B-light chain 3 |
| MAPK | Mitogen-activated protein kinase |
| mTOR | Mammalian target of rapamycin |
| MCT4 | Monocarboxylate transporter-4 |
| Mcl-1 | Myeloid cell leukemia 1 |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| ω3-PUFAs | Omega-3-polyunsaturated fatty acids |
| OS | Overall survival |
| PXN | Paxillin |
| PAS | Phagophore assembly site |
| NOXA | Phorbol-12-myristate-13-acetate-induced protein 1 |
| PI3P | Phosphatidylinositol 3-phosphate |
| PE | Phosphatidylethanolamine |
| PI3K | Phosphoinositide 3-kinase |
| PDGFR | Platelet-derived growth factor receptor |
| PHD2 | Prolyl hydroxylase domain protein 2 |
| PFS | Progression-free survival |
| PERK | Protein endoplasmic reticulum kinase |
| PUMA | p53 upregulated modulator of apoptosis |
| RAC1 | RAS-related C3 botulinum toxin substrate 1 |
| ROS | Reactive oxygen species |
| RB | Retinoblastoma |
| SMAC | Second Mitochondria-derived Activator of Caspases |
| SQSTM1 | Sequestosome 1 |
| AKT | Serine/threonine kinase |
| SE | Sinomenine hydrochloride |
| SNARE | Soluble NSF Attachment Protein |
| Kit | Stem cell-factor |
| Stx17 | Syntaxin 17 |
| TORC1 | Target of rapamycin complex 1 |
| TMZ | Temozolomide |
| TKI | Tyrosine-kinase inhibitors |
| TFEB | Transcription factor EB |
| TAK1 | Transforming growth factor-β-activating kinase 1 |
| TRIM16 | Tripartite motif 16 |
| PTEN | Tensin homologue on chromosome ten |
| TNF | Tumor Necrosis Factor receptors |
| TRAIL | Tumor necrosis factor-related apoptosis-inducing ligand |
| ULK1/ULK2 | Unc-51-Like Kinase 1 and 2 |
| UVRAG | UV irradiation resistance-associated gene |
| Vps34 | Vacuolar protein sorting 34 |
| VEGFR | Vascular endothelial growth factor receptor |
| VHL | Von Hippel Lindau protein |
| XIAP | X-linked inhibitor of apoptosis protein |
References
- Deangelis, L.M. Brain Tumors. N. Engl. J. Med. 2001, 344, 114–123. [Google Scholar] [CrossRef] [Green Version]
- Kleihues, P.; Ohgaki, H. Primary And Secondary Glioblastomas: From Concept To Clinical Diagnosis. Neuro Oncol. 1999, 1, 44–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budke, M.; Isla-Guerrero, A.; Perez-Lopez, C.; Perez-Alvarez, M.; Garcia-Grande, A.; Bello, M.J.; Rey, J. A Comparative Study Of The Treatment Of High Grade Gliomas. Rev. Neurolog. 2003, 37, 912–916. [Google Scholar] [CrossRef]
- Biernat, W.; Tohma, Y.; Yonekawa, Y.; Kleihues, P.; Ohgaki, H. Alterations Of Cell Cycle Regulatory Genes In Primary (De Novo) And Secondary Glioblastomas. Acta Neuropathol. 1997, 94, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Gramatzki, D.; Dehler, S.; Rushing, E.J.; Zaugg, K.; Hofer, S.; Yonekawa, Y.; Bertalanffy, H.; Valavanis, A.; Korol, D.; Rohrmann, S.; et al. Glioblastoma In The Canton Of Zurich, Switzerland Revisited: 2005 To 2009. Cancer 2016, 122, 2206–2215. [Google Scholar] [CrossRef] [PubMed]
- Voldborg, B.R.; Damstrup, L.; Spang-Thomsen, M.; Poulsen, H.S. Epidermal Growth Factor Receptor (Egfr) And Egfr Mutations, Function And Possible Role In Clinical Trials. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 1997, 8, 1197–1206. [Google Scholar] [CrossRef]
- Katz, A.M.; Amankulor, N.M.; Pitter, K.; Helmy, K.; Squatrito, M.; Holland, E.C. Astrocyte-Specific Expression Patterns Associated With The Pdgf-Induced Glioma Microenvironment. PLoS ONE 2012, 7, E32453. [Google Scholar] [CrossRef] [Green Version]
- Sjostrom, S.; Wibom, C.; Andersson, U.; Brannstrom, T.; Broholm, H.; Johansen, C.; Collatz-Laier, H.; Liu, Y.; Bondy, M.; Henriksson, R.; et al. Genetic Variations In Vegf And Vegfr2 And Glioblastoma Outcome. J. Neuro Oncol. 2011, 104, 523–527. [Google Scholar] [CrossRef] [Green Version]
- Sathornsumetee, S.; Cao, Y.; Marcello, J.E.; Herndon, J.E., 2nd; Mclendon, R.E.; Desjardins, A.; Friedman, H.S.; Dewhirst, M.W.; Vredenburgh, J.J.; Rich, J.N. Tumor Angiogenic And Hypoxic Profiles Predict Radiographic Response And Survival In Malignant Astrocytoma Patients Treated With Bevacizumab And Irinotecan. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 271–278. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive Genomic Characterization Defines Human Glioblastoma Genes And Core Pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Soomro, S.H.; Ting, L.R.; Qing, Y.Y.; Ren, M. Molecular Biology Of Glioblastoma: Classification And Mutational Locations. JPMA J. Pak. Med. Assoc. 2017, 67, 1410–1414. [Google Scholar] [PubMed]
- Fueyo, J.; Gomez-Manzano, C.; Yung, W.K.; Kyritsis, A.P. The Functional Role Of Tumor Suppressor Genes In Gliomas: Clues For Future Therapeutic Strategies. Neurology 1998, 51, 1250–1255. [Google Scholar] [CrossRef] [PubMed]
- Besson, A.; Yong, V.W. Mitogenic Signaling And The Relationship To Cell Cycle Regulation In Astrocytomas. J. Neuro Oncol. 2001, 51, 245–264. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Yang, F.; Fujisawa, H.; Yonekawa, Y.; Kleihues, P.; Ohgaki, H. Loss Of Heterozygosity On Chromosome 19 In Secondary Glioblastomas. J. Neuropathol. Exp. Neurol. 2000, 59, 539–543. [Google Scholar] [CrossRef] [Green Version]
- Trejo-Solis, C.; Serrano-Garcia, N.; Escamilla-Ramirez, A.; Castillo-Rodriguez, R.A.; Jimenez-Farfan, D.; Palencia, G.; Calvillo, M.; Alvarez-Lemus, M.A.; Flores-Najera, A.; Cruz-Salgado, A.; et al. Autophagic And Apoptotic Pathways As Targets For Chemotherapy In Glioblastoma. Int. J. Mol. Sci. 2018, 19, 3773. [Google Scholar] [CrossRef] [Green Version]
- Wick, W.; Wagner, S.; Kerkau, S.; Dichgans, J.; Tonn, J.C.; Weller, M. Bcl-2 Promotes Migration And Invasiveness Of Human Glioma Cells. Febs Lett. 1998, 440, 419–424. [Google Scholar] [CrossRef] [Green Version]
- Steinbach, J.P.; Weller, M. Apoptosis In Gliomas: Molecular Mechanisms And Therapeutic Implications. J. Neuro Oncol. 2004, 70, 247–256. [Google Scholar] [CrossRef]
- Wang, H.B.; Li, T.; Ma, D.Z.; Ji, Y.X.; Zhi, H. Overexpression Of Fadd And Caspase-8 Inhibits Proliferation And Promotes Apoptosis Of Human Glioblastoma Cells. Biomed. Pharmacother. Biomed. Pharmacother. 2017, 93, 1–7. [Google Scholar] [CrossRef]
- Blahovcova, E.; Richterova, R.; Kolarovszki, B.; Dobrota, D.; Racay, P.; Hatok, J. Apoptosis-Related Gene Expression In Tumor Tissue Samples Obtained From Patients Diagnosed With Glioblastoma Multiforme. Int. J. Mol. Med. 2015, 36, 1677–1684. [Google Scholar] [CrossRef] [Green Version]
- Wagenknecht, B.; Glaser, T.; Naumann, U.; Kugler, S.; Isenmann, S.; Bahr, M.; Korneluk, R.; Liston, P.; Weller, M. Expression And Biological Activity Of X-Linked Inhibitor Of Apoptosis (Xiap) In Human Malignant Glioma. Cell Death Differ. 1999, 6, 370–376. [Google Scholar] [CrossRef] [Green Version]
- Trejo-Solis, C.; Jimenez-Farfan, D.; Rodriguez-Enriquez, S.; Fernandez-Valverde, F.; Cruz-Salgado, A.; Ruiz-Azuara, L.; Sotelo, J. Copper Compound Induces Autophagy And Apoptosis Of Glioma Cells By Reactive Oxygen Species And Jnk Activation. BMC Cancer 2012, 12, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, M.; Bajpai, V.K.; Sahasrabuddhe, A.A.; Kumar, A.; Sinha, R.A.; Behari, S.; Godbole, M.M. Inhibition Of N-(4-Hydroxyphenyl)Retinamide-Induced Autophagy At A Lower Dose Enhances Cell Death In Malignant Glioma Cells. Carcinogenesis 2008, 29, 600–609. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Vicencio, J.M.; Kepp, O.; Tasdemir, E.; Maiuri, M.C.; Kroemer, G. To Die Or Not To Die: That Is The Autophagic Question. Curr. Mol. Med. 2008, 8, 78–91. [Google Scholar] [CrossRef]
- Thayyullathil, F.; Rahman, A.; Pallichankandy, S.; Patel, M.; Galadari, S. Ros-Dependent Prostate Apoptosis Response-4 (Par-4) Up-Regulation And Ceramide Generation Are The Prime Signaling Events Associated With Curcumin-Induced Autophagic Cell Death In Human Malignant Glioma. Febs Open Bio 2014, 4, 763–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keshmiri-Neghab, H.; Goliaei, B.; Nikoofar, A. Gossypol Enhances Radiation Induced Autophagy In Glioblastoma Multiforme. Gen. Physiol. Biophys. 2014, 33, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Debnath, J. Autophagy At The Crossroads Of Catabolism And Anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy In Yeast Demonstrated With Proteinase-Deficient Mutants And Conditions For Its Induction. J. Cell Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Kamada, Y.; Funakoshi, T.; Shintani, T.; Nagano, K.; Ohsumi, M.; Ohsumi, Y. Tor-Mediated Induction Of Autophagy Via An Apg1 Protein Kinase Complex. J. Cell Biol. 2000, 150, 1507–1513. [Google Scholar] [CrossRef] [Green Version]
- Ragusa, M.J.; Stanley, R.E.; Hurley, J.H. Architecture Of The Atg17 Complex As A Scaffold For Autophagosome Biogenesis. Cell 2012, 151, 1501–1512. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.B.; Kim, S.; Lee, J.; Park, J.; Lee, G.; Kim, Y.; Kim, J.M.; Chung, J. Atg1, An Autophagy Regulator, Inhibits Cell Growth By Negatively Regulating S6 Kinase. Embo Rep. 2007, 8, 360–365. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. Lc3, A Mammalian Homologue Of Yeast Apg8p, Is Localized In Autophagosome Membranes After Processing. Embo J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Cheong, H.; Klionsky, D.J. Dual Role Of Atg1 In Regulation Of Autophagy-Specific Pas Assembly In Saccharomyces Cerevisiae. Autophagy 2008, 4, 724–726. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Kubota, Y.; Sekito, T.; Ohsumi, Y. Hierarchy Of Atg Proteins In Pre-Autophagosomal Structure Organization. Genes Cells Devot. Mol. Cell. Mech. 2007, 12, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Longatti, A.; Tooze, S.A. Vesicular Trafficking And Autophagosome Formation. Cell Death Differ. 2009, 16, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Budovskaya, Y.V.; Stephan, J.S.; Deminoff, S.J.; Herman, P.K. An Evolutionary Proteomics Approach Identifies Substrates Of The Camp-Dependent Protein Kinase. Proc. Natl. Acad. Sci. USA 2005, 102, 13933–13938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Codogno, P.; Mehrpour, M.; Proikas-Cezanne, T. Canonical And Non-Canonical Autophagy: Variations On A Common Theme Of Self-Eating? Nat. Rev. Mol. Cell Biol. 2011, 13, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.; Subramanian, V.; Acharya, K.R. C9orf72, A Protein Associated With Amyotrophic Lateral Sclerosis (Als) Is A Guanine Nucleotide Exchange Factor. PEERJ 2018, 6, E5815. [Google Scholar] [CrossRef] [Green Version]
- Webster, C.P.; Smith, E.F.; Grierson, A.J.; De Vos, K.J. C9orf72 Plays A Central Role In Rab Gtpase-Dependent Regulation Of Autophagy. Small Gtpases 2018, 9, 399–408. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, H.; Kakuta, S.; Watanabe, T.M.; Kitamura, A.; Sekito, T.; Kondo-Kakuta, C.; Ichikawa, R.; Kinjo, M.; Ohsumi, Y. Atg9 Vesicles Are An Important Membrane Source During Early Steps Of Autophagosome Formation. J. Cell Biol. 2012, 198, 219–233. [Google Scholar] [CrossRef] [Green Version]
- Kihara, A.; Noda, T.; Ishihara, N.; Ohsumi, Y. Two Distinct Vps34 Phosphatidylinositol 3-Kinase Complexes Function In Autophagy And Carboxypeptidase Y Sorting In Saccharomyces Cerevisiae. J. Cell Biol. 2001, 152, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Wurmser, A.E.; Emr, S.D. Novel Ptdins(3)P-Binding Protein Etf1 Functions As An Effector Of The Vps34 Ptdins 3-Kinase In Autophagy. J. Cell Biol. 2002, 158, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Nice, D.C.; Sato, T.K.; Stromhaug, P.E.; Emr, S.D.; Klionsky, D.J. Cooperative Binding Of The Cytoplasm To Vacuole Targeting Pathway Proteins, Cvt13 And Cvt20, To Phosphatidylinositol 3-Phosphate At The Pre-Autophagosomal Structure Is Required For Selective Autophagy. J. Biol. Chem. 2002, 277, 30198–30207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotani, T.; Kirisako, H.; Koizumi, M.; Ohsumi, Y.; Nakatogawa, H. The Atg2-Atg18 Complex Tethers Pre-Autophagosomal Membranes To The Endoplasmic Reticulum For Autophagosome Formation. Proc. Natl. Acad. Sci. USA 2018, 115, 10363–10368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, C.; Feng, P.; Ku, B.; Dotan, I.; Canaani, D.; Oh, B.H.; Jung, J.U. Autophagic And Tumour Suppressor Activity Of A Novel Beclin1-Binding Protein Uvrag. Nat. Cell Biol. 2006, 8, 688–699. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Coppola, D.; Matsushita, N.; Cualing, H.D.; Sun, M.; Sato, Y.; Liang, C.; Jung, J.U.; Cheng, J.Q.; Mule, J.J.; et al. Bif-1 Interacts With Beclin 1 Through Uvrag And Regulates Autophagy And Tumorigenesis. Nat. Cell Biol. 2007, 9, 1142–1151. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cui, D.; Gu, S.; Chen, X.; Bi, Y.; Xiong, X.; Zhao, Y. Autophagy Regulates Apoptosis By Targeting Noxa For Degradation. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1105–1113. [Google Scholar] [CrossRef]
- Ouyang, L.; Shi, Z.; Zhao, S.; Wang, F.T.; Zhou, T.T.; Liu, B.; Bao, J.K. Programmed Cell Death Pathways In Cancer: A Review Of Apoptosis, Autophagy And Programmed Necrosis. Cell Prolif. 2012, 45, 487–498. [Google Scholar] [CrossRef]
- He, C.; Klionsky, D.J. Regulation Mechanisms And Signaling Pathways Of Autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [Green Version]
- Morselli, E.; Galluzzi, L.; Kepp, O.; Vicencio, J.M.; Criollo, A.; Maiuri, M.C.; Kroemer, G. Anti- And Pro-Tumor Functions Of Autophagy. Biochim. Biophys. Acta 2009, 1793, 1524–1532. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Klionsky, D.J. Mammalian Autophagy: Core Molecular Machinery And Signaling Regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Klionsky, D.J. Eaten Alive: A History Of Macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.; Zhou, X.J.; Zhang, H. Exploring The Role Of Autophagy-Related Gene 5 (Atg5) Yields Important Insights Into Autophagy In Autoimmune/Autoinflammatory Diseases. Front. Immunol. 2018, 9, 2334. [Google Scholar] [CrossRef] [PubMed]
- Palanisamy, K.; Tsai, T.H.; Yu, T.M.; Sun, K.T.; Yu, S.H.; Lin, F.Y.; Wang, I.K.; Li, C.Y. Rna-Binding Protein, Human Antigen R Regulates Hypoxia-Induced Autophagy By Targeting Atg7/Atg16l1 Expressions And Autophagosome Formation. J. Cell. Physiol. 2019, 234, 7448–7458. [Google Scholar] [CrossRef] [PubMed]
- Nakatogawa, H.; Suzuki, K.; Kamada, Y.; Ohsumi, Y. Dynamics And Diversity In Autophagy Mechanisms: Lessons From Yeast. Nat. Rev. Mol. Cell Biol. 2009, 10, 458–467. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Klionsky, D.J. Autophagosome Formation: Core Machinery And Adaptations. Nat. Cell Biol. 2007, 9, 1102–1109. [Google Scholar] [CrossRef]
- Takahashi, Y.; He, H.; Tang, Z.; Hattori, T.; Liu, Y.; Young, M.M.; Serfass, J.M.; Chen, L.; Gebru, M.; Chen, C.; et al. An Autophagy Assay Reveals The Escrt-Iii Component Chmp2a As A Regulator Of Phagophore Closure. Nat. Commun. 2018, 9, 2855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.; Bruzek, L.M.; Meng, X.W.; Gores, G.J.; Carter, C.A.; Kaufmann, S.H.; Adjei, A.A. The Role Of Mcl-1 Downregulation In The Proapoptotic Activity Of The Multikinase Inhibitor Bay 43-9006. Oncogene 2005, 24, 6861–6869. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, O.; Teis, D. The Escrt Machinery. Curr. Biol. 2012, 22, R116–R120. [Google Scholar] [CrossRef] [Green Version]
- Rusten, T.E.; Stenmark, H. How Do Escrt Proteins Control Autophagy? J. Cell Sci. 2009, 122, 2179–2183. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, M.G.; Munafo, D.B.; Beron, W.; Colombo, M.I. Rab7 Is Required For The Normal Progression Of The Autophagic Pathway In Mammalian Cells. J. Cell Sci. 2004, 117, 2687–2697. [Google Scholar] [CrossRef] [Green Version]
- Jager, S.; Bucci, C.; Tanida, I.; Ueno, T.; Kominami, E.; Saftig, P.; Eskelinen, E.L. Role For Rab7 In Maturation Of Late Autophagic Vacuoles. J. Cell Sci. 2004, 117, 4837–4848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Jain, A.; Farzam, F.; Jia, J.; Gu, Y.; Choi, S.W.; Mudd, M.H.; Claude-Taupin, A.; Wester, M.J.; Lidke, K.A.; et al. Mechanism Of Stx17 Recruitment To Autophagosomes Via Irgm And Mammalian Atg8 Proteins. J. Cell Biol. 2018, 217, 997–1013. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Lee, J.S.; Inn, K.S.; Gack, M.U.; Li, Q.; Roberts, E.A.; Vergne, I.; Deretic, V.; Feng, P.; Akazawa, C.; et al. Beclin1-Binding Uvrag Targets The Class C Vps Complex To Coordinate Autophagosome Maturation And Endocytic Trafficking. Nat. Cell Biol. 2008, 10, 776–787. [Google Scholar] [CrossRef] [Green Version]
- Johansen, T.; Lamark, T. Selective Autophagy Mediated By Autophagic Adapter Proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Jena, K.K.; Kolapalli, S.P.; Mehto, S.; Nath, P.; Das, B.; Sahoo, P.K.; Ahad, A.; Syed, G.H.; Raghav, S.K.; Senapati, S.; et al. Trim16 Controls Assembly And Degradation Of Protein Aggregates By Modulating The P62-Nrf2 Axis And Autophagy. Embo J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Kinsella, T.J. Mammalian Target Of Rapamycin And S6 Kinase 1 Positively Regulate 6-Thioguanine-Induced Autophagy. Cancer Res. 2008, 68, 2384–2390. [Google Scholar] [CrossRef] [Green Version]
- Loewith, R.; Jacinto, E.; Wullschleger, S.; Lorberg, A.; Crespo, J.L.; Bonenfant, D.; Oppliger, W.; Jenoe, P.; Hall, M.N. Two Tor Complexes, Only One Of Which Is Rapamycin Sensitive, Have Distinct Roles In Cell Growth Control. Mol. Cell 2002, 10, 457–468. [Google Scholar] [CrossRef]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Ruegg, M.A.; Hall, A.; Hall, M.N. Mammalian Tor Complex 2 Controls The Actin Cytoskeleton And Is Rapamycin Insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef]
- Pearce, L.R.; Huang, X.; Boudeau, J.; Pawlowski, R.; Wullschleger, S.; Deak, M.; Ibrahim, A.F.; Gourlay, R.; Magnuson, M.A.; Alessi, D.R. Identification Of Protor As A Novel Rictor-Binding Component Of Mtor Complex-2. Biochem. J. 2007, 405, 513–522. [Google Scholar] [CrossRef] [Green Version]
- Wan, W.; You, Z.; Zhou, L.; Xu, Y.; Peng, C.; Zhou, T.; Yi, C.; Shi, Y.; Liu, W. Mtorc1-Regulated And Huwe1-Mediated Wipi2 Degradation Controls Autophagy Flux. Mol. Cell 2018, 72, 303–315.e6. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. Tsc2 Is Phosphorylated And Inhibited By Akt And Suppresses Mtor Signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Liu, H.; Foyil, S.R.; Godar, R.J.; Weinheimer, C.J.; Hill, J.A.; Diwan, A. Impaired Autophagosome Clearance Contributes To Cardiomyocyte Death In Ischemia/Reperfusion Injury. Circulation 2012, 125, 3170–3181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godlewski, J.; Nowicki, M.O.; Bronisz, A.; Nuovo, G.; Palatini, J.; De Lay, M.; Van Brocklyn, J.; Ostrowski, M.C.; Chiocca, E.A.; Lawler, S.E. Microrna-451 Regulates Lkb1/Ampk Signaling And Allows Adaptation To Metabolic Stress In Glioma Cells. Mol. Cell 2010, 37, 620–632. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Kang, H.; Powathil, G.; Kim, H.; Trucu, D.; Lee, W.; Lawler, S.; Chaplain, M. Role Of Extracellular Matrix And Microenvironment In Regulation Of Tumor Growth And Lar-Mediated Invasion In Glioblastoma. PLoS ONE 2018, 13, E0204865. [Google Scholar] [CrossRef]
- Herrero-Martin, G.; Hoyer-Hansen, M.; Garcia-Garcia, C.; Fumarola, C.; Farkas, T.; Lopez-Rivas, A.; Jaattela, M. Tak1 Activates Ampk-Dependent Cytoprotective Autophagy In Trail-Treated Epithelial Cells. Embo J. 2009, 28, 677–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoyer-Hansen, M.; Jaattela, M. Amp-Activated Protein Kinase: A Universal Regulator Of Autophagy? Autophagy 2007, 3, 381–383. [Google Scholar] [CrossRef] [Green Version]
- Shiloh, R.; Gilad, Y.; Ber, Y.; Eisenstein, M.; Aweida, D.; Bialik, S.; Cohen, S.; Kimchi, A. Non-Canonical Activation Of Dapk2 By Ampk Constitutes A New Pathway Linking Metabolic Stress To Autophagy. Nat. Commun. 2018, 9, 1759. [Google Scholar] [CrossRef] [Green Version]
- Bi, Y.; Li, H.; Yi, D.; Sun, Y.; Bai, Y.; Zhong, S.; Song, Y.; Zhao, G.; Chen, Y. Cordycepin Augments The Chemosensitivity Of Human Glioma Cells To Temozolomide By Activating Ampk And Inhibiting The Akt Signaling Pathway. Mol. Pharm. 2018, 15, 4912–4925. [Google Scholar] [CrossRef]
- Bai, Y.; Chen, Y.; Hong, X.; Liu, X.; Su, X.; Li, S.; Dong, X.; Zhao, G.; Li, Y. Newcastle Disease Virus Enhances The Growth-Inhibiting And Proapoptotic Effects Of Temozolomide On Glioblastoma Cells In Vitro And In Vivo. Sci. Rep. 2018, 8, 11470. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Z.S.; Gong, S.F.; Si, W.; Jiang, T.; Li, Q.L.; Wang, T.J.; Wang, W.J.; Wu, R.Y.; Jiang, K. Effect Of Metformin On Cell Proliferation, Apoptosis, Migration And Invasion In A172 Glioma Cells And Its Mechanisms. Mol. Med. Rep. 2019, 20, 887–894. [Google Scholar] [CrossRef]
- Pan, S.J.; Ren, J.; Jiang, H.; Liu, W.; Hu, L.Y.; Pan, Y.X.; Sun, B.; Sun, Q.F.; Bian, L.G. Magea6 Promotes Human Glioma Cell Survival Via Targeting Ampkalpha1. Cancer Lett. 2018, 412, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jing, K.; Shin, S.; Jeong, S.; Han, S.H.; Oh, H.; Yoo, Y.S.; Han, J.; Jeon, Y.J.; Heo, J.Y.; et al. Omega3-Polyunsaturated Fatty Acids Induce Cell Death Through Apoptosis And Autophagy In Glioblastoma Cells: In Vitro And In Vivo. Oncol. Rep. 2018, 39, 239–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordani, M.; Butera, G.; Pacchiana, R.; Donadelli, M. Molecular Interplay Between Mutant P53 Proteins And Autophagy In Cancer Cells. Biochim. Biophys. Acta Rev. Cancer 2017, 1867, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Budanov, A.V.; Karin, M. P53 Target Genes Sestrin1 And Sestrin2 Connect Genotoxic Stress And Mtor Signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, M.; Kakudo, Y.; Takahashi, S.; Sakamoto, Y.; Kato, S.; Ishioka, C. Overexpression Of Dram Enhances P53-Dependent Apoptosis. Cancer Med. 2013, 2, 1–10. [Google Scholar] [CrossRef]
- Chollat-Namy, M.; Ben Safta-Saadoun, T.; Haferssas, D.; Meurice, G.; Chouaib, S.; Thiery, J. The Pharmalogical Reactivation Of P53 Function Improves Breast Tumor Cell Lysis By Granzyme B And Nk Cells Through Induction Of Autophagy. Cell Death Dis. 2019, 10, 695. [Google Scholar] [CrossRef]
- White, E. Autophagy And P53. Cold Spring Harb. Persp. Med. 2016, 6, A026120. [Google Scholar] [CrossRef]
- Di Malta, C.; Cinque, L.; Settembre, C. Transcriptional Regulation Of Autophagy: Mechanisms And Diseases. Front. Cell Dev. Biol. 2019, 7, 114. [Google Scholar] [CrossRef]
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. Mtorc1 Functions As A Transcriptional Regulator Of Autophagy By Preventing Nuclear Transport Of Tfeb. Autophagy 2012, 8, 903–914. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. Tfeb Links Autophagy To Lysosomal Biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [Green Version]
- Giatromanolaki, A.; Sivridis, E.; Mitrakas, A.; Kalamida, D.; Zois, C.E.; Haider, S.; Piperidou, C.; Pappa, A.; Gatter, K.C.; Harris, A.L.; et al. Autophagy And Lysosomal Related Protein Expression Patterns In Human Glioblastoma. Cancer Biol. Ther. 2014, 15, 1468–1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nager, M.; Sallan, M.C.; Visa, A.; Pushparaj, C.; Santacana, M.; Macia, A.; Yeramian, A.; Canti, C.; Herreros, J. Inhibition Of Wnt-Ctnnb1 Signaling Upregulates Sqstm1 And Sensitizes Glioblastoma Cells To Autophagy Blockers. Autophagy 2018, 14, 619–636. [Google Scholar] [CrossRef] [PubMed]
- Webb, A.E.; Brunet, A. Foxo Transcription Factors: Key Regulators Of Cellular Quality Control. Trends Biochem. Sci. 2014, 39, 159–169. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z. The Foxo-Autophagy Axis In Health And Disease. Trends Endocrinol. Metab. 2019, 30, 658–671. [Google Scholar] [CrossRef]
- Hou, T.; Li, Z.; Zhao, Y.; Zhu, W.G. Mechanisms Controlling The Anti-Neoplastic Functions Of Foxo Proteins. Sem. Cancer Biol. 2018, 50, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ng, S.; Wang, J.; Zhou, J.; Tan, S.H.; Yang, N.; Lin, Q.; Xia, D.; Shen, H.M. Histone Deacetylase Inhibitors Induce Autophagy Through Foxo1-Dependent Pathways. Autophagy 2015, 11, 629–642. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Hif-1: Upstream And Downstream Of Cancer Metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Hypoxia-Induced Autophagy Is Mediated Through Hypoxia-Inducible Factor Induction Of Bnip3 And Bnip3l Via Their Bh3 Domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial Autophagy Is An Hif-1-Dependent Adaptive Metabolic Response To Hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [Green Version]
- Mazure, N.M.; Pouyssegur, J. Atypical Bh3-Domains Of Bnip3 And Bnip3l Lead To Autophagy In Hypoxia. Autophagy 2009, 5, 868–869. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Kim, H.P.; Jin, Y.; Choi, A.M.; Ryter, S.W. Beclin 1 Deficiency Is Associated With Increased Hypoxia-Induced Angiogenesis. Autophagy 2011, 7, 829–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papandreou, I.; Lim, A.L.; Laderoute, K.; Denko, N.C. Hypoxia Signals Autophagy In Tumor Cells Via Ampk Activity, Independent Of Hif-1, Bnip3, And Bnip3l. Cell Death Differ. 2008, 15, 1572–1581. [Google Scholar] [CrossRef]
- Turcotte, S.; Chan, D.A.; Sutphin, P.D.; Hay, M.P.; Denny, W.A.; Giaccia, A.J. A Molecule Targeting Vhl-Deficient Renal Cell Carcinoma That Induces Autophagy. Cancer Cell 2008, 14, 90–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbi, M.E.; Hu, H.; Kshitiz; Ahmed, I.; Levchenko, A.; Semenza, G.L. Chaperone-Mediated Autophagy Targets Hypoxia-Inducible Factor-1alpha (Hif-1alpha) For Lysosomal Degradation. J. Biol. Chem. 2013, 288, 10703–10714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.H.; Zhang, P.; Chen, W.D.; Li, D.D.; Wu, X.Q.; Deng, R.; Jiao, L.; Li, X.; Ji, J.; Feng, G.K.; et al. Atm-Mediated Pten Phosphorylation Promotes Pten Nuclear Translocation And Autophagy In Response To Dna-Damaging Agents In Cancer Cells. Autophagy 2015, 11, 239–252. [Google Scholar] [CrossRef] [Green Version]
- Trocoli, A.; Djavaheri-Mergny, M. The Complex Interplay Between Autophagy And Nf-Kappab Signaling Pathways In Cancer Cells. Am. J. Cancer Res. 2011, 1, 629–649. [Google Scholar]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy In Human Health And Disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef]
- Kimmelman, A.C. The Dynamic Nature Of Autophagy In Cancer. Genes Dev. 2011, 25, 1999–2010. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.Y.; Xia, B.; White, E. Autophagy-Mediated Tumor Promotion. Cell 2013, 155, 1216–1219. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction Of Autophagy And Inhibition Of Tumorigenesis By Beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Vial, D.; Mckeown-Longo, P.J. Role Of Egfr Expression Levels In The Regulation Of Integrin Function By Egf. Mol. Carcinog. 2016, 55, 1118–1123. [Google Scholar] [CrossRef] [Green Version]
- Magnus, N.; Garnier, D.; Meehan, B.; Mcgraw, S.; Lee, T.H.; Caron, M.; Bourque, G.; Milsom, C.; Jabado, N.; Trasler, J.; et al. Tissue Factor Expression Provokes Escape From Tumor Dormancy And Leads To Genomic Alterations. Proc. Natl. Acad. Sci. USA 2014, 111, 3544–3549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirtoli, L.; Cevenini, G.; Tini, P.; Vannini, M.; Oliveri, G.; Marsili, S.; Mourmouras, V.; Rubino, G.; Miracco, C. The Prognostic Role Of Beclin 1 Protein Expression In High-Grade Gliomas. Autophagy 2009, 5, 930–936. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Bai, H.M.; Chen, L.; Li, B.; Lu, Y.C. Reduced Expression Of Lc3b-Ii And Beclin 1 In Glioblastoma Multiforme Indicates A Down-Regulated Autophagic Capacity That Relates To The Progression Of Astrocytic Tumors. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2010, 17, 1515–1519. [Google Scholar] [CrossRef]
- Shukla, S.; Patric, I.R.; Patil, V.; Shwetha, S.D.; Hegde, A.S.; Chandramouli, B.A.; Arivazhagan, A.; Santosh, V.; Somasundaram, K. Methylation Silencing Of Ulk2, An Autophagy Gene, Is Essential For Astrocyte Transformation And Tumor Growth. J. Biol. Chem. 2014, 289, 22306–22318. [Google Scholar] [CrossRef] [Green Version]
- Miracco, C.; Cosci, E.; Oliveri, G.; Luzi, P.; Pacenti, L.; Monciatti, I.; Mannucci, S.; De Nisi, M.C.; Toscano, M.; Malagnino, V.; et al. Protein And Mrna Expression Of Autophagy Gene Beclin 1 In Human Brain Tumours. Int. J. Oncol. 2007, 30, 429–436. [Google Scholar]
- Aoki, H.; Kondo, Y.; Aldape, K.; Yamamoto, A.; Iwado, E.; Yokoyama, T.; Hollingsworth, E.F.; Kobayashi, R.; Hess, K.; Shinojima, N.; et al. Monitoring Autophagy In Glioblastoma With Antibody Against Isoform B Of Human Microtubule-Associated Protein 1 Light Chain 3. Autophagy 2008, 4, 467–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galavotti, S.; Bartesaghi, S.; Faccenda, D.; Shaked-Rabi, M.; Sanzone, S.; Mcevoy, A.; Dinsdale, D.; Condorelli, F.; Brandner, S.; Campanella, M.; et al. The Autophagy-Associated Factors Dram1 And P62 Regulate Cell Migration And Invasion In Glioblastoma Stem Cells. Oncogene 2013, 32, 699–712. [Google Scholar] [CrossRef] [Green Version]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Targeting Mtor In Glioblastoma: Rationale And Preclinical/Clinical Evidence. Dis. Mark. 2018, 2018, 9230479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.Y.; Zhang, L.Q.; Zhang, X.G.; Li, X.; Ren, Y.B.; Ma, X.Y.; Li, X.G.; Wang, L.X. Association Between Akt/Mtor Signalling Pathway And Malignancy Grade Of Human Gliomas. J. Neuro Oncol. 2011, 103, 453–458. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; Mclendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma Stem Cells Promote Radioresistance By Preferential Activation Of The Dna Damage Response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Jhanwar-Uniyal, M.; Jeevan, D.; Neil, J.; Shannon, C.; Albert, L.; Murali, R. Deconstructing Mtor Complexes In Regulation Of Glioblastoma Multiforme And Its Stem Cells. Adv. Biol. Regul. 2013, 53, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Tini, P.; Belmonte, G.; Toscano, M.; Miracco, C.; Palumbo, S.; Pastina, P.; Battaglia, G.; Nardone, V.; Butorano, M.A.; Masucci, A.; et al. Combined Epidermal Growth Factor Receptor And Beclin1 Autophagic Protein Expression Analysis Identifies Different Clinical Presentations, Responses To Chemo- And Radiotherapy, And Prognosis In Glioblastoma. Biomed. Res. Int. 2015, 2015, 208076. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, T.; Hu, J.; Zhang, R.; Rao, Y.; Wang, S.; Chen, R.; Mclendon, R.E.; Friedman, A.H.; Keir, S.T.; et al. Mir-33a Promotes Glioma-Initiating Cell Self-Renewal Via Pka And Notch Pathways. J. Clin. Investig. 2014, 124, 4489–4502. [Google Scholar] [CrossRef]
- Guo, X.; Xue, H.; Guo, X.; Gao, X.; Xu, S.; Yan, S.; Han, X.; Li, T.; Shen, J.; Li, G. Mir224-3p Inhibits Hypoxia-Induced Autophagy By Targeting Autophagy-Related Genes In Human Glioblastoma Cells. Oncotarget 2015, 6, 41620–41637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azad, M.B.; Chen, Y.; Henson, E.S.; Cizeau, J.; Mcmillan-Ward, E.; Israels, S.J.; Gibson, S.B. Hypoxia Induces Autophagic Cell Death In Apoptosis-Competent Cells Through A Mechanism Involving Bnip3. Autophagy 2008, 4, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.L.; Jahangiri, A.; Delay, M.; Aghi, M.K. Tumor Cell Autophagy As An Adaptive Response Mediating Resistance To Treatments Such As Antiangiogenic Therapy. Cancer Res. 2012, 72, 4294–4299. [Google Scholar] [CrossRef] [Green Version]
- Mathew, R.; Kongara, S.; Beaudoin, B.; Karp, C.M.; Bray, K.; Degenhardt, K.; Chen, G.; Jin, S.; White, E. Autophagy Suppresses Tumor Progression By Limiting Chromosomal Instability. Genes Dev. 2007, 21, 1367–1381. [Google Scholar] [CrossRef] [Green Version]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy Suppresses Tumorigenesis Through Elimination Of P62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef] [Green Version]
- Qiang, L.; Zhao, B.; Ming, M.; Wang, N.; He, T.C.; Hwang, S.; Thorburn, A.; He, Y.Y. Regulation Of Cell Proliferation And Migration By P62 Through Stabilization Of Twist1. Proc. Natl. Acad. Sci. USA 2014, 111, 9241–9246. [Google Scholar] [CrossRef] [Green Version]
- Duran, A.; Linares, J.F.; Galvez, A.S.; Wikenheiser, K.; Flores, J.M.; Diaz-Meco, M.T.; Moscat, J. The Signaling Adaptor P62 Is An Important Nf-Kappab Mediator In Tumorigenesis. Cancer Cell 2008, 13, 343–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wooten, M.W.; Geetha, T.; Seibenhener, M.L.; Babu, J.R.; Diaz-Meco, M.T.; Moscat, J. The P62 Scaffold Regulates Nerve Growth Factor-Induced Nf-Kappab Activation By Influencing Traf6 Polyubiquitination. J. Biol. Chem. 2005, 280, 35625–35629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz, L.; Sanchez, P.; Lallena, M.J.; Diaz-Meco, M.T.; Moscat, J. The Interaction Of P62 With Rip Links The Atypical Pkcs To Nf-Kappab Activation. EMBO J. 1999, 18, 3044–3053. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Liu, F.; Xia, M.; Xu, Y.; Li, X.; Kang, J.; Li, Y.; Sun, L. P62 Participates In The Inhibition Of Nf-Kappab Signaling And Apoptosis Induced By Sulfasalazine In Human Glioma U251 Cells. Oncol. Rep. 2015, 34, 235–243. [Google Scholar] [CrossRef]
- Xu, H.; Sun, L.; Zheng, Y.; Yu, S.; Ou-Yang, J.; Han, H.; Dai, X.; Yu, X.; Li, M.; Lan, Q. Gbp3 Promotes Glioma Cell Proliferation Via Sqstm1/P62-Erk1/2 Axis. Biochem. Biophys. Res. Commun. 2018, 495, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, S.; Xu, B.; Wang, T.; Zheng, Y.; Liu, F.; Ren, F.; Jiang, J.; Shi, H.; Zou, B.; et al. P62 Functions As An Oncogene In Colorectal Cancer Through Inhibiting Apoptosis And Promoting Cell Proliferation By Interacting With The Vitamin D Receptor. Cell Prolif. 2019, 52, E12585. [Google Scholar] [CrossRef]
- Lv, Q.; Wang, W.; Xue, J.; Hua, F.; Mu, R.; Lin, H.; Yan, J.; Lv, X.; Chen, X.; Hu, Z.W. Dedd Interacts With Pi3kc3 To Activate Autophagy And Attenuate Epithelial-Mesenchymal Transition In Human Breast Cancer. Cancer Res. 2012, 72, 3238–3250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Craene, B.; Berx, G. Regulatory Networks Defining Emt During Cancer Initiation And Progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef]
- Jiang, Y.; Jiao, Y.; Wang, Z.; Li, T.; Liu, Y.; Li, Y.; Zhao, X.; Wang, D. Sinomenine Hydrochloride Inhibits Human Glioblastoma Cell Growth Through Reactive Oxygen Species Generation And Autophagy-Lysosome Pathway Activation: An In Vitro And In Vivo Study. Int. J. Mol. Sci. 2017, 18, 1945. [Google Scholar] [CrossRef] [Green Version]
- Catalano, M.; D’alessandro, G.; Lepore, F.; Corazzari, M.; Caldarola, S.; Valacca, C.; Faienza, F.; Esposito, V.; Limatola, C.; Cecconi, F.; et al. Autophagy Induction Impairs Migration And Invasion By Reversing Emt In Glioblastoma Cells. Mol. Oncol. 2015, 9, 1612–1625. [Google Scholar] [CrossRef] [Green Version]
- Young, A.R.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.; Tavare, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy Mediates The Mitotic Senescence Transition. Genes Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knizhnik, A.V.; Roos, W.P.; Nikolova, T.; Quiros, S.; Tomaszowski, K.H.; Christmann, M.; Kaina, B. Survival And Death Strategies In Glioma Cells: Autophagy, Senescence And Apoptosis Triggered By A Single Type Of Temozolomide-Induced Dna Damage. PLoS ONE 2013, 8, E55665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippi-Chiela, E.C.; Thome, M.P.; Bueno, E.; Silva, M.M.; Pelegrini, A.L.; Ledur, P.F.; Garicochea, B.; Zamin, L.L.; Lenz, G. Resveratrol Abrogates The Temozolomide-Induced G2 Arrest Leading To Mitotic Catastrophe And Reinforces The Temozolomide-Induced Senescence In Glioma Cells. BMC Cancer 2013, 13, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Xue, X.; Guo, R.B.; Sun, X.L.; Hu, G. Resveratrol Enhances The Antitumor Effects Of Temozolomide In Glioblastoma Via Ros-Dependent Ampk-Tsc-Mtor Signaling Pathway. CNS Neurosci. Ther. 2012, 18, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Qi, Q.; Zhou, W.; Feng, Z.; Huang, B.; Chen, A.; Zhang, D.; Li, W.; Zhang, Q.; Jiang, Z.; et al. Inhibition Of Glioma Growth By Flavokawain B Is Mediated Through Endoplasmic Reticulum Stress Induced Autophagy. Autophagy 2018, 14, 2007–2022. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Wang, J.; Qi, Q.; Feng, Z.; Huang, B.; Chen, A.; Zhang, D.; Li, W.; Zhang, Q.; Bjerkvig, R.; et al. Matrine Induces Senescence Of Human Glioblastoma Cells Through Suppression Of The Igf1/Pi3k/Akt/P27 Signaling Pathway. Cancer Med. 2018, 7, 4729–4743. [Google Scholar] [CrossRef] [Green Version]
- Lepine, S.; Allegood, J.C.; Edmonds, Y.; Milstien, S.; Spiegel, S. Autophagy Induced By Deficiency Of Sphingosine-1-Phosphate Phosphohydrolase 1 Is Switched To Apoptosis By Calpain-Mediated Autophagy-Related Gene 5 (Atg5) Cleavage. J. Biol. Chem. 2011, 286, 44380–44390. [Google Scholar] [CrossRef] [Green Version]
- Yousefi, S.; Perozzo, R.; Schmid, I.; Ziemiecki, A.; Schaffner, T.; Scapozza, L.; Brunner, T.; Simon, H.U. Calpain-Mediated Cleavage Of Atg5 Switches Autophagy To Apoptosis. Nat. Cell Biol. 2006, 8, 1124–1132. [Google Scholar] [CrossRef]
- Huang, X.; Qi, Q.; Hua, X.; Li, X.; Zhang, W.; Sun, H.; Li, S.; Wang, X.; Li, B. Beclin 1, An Autophagy-Related Gene, Augments Apoptosis In U87 Glioblastoma Cells. Oncol. Rep. 2014, 31, 1761–1767. [Google Scholar] [CrossRef] [Green Version]
- Levin-Salomon, V.; Bialik, S.; Kimchi, A. Dap-Kinase And Autophagy. Apoptosis Int. J. Program. Cell Death 2014, 19, 346–356. [Google Scholar] [CrossRef]
- Jo, G.H.; Bogler, O.; Chwae, Y.J.; Yoo, H.; Lee, S.H.; Park, J.B.; Kim, Y.J.; Kim, J.H.; Gwak, H.S. Radiation-Induced Autophagy Contributes To Cell Death And Induces Apoptosis Partly In Malignant Glioma Cells. Cancer Res. Treat. Off. J. Korean Cancer Assoc. 2015, 47, 221–241. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Okamoto, K.; Yu, C.; Sinicrope, F.A. P62/Sequestosome-1 Up-Regulation Promotes Abt-263-Induced Caspase-8 Aggregation/Activation On The Autophagosome. J. Biol. Chem. 2013, 288, 33654–33666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.B.; Gong, J.L.; Xing, T.Y.; Zheng, S.P.; Ding, W. Autophagy Protein P62/Sqstm1 Is Involved In Hamlet-Induced Cell Death By Modulating Apotosis In U87mg Cells. Cell Death Dis. 2013, 4, E550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, R.X.; Zhang, Y.B.; Fan, Y.; Wu, G.L. P62/Sqstm1 Is Involved In Caspase-8 Associated Cell Death Induced By Proteasome Inhibitor Mg132 In U87mg Cells. Cell Biol. Int. 2014, 38, 1221–1226. [Google Scholar] [CrossRef]
- Pyo, J.O.; Jang, M.H.; Kwon, Y.K.; Lee, H.J.; Jun, J.I.; Woo, H.N.; Cho, D.H.; Choi, B.; Lee, H.; Kim, J.H.; et al. Essential Roles Of Atg5 And Fadd In Autophagic Cell Death: Dissection Of Autophagic Cell Death Into Vacuole Formation And Cell Death. J. Biol. Chem. 2005, 280, 20722–20729. [Google Scholar] [CrossRef] [Green Version]
- Rubinstein, A.D.; Eisenstein, M.; Ber, Y.; Bialik, S.; Kimchi, A. The Autophagy Protein Atg12 Associates With Antiapoptotic Bcl-2 Family Members To Promote Mitochondrial Apoptosis. Mol. Cell 2011, 44, 698–709. [Google Scholar] [CrossRef] [Green Version]
- Betin, V.M.; Lane, J.D. Caspase Cleavage Of Atg4d Stimulates Gabarap-L1 Processing And Triggers Mitochondrial Targeting And Apoptosis. J. Cell Sci. 2009, 122, 2554–2566. [Google Scholar] [CrossRef] [Green Version]
- Kaza, N.; Kohli, L.; Roth, K.A. Autophagy In Brain Tumors: A New Target For Therapeutic Intervention. Brain Pathol. 2012, 22, 89–98. [Google Scholar] [CrossRef] [Green Version]
- Gammoh, N.; Fraser, J.; Puente, C.; Syred, H.M.; Kang, H.; Ozawa, T.; Lam, D.; Acosta, J.C.; Finch, A.J.; Holland, E.; et al. Suppression Of Autophagy Impedes Glioblastoma Development And Induces Senescence. Autophagy 2016, 12, 1431–1439. [Google Scholar] [CrossRef]
- Tamrakar, S.; Yashiro, M.; Kawashima, T.; Uda, T.; Terakawa, Y.; Kuwae, Y.; Ohsawa, M.; Ohata, K. Clinicopathological Significance Of Autophagy-Related Proteins And Its Association With Genetic Alterations In Gliomas. Anticancer Res. 2019, 39, 1233–1242. [Google Scholar] [CrossRef]
- Jennewein, L.; Ronellenfitsch, M.W.; Antonietti, P.; Ilina, E.I.; Jung, J.; Stadel, D.; Flohr, L.M.; Zinke, J.; Von Renesse, J.; Drott, U.; et al. Diagnostic And Clinical Relevance Of The Autophago-Lysosomal Network In Human Gliomas. Oncotarget 2016, 7, 20016–20032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padmakrishnan, C.J.; Easwer, H.V.; Vijayakurup, V.; Menon, G.R.; Nair, S.; Gopala, S. High Lc3/Beclin Expression Correlates With Poor Survival In Glioma: A Definitive Role For Autophagy As Evidenced By In Vitro Autophagic Flux. Pathol. Oncol. Res. 2019, 25, 137–148. [Google Scholar] [CrossRef]
- Wen, Z.P.; Zeng, W.J.; Chen, Y.H.; Li, H.; Wang, J.Y.; Cheng, Q.; Yu, J.; Zhou, H.H.; Liu, Z.Z.; Xiao, J.; et al. Knockdown Atg4c Inhibits Gliomas Progression And Promotes Temozolomide Chemosensitivity By Suppressing Autophagic Flux. J. Exp. Clin. Cancer Res. 2019, 38, 298. [Google Scholar] [CrossRef]
- Fu, Z.; Luo, W.; Wang, J.; Peng, T.; Sun, G.; Shi, J.; Li, Z.; Zhang, B. Malat1 Activates Autophagy And Promotes Cell Proliferation By Sponging Mir-101 And Upregulating Stmn1, Rab5a And Atg4d Expression In Glioma. Biochem. Biophys. Res. Commun. 2017, 492, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Mazure, N.M.; Pouyssegur, J. Hypoxia-Induced Autophagy: Cell Death Or Cell Survival? Curr. Opin. Cell Biol. 2010, 22, 177–180. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, E.M.; Pellegrino, M.; Santolla, M.F.; Lappano, R.; Ricchio, E.; Abonante, S.; Maggiolini, M. Gper Mediates Activation Of Hif1alpha/Vegf Signaling By Estrogens. Cancer Res. 2014, 74, 4053–4064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jawhari, S.; Ratinaud, M.H.; Verdier, M. Glioblastoma, Hypoxia And Autophagy: A Survival-Prone ’Menage-A-Trois’. Cell Death Dis. 2016, 7, E2434. [Google Scholar] [CrossRef]
- Yang, L.; Lin, C.; Wang, L.; Guo, H.; Wang, X. Hypoxia And Hypoxia-Inducible Factors In Glioblastoma Multiforme Progression And Therapeutic Implications. Exp. Cell Res. 2012, 318, 2417–2426. [Google Scholar] [CrossRef]
- Hu, Y.L.; Delay, M.; Jahangiri, A.; Molinaro, A.M.; Rose, S.D.; Carbonell, W.S.; Aghi, M.K. Hypoxia-Induced Autophagy Promotes Tumor Cell Survival And Adaptation To Antiangiogenic Treatment In Glioblastoma. Cancer Res. 2012, 72, 1773–1783. [Google Scholar] [CrossRef] [Green Version]
- Duan, S. Silencing The Autophagy-Specific Gene Beclin-1 Contributes To Attenuated Hypoxia-Induced Vasculogenic Mimicry Formation In Glioma. Cancer Biomark. Sect. A Dis. Mark. 2018, 21, 565–574. [Google Scholar] [CrossRef]
- Huang, M.; Ke, Y.; Sun, X.; Yu, L.; Yang, Z.; Zhang, Y.; Du, M.; Wang, J.; Liu, X.; Huang, S. Mammalian Target Of Rapamycin Signaling Is Involved In The Vasculogenic Mimicry Of Glioma Via Hypoxia-Inducible Factor-1alpha. Oncol. Rep. 2014, 32, 1973–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.B.; Yang, S.; Weng, H.Y.; Chen, Q.; Zhao, X.L.; Fu, W.J.; Niu, Q.; Ping, Y.F.; Wang, J.M.; Zhang, X.; et al. Autophagy-Induced Kdr/Vegfr-2 Activation Promotes The Formation Of Vasculogenic Mimicry By Glioma Stem Cells. Autophagy 2017, 13, 1528–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul Rahim, S.A.; Dirkse, A.; Oudin, A.; Schuster, A.; Bohler, J.; Barthelemy, V.; Muller, A.; Vallar, L.; Janji, B.; Golebiewska, A.; et al. Regulation Of Hypoxia-Induced Autophagy In Glioblastoma Involves Atg9a. Br. J. Cancer 2017, 117, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Pavlides, S.; Vera, I.; Gandara, R.; Sneddon, S.; Pestell, R.G.; Mercier, I.; Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; et al. Warburg Meets Autophagy: Cancer-Associated Fibroblasts Accelerate Tumor Growth And Metastasis Via Oxidative Stress, Mitophagy, And Aerobic Glycolysis. Antioxid. Redox Signal. 2012, 16, 1264–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisanti, M.P.; Martinez-Outschoorn, U.E.; Chiavarina, B.; Pavlides, S.; Whitaker-Menezes, D.; Tsirigos, A.; Witkiewicz, A.; Lin, Z.; Balliet, R.; Howell, A.; et al. Understanding The “Lethal” Drivers Of Tumor-Stroma Co-Evolution: Emerging Role(S) For Hypoxia, Oxidative Stress And Autophagy/Mitophagy In The Tumor Micro-Environment. Cancer Biol. Ther. 2010, 10, 537–542. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Outschoorn, U.E.; Trimmer, C.; Lin, Z.; Whitaker-Menezes, D.; Chiavarina, B.; Zhou, J.; Wang, C.; Pavlides, S.; Martinez-Cantarin, M.P.; Capozza, F.; et al. Autophagy In Cancer Associated Fibroblasts Promotes Tumor Cell Survival: Role Of Hypoxia, Hif1 Induction And Nfkappab Activation In The Tumor Stromal Microenvironment. Cell Cycle 2010, 9, 3515–3533. [Google Scholar] [CrossRef]
- Chen, Z.H.; Cao, J.F.; Zhou, J.S.; Liu, H.; Che, L.Q.; Mizumura, K.; Li, W.; Choi, A.M.; Shen, H.H. Interaction Of Caveolin-1 With Atg12-Atg5 System Suppresses Autophagy In Lung Epithelial Cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L1016–L1025. [Google Scholar] [CrossRef]
- Regina, A.; Jodoin, J.; Khoueir, P.; Rolland, Y.; Berthelet, F.; Moumdjian, R.; Fenart, L.; Cecchelli, R.; Demeule, M.; Beliveau, R. Down-Regulation Of Caveolin-1 In Glioma Vasculature: Modulation By Radiotherapy. J. Neurosci. Res. 2004, 75, 291–299. [Google Scholar] [CrossRef]
- Martins, D.; Beca, F.F.; Sousa, B.; Baltazar, F.; Paredes, J.; Schmitt, F. Loss Of Caveolin-1 And Gain Of Mct4 Expression In The Tumor Stroma: Key Events In The Progression From An In Situ To An Invasive Breast Carcinoma. Cell Cycle 2013, 12, 2684–2690. [Google Scholar] [CrossRef] [Green Version]
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Lin, Z.; Ertel, A.; Flomenberg, N.; Witkiewicz, A.K.; Birbe, R.C.; Howell, A.; Pavlides, S.; Gandara, R.; et al. Evidence For A Stromal-Epithelial “Lactate Shuttle” In Human Tumors: Mct4 Is A Marker Of Oxidative Stress In Cancer-Associated Fibroblasts. Cell Cycle 2011, 10, 1772–1783. [Google Scholar] [CrossRef] [Green Version]
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Flomenberg, N.; Birbe, R.C.; Witkiewicz, A.K.; Howell, A.; Pavlides, S.; Tsirigos, A.; Ertel, A.; Pestell, R.G.; et al. Hyperactivation Of Oxidative Mitochondrial Metabolism In Epithelial Cancer Cells In Situ: Visualizing The Therapeutic Effects Of Metformin In Tumor Tissue. Cell Cycle 2011, 10, 4047–4064. [Google Scholar] [CrossRef] [Green Version]
- Miranda-Goncalves, V.; Honavar, M.; Pinheiro, C.; Martinho, O.; Pires, M.M.; Pinheiro, C.; Cordeiro, M.; Bebiano, G.; Costa, P.; Palmeirim, I.; et al. Monocarboxylate Transporters (Mcts) In Gliomas: Expression And Exploitation As Therapeutic Targets. Neuro Oncol. 2013, 15, 172–188. [Google Scholar] [CrossRef]
- Zhou, H.G.; Zhang, J.D.; Zhang, Y.F. The Effect Of Downregulation Of Mct1 On The Proliferation Of Glioma Cells. Zhonghua Zhong Liu Za Zhi Chin. J. Oncol. 2019, 41, 208–213. [Google Scholar] [CrossRef]
- Miranda-Goncalves, V.; Granja, S.; Martinho, O.; Honavar, M.; Pojo, M.; Costa, B.M.; Pires, M.M.; Pinheiro, C.; Cordeiro, M.; Bebiano, G.; et al. Hypoxia-Mediated Upregulation Of Mct1 Expression Supports The Glycolytic Phenotype Of Glioblastomas. Oncotarget 2016, 7, 46335–46353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda-Goncalves, V.; Reis, R.M.; Baltazar, F. Lactate Transporters And Ph Regulation: Potential Therapeutic Targets In Glioblastomas. Curr. Cancer Drug Targets 2016, 16, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Duan, K.; Liu, Z.J.; Hu, S.Q.; Huo, H.Y.; Xu, Z.R.; Ruan, J.F.; Sun, Y.; Dai, L.P.; Yan, C.B.; Xiong, W.; et al. Lactic Acid Induces Lactate Transport And Glycolysis/Oxphos Interconversion In Glioblastoma. Biochem. Biophys. Res. Commun. 2018, 503, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Debnath, J. Detachment-Induced Autophagy During Anoikis And Lumen Formation In Epithelial Acini. Autophagy 2008, 4, 351–353. [Google Scholar] [CrossRef] [Green Version]
- Dey, S.; Sayers, C.M.; Verginadis, I.I.; Lehman, S.L.; Cheng, Y.; Cerniglia, G.J.; Tuttle, S.W.; Feldman, M.D.; Zhang, P.J.; Fuchs, S.Y.; et al. Atf4-Dependent Induction Of Heme Oxygenase 1 Prevents Anoikis And Promotes Metastasis. J. Clin. Investig. 2015, 125, 2592–2608. [Google Scholar] [CrossRef] [Green Version]
- Fung, C.; Lock, R.; Gao, S.; Salas, E.; Debnath, J. Induction Of Autophagy During Extracellular Matrix Detachment Promotes Cell Survival. Mol. Biol. Cell 2008, 19, 797–806. [Google Scholar] [CrossRef] [Green Version]
- Talukdar, S.; Pradhan, A.K.; Bhoopathi, P.; Shen, X.N.; August, L.A.; Windle, J.J.; Sarkar, D.; Furnari, F.B.; Cavenee, W.K.; Das, S.K.; et al. Mda-9/Syntenin Regulates Protective Autophagy In Anoikis-Resistant Glioma Stem Cells. Proc. Natl. Acad. Sci. USA 2018, 115, 5768–5773. [Google Scholar] [CrossRef] [Green Version]
- Bacolod, M.D.; Das, S.K.; Sokhi, U.K.; Bradley, S.; Fenstermacher, D.A.; Pellecchia, M.; Emdad, L.; Sarkar, D.; Fisher, P.B. Examination Of Epigenetic And Other Molecular Factors Associated With Mda-9/Syntenin Dysregulation In Cancer Through Integrated Analyses Of Public Genomic Datasets. Adv. Cancer Res. 2015, 127, 49–121. [Google Scholar] [CrossRef] [Green Version]
- Lum, J.J.; Bauer, D.E.; Kong, M.; Harris, M.H.; Li, C.; Lindsten, T.; Thompson, C.B. Growth Factor Regulation Of Autophagy And Cell Survival In The Absence Of Apoptosis. Cell 2005, 120, 237–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, L.; Yi, L.; Liu, P.; Abeysekera, I.R.; Hai, L.; Li, T.; Tao, Z.; Ma, H.; Xie, Y.; Huang, Y.; et al. Tumour Cell Dormancy As A Contributor To The Reduced Survival Of Gbm Patients Who Received Standard Therapy. Oncol. Rep. 2018, 40, 463–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, A.; Kimura, T.; Sadahiro, H.; Kawano, H.; Takubo, K.; Suzuki, M.; Ikeda, E. Histological Characterization Of The Tumorigenic “Peri-Necrotic Niche” Harboring Quiescent Stem-Like Tumor Cells In Glioblastoma. PLoS ONE 2016, 11, E0147366. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. Chronic Inflammation As A Promotor Of Mutagenesis In Essential Thrombocythemia, Polycythemia Vera And Myelofibrosis. A Human Inflammation Model For Cancer Development? Leuk. Res. 2013, 37, 214–220. [Google Scholar] [CrossRef]
- Lamoral-Theys, D.; Le Mercier, M.; Le Calve, B.; Rynkowski, M.A.; Bruyere, C.; Decaestecker, C.; Haibe-Kains, B.; Bontempi, G.; Dubois, J.; Lefranc, F.; et al. Long-Term Temozolomide Treatment Induces Marked Amino Metabolism Modifications And An Increase In Tmz Sensitivity In Hs683 Oligodendroglioma Cells. Neoplasia 2010, 12, 69–79. [Google Scholar] [CrossRef]
- Tiram, G.; Ferber, S.; Ofek, P.; Eldar-Boock, A.; Ben-Shushan, D.; Yeini, E.; Krivitsky, A.; Blatt, R.; Almog, N.; Henkin, J.; et al. Reverting The Molecular Fingerprint Of Tumor Dormancy As A Therapeutic Strategy For Glioblastoma. Faseb J. Off. Publ. Fed. Am. Soc. For. Exp. Biol. 2018, Fj201701568r. [Google Scholar] [CrossRef] [PubMed]
- Kanzawa, T.; Germano, I.M.; Komata, T.; Ito, H.; Kondo, Y.; Kondo, S. Role Of Autophagy In Temozolomide-Induced Cytotoxicity For Malignant Glioma Cells. Cell Death Differ. 2004, 11, 448–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paglin, S.; Hollister, T.; Delohery, T.; Hackett, N.; Mcmahill, M.; Sphicas, E.; Domingo, D.; Yahalom, J. A Novel Response Of Cancer Cells To Radiation Involves Autophagy And Formation Of Acidic Vesicles. Cancer Res. 2001, 61, 439–444. [Google Scholar] [PubMed]
- Ito, H.; Daido, S.; Kanzawa, T.; Kondo, S.; Kondo, Y. Radiation-Induced Autophagy Is Associated With Lc3 And Its Inhibition Sensitizes Malignant Glioma Cells. Int. J. Oncol. 2005, 26, 1401–1410. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy Plus Concomitant And Adjuvant Temozolomide For Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Jiricny, J. The Multifaceted Mismatch-Repair System. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.H.; Johnson, J.R.; Pazdur, R. Food And Drug Administration Drug Approval Summary: Temozolomide Plus Radiation Therapy For The Treatment Of Newly Diagnosed Glioblastoma Multiforme. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 6767–6771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteller, M.; Garcia-Foncillas, J.; Andion, E.; Goodman, S.N.; Hidalgo, O.F.; Vanaclocha, V.; Baylin, S.B.; Herman, J.G. Inactivation Of The Dna-Repair Gene Mgmt And The Clinical Response Of Gliomas To Alkylating Agents. N. Engl. J. Med. 2000, 343, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; De Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. Mgmt Gene Silencing And Benefit From Temozolomide In Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Zhou, C.; Yi, L.; Xu, L.; Xu, M. Effect And Molecular Mechanism Of Mtor Inhibitor Rapamycin On Temozolomide-Induced Autophagic Death Of U251 Glioma Cells. Oncol. Lett. 2018, 15, 2477–2484. [Google Scholar] [CrossRef] [PubMed]
- Jakubowicz-Gil, J.; Badziul, D.; Langner, E.; Wertel, I.; Zajac, A.; Rzeski, W. Temozolomide And Sorafenib As Programmed Cell Death Inducers Of Human Glioma Cells. Pharmacol. Rep. 2017, 69, 779–787. [Google Scholar] [CrossRef]
- Li, H.; Chen, L.; Li, J.J.; Zhou, Q.; Huang, A.; Liu, W.W.; Wang, K.; Gao, L.; Qi, S.T.; Lu, Y.T. Mir-519a Enhances Chemosensitivity And Promotes Autophagy In Glioblastoma By Targeting Stat3/Bcl2 Signaling Pathway. J. Hematol. Oncol. 2018, 11, 70. [Google Scholar] [CrossRef] [Green Version]
- Pandey, V.; Ranjan, N.; Narne, P.; Babu, P.P. Roscovitine Effectively Enhances Antitumor Activity Of Temozolomide In Vitro And In Vivo Mediated By Increased Autophagy And Caspase-3 Dependent Apoptosis. Sci. Rep. 2019, 9, 5012. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Zhang, X.; Wen, Z.; Tan, Y.; Huang, N.; Cheng, S.; Zheng, H.; Cheng, Y. Asn-Gly-Arg-Modified Polydopamine-Coated Nanoparticles For Dual-Targeting Therapy Of Brain Glioma In Rats. Oncotarget 2016, 7, 73681–73696. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Tang, M.; Huang, Q.; Zhao, G.; Huang, N.; Zhang, X.; Tan, Y.; Cheng, Y. Combination Of 3-Methyladenine Therapy And Asn-Gly-Arg (Ngr)-Modified Mesoporous Silica Nanoparticles Loaded With Temozolomide For Glioma Therapy In Vitro. Biochem. Biophys. Res. Commun. 2019, 509, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Ciechomska, I.A.; Marciniak, M.P.; Jackl, J.; Kaminska, B. Pre-Treatment Or Post-Treatment Of Human Glioma Cells With Bix01294, The Inhibitor Of Histone Methyltransferase G9a, Sensitizes Cells To Temozolomide. Front. Pharmacol. 2018, 9, 1271. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Li, P. Growth Suppression Of Glioma Cells Using Hdac6 Inhibitor, Tubacin. Open Med. 2018, 13, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, R.M.; Agnes, J.P.; Delgobo, M.; De Souza, P.O.; Thome, M.P.; Heimfarth, L.; Lenz, G.; Moreira, J.C.F.; Zanotto-Filho, A. Late Autophagy Inhibitor Chloroquine Improves Efficacy Of The Histone Deacetylase Inhibitor Saha And Temozolomide In Gliomas. Biochem. Pharmacol. 2019, 163, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Kitange, G.J.; Mladek, A.C.; Carlson, B.L.; Schroeder, M.A.; Pokorny, J.L.; Cen, L.; Decker, P.A.; Wu, W.; Lomberk, G.A.; Gupta, S.K.; et al. Inhibition Of Histone Deacetylation Potentiates The Evolution Of Acquired Temozolomide Resistance Linked To Mgmt Upregulation In Glioblastoma Xenografts. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 4070–4079. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.; Wan, X.; Alvarez, A.A.; James, C.D.; Song, X.; Yang, Y.; Sastry, N.; Nakano, I.; Sulman, E.P.; Hu, B.; et al. Mir93 (Microrna -93) Regulates Tumorigenicity And Therapy Response Of Glioblastoma By Targeting Autophagy. Autophagy 2019, 15, 1100–1111. [Google Scholar] [CrossRef]
- Chen, P.H.; Cheng, C.H.; Shih, C.M.; Ho, K.H.; Lin, C.W.; Lee, C.C.; Liu, A.J.; Chang, C.K.; Chen, K.C. The Inhibition Of Microrna-128 On Igf-1-Activating Mtor Signaling Involves In Temozolomide-Induced Glioma Cell Apoptotic Death. PLoS ONE 2016, 11, E0167096. [Google Scholar] [CrossRef]
- Jiang, C.; Shen, F.; Du, J.; Fang, X.; Li, X.; Su, J.; Wang, X.; Huang, X.; Liu, Z. Upregulation Of Casc2 Sensitized Glioma To Temozolomide Cytotoxicity Through Autophagy Inhibition By Sponging Mir-193a-5p And Regulating Mtor Expression. Biomed. Pharmacother. Biom. Pharmacother. 2018, 97, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, L.; Xue, Y.; Ma, Y.; Liu, X.; Li, Z.; Li, Z.; Liu, Y. Endothelial Monocyte-Activating Polypeptide-Ii Induces Bnip3-Mediated Mitophagy To Enhance Temozolomide Cytotoxicity Of Glioma Stem Cells Via Down-Regulating Mir-24-3p. Front. Mol. Neurosci. 2018, 11, 92. [Google Scholar] [CrossRef]
- Huang, S.; Qi, P.; Zhang, T.; Li, F.; He, X. The Hif1alpha/Mir2243p/Atg5 Axis Affects Cell Mobility And Chemosensitivity By Regulating Hypoxiainduced Protective Autophagy In Glioblastoma And Astrocytoma. Oncol. Rep. 2019, 41, 1759–1768. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.H.; Shen, W.L.; Shih, C.M.; Ho, K.H.; Cheng, C.H.; Lin, C.W.; Lee, C.C.; Liu, A.J.; Chen, K.C. The Chac1-Inhibited Notch3 Pathway Is Involved In Temozolomide-Induced Glioma Cytotoxicity. Neuropharmacology 2017, 116, 300–314. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Guo, H.; Zhang, R.; Liu, H.; Wu, L.; Wu, Q.; Liu, J.; Liu, T.; Zhang, Q. The Pi3k Inhibitor Gdc-0941 Enhances Radiosensitization And Reduces Chemoresistance To Temozolomide In Gbm Cell Lines. Neuroscience 2017, 346, 298–308. [Google Scholar] [CrossRef]
- Oliveira, K.A.; Dal-Cim, T.A.; Lopes, F.G.; Nedel, C.B.; Tasca, C.I. Guanosine Promotes Cytotoxicity Via Adenosine Receptors And Induces Apoptosis In Temozolomide-Treated A172 Glioma Cells. Purinergic Signal. 2017, 13, 305–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, C.W.; Ko, H.J.; Chou, C.H.; Cheng, T.S.; Cheng, H.W.; Liang, Y.H.; Lai, Y.L.; Lin, C.Y.; Wang, C.; Loh, J.K.; et al. Thioridazine Enhances P62-Mediated Autophagy And Apoptosis Through Wnt/Beta-Catenin Signaling Pathway In Glioma Cells. Int. J. Mol. Sci. 2019, 20, 473. [Google Scholar] [CrossRef] [Green Version]
- Elmaci, I.; Ozpinar, A.; Bilir, A.; Altinoz, M.A. Medroxyprogesterone Effects On Colony Growth, Autophagy And Mitochondria Of C6 Glioma Cells Are Augmented With Tibolone And Temozolomide: Cell Kinetic And Electron Microscopical Studies With A Broad Review Of The Literature. Clin. Neurol. Neurosurg. 2019, 177, 77–85. [Google Scholar] [CrossRef]
- Wang, N.; Zhang, Q.; Luo, L.; Ning, B.; Fang, Y. Beta-Asarone Inhibited Cell Growth And Promoted Autophagy Via P53/Bcl-2/Bclin-1 And P53/Ampk/Mtor Pathways In Human Glioma U251 Cells. J. Cell. Physiol. 2018, 233, 2434–2443. [Google Scholar] [CrossRef]
- Lin, C.J.; Chang, Y.A.; Lin, Y.L.; Liu, S.H.; Chang, C.K.; Chen, R.M. Preclinical Effects Of Honokiol On Treating Glioblastoma Multiforme Via G1 Phase Arrest And Cell Apoptosis. Phytomed. Int. J. Phytother. Phytopharmacol. 2016, 23, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Chen, T.L.; Tseng, Y.Y.; Wu, G.J.; Hsieh, M.H.; Lin, Y.W.; Chen, R.M. Honokiol Induces Autophagic Cell Death In Malignant Glioma Through Reactive Oxygen Species-Mediated Regulation Of The P53/Pi3k/Akt/Mtor Signaling Pathway. Toxicol. Appl. Pharmacol. 2016, 304, 59–69. [Google Scholar] [CrossRef]
- Chio, C.C.; Chen, K.Y.; Chang, C.K.; Chuang, J.Y.; Liu, C.C.; Liu, S.H.; Chen, R.M. Improved Effects Of Honokiol On Temozolomide-Induced Autophagy And Apoptosis Of Drug-Sensitive And -Tolerant Glioma Cells. BMC Cancer 2018, 18, 379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, N.; Mao, J.; Xue, L.; Wang, R.; Zhi, F.; Lan, Q. Carnosic Acid Potentiates The Anticancer Effect Of Temozolomide By Inducing Apoptosis And Autophagy In Glioma. J. Neuro Oncol. 2019, 141, 277–288. [Google Scholar] [CrossRef] [Green Version]
- Luthra, P.M.; Lal, N. Prospective Of Curcumin, A Pleiotropic Signalling Molecule From Curcuma Longa In The Treatment Of Glioblastoma. Eur. J. Med. Chem. 2016, 109, 23–35. [Google Scholar] [CrossRef]
- Grek, C.L.; Sheng, Z.; Naus, C.C.; Sin, W.C.; Gourdie, R.G.; Ghatnekar, G.G. Novel Approach To Temozolomide Resistance In Malignant Glioma: Connexin43-Directed Therapeutics. Curr. Opin. Pharmacol. 2018, 41, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.R.; Tsai, C.H.; Chen, C.C.; Way, T.D.; Kao, J.Y.; Liu, Y.S.; Lin, H.Y.; Lai, S.W.; Lu, D.Y. Curcumin Promotes Connexin 43 Degradation And Temozolomide-Induced Apoptosis In Glioblastoma Cells. Am. J. Chin. Med. 2019, 47, 657–674. [Google Scholar] [CrossRef] [PubMed]
- Silva, V.A.O.; Rosa, M.N.; Miranda-Goncalves, V.; Costa, A.M.; Tansini, A.; Evangelista, A.F.; Martinho, O.; Carloni, A.C.; Jones, C.; Lima, J.P.; et al. Euphol, A Tetracyclic Triterpene, From Euphorbia Tirucalli Induces Autophagy And Sensitizes Temozolomide Cytotoxicity On Glioblastoma Cells. Investig. N. Drugs 2019, 37, 223–237. [Google Scholar] [CrossRef]
- López-Valero, I.; Torres, S.; Salazar-Roa, M.; Garcia-Taboada, E.; Hernandez-Tiedra, S.; Guzman, M.; Sepulveda, J.M.; Velasco, G.; Lorente, M. Optimization Of A Preclinical Therapy Of Cannabinoids In Combination With Temozolomide Against Glioma. Biochem. Pharmacol. 2018, 157, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Soignet, S.L.; Maslak, P.; Wang, Z.G.; Jhanwar, S.; Calleja, E.; Dardashti, L.J.; Corso, D.; Deblasio, A.; Gabrilove, J.; Scheinberg, D.A.; et al. Complete Remission After Treatment Of Acute Promyelocytic Leukemia With Arsenic Trioxide. N. Engl. J. Med. 1998, 339, 1341–1348. [Google Scholar] [CrossRef]
- Zhong, F.; Zhang, S.; Shao, C.; Yang, J.; Wu, X. Arsenic Trioxide Inhibits Cholangiocarcinoma Cell Growth And Induces Apoptosis. Pathol. Oncol. Res. 2010, 16, 413–420. [Google Scholar] [CrossRef]
- Cai, X.; Yu, K.; Zhang, L.; Li, Y.; Li, Q.; Yang, Z.; Shen, T.; Duan, L.; Xiong, W.; Wang, W. Synergistic Inhibition Of Colon Carcinoma Cell Growth By Hedgehog-Gli1 Inhibitor Arsenic Trioxide And Phosphoinositide 3-Kinase Inhibitor Ly294002. Oncotarg. Ther. 2015, 8, 877–883. [Google Scholar] [CrossRef] [Green Version]
- Luo, Q.; Li, Y.; Lai, Y.; Zhang, Z. The Role Of Nf-Kappab In Parp-Inhibitor-Mediated Sensitization And Detoxification Of Arsenic Trioxide In Hepatocellular Carcinoma Cells. J. Toxicol. Sci. 2015, 40, 349–363. [Google Scholar] [CrossRef] [Green Version]
- Miller, W.H., Jr.; Schipper, H.M.; Lee, J.S.; Singer, J.; Waxman, S. Mechanisms Of Action Of Arsenic Trioxide. Cancer Res. 2002, 62, 3893–3903. [Google Scholar]
- Grimm, S.A.; Marymont, M.; Chandler, J.P.; Muro, K.; Newman, S.B.; Levy, R.M.; Jovanovic, B.; Mccarthy, K.; Raizer, J.J. Phase I Study Of Arsenic Trioxide And Temozolomide In Combination With Radiation Therapy In Patients With Malignant Gliomas. J. Neuro Oncol. 2012, 110, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Cohen, K.J.; Gibbs, I.C.; Fisher, P.G.; Hayashi, R.J.; Macy, M.E.; Gore, L. A Phase I Trial Of Arsenic Trioxide Chemoradiotherapy For Infiltrating Astrocytomas Of Childhood. Neuro Oncol. 2013, 15, 783–787. [Google Scholar] [CrossRef] [Green Version]
- Kumthekar, P.; Grimm, S.; Chandler, J.; Mehta, M.; Marymont, M.; Levy, R.; Muro, K.; Helenowski, I.; Mccarthy, K.; Fountas, L.; et al. A Phase Ii Trial Of Arsenic Trioxide And Temozolomide In Combination With Radiation Therapy For Patients With Malignant Gliomas. J. Neuro Oncol. 2017, 133, 589–594. [Google Scholar] [CrossRef]
- Gupta, S.; Yel, L.; Kim, D.; Kim, C.; Chiplunkar, S.; Gollapudi, S. Arsenic Trioxide Induces Apoptosis In Peripheral Blood T Lymphocyte Subsets By Inducing Oxidative Stress: A Role Of Bcl-2. Mol. Cancer Ther. 2003, 2, 711–719. [Google Scholar] [PubMed]
- Sanchez, Y.; Amran, D.; De Blas, E.; Aller, P. Regulation Of Genistein-Induced Differentiation In Human Acute Myeloid Leukaemia Cells (Hl60, Nb4) Protein Kinase Modulation And Reactive Oxygen Species Generation. Biochem. Pharmacol. 2009, 77, 384–396. [Google Scholar] [CrossRef] [Green Version]
- Chou, W.C.; Jie, C.; Kenedy, A.A.; Jones, R.J.; Trush, M.A.; Dang, C.V. Role Of Nadph Oxidase In Arsenic-Induced Reactive Oxygen Species Formation And Cytotoxicity In Myeloid Leukemia Cells. Proc. Natl. Acad. Sci. USA 2004, 101, 4578–4583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davison, K.; Mann, K.K.; Waxman, S.; Miller, W.H., Jr. Jnk Activation Is A Mediator Of Arsenic Trioxide-Induced Apoptosis In Acute Promyelocytic Leukemia Cells. Blood 2004, 103, 3496–3502. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Yamaguchi, H.; Tian, C.; Lee, M.W.; Tang, H.; Wang, H.G.; Chen, Q. Arsenic Trioxide (As(2)O(3)) Induces Apoptosis Through Activation Of Bax In Hematopoietic Cells. Oncogene 2005, 24, 3339–3347. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Wang, C.; Wang, L.; Dai, Z.; Yang, K. Arsenic Trioxide Induces Apoptosis And The Formation Of Reactive Oxygen Species In Rat Glioma Cells. Cell. Mol. Biol. Lett. 2018, 23, 13. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.B.; Liu, J.H.; Hu, J.; Xue, K. Mechanism Of As2o3 Induces Apoptosis Of Glioma U87 Cells. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 4875–4881. [Google Scholar]
- Zhen, Y.; Zhao, S.; Li, Q.; Li, Y.; Kawamoto, K. Arsenic Trioxide-Mediated Notch Pathway Inhibition Depletes The Cancer Stem-Like Cell Population In Gliomas. Cancer Lett. 2010, 292, 64–72. [Google Scholar] [CrossRef]
- Linder, B.; Wehle, A.; Hehlgans, S.; Bonn, F.; Dikic, I.; Rodel, F.; Seifert, V.; Kogel, D. Arsenic Trioxide And (-)-Gossypol Synergistically Target Glioma Stem-Like Cells Via Inhibition Of Hedgehog And Notch Signaling. Cancers 2019, 11, 350. [Google Scholar] [CrossRef] [Green Version]
- Ding, D.; Lim, K.S.; Eberhart, C.G. Arsenic Trioxide Inhibits Hedgehog, Notch And Stem Cell Properties In Glioblastoma Neurospheres. Acta Neuropathol. Commun. 2014, 2, 31. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, Y.; Jiang, C. Mir-133b Contributes To Arsenic-Induced Apoptosis In U251 Glioma Cells By Targeting The Herg Channel. J. Mol. Neurosci. 2015, 55, 985–994. [Google Scholar] [CrossRef]
- Li, Y.; Qu, X.; Qu, J.; Zhang, Y.; Liu, J.; Teng, Y.; Hu, X.; Hou, K.; Liu, Y. Arsenic Trioxide Induces Apoptosis And G2/M Phase Arrest By Inducing Cbl To Inhibit Pi3k/Akt Signaling And Thereby Regulate P53 Activation. Cancer Lett. 2009, 284, 208–215. [Google Scholar] [CrossRef]
- Mathieu, J.; Besancon, F. Clinically Tolerable Concentrations Of Arsenic Trioxide Induce P53-Independent Cell Death And Repress Nf-Kappa B Activation In Ewing Sarcoma Cells. Int. J. Cancer 2006, 119, 1723–1727. [Google Scholar] [CrossRef]
- Ghaffari, S.H.; Yousefi, M.; Dizaji, M.Z.; Momeny, M.; Bashash, D.; Zekri, A.; Alimoghaddam, K.; Ghavamzadeh, A. Arsenic Trioxide Induces Apoptosis And Incapacitates Proliferation And Invasive Properties Of U87mg Glioblastoma Cells Through A Possible Nf-Kappab-Mediated Mechanism. Asian Pac. J. Cancer Prev. APJCP 2016, 17, 1553–1564. [Google Scholar] [CrossRef] [Green Version]
- Scholz, C.; Wieder, T.; Starck, L.; Essmann, F.; Schulze-Osthoff, K.; Dorken, B.; Daniel, P.T. Arsenic Trioxide Triggers A Regulated Form Of Caspase-Independent Necrotic Cell Death Via The Mitochondrial Death Pathway. Oncogene 2005, 24, 1904–1913. [Google Scholar] [CrossRef] [Green Version]
- Mccafferty-Grad, J.; Bahlis, N.J.; Krett, N.; Aguilar, T.M.; Reis, I.; Lee, K.P.; Boise, L.H. Arsenic Trioxide Uses Caspase-Dependent And Caspase-Independent Death Pathways In Myeloma Cells. Mol. Cancer Ther. 2003, 2, 1155–1164. [Google Scholar]
- Karlsson, J.; Ora, I.; Porn-Ares, I.; Pahlman, S. Arsenic Trioxide-Induced Death Of Neuroblastoma Cells Involves Activation Of Bax And Does Not Require P53. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 3179–3188. [Google Scholar] [CrossRef] [Green Version]
- Kanzawa, T.; Zhang, L.; Xiao, L.; Germano, I.M.; Kondo, Y.; Kondo, S. Arsenic Trioxide Induces Autophagic Cell Death In Malignant Glioma Cells By Upregulation Of Mitochondrial Cell Death Protein Bnip3. Oncogene 2005, 24, 980–991. [Google Scholar] [CrossRef] [Green Version]
- Chiu, H.W.; Yeh, Y.L.; Wang, Y.C.; Huang, W.J.; Ho, S.Y.; Lin, P.; Wang, Y.J. Combination Of The Novel Histone Deacetylase Inhibitor Ycw1 And Radiation Induces Autophagic Cell Death Through The Downregulation Of Bnip3 In Triple-Negative Breast Cancer Cells In Vitro And In An Orthotopic Mouse Model. Mol. Cancer 2016, 15, 46. [Google Scholar] [CrossRef] [Green Version]
- Altieri, D.C. Survivin, Cancer Networks And Pathway-Directed Drug Discovery. Nat. Rev. Cancer 2008, 8, 61–70. [Google Scholar] [CrossRef]
- Li, C.; Liu, Y.; Liu, H.; Zhang, W.; Shen, C.; Cho, K.; Chen, X.; Peng, F.; Bi, Y.; Hou, X.; et al. Impact Of Autophagy Inhibition At Different Stages On Cytotoxic Effect Of Autophagy Inducer In Glioblastoma Cells. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2015, 35, 1303–1316. [Google Scholar] [CrossRef]
- Krishna, S.; White, N.J. Pharmacokinetics Of Quinine, Chloroquine And Amodiaquine. Clinical Implications. Clin. Pharmacokinet. 1996, 30, 263–299. [Google Scholar] [CrossRef]
- Al-Bari, M.A. Chloroquine Analogues In Drug Discovery: New Directions Of Uses, Mechanisms Of Actions And Toxic Manifestations From Malaria To Multifarious Diseases. J. Antimicrob. Chemother. 2015, 70, 1608–1621. [Google Scholar] [CrossRef] [Green Version]
- Geng, Y.; Kohli, L.; Klocke, B.J.; Roth, K.A. Chloroquine-Induced Autophagic Vacuole Accumulation And Cell Death In Glioma Cells Is P53 Independent. Neuro Oncol. 2010, 12, 473–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jutten, B.; Keulers, T.G.; Peeters, H.J.M.; Schaaf, M.B.E.; Savelkouls, K.G.M.; Compter, I.; Clarijs, R.; Schijns, O.; Ackermans, L.; Teernstra, O.P.M.; et al. Egfrviii Expression Triggers A Metabolic Dependency And Therapeutic Vulnerability Sensitive To Autophagy Inhibition. Autophagy 2018, 14, 283–295. [Google Scholar] [CrossRef]
- Briceno, E.; Reyes, S.; Sotelo, J. Therapy Of Glioblastoma Multiforme Improved By The Antimutagenic Chloroquine. Neurosurg. Focus 2003, 14, E3. [Google Scholar] [CrossRef]
- Sotelo, J.; Briceno, E.; Lopez-Gonzalez, M.A. Adding Chloroquine To Conventional Treatment For Glioblastoma Multiforme: A Randomized, Double-Blind, Placebo-Controlled Trial. Ann. Intern. Med. 2006, 144, 337–343. [Google Scholar] [CrossRef]
- Briceno, E.; Calderon, A.; Sotelo, J. Institutional Experience With Chloroquine As An Adjuvant To The Therapy For Glioblastoma Multiforme. Surg. Neurol. 2007, 67, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Golden, E.B.; Cho, H.Y.; Jahanian, A.; Hofman, F.M.; Louie, S.G.; Schonthal, A.H.; Chen, T.C. Chloroquine Enhances Temozolomide Cytotoxicity In Malignant Gliomas By Blocking Autophagy. Neurosurg. Focus 2014, 37, E12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, H.; Chen, M.; Cao, F.; Huang, H.; Zhan, R.; Zheng, X. Chloroquine, An Autophagy Inhibitor, Potentiates The Radiosensitivity Of Glioma Initiating Cells By Inhibiting Autophagy And Activating Apoptosis. BMC Neurol. 2016, 16, 178. [Google Scholar] [CrossRef] [Green Version]
- Firat, E.; Weyerbrock, A.; Gaedicke, S.; Grosu, A.L.; Niedermann, G. Chloroquine Or Chloroquine-Pi3k/Akt Pathway Inhibitor Combinations Strongly Promote Gamma-Irradiation-Induced Cell Death In Primary Stem-Like Glioma Cells. PLoS ONE 2012, 7, E47357. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhang, M.; Cheng, K.; Li, P.; Han, S.; Li, R.; Su, M.; Zeng, W.; Liu, J.; Guo, J.; et al. Resistance Of Glioma Cells To Nutrient-Deprived Microenvironment Can Be Enhanced By Cd133-Mediated Autophagy. Oncotarget 2016, 7, 76238–76249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griguer, C.E.; Oliva, C.R.; Gobin, E.; Marcorelles, P.; Benos, D.J.; Lancaster, J.R., Jr.; Gillespie, G.Y. Cd133 Is A Marker Of Bioenergetic Stress In Human Glioma. PLoS ONE 2008, 3, E3655. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Sun, K.; Wang, H.; Dai, Y. Inhibition Of Autophagy By Chloroquine Enhances The Antitumor Efficacy Of Sorafenib In Glioblastoma. Cell. Mol. Neurobiol. 2016, 36, 1197–1208. [Google Scholar] [CrossRef]
- Pernicova, I.; Korbonits, M. Metformin—Mode Of Action And Clinical Implications For Diabetes And Cancer. Nat. Rev. Endocrinol. 2014, 10, 143–156. [Google Scholar] [CrossRef]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin And Reduced Risk Of Cancer In Diabetic Patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [Green Version]
- Sesen, J.; Dahan, P.; Scotland, S.J.; Saland, E.; Dang, V.T.; Lemarie, A.; Tyler, B.M.; Brem, H.; Toulas, C.; Cohen-Jonathan Moyal, E.; et al. Metformin Inhibits Growth Of Human Glioblastoma Cells And Enhances Therapeutic Response. PLoS ONE 2015, 10, E0123721. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.B.; Tian, S.; Gao, H.H.; Xu, Y.Y. Metformin Inhibits Glioma Cell U251 Invasion By Downregulation Of Fibulin-3. Neuroreport 2013, 24, 504–508. [Google Scholar] [CrossRef] [PubMed]
- Leidgens, V.; Proske, J.; Rauer, L.; Moeckel, S.; Renner, K.; Bogdahn, U.; Riemenschneider, M.J.; Proescholdt, M.; Vollmann-Zwerenz, A.; Hau, P.; et al. Stattic And Metformin Inhibit Brain Tumor Initiating Cells By Reducing Stat3-Phosphorylation. Oncotarget 2017, 8, 8250–8263. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.H.; Li, S.; Lu, G.; Xue, H.; Kim, D.H.; Zhu, J.J.; Liu, Y. Metformin Treatment Reduces Temozolomide Resistance Of Glioblastoma Cells. Oncotarget 2016, 7, 78787–78803. [Google Scholar] [CrossRef] [Green Version]
- Aldea, M.D.; Petrushev, B.; Soritau, O.; Tomuleasa, C.I.; Berindan-Neagoe, I.; Filip, A.G.; Chereches, G.; Cenariu, M.; Craciun, L.; Tatomir, C.; et al. Metformin Plus Sorafenib Highly Impacts Temozolomide Resistant Glioblastoma Stem-Like Cells. J. B.U. On. Off. J. Balk. Union Of. 2014, 19, 502–511. [Google Scholar]
- Kim, H.G.; Hien, T.T.; Han, E.H.; Hwang, Y.P.; Choi, J.H.; Kang, K.W.; Kwon, K.I.; Kim, B.H.; Kim, S.K.; Song, G.Y.; et al. Metformin Inhibits P-Glycoprotein Expression Via The Nf-Kappab Pathway And Cre Transcriptional Activity Through Ampk Activation. Br. J. Pharmacol. 2011, 162, 1096–1108. [Google Scholar] [CrossRef] [Green Version]
- Sato, A.; Sunayama, J.; Okada, M.; Watanabe, E.; Seino, S.; Shibuya, K.; Suzuki, K.; Narita, Y.; Shibui, S.; Kayama, T.; et al. Glioma-Initiating Cell Elimination By Metformin Activation Of Foxo3 Via Ampk. Stem Cells Transl. Med. 2012, 1, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Regan, L.S. Screening For Hepatocellular Carcinoma In High-Risk Individuals. A Clinical Review. Arch. Int. Med. 1989, 149, 1741–1744. [Google Scholar] [CrossRef]
- Seliger, C.; Ricci, C.; Meier, C.R.; Bodmer, M.; Jick, S.S.; Bogdahn, U.; Hau, P.; Leitzmann, M.F. Diabetes, Use Of Antidiabetic Drugs, And The Risk Of Glioma. Neuro Oncol. 2016, 18, 340–349. [Google Scholar] [CrossRef] [Green Version]
- Adeberg, S.; Bernhardt, D.; Foerster, R.; Bostel, T.; Koerber, S.A.; Mohr, A.; Koelsche, C.; Rieken, S.; Debus, J. The Influence Of Hyperglycemia During Radiotherapy On Survival In Patients With Primary Glioblastoma. Acta Oncol. 2016, 55, 201–207. [Google Scholar] [CrossRef] [Green Version]
- Seliger, C.; Luber, C.; Gerken, M.; Schaertl, J.; Proescholdt, M.; Riemenschneider, M.J.; Meier, C.R.; Bogdahn, U.; Leitzmann, M.F.; Klinkhammer-Schalke, M.; et al. Use Of Metformin And Survival Of Patients With High-Grade Glioma. Int. J. Cancer 2019, 144, 273–280. [Google Scholar] [CrossRef] [Green Version]
- Seliger, C.; Genbrugge, E.; Gorlia, T.; Chinot, O.; Stupp, R.; Nabors, B.; Weller, M.; Hau, P. EORTC Brain Tumor Group. Use Of Metformin And Outcome Of Patients With Newly Diagnosed Glioblastoma: Pooled Analysis. Int. J. Cancer 2019. [Google Scholar] [CrossRef]
- Ohgaki, H.; Kleihues, P. Population-Based Studies On Incidence, Survival Rates, And Genetic Alterations In Astrocytic And Oligodendroglial Gliomas. J. Neuropathol. Exp. Neurol. 2005, 64, 479–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quatrale, A.E.; Porcelli, L.; Silvestris, N.; Colucci, G.; Angelo, A.; Azzariti, A. Egfr Tyrosine Kinases Inhibitors In Cancer Treatment: In Vitro And In Vivo Evidence. Front. Biosci. 2011, 16, 1962–1972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eimer, S.; Belaud-Rotureau, M.A.; Airiau, K.; Jeanneteau, M.; Laharanne, E.; Veron, N.; Vital, A.; Loiseau, H.; Merlio, J.P.; Belloc, F. Autophagy Inhibition Cooperates With Erlotinib To Induce Glioblastoma Cell Death. Cancer Biol. Ther. 2011, 11, 1017–1027. [Google Scholar] [CrossRef] [Green Version]
- Shingu, T.; Holmes, L.; Henry, V.; Wang, Q.; Latha, K.; Gururaj, A.E.; Gibson, L.A.; Doucette, T.; Lang, F.F.; Rao, G.; et al. Suppression Of Raf/Mek Or Pi3k Synergizes Cytotoxicity Of Receptor Tyrosine Kinase Inhibitors In Glioma Tumor-Initiating Cells. J. Transl. Med. 2016, 14, 46. [Google Scholar] [CrossRef] [Green Version]
- Mazurek, S.; Boschek, C.B.; Hugo, F.; Eigenbrodt, E. Pyruvate Kinase Type M2 And Its Role In Tumor Growth And Spreading. Sem. Cancer Biol. 2005, 15, 300–308. [Google Scholar] [CrossRef]
- Yang, W.; Xia, Y.; Ji, H.; Zheng, Y.; Liang, J.; Huang, W.; Gao, X.; Aldape, K.; Lu, Z. Nuclear Pkm2 Regulates Beta-Catenin Transactivation Upon Egfr Activation. Nature 2011, 480, 118–122. [Google Scholar] [CrossRef]
- Karpel-Massler, G.; Westhoff, M.A.; Kast, R.E.; Dwucet, A.; Karpel-Massler, S.; Nonnenmacher, L.; Siegelin, M.D.; Wirtz, C.R.; Debatin, K.M.; Halatsch, M.E. Simultaneous Interference With Her1/Egfr And Rac1 Signaling Drives Cytostasis And Suppression Of Survivin In Human Glioma Cells In Vitro. Neurochem. Res. 2017, 42, 1543–1554. [Google Scholar] [CrossRef]
- Van Den Bent, M.J.; Brandes, A.A.; Rampling, R.; Kouwenhoven, M.C.; Kros, J.M.; Carpentier, A.F.; Clement, P.M.; Frenay, M.; Campone, M.; Baurain, J.F.; et al. Randomized Phase Ii Trial Of Erlotinib Versus Temozolomide Or Carmustine In Recurrent Glioblastoma: Eortc Brain Tumor Group Study 26034. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 1268–1274. [Google Scholar] [CrossRef] [Green Version]
- Prados, M.D.; Chang, S.M.; Butowski, N.; Deboer, R.; Parvataneni, R.; Carliner, H.; Kabuubi, P.; Ayers-Ringler, J.; Rabbitt, J.; Page, M.; et al. Phase Ii Study Of Erlotinib Plus Temozolomide During And After Radiation Therapy In Patients With Newly Diagnosed Glioblastoma Multiforme Or Gliosarcoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 579–584. [Google Scholar] [CrossRef] [Green Version]
- Raizer, J.J.; Giglio, P.; Hu, J.; Groves, M.; Merrell, R.; Conrad, C.; Phuphanich, S.; Puduvalli, V.K.; Loghin, M.; Paleologos, N.; et al. A Phase Ii Study Of Bevacizumab And Erlotinib After Radiation And Temozolomide In Mgmt Unmethylated Gbm Patients. J. Neuro Oncol. 2016, 126, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, J.L.; Molinaro, A.M.; Phillips, J.J.; Butowski, N.A.; Chang, S.M.; Perry, A.; Costello, J.F.; Desilva, A.A.; Rabbitt, J.E.; Prados, M.D. A Single-Institution Phase Ii Trial Of Radiation, Temozolomide, Erlotinib, And Bevacizumab For Initial Treatment Of Glioblastoma. Neuro Oncol. 2014, 16, 984–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qaddoumi, I.; Kocak, M.; Pai Panandiker, A.S.; Armstrong, G.T.; Wetmore, C.; Crawford, J.R.; Lin, T.; Boyett, J.M.; Kun, L.E.; Boop, F.A.; et al. Phase Ii Trial Of Erlotinib During And After Radiotherapy In Children With Newly Diagnosed High-Grade Gliomas. Front. Oncol. 2014, 4, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, P.Y.; Chang, S.M.; Lamborn, K.R.; Kuhn, J.G.; Norden, A.D.; Cloughesy, T.F.; Robins, H.I.; Lieberman, F.S.; Gilbert, M.R.; Mehta, M.P.; et al. Phase I/Ii Study Of Erlotinib And Temsirolimus For Patients With Recurrent Malignant Gliomas: North American Brain Tumor Consortium Trial 04-02. Neuro Oncol. 2014, 16, 567–578. [Google Scholar] [CrossRef] [Green Version]
- Carrasco-Garcia, E.; Saceda, M.; Grasso, S.; Rocamora-Reverte, L.; Conde, M.; Gomez-Martinez, A.; Garcia-Morales, P.; Ferragut, J.A.; Martinez-Lacaci, I. Small Tyrosine Kinase Inhibitors Interrupt Egfr Signaling By Interacting With Erbb3 And Erbb4 In Glioblastoma Cell Lines. Exp. Cell Res. 2011, 317, 1476–1489. [Google Scholar] [CrossRef]
- Parker, J.J.; Dionne, K.R.; Massarwa, R.; Klaassen, M.; Foreman, N.K.; Niswander, L.; Canoll, P.; Kleinschmidt-Demasters, B.K.; Waziri, A. Gefitinib Selectively Inhibits Tumor Cell Migration In Egfr-Amplified Human Glioblastoma. Neuro Oncol. 2013, 15, 1048–1057. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.Y.; Shen, C.C.; Su, H.L.; Chen, C.J. Gefitinib Induces Apoptosis In Human Glioma Cells By Targeting Bad Phosphorylation. J. Neuro Oncol. 2011, 105, 507–522. [Google Scholar] [CrossRef]
- Chang, C.Y.; Kuan, Y.H.; Ou, Y.C.; Li, J.R.; Wu, C.C.; Pan, P.H.; Chen, W.Y.; Huang, H.Y.; Chen, C.J. Autophagy Contributes To Gefitinib-Induced Glioma Cell Growth Inhibition. Exp. Cell Res. 2014, 327, 102–112. [Google Scholar] [CrossRef]
- Cheng, Y.; Zhang, Y.; Zhang, L.; Ren, X.; Huber-Keener, K.J.; Liu, X.; Zhou, L.; Liao, J.; Keihack, H.; Yan, L.; et al. Mk-2206, A Novel Allosteric Inhibitor Of Akt, Synergizes With Gefitinib Against Malignant Glioma Via Modulating Both Autophagy And Apoptosis. Mol. Cancer Ther. 2012, 11, 154–164. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.Y.; Li, J.R.; Wu, C.C.; Ou, Y.C.; Chen, W.Y.; Kuan, Y.H.; Wang, W.Y.; Chen, C.J. Valproic Acid Sensitizes Human Glioma Cells To Gefitinib-Induced Autophagy. IUBMB Life 2015, 67, 869–879. [Google Scholar] [CrossRef] [Green Version]
- Mu, L.; Wang, T.; Chen, Y.; Tang, X.; Yuan, Y.; Zhao, Y. Beta-Elemene Enhances The Efficacy Of Gefitinib On Glioblastoma Multiforme Cells Through The Inhibition Of The Egfr Signaling Pathway. Int. J. Oncol. 2016, 49, 1427–1436. [Google Scholar] [CrossRef] [PubMed]
- Doherty, L.; Gigas, D.C.; Kesari, S.; Drappatz, J.; Kim, R.; Zimmerman, J.; Ostrowsky, L.; Wen, P.Y. Pilot Study Of The Combination Of Egfr And Mtor Inhibitors In Recurrent Malignant Gliomas. Neurology 2006, 67, 156–158. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, A.; Wang, M.; Robins, H.I.; Lautenschlaeger, T.; Curran, W.J.; Brachman, D.G.; Schultz, C.J.; Choucair, A.; Dolled-Filhart, M.; Christiansen, J.; et al. Rtog 0211: A Phase 1/2 Study Of Radiation Therapy With Concurrent Gefitinib For Newly Diagnosed Glioblastoma Patients. Int. J. Radiat. Oncol. Biol. Phys. 2013, 85, 1206–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhm, J.H.; Ballman, K.V.; Wu, W.; Giannini, C.; Krauss, J.C.; Buckner, J.C.; James, C.D.; Scheithauer, B.W.; Behrens, R.J.; Flynn, P.J.; et al. Phase Ii Evaluation Of Gefitinib In Patients With Newly Diagnosed Grade 4 Astrocytoma: Mayo/North Central Cancer Treatment Group Study N0074. Int. J. Radiat. Oncol. Biol. Phys. 2011, 80, 347–353. [Google Scholar] [CrossRef] [Green Version]
- Rich, J.N.; Reardon, D.A.; Peery, T.; Dowell, J.M.; Quinn, J.A.; Penne, K.L.; Wikstrand, C.J.; Van Duyn, L.B.; Dancey, J.E.; Mclendon, R.E.; et al. Phase Ii Trial Of Gefitinib In Recurrent Glioblastoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2004, 22, 133–142. [Google Scholar] [CrossRef]
- Engelman, J.A.; Zejnullahu, K.; Gale, C.M.; Lifshits, E.; Gonzales, A.J.; Shimamura, T.; Zhao, F.; Vincent, P.W.; Naumov, G.N.; Bradner, J.E.; et al. Pf00299804, An Irreversible Pan-Erbb Inhibitor, Is Effective In Lung Cancer Models With Egfr And Erbb2 Mutations That Are Resistant To Gefitinib. Cancer Res. 2007, 67, 11924–11932. [Google Scholar] [CrossRef] [Green Version]
- Endersby, R.; Whitehouse, J.; Hii, H.; Greenall, S.A.; Johns, T.G.; Gottardo, N.G. A Pre-Clinical Assessment Of The Pan-Erbb Inhibitor Dacomitinib In Pediatric And Adult Brain Tumors. Neoplasia 2018, 20, 432–442. [Google Scholar] [CrossRef]
- Zahonero, C.; Aguilera, P.; Ramirez-Castillejo, C.; Pajares, M.; Bolos, M.V.; Cantero, D.; Perez-Nunez, A.; Hernandez-Lain, A.; Sanchez-Gomez, P.; Sepulveda, J.M. Preclinical Test Of Dacomitinib, An Irreversible Egfr Inhibitor, Confirms Its Effectiveness For Glioblastoma. Mol. Cancer Ther. 2015, 14, 1548–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Shah, K. Multiple Lesions In Receptor Tyrosine Kinase Pathway Determine Glioblastoma Response To Pan-Erbb Inhibitor Pf-00299804 And Pi3k/Mtor Dual Inhibitor Pf-05212384. Cancer Biol. Ther. 2014, 15, 815–822. [Google Scholar] [CrossRef] [Green Version]
- Sepulveda, J.M.; Sanchez-Gomez, P.; Vaz Salgado, M.A.; Gargini, R.; Balana, C. Dacomitinib: An Investigational Drug For The Treatment Of Glioblastoma. Exp. Opin. Investig. Drugs 2018, 27, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda-Sanchez, J.M.; Vaz, M.A.; Balana, C.; Gil-Gil, M.; Reynes, G.; Gallego, O.; Martinez-Garcia, M.; Vicente, E.; Quindos, M.; Luque, R.; et al. Phase Ii Trial Of Dacomitinib, A Pan-Human Egfr Tyrosine Kinase Inhibitor, In Recurrent Glioblastoma Patients With Egfr Amplification. Neuro Oncol. 2017, 19, 1522–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Zou, Z.; Becker, N.; Anderson, M.; Sumpter, R.; Xiao, G.; Kinch, L.; Koduru, P.; Christudass, C.S.; Veltri, R.W.; et al. Egfr-Mediated Beclin 1 Phosphorylation In Autophagy Suppression, Tumor Progression, And Tumor Chemoresistance. Cell 2013, 154, 1269–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.; Jia, L.; Wang, X.; Tan, X.; Xu, J.; Deng, Z.; Jiang, T.; Rainov, N.G.; Li, B.; Ren, H. Selective Inhibition Of Pdgfr By Imatinib Elicits The Sustained Activation Of Erk And Downstream Receptor Signaling In Malignant Glioma Cells. Int. J. Oncol. 2011, 38, 555–569. [Google Scholar] [CrossRef] [PubMed]
- Shingu, T.; Fujiwara, K.; Bogler, O.; Akiyama, Y.; Moritake, K.; Shinojima, N.; Tamada, Y.; Yokoyama, T.; Kondo, S. Inhibition Of Autophagy At A Late Stage Enhances Imatinib-Induced Cytotoxicity In Human Malignant Glioma Cells. Int. J. Cancer 2009, 124, 1060–1071. [Google Scholar] [CrossRef] [PubMed]
- Bilir, A.; Erguven, M.; Oktem, G.; Ozdemir, A.; Uslu, A.; Aktas, E.; Bonavida, B. Potentiation Of Cytotoxicity By Combination Of Imatinib And Chlorimipramine In Glioma. Int. J. Oncol. 2008, 32, 829–839. [Google Scholar]
- Xia, Z.; Lundgren, B.; Bergstrand, A.; Depierre, J.W.; Nassberger, L. Changes In The Generation Of Reactive Oxygen Species And In Mitochondrial Membrane Potential During Apoptosis Induced By The Antidepressants Imipramine, Clomipramine, And Citalopram And The Effects On These Changes By Bcl-2 And Bcl-X(L). Biochem. Pharmacol. 1999, 57, 1199–1208. [Google Scholar] [CrossRef]
- Erguven, M.; Yazihan, N.; Aktas, E.; Sabanci, A.; Li, C.J.; Oktem, G.; Bilir, A. Carvedilol In Glioma Treatment Alone And With Imatinib In Vitro. Int. J. Oncol. 2010, 36, 857–866. [Google Scholar] [CrossRef] [Green Version]
- Frolov, A.; Evans, I.M.; Li, N.; Sidlauskas, K.; Paliashvili, K.; Lockwood, N.; Barrett, A.; Brandner, S.; Zachary, I.C.; Frankel, P. Imatinib And Nilotinib Increase Glioblastoma Cell Invasion Via Abl-Independent Stimulation Of P130cas And Fak Signalling. Sci. Rep. 2016, 6, 27378. [Google Scholar] [CrossRef]
- Au, K.; Singh, S.K.; Burrell, K.; Sabha, N.; Hawkins, C.; Huang, A.; Zadeh, G. A Preclinical Study Demonstrating The Efficacy Of Nilotinib In Inhibiting The Growth Of Pediatric High-Grade Glioma. J. Neuro Oncol. 2015, 122, 471–480. [Google Scholar] [CrossRef] [Green Version]
- Raymond, E.; Brandes, A.A.; Dittrich, C.; Fumoleau, P.; Coudert, B.; Clement, P.M.; Frenay, M.; Rampling, R.; Stupp, R.; Kros, J.M.; et al. Phase Ii Study Of Imatinib In Patients With Recurrent Gliomas Of Various Histologies: A European Organisation For Research And Treatment Of Cancer Brain Tumor Group Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 4659–4665. [Google Scholar] [CrossRef] [Green Version]
- Wen, P.Y.; Yung, W.K.; Lamborn, K.R.; Dahia, P.L.; Wang, Y.; Peng, B.; Abrey, L.E.; Raizer, J.; Cloughesy, T.F.; Fink, K.; et al. Phase I/Ii Study Of Imatinib Mesylate For Recurrent Malignant Gliomas: North American Brain Tumor Consortium Study 99-08. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 4899–4907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reardon, D.A.; Egorin, M.J.; Quinn, J.A.; Rich, J.N.; Gururangan, S.; Vredenburgh, J.J.; Desjardins, A.; Sathornsumetee, S.; Provenzale, J.M.; Herndon, J.E., 2nd; et al. Phase Ii Study Of Imatinib Mesylate Plus Hydroxyurea In Adults With Recurrent Glioblastoma Multiforme. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 9359–9368. [Google Scholar] [CrossRef] [PubMed]
- Faivre, S.; Demetri, G.; Sargent, W.; Raymond, E. Molecular Basis For Sunitinib Efficacy And Future Clinical Development. Nat. Rev. Drug Discov. 2007, 6, 734–745. [Google Scholar] [CrossRef]
- De Bouard, S.; Herlin, P.; Christensen, J.G.; Lemoisson, E.; Gauduchon, P.; Raymond, E.; Guillamo, J.S. Antiangiogenic And Anti-Invasive Effects Of Sunitinib On Experimental Human Glioblastoma. Neuro Oncol. 2007, 9, 412–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmaksiz, G.; Czabanka, M.; Vinci, M.; Vajkoczy, P. Antiangiogenic Therapy Inhibits The Recruitment Of Vascular Accessory Cells To The Perivascular Niche In Glioma Angiogenesis. J. Vasc. Res. 2014, 51, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, M.; Avraham, I.; Dor, Y.; Bachar-Lustig, E.; Itin, A.; Jung, S.; Chimenti, S.; Landsman, L.; Abramovitch, R.; Keshet, E. Vegf-Induced Adult Neovascularization: Recruitment, Retention, And Role Of Accessory Cells. Cell 2006, 124, 175–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiedmer, T.; Blank, A.; Pantasis, S.; Normand, L.; Bill, R.; Krebs, P.; Tschan, M.P.; Marinoni, I.; Perren, A. Autophagy Inhibition Improves Sunitinib Efficacy In Pancreatic Neuroendocrine Tumors Via A Lysosome-Dependent Mechanism. Mol. Cancer Ther. 2017, 16, 2502–2515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Aziz, A.K.; Shouman, S.; El-Demerdash, E.; Elgendy, M.; Abdel-Naim, A.B. Chloroquine Synergizes Sunitinib Cytotoxicity Via Modulating Autophagic, Apoptotic And Angiogenic Machineries. Chem. Biol. Interact. 2014, 217, 28–40. [Google Scholar] [CrossRef]
- Li, M.L.; Xu, Y.Z.; Lu, W.J.; Li, Y.H.; Tan, S.S.; Lin, H.J.; Wu, T.M.; Li, Y.; Wang, S.Y.; Zhao, Y.L. Chloroquine Potentiates The Anticancer Effect Of Sunitinib On Renal Cell Carcinoma By Inhibiting Autophagy And Inducing Apoptosis. Oncol. Lett. 2018, 15, 2839–2846. [Google Scholar] [CrossRef]
- Navone, S.E.; Guarnaccia, L.; Cordiglieri, C.; Crisa, F.M.; Caroli, M.; Locatelli, M.; Schisano, L.; Rampini, P.; Miozzo, M.; La Verde, N.; et al. Aspirin Affects Tumor Angiogenesis And Sensitizes Human Glioblastoma Endothelial Cells To Temozolomide, Bevacizumab, And Sunitinib, Impairing Vascular Endothelial Growth Factor-Related Signaling. World Neurosurg. 2018, 120, E380–E391. [Google Scholar] [CrossRef]
- Joshi, A.D.; Loilome, W.; Siu, I.M.; Tyler, B.; Gallia, G.L.; Riggins, G.J. Evaluation Of Tyrosine Kinase Inhibitor Combinations For Glioblastoma Therapy. PLoS ONE 2012, 7, E44372. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liu, Z.; Jiang, B.; Huo, L.; Liu, J.; Lu, J. Synthetic Mir-145 Mimic Enhances The Cytotoxic Effect Of The Antiangiogenic Drug Sunitinib In Glioblastoma. Cell Biochem. Biophys. 2015, 72, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Czabanka, M.; Bruenner, J.; Parmaksiz, G.; Broggini, T.; Topalovic, M.; Bayerl, S.H.; Auf, G.; Kremenetskaia, I.; Nieminen, M.; Jabouille, A.; et al. Combined Temozolomide And Sunitinib Treatment Leads To Better Tumour Control But Increased Vascular Resistance In O6-Methylguanine Methyltransferase-Methylated Gliomas. Eur. J. Cancer 2013, 49, 2243–2252. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Guo, P.; Gallo, J.M. Impact Of Angiogenesis Inhibition By Sunitinib On Tumor Distribution Of Temozolomide. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 1540–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhl, C.; Markel, M.; Broggini, T.; Nieminen, M.; Kremenetskaia, I.; Vajkoczy, P.; Czabanka, M. Ephb4 Mediates Resistance To Antiangiogenic Therapy In Experimental Glioma. Angiogenesis 2018, 21, 873–881. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Liu, X.; Yi, S.; Zhang, J.; Ge, J.; Liu, Z. Ephb4 Is Overexpressed In Gliomas And Promotes The Growth Of Glioma Cells. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2013, 34, 379–385. [Google Scholar] [CrossRef]
- Xu, L.; Duda, D.G.; Di Tomaso, E.; Ancukiewicz, M.; Chung, D.C.; Lauwers, G.Y.; Samuel, R.; Shellito, P.; Czito, B.G.; Lin, P.C.; et al. Direct Evidence That Bevacizumab, An Anti-Vegf Antibody, Up-Regulates Sdf1alpha, Cxcr4, Cxcl6, And Neuropilin 1 In Tumors From Patients With Rectal Cancer. Cancer Res. 2009, 69, 7905–7910. [Google Scholar] [CrossRef] [Green Version]
- Gravina, G.L.; Mancini, A.; Marampon, F.; Colapietro, A.; Delle Monache, S.; Sferra, R.; Vitale, F.; Richardson, P.J.; Patient, L.; Burbidge, S.; et al. The Brain-Penetrating Cxcr4 Antagonist, Prx177561, Increases The Antitumor Effects Of Bevacizumab And Sunitinib In Preclinical Models Of Human Glioblastoma. J. Hematol. Oncol. 2017, 10, 5. [Google Scholar] [CrossRef] [Green Version]
- Hutterer, M.; Nowosielski, M.; Haybaeck, J.; Embacher, S.; Stockhammer, F.; Gotwald, T.; Holzner, B.; Capper, D.; Preusser, M.; Marosi, C.; et al. A Single-Arm Phase Ii Austrian/German Multicenter Trial On Continuous Daily Sunitinib In Primary Glioblastoma At First Recurrence (Surge 01-07). Neuro Oncol. 2014, 16, 92–102. [Google Scholar] [CrossRef] [Green Version]
- Reardon, D.A.; Vredenburgh, J.J.; Coan, A.; Desjardins, A.; Peters, K.B.; Gururangan, S.; Sathornsumetee, S.; Rich, J.N.; Herndon, J.E.; Friedman, H.S. Phase I Study Of Sunitinib And Irinotecan For Patients With Recurrent Malignant Glioma. J. Neuro Oncol. 2011, 105, 621–627. [Google Scholar] [CrossRef] [Green Version]
- Reardon, D.A.; Vredenburgh, J.J.; Desjardins, A.; Peters, K.; Gururangan, S.; Sampson, J.H.; Marcello, J.; Herndon, J.E., 2nd; Mclendon, R.E.; Janney, D.; et al. Effect Of Cyp3a-Inducing Anti-Epileptics On Sorafenib Exposure: Results Of A Phase Ii Study Of Sorafenib Plus Daily Temozolomide In Adults With Recurrent Glioblastoma. J. Neuro Oncol. 2011, 101, 57–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Xia, L.; Gabrilove, J.; Waxman, S.; Jing, Y. Sorafenib Inhibition Of Mcl-1 Accelerates Atra-Induced Apoptosis In Differentiation-Responsive Aml Cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 1211–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Cao, Y.; Chen, C.; Zhang, X.; Mcnabola, A.; Wilkie, D.; Wilhelm, S.; Lynch, M.; Carter, C. Sorafenib Blocks The Raf/Mek/Erk Pathway, Inhibits Tumor Angiogenesis, And Induces Tumor Cell Apoptosis In Hepatocellular Carcinoma Model Plc/Prf/5. Cancer Res. 2006, 66, 11851–11858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clavreul, A.; Roger, E.; Pourbaghi-Masouleh, M.; Lemaire, L.; Tetaud, C.; Menei, P. Development And Characterization Of Sorafenib-Loaded Lipid Nanocapsules For The Treatment Of Glioblastoma. Drug Deliv. 2018, 25, 1756–1765. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Zhan, S.; Zhou, D.; Wang, P.; Chen, G.; Qin, K.; Lin, X. Characterizing Nanoscale Changes In The Activity Of Vegfr-2 On Glioma Microvascular Endothelial Cell Membranes Using Atomic Force Microscopy. Exp. Ther. Med. 2017, 13, 483–488. [Google Scholar] [CrossRef] [Green Version]
- Carra, E.; Barbieri, F.; Marubbi, D.; Pattarozzi, A.; Favoni, R.E.; Florio, T.; Daga, A. Sorafenib Selectively Depletes Human Glioblastoma Tumor-Initiating Cells From Primary Cultures. Cell Cycle 2013, 12, 491–500. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Brown, C.; Buettner, R.; Hedvat, M.; Starr, R.; Scuto, A.; Schroeder, A.; Jensen, M.; Jove, R. Sorafenib Induces Growth Arrest And Apoptosis Of Human Glioblastoma Cells Through The Dephosphorylation Of Signal Transducers And Activators Of Transcription 3. Mol. Cancer Ther. 2010, 9, 953–962. [Google Scholar] [CrossRef] [Green Version]
- Kiprianova, I.; Remy, J.; Milosch, N.; Mohrenz, I.V.; Seifert, V.; Aigner, A.; Kogel, D. Sorafenib Sensitizes Glioma Cells To The Bh3 Mimetic Abt-737 By Targeting Mcl1 In A Stat3-Dependent Manner. Neoplasia 2015, 17, 564–573. [Google Scholar] [CrossRef] [Green Version]
- Jo, Y.; Kim, E.H.; Sai, S.; Kim, J.S.; Cho, J.M.; Kim, H.; Baek, J.H.; Kim, J.Y.; Hwang, S.G.; Yoon, M. Functional Biological Activity Of Sorafenib As A Tumor-Treating Field Sensitizer For Glioblastoma Therapy. Int. J. Mol. Sci. 2018, 19, 3684. [Google Scholar] [CrossRef] [Green Version]
- Hamed, H.A.; Tavallai, S.; Grant, S.; Poklepovic, A.; Dent, P. Sorafenib/Regorafenib And Lapatinib Interact To Kill Cns Tumor Cells. J. Cell. Physiol. 2015, 230, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.Q.; Kuhn, J.; Lamborn, K.R.; Abrey, L.; Deangelis, L.M.; Lieberman, F.; Robins, H.I.; Chang, S.M.; Yung, W.K.; Drappatz, J.; et al. Phase I/Ii Study Of Sorafenib In Combination With Temsirolimus For Recurrent Glioblastoma Or Gliosarcoma: North American Brain Tumor Consortium Study 05-02. Neuro Oncol. 2012, 14, 1511–1518. [Google Scholar] [CrossRef] [Green Version]
- Schiff, D.; Jaeckle, K.A.; Anderson, S.K.; Galanis, E.; Giannini, C.; Buckner, J.C.; Stella, P.; Flynn, P.J.; Erickson, B.J.; Schwerkoske, J.F.; et al. Phase 1/2 Trial Of Temsirolimus And Sorafenib In The Treatment Of Patients With Recurrent Glioblastoma: North Central Cancer Treatment Group Study/Alliance N0572. Cancer 2018, 124, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Hottinger, A.F.; Ben Aissa, A.; Espeli, V.; Squiban, D.; Dunkel, N.; Vargas, M.I.; Hundsberger, T.; Mach, N.; Schaller, K.; Weber, D.C.; et al. Phase I Study Of Sorafenib Combined With Radiation Therapy And Temozolomide As First-Line Treatment Of High-Grade Glioma. Br. J. Cancer 2014, 110, 2655–2661. [Google Scholar] [CrossRef] [PubMed]
- Zustovich, F.; Landi, L.; Lombardi, G.; Porta, C.; Galli, L.; Fontana, A.; Amoroso, D.; Galli, C.; Andreuccetti, M.; Falcone, A.; et al. Sorafenib Plus Daily Low-Dose Temozolomide For Relapsed Glioblastoma: A Phase Ii Study. Anticancer Res. 2013, 33, 3487–3494. [Google Scholar] [CrossRef] [PubMed]
- Galanis, E.; Anderson, S.K.; Lafky, J.M.; Uhm, J.H.; Giannini, C.; Kumar, S.K.; Kimlinger, T.K.; Northfelt, D.W.; Flynn, P.J.; Jaeckle, K.A.; et al. Phase Ii Study Of Bevacizumab In Combination With Sorafenib In Recurrent Glioblastoma (N0776): A North Central Cancer Treatment Group Trial. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 4816–4823. [Google Scholar] [CrossRef] [Green Version]
- Nghiemphu, P.L.; Ebiana, V.A.; Wen, P.; Gilbert, M.; Abrey, L.E.; Lieberman, F.; Deangelis, L.M.; Robins, H.I.; Yung, W.K.A.; Chang, S.; et al. Phase I Study Of Sorafenib And Tipifarnib For Recurrent Glioblastoma: Nabtc 05-02. J. Neuro Oncol. 2018, 136, 79–86. [Google Scholar] [CrossRef]
- Kreisl, T.N.; Mcneill, K.A.; Sul, J.; Iwamoto, F.M.; Shih, J.; Fine, H.A. A Phase I/Ii Trial Of Vandetanib For Patients With Recurrent Malignant Glioma. Neuro Oncol. 2012, 14, 1519–1526. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Chen, B.; Su, C.H.; Zhao, R.; Xu, Z.X.; Sun, L.; Lee, M.H.; Yeung, S.C. Autophagy Induced By Farnesyltransferase Inhibitors In Cancer Cells. Cancer Biol. Ther. 2008, 7, 1679–1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takada, Y.; Khuri, F.R.; Aggarwal, B.B. Protein Farnesyltransferase Inhibitor (Sch 66336) Abolishes Nf-Kappab Activation Induced By Various Carcinogens And Inflammatory Stimuli Leading To Suppression Of Nf-Kappab-Regulated Gene Expression And Up-Regulation Of Apoptosis. J. Biol. Chem. 2004, 279, 26287–26299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.Y.; Liu, X.; Zou, W.; Yue, P.; Marcus, A.I.; Khuri, F.R. The Farnesyltransferase Inhibitor Lonafarnib Induces Ccaat/Enhancer-Binding Protein Homologous Protein-Dependent Expression Of Death Receptor 5, Leading To Induction Of Apoptosis In Human Cancer Cells. J. Biol. Chem. 2007, 282, 18800–18809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basso, A.D.; Mirza, A.; Liu, G.; Long, B.J.; Bishop, W.R.; Kirschmeier, P. The Farnesyl Transferase Inhibitor (Fti) Sch66336 (Lonafarnib) Inhibits Rheb Farnesylation And Mtor Signaling. Role In Fti Enhancement Of Taxane And Tamoxifen Anti-Tumor Activity. J. Biol. Chem. 2005, 280, 31101–31108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niessner, H.; Beck, D.; Sinnberg, T.; Lasithiotakis, K.; Maczey, E.; Gogel, J.; Venturelli, S.; Berger, A.; Mauthe, M.; Toulany, M.; et al. The Farnesyl Transferase Inhibitor Lonafarnib Inhibits Mtor Signaling And Enforces Sorafenib-Induced Apoptosis In Melanoma Cells. J. Invest. Dermatol. 2011, 131, 468–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldkamp, M.M.; Lau, N.; Roncari, L.; Guha, A. Isotype-Specific Ras.Gtp-Levels Predict The Efficacy Of Farnesyl Transferase Inhibitors Against Human Astrocytomas Regardless Of Ras Mutational Status. Cancer Res. 2001, 61, 4425–4431. [Google Scholar] [PubMed]
- Glass, T.L.; Liu, T.J.; Yung, W.K. Inhibition Of Cell Growth In Human Glioblastoma Cell Lines By Farnesyltransferase Inhibitor Sch66336. Neuro Oncol. 2000, 2, 151–158. [Google Scholar] [CrossRef] [Green Version]
- Adjei, A.A. Ras Signaling Pathway Proteins As Therapeutic Targets. Curr. Pharm. Des. 2001, 7, 1581–1594. [Google Scholar] [CrossRef]
- Chaponis, D.; Barnes, J.W.; Dellagatta, J.L.; Kesari, S.; Fast, E.; Sauvageot, C.; Panagrahy, D.; Greene, E.R.; Ramakrishna, N.; Wen, P.Y.; et al. Lonafarnib (Sch66336) Improves The Activity Of Temozolomide And Radiation For Orthotopic Malignant Gliomas. J. Neuro Oncol. 2011, 104, 179–189. [Google Scholar] [CrossRef] [Green Version]
- Kieran, M.W.; Packer, R.J.; Onar, A.; Blaney, S.M.; Phillips, P.; Pollack, I.F.; Geyer, J.R.; Gururangan, S.; Banerjee, A.; Goldman, S.; et al. Phase I And Pharmacokinetic Study Of The Oral Farnesyltransferase Inhibitor Lonafarnib Administered Twice Daily To Pediatric Patients With Advanced Central Nervous System Tumors Using A Modified Continuous Reassessment Method: A Pediatric Brain Tumor Consortium Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2007, 25, 3137–3143. [Google Scholar] [CrossRef]
- Desjardins, A.; Reardon, D.A.; Peters, K.B.; Threatt, S.; Coan, A.D.; Herndon, J.E., 2nd; Friedman, A.H.; Friedman, H.S.; Vredenburgh, J.J. A Phase I Trial Of The Farnesyl Transferase Inhibitor, Sch 66336, With Temozolomide For Patients With Malignant Glioma. J. Neuro Oncol. 2011, 105, 601–606. [Google Scholar] [CrossRef]
- Yust-Katz, S.; Liu, D.; Yuan, Y.; Liu, V.; Kang, S.; Groves, M.; Puduvalli, V.; Levin, V.; Conrad, C.; Colman, H.; et al. Phase 1/1b Study Of Lonafarnib And Temozolomide In Patients With Recurrent Or Temozolomide Refractory Glioblastoma. Cancer 2013, 119, 2747–2753. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.H.; Jin, Q.; Kim, E.S.; Khuri, F.R.; Lee, H.Y. Insulin-Like Growth Factor-I Receptor Signaling Pathway Induces Resistance To The Apoptotic Activities Of Sch66336 (Lonafarnib) Through Akt/Mammalian Target Of Rapamycin-Mediated Increases In Survivin Expression. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 1581–1589. [Google Scholar] [CrossRef] [Green Version]
- Halaban, R.; Zhang, W.; Bacchiocchi, A.; Cheng, E.; Parisi, F.; Ariyan, S.; Krauthammer, M.; Mccusker, J.P.; Kluger, Y.; Sznol, M. Plx4032, A Selective Braf(V600e) Kinase Inhibitor, Activates The Erk Pathway And Enhances Cell Migration And Proliferation Of Braf Melanoma Cells. Pigment Cell Melanoma Res. 2010, 23, 190–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, J.M.; Thompson, J.C.; Griesinger, A.M.; Amani, V.; Donson, A.M.; Birks, D.K.; Morgan, M.J.; Mirsky, D.M.; Handler, M.H.; Foreman, N.K.; et al. Autophagy Inhibition Improves Chemosensitivity In Braf(V600e) Brain Tumors. Cancer Discov. 2014, 4, 773–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulcahy Levy, J.M.; Zahedi, S.; Griesinger, A.M.; Morin, A.; Davies, K.D.; Aisner, D.L.; Kleinschmidt-Demasters, B.K.; Fitzwalter, B.E.; Goodall, M.L.; Thorburn, J.; et al. Autophagy Inhibition Overcomes Multiple Mechanisms Of Resistance To Braf Inhibition In Brain Tumors. ELIFE 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Festa, M.; Del Valle, L.; Khalili, K.; Franco, R.; Scognamiglio, G.; Graziano, V.; De Laurenzi, V.; Turco, M.C.; Rosati, A. Bag3 Protein Is Overexpressed In Human Glioblastoma And Is A Potential Target For Therapy. Am. J. Pathol. 2011, 178, 2504–2512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerriero, L.; Palmieri, G.; De Marco, M.; Cossu, A.; Remondelli, P.; Capunzo, M.; Turco, M.C.; Rosati, A. The Anti-Apoptotic Bag3 Protein Is Involved In Braf Inhibitor Resistance In Melanoma Cells. Oncotarget 2017, 8, 80393–80404. [Google Scholar] [CrossRef] [Green Version]
- Cvetanova, B.; Shen, Y.C.; Shyur, L.F. Cumingianoside A, A Phyto-Triterpenoid Saponin Inhibits Acquired Braf Inhibitor Resistant Melanoma Growth Via Programmed Cell Death. Front. Pharmacol. 2019, 10, 30. [Google Scholar] [CrossRef]
- Behling, F.; Barrantes-Freer, A.; Skardelly, M.; Nieser, M.; Christians, A.; Stockhammer, F.; Rohde, V.; Tatagiba, M.; Hartmann, C.; Stadelmann, C.; et al. Frequency Of Braf V600e Mutations In 969 Central Nervous System Neoplasms. Diagn. Pathol. 2016, 11, 55. [Google Scholar] [CrossRef] [Green Version]
- Kaley, T.; Touat, M.; Subbiah, V.; Hollebecque, A.; Rodon, J.; Lockhart, A.C.; Keedy, V.; Bielle, F.; Hofheinz, R.D.; Joly, F.; et al. Braf Inhibition In Braf(V600)-Mutant Gliomas: Results From The Ve-Basket Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 3477. [Google Scholar] [CrossRef]
- Koh, H.Y.; Kim, S.H.; Jang, J.; Kim, H.; Han, S.; Lim, J.S.; Son, G.; Choi, J.; Park, B.O.; Do Heo, W.; et al. Braf Somatic Mutation Contributes To Intrinsic Epileptogenicity In Pediatric Brain Tumors. Nat. Med. 2018, 24, 1662–1668. [Google Scholar] [CrossRef]
- Lee, E.Q.; Ruland, S.; Leboeuf, N.R.; Wen, P.Y.; Santagata, S. Successful Treatment Of A Progressive Braf V600e-Mutated Anaplastic Pleomorphic Xanthoastrocytoma With Vemurafenib Monotherapy. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, E87–E89. [Google Scholar] [CrossRef] [Green Version]
- Rush, S.; Foreman, N.; Liu, A. Brainstem Ganglioglioma Successfully Treated With Vemurafenib. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, E159–E160. [Google Scholar] [CrossRef] [PubMed]
- Touat, M.; Gratieux, J.; Condette Auliac, S.; Sejean, K.; Aldea, S.; Savatovsky, J.; Perkins, G.; Blons, H.; Ligon, K.L.; Idbaih, A.; et al. Vemurafenib And Cobimetinib Overcome Resistance To Vemurafenib In Braf-Mutant Ganglioglioma. Neurology 2018, 91, 523–525. [Google Scholar] [CrossRef]
- Leaver, K.E.; Zhang, N.; Ziskin, J.L.; Vogel, H.; Recht, L.; Thomas, R.P. Response Of Metastatic Glioma To Vemurafenib. Neuro Oncol. Pract. 2016, 3, 268–271. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.M.; Wen, P.; Cloughesy, T.; Greenberg, H.; Schiff, D.; Conrad, C.; Fink, K.; Robins, H.I.; De Angelis, L.; Raizer, J.; et al. Phase Ii Study Of Cci-779 In Patients With Recurrent Glioblastoma Multiforme. Investig. N. Drugs 2005, 23, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Yazbeck, V.Y.; Buglio, D.; Georgakis, G.V.; Li, Y.; Iwado, E.; Romaguera, J.E.; Kondo, S.; Younes, A. Temsirolimus Downregulates P21 Without Altering Cyclin D1 Expression And Induces Autophagy And Synergizes With Vorinostat In Mantle Cell Lymphoma. Exp. Hematol. 2008, 36, 443–450. [Google Scholar] [CrossRef]
- Jhanwar-Uniyal, M.; Gillick, J.L.; Neil, J.; Tobias, M.; Thwing, Z.E.; Murali, R. Distinct Signaling Mechanisms Of Mtorc1 And Mtorc2 In Glioblastoma Multiforme: A Tale Of Two Complexes. Adv. Biol. Regul. 2015, 57, 64–74. [Google Scholar] [CrossRef]
- Yu, K.; Shi, C.; Toral-Barza, L.; Lucas, J.; Shor, B.; Kim, J.E.; Zhang, W.G.; Mahoney, R.; Gaydos, C.; Tardio, L.; et al. Beyond Rapalog Therapy: Preclinical Pharmacology And Antitumor Activity Of Wye-125132, An Atp-Competitive And Specific Inhibitor Of Mtorc1 And Mtorc2. Cancer Res. 2010, 70, 621–631. [Google Scholar] [CrossRef] [Green Version]
- Chandrika, G.; Natesh, K.; Ranade, D.; Chugh, A.; Shastry, P. Suppression Of The Invasive Potential Of Glioblastoma Cells By Mtor Inhibitors Involves Modulation Of Nfkappab And Pkc-Alpha Signaling. Sci. Rep. 2016, 6, 22455. [Google Scholar] [CrossRef] [Green Version]
- Chandrika, G.; Natesh, K.; Ranade, D.; Chugh, A.; Shastry, P. Mammalian Target Of Rapamycin Inhibitors, Temsirolimus And Torin 1, Attenuate Stemness-Associated Properties And Expression Of Mesenchymal Markers Promoted By Phorbol-Myristate-Acetate And Oncostatin-M In Glioblastoma Cells. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2017, 39, 1010428317695921. [Google Scholar] [CrossRef] [Green Version]
- Pitter, K.L.; Galban, C.J.; Galban, S.; Tehrani, O.S.; Li, F.; Charles, N.; Bradbury, M.S.; Becher, O.J.; Chenevert, T.L.; Rehemtulla, A.; et al. Perifosine And Cci 779 Co-Operate To Induce Cell Death And Decrease Proliferation In Pten-Intact And Pten-Deficient Pdgf-Driven Murine Glioblastoma. PLoS ONE 2011, 6, E14545. [Google Scholar] [CrossRef]
- Becher, O.J.; Gilheeney, S.W.; Khakoo, Y.; Lyden, D.C.; Haque, S.; De Braganca, K.C.; Kolesar, J.M.; Huse, J.T.; Modak, S.; Wexler, L.H.; et al. A Phase I Study Of Perifosine With Temsirolimus For Recurrent Pediatric Solid Tumors. Pediatr. Blood Cancer 2017, 64. [Google Scholar] [CrossRef]
- Tsoli, M.; Liu, J.; Franshaw, L.; Shen, H.; Cheng, C.; Jung, M.; Joshi, S.; Ehteda, A.; Khan, A.; Montero-Carcabosso, A.; et al. Dual Targeting Of Mitochondrial Function And Mtor Pathway As A Therapeutic Strategy For Diffuse Intrinsic Pontine Glioma. Oncotarget 2018, 9, 7541–7556. [Google Scholar] [CrossRef] [Green Version]
- Asby, D.J.; Killick-Cole, C.L.; Boulter, L.J.; Singleton, W.G.; Asby, C.A.; Wyatt, M.J.; Barua, N.U.; Bienemann, A.S.; Gill, S.S. Combined Use Of Cdk4/6 And Mtor Inhibitors Induce Synergistic Growth Arrest Of Diffuse Intrinsic Pontine Glioma Cells Via Mutual Downregulation Of Mtorc1 Activity. Cancer Manag. Res. 2018, 10, 3483–3500. [Google Scholar] [CrossRef] [Green Version]
- Weiler, M.; Pfenning, P.N.; Thiepold, A.L.; Blaes, J.; Jestaedt, L.; Gronych, J.; Dittmann, L.M.; Berger, B.; Jugold, M.; Kosch, M.; et al. Suppression Of Proinvasive Rgs4 By Mtor Inhibition Optimizes Glioma Treatment. Oncogene 2013, 32, 1099–1109. [Google Scholar] [CrossRef] [Green Version]
- Galanis, E.; Buckner, J.C.; Maurer, M.J.; Kreisberg, J.I.; Ballman, K.; Boni, J.; Peralba, J.M.; Jenkins, R.B.; Dakhil, S.R.; Morton, R.F.; et al. Phase Ii Trial Of Temsirolimus (Cci-779) In Recurrent Glioblastoma Multiforme: A North Central Cancer Treatment Group Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 5294–5304. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Galanis, E.; Wu, W.; Dietz, A.B.; Kaufmann, T.J.; Gustafson, M.P.; Brown, P.D.; Uhm, J.H.; Rao, R.D.; Doyle, L.; et al. Combination Of Temsirolimus (Cci-779) With Chemoradiation In Newly Diagnosed Glioblastoma Multiforme (Gbm) (Ncctg Trial N027d) Is Associated With Increased Infectious Risks. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 5573–5580. [Google Scholar] [CrossRef] [Green Version]
- Wick, W.; Gorlia, T.; Bady, P.; Platten, M.; Van Den Bent, M.J.; Taphoorn, M.J.; Steuve, J.; Brandes, A.A.; Hamou, M.F.; Wick, A.; et al. Phase Ii Study Of Radiotherapy And Temsirolimus Versus Radiochemotherapy With Temozolomide In Patients With Newly Diagnosed Glioblastoma Without Mgmt Promoter Hypermethylation (Eortc 26082). Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 4797–4806. [Google Scholar] [CrossRef] [Green Version]
- Pachow, D.; Wick, W.; Gutmann, D.H.; Mawrin, C. The Mtor Signaling Pathway As A Treatment Target For Intracranial Neoplasms. Neuro Oncol. 2015, 17, 189–199. [Google Scholar] [CrossRef] [Green Version]
- Lassen, U.; Sorensen, M.; Gaziel, T.B.; Hasselbalch, B.; Poulsen, H.S. Phase Ii Study Of Bevacizumab And Temsirolimus Combination Therapy For Recurrent Glioblastoma Multiforme. Anticancer Res. 2013, 33, 1657–1660. [Google Scholar]
- Josset, E.; Burckel, H.; Noel, G.; Bischoff, P. The Mtor Inhibitor Rad001 Potentiates Autophagic Cell Death Induced By Temozolomide In A Glioblastoma Cell Line. Anticancer Res. 2013, 33, 1845–1851. [Google Scholar]
- Piha-Paul, S.A.; Shin, S.J.; Vats, T.; Guha-Thakurta, N.; Aaron, J.; Rytting, M.; Kleinerman, E.; Kurzrock, R. Pediatric Patients With Refractory Central Nervous System Tumors: Experiences Of A Clinical Trial Combining Bevacizumab And Temsirolimus. Anticancer Res. 2014, 34, 1939–1945. [Google Scholar]
- Stepanenko, A.A.; Andreieva, S.V.; Korets, K.V.; Mykytenko, D.O.; Baklaushev, V.P.; Chekhonin, V.P.; Dmitrenko, V.V. Mtor Inhibitor Temsirolimus And Mek1/2 Inhibitor U0126 Promote Chromosomal Instability And Cell Type-Dependent Phenotype Changes Of Glioblastoma Cells. Gene 2016, 579, 58–68. [Google Scholar] [CrossRef]
- Moreno-Smith, M.; Lakoma, A.; Chen, Z.; Tao, L.; Scorsone, K.A.; Schild, L.; Aviles-Padilla, K.; Nikzad, R.; Zhang, Y.; Chakraborty, R.; et al. P53 Nongenotoxic Activation And Mtorc1 Inhibition Lead To Effective Combination For Neuroblastoma Therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 6629–6639. [Google Scholar] [CrossRef] [Green Version]
- Carracedo, A.; Pandolfi, P.P. The Pten-Pi3k Pathway: Of Feedbacks And Cross-Talks. Oncogene 2008, 27, 5527–5541. [Google Scholar] [CrossRef] [Green Version]
- Wick, W.; Dettmer, S.; Berberich, A.; Kessler, T.; Karapanagiotou-Schenkel, I.; Wick, A.; Winkler, F.; Pfaff, E.; Brors, B.; Debus, J.; et al. N2m2 (Noa-20) Phase I/Ii Trial Of Molecularly Matched Targeted Therapies Plus Radiotherapy In Patients With Newly Diagnosed Non-Mgmt Hypermethylated Glioblastoma. Neuro Oncol. 2019, 21, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Pfaff, E.; Kessler, T.; Balasubramanian, G.P.; Berberich, A.; Schrimpf, D.; Wick, A.; Debus, J.; Unterberg, A.; Bendszus, M.; Herold-Mende, C.; et al. Feasibility Of Real-Time Molecular Profiling For Patients With Newly Diagnosed Glioblastoma Without Mgmt Promoter Hypermethylation-The Nct Neuro Master Match (N2m2) Pilot Study. Neuro Oncol. 2018, 20, 826–837. [Google Scholar] [CrossRef]
- Jhanwar-Uniyal, M.; Labagnara, M.; Friedman, M.; Kwasnicki, A.; Murali, R. Glioblastoma: Molecular Pathways, Stem Cells And Therapeutic Targets. Cancers 2015, 7, 538–555. [Google Scholar] [CrossRef] [Green Version]
- Venkatesh, H.S.; Chaumeil, M.M.; Ward, C.S.; Haas-Kogan, D.A.; James, C.D.; Ronen, S.M. Reduced Phosphocholine And Hyperpolarized Lactate Provide Magnetic Resonance Biomarkers Of Pi3k/Akt/Mtor Inhibition In Glioblastoma. Neuro Oncol. 2012, 14, 315–325. [Google Scholar] [CrossRef] [Green Version]
- Day, B.W.; Stringer, B.W.; Spanevello, M.D.; Charmsaz, S.; Jamieson, P.R.; Ensbey, K.S.; Carter, J.C.; Cox, J.M.; Ellis, V.J.; Brown, C.L.; et al. Elk4 Neutralization Sensitizes Glioblastoma To Apoptosis Through Downregulation Of The Anti-Apoptotic Protein Mcl-1. Neuro Oncol. 2011, 13, 1202–1212. [Google Scholar] [CrossRef] [Green Version]
- Mattoo, A.R.; Joun, A.; Jessup, J.M. Repurposing Of Mtor Complex Inhibitors Attenuates Mcl-1 And Sensitizes To Parp Inhibition. Mol. Cancer Res. 2019, 17, 42–53. [Google Scholar] [CrossRef] [Green Version]
- Berney, E.; Sabnis, N.; Panchoo, M.; Raut, S.; Dickerman, R.; Lacko, A.G. The Sr-B1 Receptor As A Potential Target For Treating Glioblastoma. J. Oncol. 2019, 2019, 1805841. [Google Scholar] [CrossRef] [Green Version]
- Olmez, I.; Brenneman, B.; Xiao, A.; Serbulea, V.; Benamar, M.; Zhang, Y.; Manigat, L.; Abbas, T.; Lee, J.; Nakano, I.; et al. Combined Cdk4/6 And Mtor Inhibition Is Synergistic Against Glioblastoma Via Multiple Mechanisms. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 6958–6968. [Google Scholar] [CrossRef] [Green Version]
- Hjelmeland, A.B.; Lattimore, K.P.; Fee, B.E.; Shi, Q.; Wickman, S.; Keir, S.T.; Hjelmeland, M.D.; Batt, D.; Bigner, D.D.; Friedman, H.S.; et al. The Combination Of Novel Low Molecular Weight Inhibitors Of Raf (Lbt613) And Target Of Rapamycin (Rad001) Decreases Glioma Proliferation And Invasion. Mol. Cancer Ther. 2007, 6, 2449–2457. [Google Scholar] [CrossRef] [Green Version]
- Goudar, R.K.; Shi, Q.; Hjelmeland, M.D.; Keir, S.T.; Mclendon, R.E.; Wikstrand, C.J.; Reese, E.D.; Conrad, C.A.; Traxler, P.; Lane, H.A.; et al. Combination Therapy Of Inhibitors Of Epidermal Growth Factor Receptor/Vascular Endothelial Growth Factor Receptor 2 (Aee788) And The Mammalian Target Of Rapamycin (Rad001) Offers Improved Glioblastoma Tumor Growth Inhibition. Mol. Cancer Ther. 2005, 4, 101–112. [Google Scholar]
- Alonso, M.M.; Jiang, H.; Yokoyama, T.; Xu, J.; Bekele, N.B.; Lang, F.F.; Kondo, S.; Gomez-Manzano, C.; Fueyo, J. Delta-24-Rgd In Combination With Rad001 Induces Enhanced Anti-Glioma Effect Via Autophagic Cell Death. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 487–493. [Google Scholar] [CrossRef]
- Chinnaiyan, P.; Won, M.; Wen, P.Y.; Rojiani, A.M.; Wendland, M.; Dipetrillo, T.A.; Corn, B.W.; Mehta, M.P. Rtog 0913: A Phase 1 Study Of Daily Everolimus (Rad001) In Combination With Radiation Therapy And Temozolomide In Patients With Newly Diagnosed Glioblastoma. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 880–884. [Google Scholar] [CrossRef] [Green Version]
- Ma, D.J.; Galanis, E.; Anderson, S.K.; Schiff, D.; Kaufmann, T.J.; Peller, P.J.; Giannini, C.; Brown, P.D.; Uhm, J.H.; Mcgraw, S.; et al. A Phase Ii Trial Of Everolimus, Temozolomide, And Radiotherapy In Patients With Newly Diagnosed Glioblastoma: Ncctg N057k. Neuro Oncol. 2015, 17, 1261–1269. [Google Scholar] [CrossRef]
- Chinnaiyan, P.; Won, M.; Wen, P.Y.; Rojiani, A.M.; Werner-Wasik, M.; Shih, H.A.; Ashby, L.S.; Michael Yu, H.H.; Stieber, V.W.; Malone, S.C.; et al. A Randomized Phase Ii Study Of Everolimus In Combination With Chemoradiation In Newly Diagnosed Glioblastoma: Results Of Nrg Oncology Rtog 0913. Neuro Oncol. 2018, 20, 666–673. [Google Scholar] [CrossRef] [Green Version]
- Zureick, A.H.; Mcfadden, K.A.; Mody, R.; Koschmann, C. Successful Treatment Of A Tsc2-Mutant Glioblastoma With Everolimus. BMJ Case Rep. 2019, 12. [Google Scholar] [CrossRef]
- Hainsworth, J.D.; Shih, K.C.; Shepard, G.C.; Tillinghast, G.W.; Brinker, B.T.; Spigel, D.R. Phase Ii Study Of Concurrent Radiation Therapy, Temozolomide, And Bevacizumab Followed By Bevacizumab/Everolimus As First-Line Treatment For Patients With Glioblastoma. Clin. Adv. Hemato. Oncol. H&O 2012, 10, 240–246. [Google Scholar]
- Kreisl, T.N.; Lassman, A.B.; Mischel, P.S.; Rosen, N.; Scher, H.I.; Teruya-Feldstein, J.; Shaffer, D.; Lis, E.; Abrey, L.E. A Pilot Study Of Everolimus And Gefitinib In The Treatment Of Recurrent Glioblastoma (Gbm). J. Neuro Oncol. 2009, 92, 99–105. [Google Scholar] [CrossRef]
- Gebert, L.F.R.; Macrae, I.J. Regulation Of Microrna Function In Animals. Nat. Rev. Mol. Cell Biol 2019, 20, 21–37. [Google Scholar] [CrossRef]
- Ling, H.; Zhang, W.; Calin, G.A. Principles Of Microrna Involvement In Human Cancers. Chin. J. Cancer 2011, 30, 739–748. [Google Scholar] [CrossRef]
- Xue, H.; Yuan, G.; Guo, X.; Liu, Q.; Zhang, J.; Gao, X.; Xu, S.; Li, T.; Shao, Q.; Yan, S.; et al. A Novel Tumor-Promoting Mechanism Of Il6 And The Therapeutic Efficacy Of Tocilizumab: Hypoxia-Induced Il6 Is A Potent Autophagy Initiator In Glioblastoma Via The P-Stat3-Mir155-3p-Crebrf Pathway. Autophagy 2016, 12, 1129–1152. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Xiao, L.; Liu, Y.; Wang, H.; Li, H.; Zhou, Q.; Pan, J.; Lei, B.; Huang, A.; Qi, S. Mir517c Inhibits Autophagy And The Epithelial-To-Mesenchymal (-Like) Transition Phenotype In Human Glioblastoma Through Kpna2-Dependent Disruption Of Tp53 Nuclear Translocation. Autophagy 2015, 11, 2213–2232. [Google Scholar] [CrossRef] [Green Version]
- Sana, J.; Hajduch, M.; Michalek, J.; Vyzula, R.; Slaby, O. Micrornas And Glioblastoma: Roles In Core Signalling Pathways And Potential Clinical Implications. J. Cell Mol. Med. 2011, 15, 1636–1644. [Google Scholar] [CrossRef] [Green Version]
- Coyle, T.; Levante, S.; Shetler, M.; Winfield, J. In Vitro And In Vivo Cytotoxicity Of Gossypol Against Central Nervous System Tumor Cell Lines. J. Neurooncol. 1994, 19, 25–35. [Google Scholar] [CrossRef]
- Kim, H.Y.; Lee, B.I.; Jeon, J.H.; Kim, D.K.; Kang, S.G.; Shim, J.K.; Kim, S.Y.; Kang, S.W.; Jang, H. Gossypol Suppresses Growth Of Temozolomide-Resistant Glioblastoma Tumor Spheres. Biomolecules 2019, 9, 595. [Google Scholar] [CrossRef] [Green Version]
- Voss, V.; Senft, C.; Lang, V.; Ronellenfitsch, M.W.; Steinbach, J.P.; Seifert, V.; Kögel, D. The Pan-Bcl-2 Inhibitor (-)-Gossypol Triggers Autophagic Cell Death In Malignant Glioma. Mol. Cancer Res. 2010, 8, 1002–1016. [Google Scholar] [CrossRef] [Green Version]
- Jarzabek, M.A.; Amberger-Murphy, V.; Callanan, J.J.; Gao, C.; Zagozdzon, A.M.; Shiels, L.; Wang, J.; Ligon, K.L.; Rich, B.E.; Dicker, P.; et al. Interrogation Of Gossypol Therapy In Glioblastoma Implementing Cell Line And Patient-Derived Tumour Models. Br. J. Cancer 2014, 111, 2275–2286. [Google Scholar] [CrossRef] [Green Version]
- Meyer, N.; Zielke, S.; Michaelis, J.B.; Linder, B.; Warnsmann, V.; Rakel, S.; Osiewacz, H.D.; Fulda, S.; Mittelbronn, M.; Münch, C.; et al. At 101 Induces Early Mitochondrial Dysfunction And Hmox1 (Heme Oxygenase 1) To Trigger Mitophagic Cell Death In Glioma Cells. Autophagy 2018, 14, 1693–1709. [Google Scholar] [CrossRef]
- Hernández-Tiedra, S.; Fabriàs, G.; Dávila, D.; Salanueva, Í.; Casas, J.; Montes, L.R.; Antón, Z.; García-Taboada, E.; Salazar-Roa, M.; Lorente, M.; et al. Dihydroceramide Accumulation Mediates Cytotoxic Autophagy Of Cancer Cells Via Autolysosome Destabilization. Autophagy 2016, 12, 2213–2229. [Google Scholar] [CrossRef] [Green Version]
- Salazar, M.; Carracedo, A.; Salanueva, I.J.; Hernández-Tiedra, S.; Lorente, M.; Egia, A.; Vázquez, P.; Blázquez, C.; Torres, S.; García, S.; et al. Cannabinoid Action Induces Autophagy-Mediated Cell Death Through Stimulation Of Er Stress In Human Glioma Cells. J. Clin. Invest. 2009, 119, 1359–1372. [Google Scholar] [CrossRef] [Green Version]
- Ellert-Miklaszewska, A.; Ciechomska, I.A.; Kaminska, B. Cannabinoid Signaling In Glioma Cells. Adv. Exp. Med. Biol. 2020, 1202, 223–241. [Google Scholar] [CrossRef]
- Lorente, M.; Torres, S.; Salazar, M.; Carracedo, A.; Hernández-Tiedra, S.; Rodríguez-Fornés, F.; García-Taboada, E.; Meléndez, B.; Mollejo, M.; Campos-Martín, Y.; et al. Stimulation Of The Midkine/Alk Axis Renders Glioma Cells Resistant To Cannabinoid Antitumoral Action. Cell Death Differ. 2011, 18, 959–973. [Google Scholar] [CrossRef]
- Lorente, M.; Torres, S.; Salazar, M.; Carracedo, A.; Hernández-Tiedra, S.; Rodríguez-Fornés, F.; García-Taboada, E.; Meléndez, B.; Mollejo, M.; Campos-Martín, Y.; et al. Stimulation Of Alk By The Growth Factor Midkine Renders Glioma Cells Resistant To Autophagy-Mediated Cell Death. Autophagy 2011, 7, 1071–1073. [Google Scholar] [CrossRef] [Green Version]
- Torres, S.; Lorente, M.; Rodríguez-Fornés, F.; Hernández-Tiedra, S.; Salazar, M.; García-Taboada, E.; Barcia, J.; Guzmán, M.; Velasco, G. A Combined Preclinical Therapy Of Cannabinoids And Temozolomide Against Glioma. Mol. Cancer Ther. 2011, 10, 90–103. [Google Scholar] [CrossRef] [Green Version]
- Irrera, N.; D’ascola, A.; Pallio, G.; Bitto, A.; Mannino, F.; Arcoraci, V.; Rottura, M.; Ieni, A.; Minutoli, L.; Metro, D.; et al. Β-Caryophyllene Inhibits Cell Proliferation Through A Direct Modulation Of Cb2 Receptors In Glioblastoma Cells. Cancers (Basel) 2020, 12, 1038. [Google Scholar] [CrossRef] [Green Version]
- Nabissi, M.; Morelli, M.B.; Amantini, C.; Liberati, S.; Santoni, M.; Ricci-Vitiani, L.; Pallini, R.; Santoni, G. Cannabidiol Stimulates Aml-1a-Dependent Glial Differentiation And Inhibits Glioma Stem-Like Cells Proliferation By Inducing Autophagy In A Trpv2-Dependent Manner. Int. J. Cancer 2015, 137, 1855–1869. [Google Scholar] [CrossRef] [Green Version]
- Lohitesh, K.; Saini, H.; Srivastava, A.; Mukherjee, S.; Roy, A.; Chowdhury, R. Autophagy Inhibition Potentiates Saha-Mediated Apoptosis In Glioblastoma Cells By Accumulation Of Damaged Mitochondria. Oncol. Rep. 2018, 39, 2787–2796. [Google Scholar] [CrossRef]
- Hrzenjak, A.; Kremser, M.L.; Strohmeier, B.; Moinfar, F.; Zatloukal, K.; Denk, H. Saha Induces Caspase-Independent, Autophagic Cell Death Of Endometrial Stromal Sarcoma Cells By Influencing The Mtor Pathway. J. Pathol. 2008, 216, 495–504. [Google Scholar] [CrossRef]
- Chiao, M.T.; Cheng, W.Y.; Yang, Y.C.; Shen, C.C.; Ko, J.L. Suberoylanilide Hydroxamic Acid (Saha) Causes Tumor Growth Slowdown And Triggers Autophagy In Glioblastoma Stem Cells. Autophagy 2013, 9, 1509–1526. [Google Scholar] [CrossRef] [Green Version]
- Gammoh, N.; Lam, D.; Puente, C.; Ganley, I.; Marks, P.A.; Jiang, X. Role Of Autophagy In Histone Deacetylase Inhibitor-Induced Apoptotic And Nonapoptotic Cell Death. Proc. Natl. Acad. Sci. USA 2012, 109, 6561–6565. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.R.; Yu, C.W.; Hung, P.Y.; Hsin, L.W.; Chern, J.W. High-Selective Hdac6 Inhibitor Promotes Hdac6 Degradation Following Autophagy Modulation And Enhanced Antitumor Immunity In Glioblastoma. Biochem. Pharmacol. 2019, 163, 458–471. [Google Scholar] [CrossRef]
- Ge, P.F.; Zhang, J.Z.; Wang, X.F.; Meng, F.K.; Li, W.C.; Luan, Y.X.; Ling, F.; Luo, Y.N. Inhibition Of Autophagy Induced By Proteasome Inhibition Increases Cell Death In Human Shg-44 Glioma Cells. Acta Pharmacol. Sin. 2009, 30, 1046–1052. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Li, W.; Wang, C.; Leng, X.; Lian, S.; Feng, J.; Li, J.; Wang, H. Inhibition Of Autophagy Enhances Apoptosis Induced By Proteasome Inhibitor Bortezomib In Human Glioblastoma U87 And U251 Cells. Mol. Cell Biochem. 2014, 385, 265–275. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.; Rhyasen, G.; Bolanos, L.; Rasch, C.; Varney, M.; Wunderlich, M.; Goyama, S.; Jansen, G.; Cloos, J.; Rigolino, C.; et al. Cytotoxic Effects Of Bortezomib In Myelodysplastic Syndrome/Acute Myeloid Leukemia Depend On Autophagy-Mediated Lysosomal Degradation Of Traf6 And Repression Of Psma1. Blood 2012, 120, 858–867. [Google Scholar] [CrossRef]
- Han, S.H.; Korm, S.; Han, Y.G.; Choi, S.Y.; Kim, S.H.; Chung, H.J.; Park, K.; Kim, J.Y.; Myung, K.; Lee, J.Y.; et al. Gca Links Traf6-Ulk1-Dependent Autophagy Activation In Resistant Chronic Myeloid Leukemia. Autophagy 2019, 15, 2076–2090. [Google Scholar] [CrossRef]

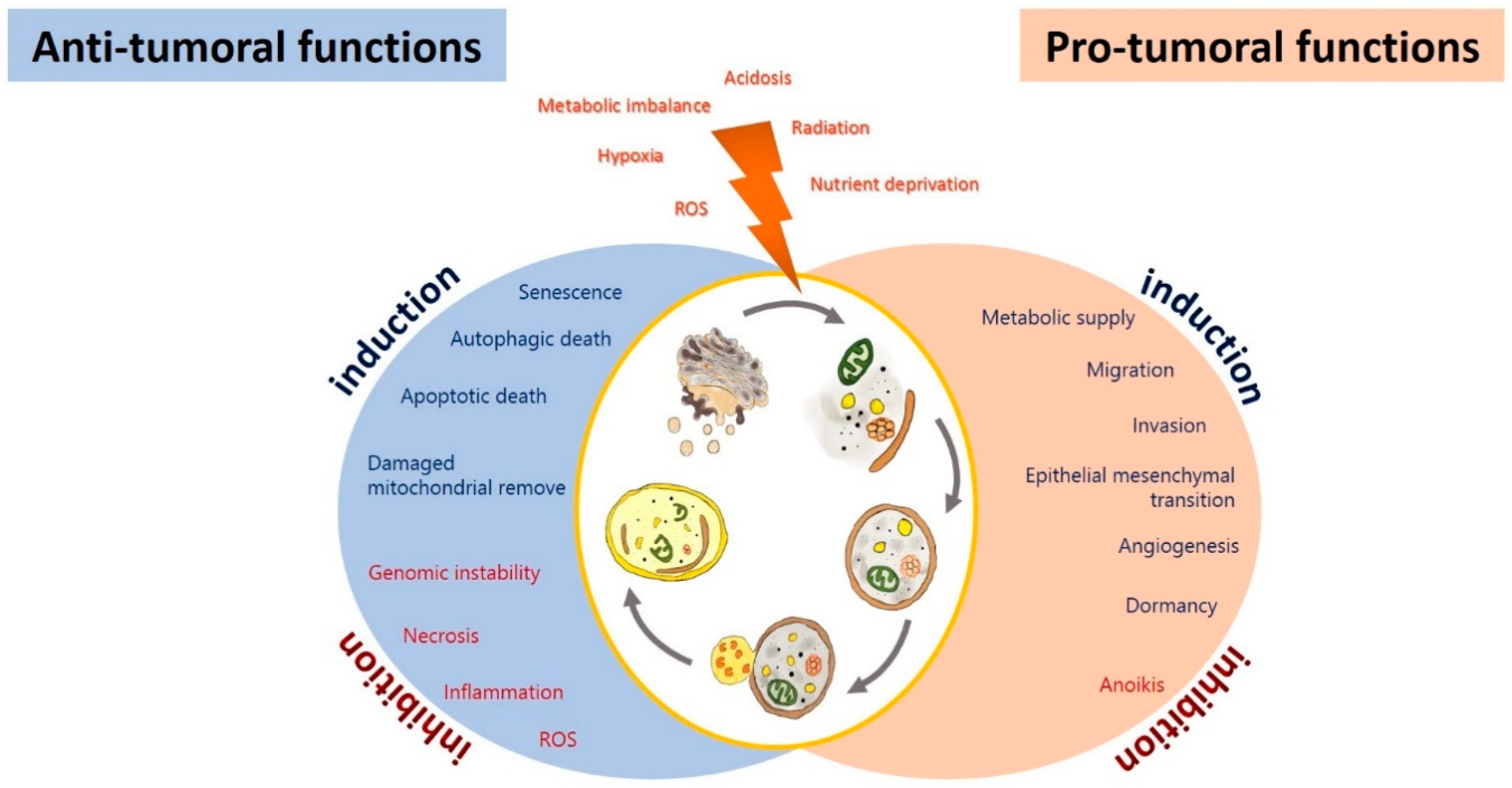
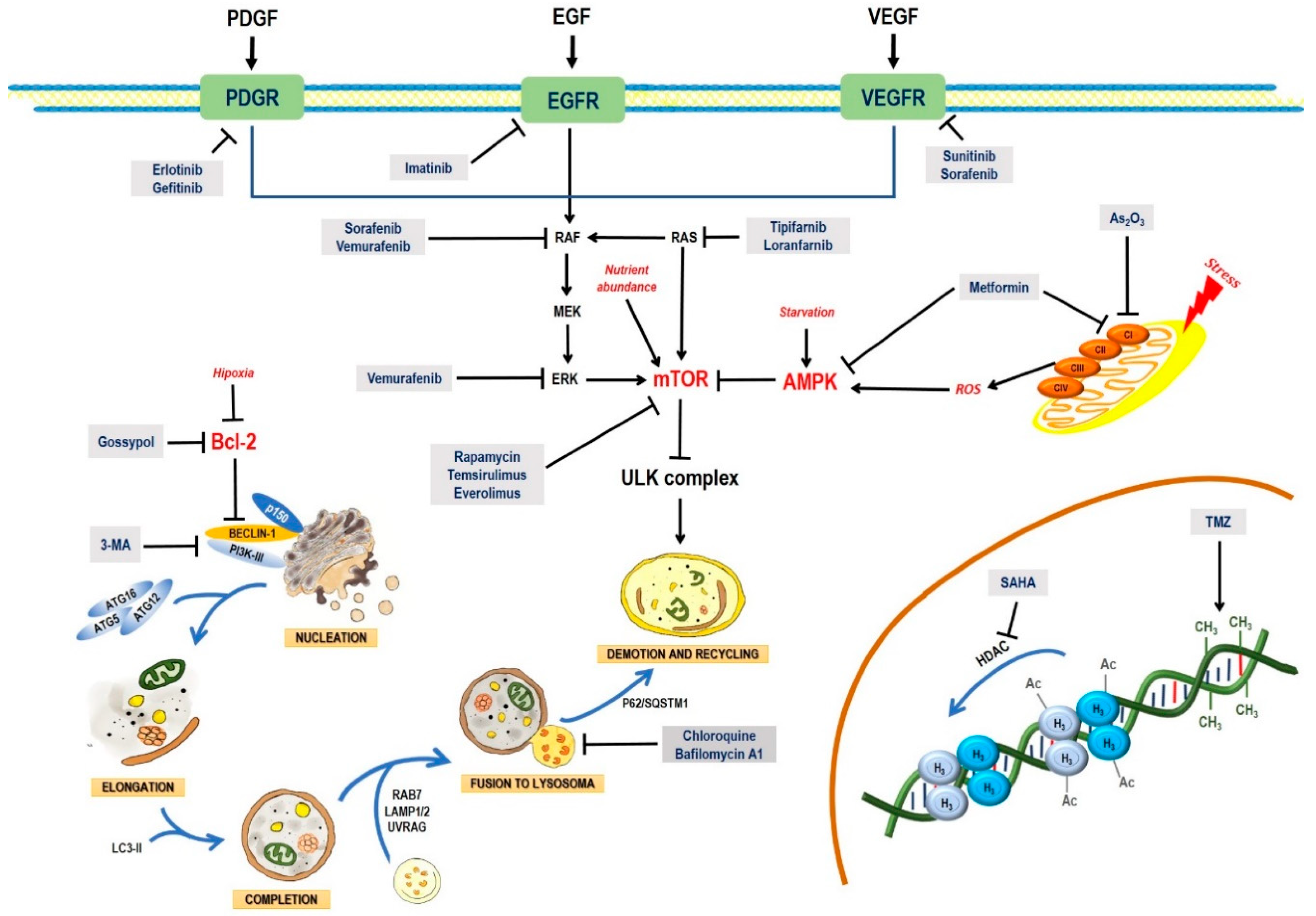

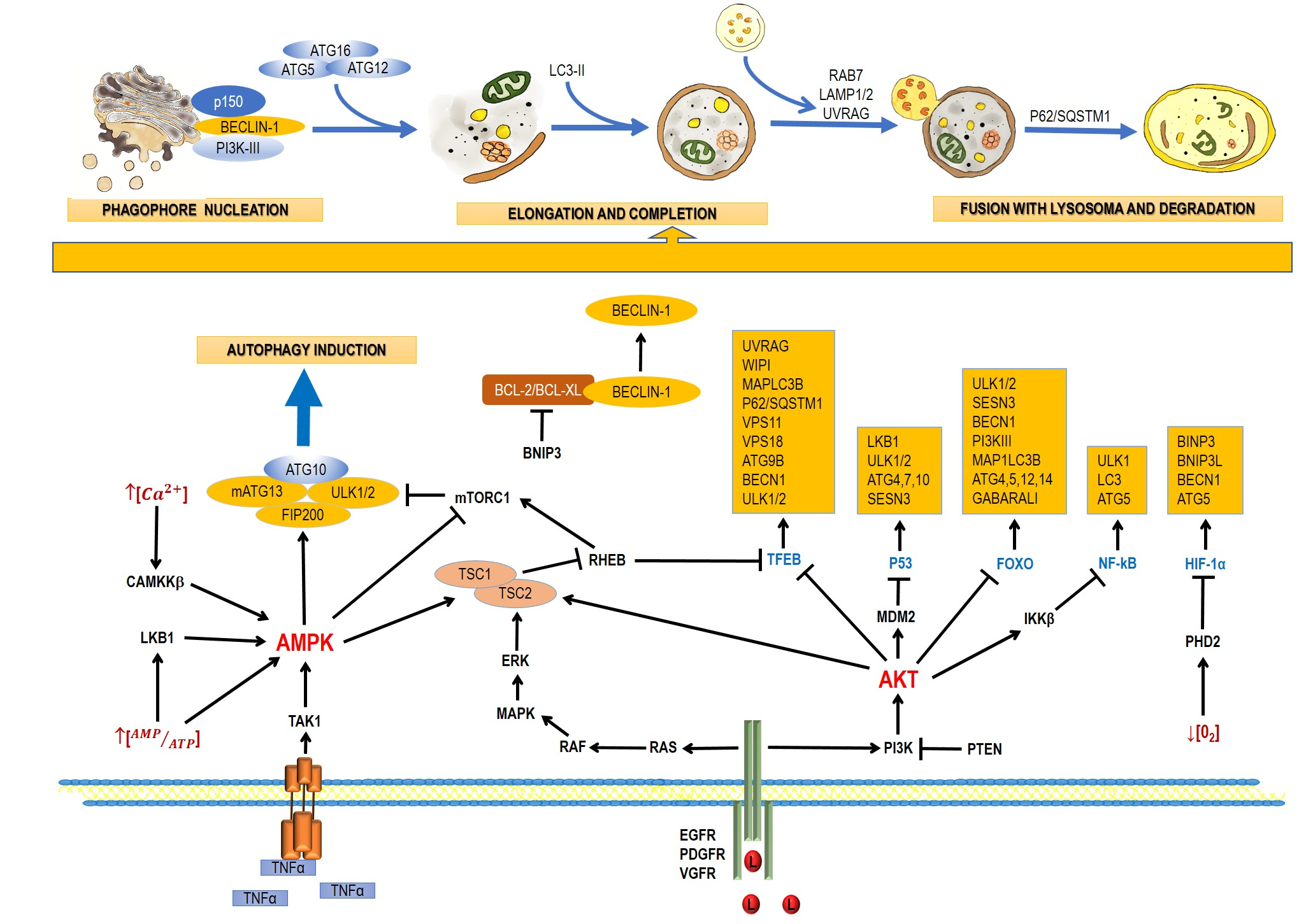{kind=link}
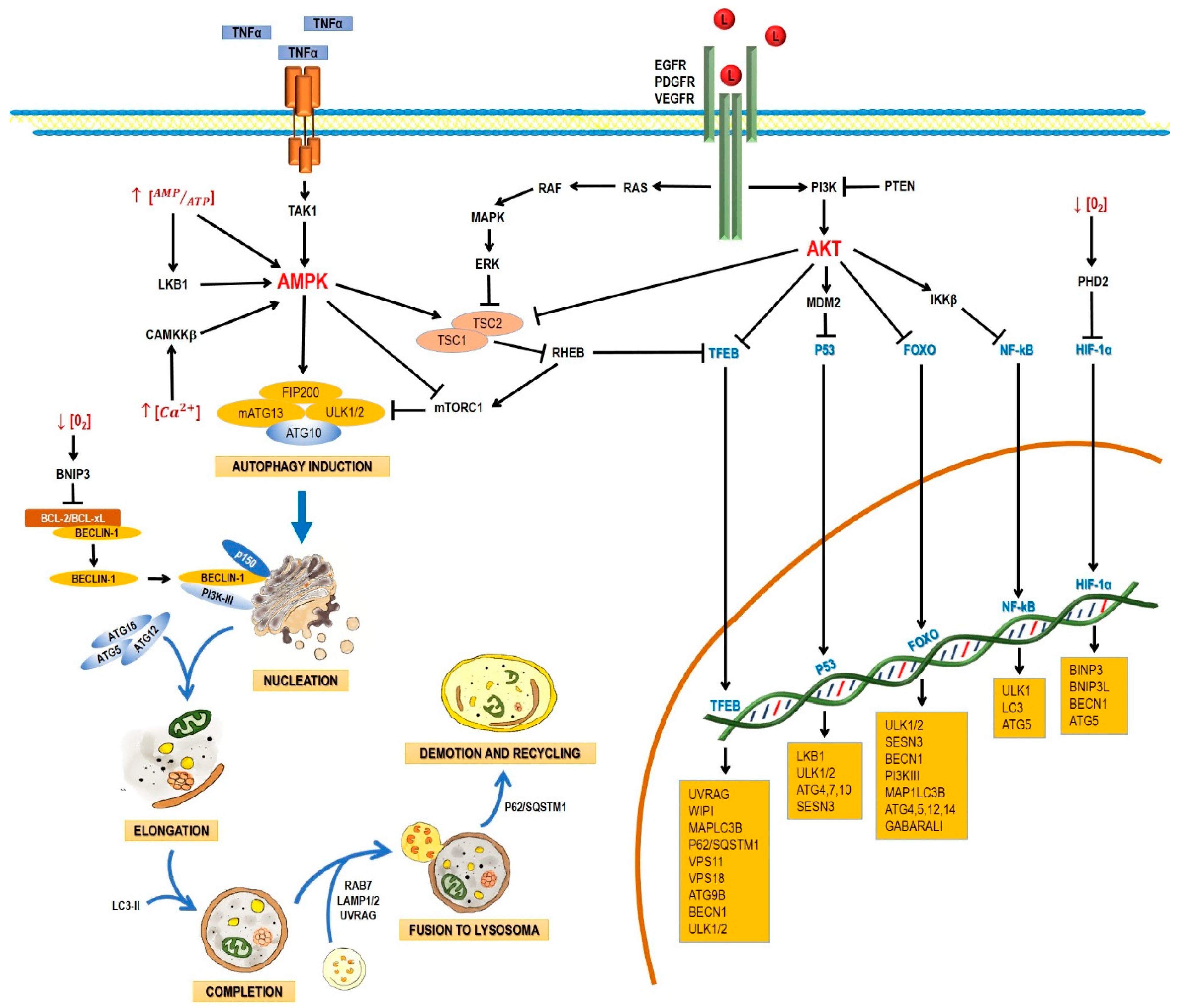{kind=link}
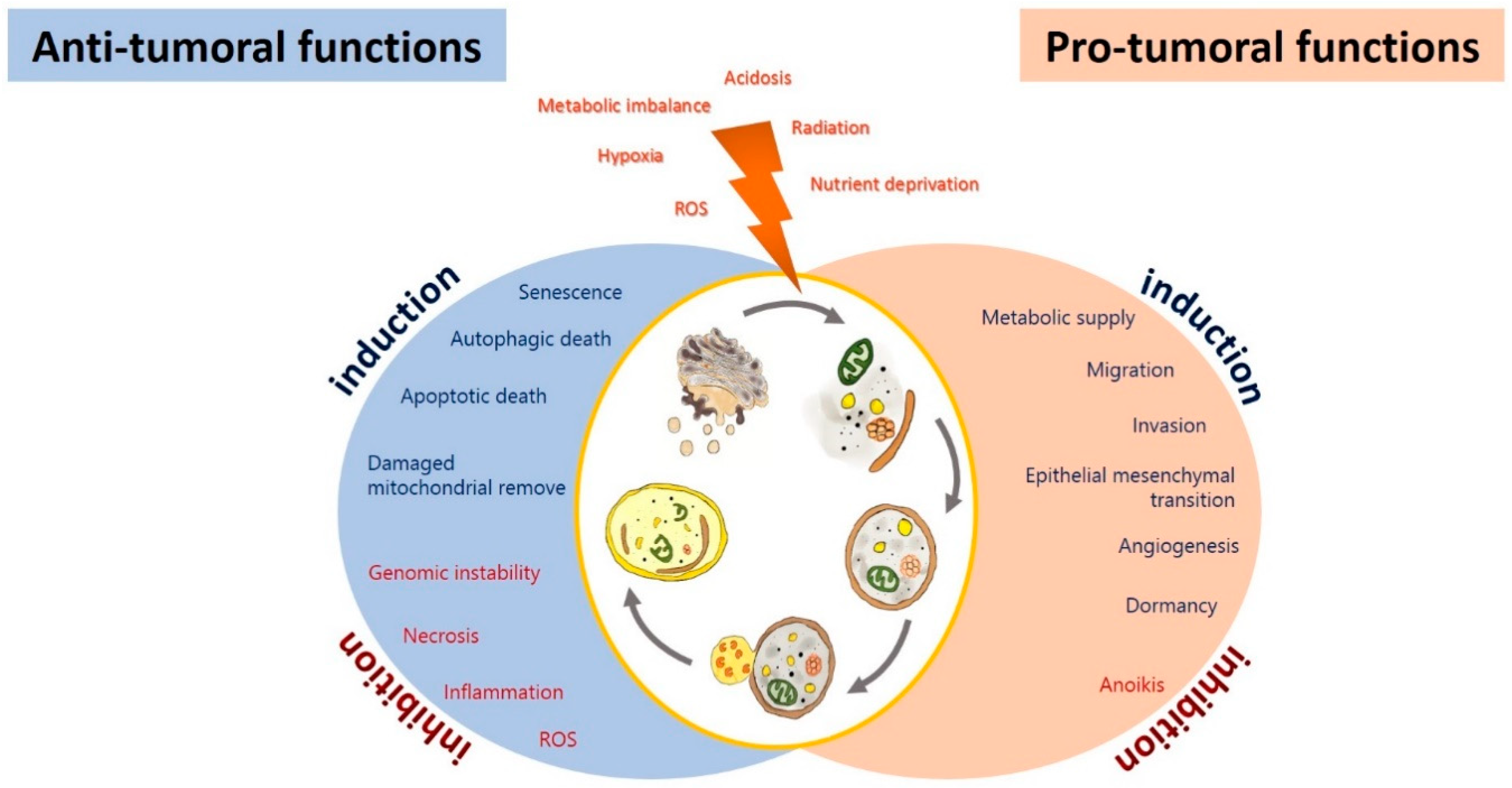{kind=link}
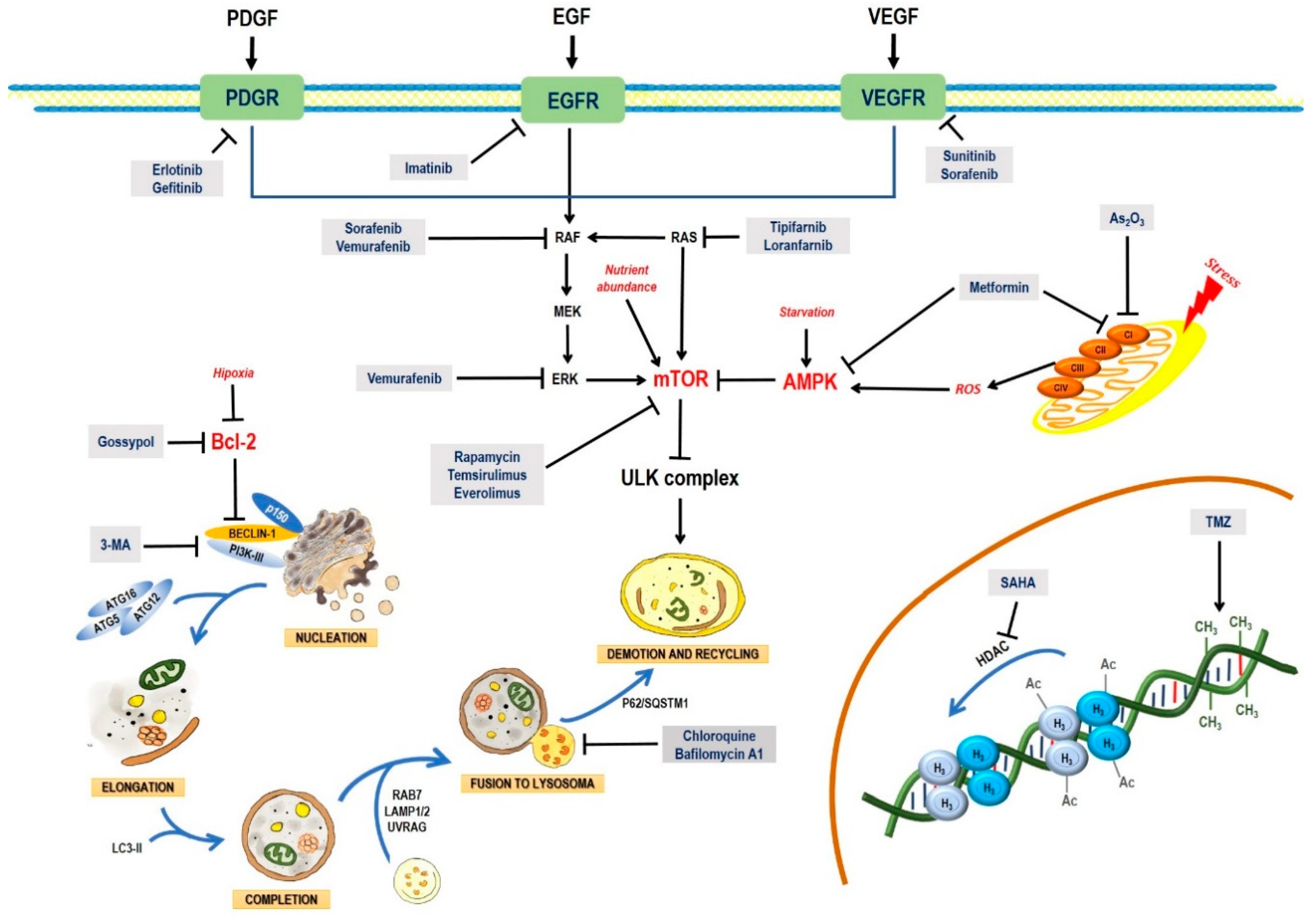{kind=link}
| Treatment/Pharmacologic Category | Mechanism of Action | Clinical and Pre-Clinical Trials | |
|---|---|---|---|
| Temozolomide (TMZ) (Second-generation alkylating agent analogue to mitozolomide) |
|
| |
| TMZ in combination with other treatments | Sorafenib (Multikinase inhibitor) |
|
|
| Momelotinib (Inhibit to Janus kinase (JAK)-1/2) |
|
| |
| Roscovitine (Cyclin-dependent kinase Cdk inhibitor) |
|
| |
| MSNP-TMZ-PDA-NGR plus 3-methyladenine (3-MA) (Controlled release of TMZ in cells with overexpression of CD13; 3-MA is an inhibitor of PI3K that avoid autophagy) | |||
| BIX01294 (Histone methyltransferase G9a inhibitor) |
|
| |
| Tubacin (Histone deacetylase 6 (HDAC6) inhibitor) |
|
| |
| Suberoylanilide hydroxamic acid (SAHA) (Histone deacetylase inhibitor) |
|
| |
| miR-93 overexpression (Downregulates the expression of proteins involved in autophagy) |
|
| |
| miR-128 (Regulates cell death and survival genes) |
|
| |
| Long non-coding RNAS cancer susceptibility candidate 2 (CASC2) (Tumor suppressor) |
|
| |
| miR-519a overexpression (Tumor suppressor by regulation of the STAT3 pathway in glioma) |
|
| |
| miR-224-3p overexpression (Regulates hypoxia-induced autophagy) |
|
| |
| Endothelial-monocyte- activating polypeptide-II (EMAPII) (Secretory polypeptide protein, with angiogenesis and pro-apoptosis effect) |
|
| |
| Cation transport regulator-like protein 1 (CHAC1) (Pro-apoptotic proprieties) |
|
| |
| GDC-0941 (Specific pan-PI3K inhibitor) |
|
| |
| Ionophores nigericin and salinomycin (Intracellular Ca2+ and K+ mobilizing) |
|
| |
| Guanosine (Neuroprotective agent with the propriety to mediate the glutamatergic system) |
|
| |
| Atorvastatin (Reduces the biosynthesis of cholesterol, regulates abnormal lipid storage, has anti-inflammatory and neuroprotective effects) |
|
| |
| Thioridazine (Antipsychotic drug with antineoplastic effects) |
|
| |
| Tuibolone and/or medroxyprogesterone acetate (MPA) (Hormone replacement therapy, analog of progesterone) |
|
| |
| Β-asarone (CIS-2,4,5-trimethoxy-1-allyl phenyl) (Polyphenols extracted from Acorus tatarinowii Schott and Guatteria gaumeri Greenman) |
|
| |
| Honokiol (2-(4-hydroxy-3-prop-2-enyl-phenyl)-4-prop-2-enyl-phenol) (Polyphenol extracted from Magnolia officinalis with anti-neoplastic effect) |
| ||
| Carnosic acid (Polyphenolic from Rosmarinus officinalis or Salvia officinalis with anticancer properties) |
|
| |
| Curcumin (Polyphenol isolated from rhizome of Curcuma longa with anti-angiogenic and anti-proliferative effects) | |||
| Euphol (A tetracyclic triterpene alcohol) (Phytopharmaceutical with analgesic, anti-inflammatory and antiviral properties, isolated from Euphorbia tirucalli) |
|
| |
| Δ9-tetrahydrocannabinol (THC) and cannabinol (CBD) (Cannabinoids extracted from Cannabis sativa) |
|
| |
| Treatment/Pharmacologic Category | Mechanism of Action In Vitro | Clinical Trials |
|---|---|---|
Arsenic trioxide Inorganic compound; molecular formula: As2O3 |
| |
Chloroquine (CQ) Synthetic antimalarial agent; molecular formula: C18H26ClN3 [4-N-(7-chloroquinolin-4-yl)-1-N,1-N-diethylpentane-1,4-diamine] |
| |
Metformin Biguanide type, obtained from the extract of Galega officinalis [276]; molecular formula: C4H11N5 [3-(diaminomethylidene)-1,1-dimethylguanidine] |
|
|
| Tyrosine kinase inhibitors | ||
Erlotinib Synthetic compound; molecular formula: C22H24ClN3O4 [6–bis(2-methoxy ethoxy)quinazoline-4-yl]-(3-ethynylphenyl)amine] |
|
|
Gefitinib Synthetic compound; molecular formula: C22H24ClFN4O3 [N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholin-4-ylpropoxy)quinazolin-4-amine] |
|
|
Dacomitinib Synthetic compound; molecular formula: C24H25ClFN5O2 [(E)-N-[4-(3-chloro-4-fluoroanilino)-7-methoxyquinazolin-6-yl]-4-piperidin-1-ylbut-2-enamide] |
| ∙ Poor antineoplastic activity in patients with EGFR amplification, with or without mutation in EGFRvIII [320]. |
Imatinib Synthetic compound; molecular formula: C29H31N7O [4-[(4-methylpiperazin-1-yl)methyl]-N-[4-methyl-3-[(4-pyridin-3- ylpyrimidin-2-yl)amino]phenyl]benzamide] |
| |
Sunitinib Synthetic compound: C22H27FN4O2 [N-[2-(diethylamino)ethyl]-5-[(Z)-(5-fluoro-2-oxo-1H-indol-3-ylidene)methyl]-2,4-dimethyl-1H-pyrrole-3-carboxamide] |
| |
Sorafenib Synthetic compound: C21H16ClF3N4O3 [4-[4-[[4-chloro-3-(trifluoromethyl)phenyl]carbamoylamino] phenoxy]-N-methylpyridine-2-carboxamide] |
|
|
| RAS/RAF/MAPK inhibitors | ||
Lonafarnib Synthetic compound: C27H31Br2ClN4O2 [4-[2-[4-[(2R)-6,15-dibromo-13-chloro-4-azatricyclo[9.4.0.03,8]pentadeca-1(11),3(8),4,6,12,14-hexaen-2-yl]piperidin-1-yl]-2-oxoethyl]piperidine-1-carboxamide] |
| |
VEMURAFENIB Synthetic compound: C23H18ClF2N3O3S [N-[3-[5-(4-chlorophenyl)-1H-pyrrolo[2-b]pyridine-3-carbonyl]-2,4-difluorophenyl]propane-1-sulfonamide] | ||
| PI3K/AKT/mTOR Inhibitors | ||
Temsirolimus Synthetic compound: C56H87NO16 [(1R,2R,4S)-4-[(2R)-2-[(1R,9S,12S,15R,16E,18R,19R,21R,23S,24E,26E,28E,30S,32S,35R)-1,18-dihydroxy-19,30-dimethoxy-15,17,21,23,29,35-hexamethyl-2,3,10,14,20-pentaoxo-11,36-dioxa-4-azatricyclo[30.3.1.04,9]hexatriaconta-16,24,26,28-tetraen-12-yl]propyl]-2-methoxycyclohexyl]-3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate] |
| |
Everolimus Synthetic compound: C53H83NO14[(1R,9S,12S,15R,16E,18R,19R,21R,23S,24E,26E,28E,30S,32S,35R)-1,18-dihydroxy-12-[(2R)-1-[(1S,3R,4R)-4-(2-hydroxyethoxy)-3-methoxycyclohexyl]propan-2-yl]-19,30-dimethoxy-15,17,21,23,29,35-hexamethyl-11,36-dioxa-4-azatricyclo[30.3.1.04,9]hexatriaconta-16,24,26,28-tetraene-2,3,10,14,20-pentone] |
| |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Escamilla-Ramírez, A.; Castillo-Rodríguez, R.A.; Zavala-Vega, S.; Jimenez-Farfan, D.; Anaya-Rubio, I.; Briseño, E.; Palencia, G.; Guevara, P.; Cruz-Salgado, A.; Sotelo, J.; et al. Autophagy as a Potential Therapy for Malignant Glioma. Pharmaceuticals 2020, 13, 156. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13070156
Escamilla-Ramírez A, Castillo-Rodríguez RA, Zavala-Vega S, Jimenez-Farfan D, Anaya-Rubio I, Briseño E, Palencia G, Guevara P, Cruz-Salgado A, Sotelo J, et al. Autophagy as a Potential Therapy for Malignant Glioma. Pharmaceuticals. 2020; 13(7):156. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13070156
Chicago/Turabian StyleEscamilla-Ramírez, Angel, Rosa A. Castillo-Rodríguez, Sergio Zavala-Vega, Dolores Jimenez-Farfan, Isabel Anaya-Rubio, Eduardo Briseño, Guadalupe Palencia, Patricia Guevara, Arturo Cruz-Salgado, Julio Sotelo, and et al. 2020. "Autophagy as a Potential Therapy for Malignant Glioma" Pharmaceuticals 13, no. 7: 156. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13070156