Current State of Radiolabeled Heterobivalent Peptidic Ligands in Tumor Imaging and Therapy

 ,
,
Abstract
:1. Introduction
1.1. General Advantages of Radiolabeled Peptide Heterodimers for Tumor Imaging
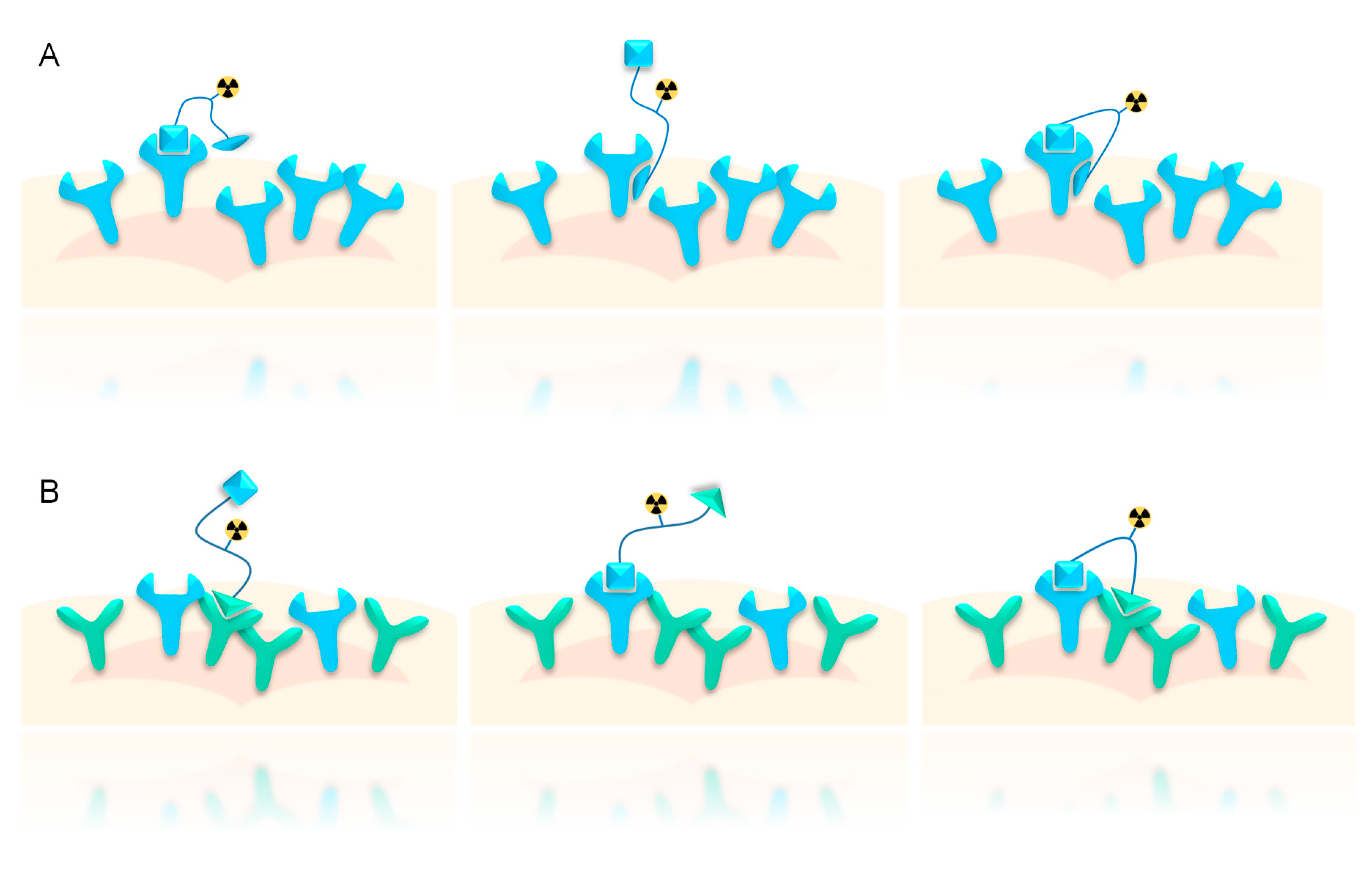
1.2. Influence of Receptor Density on Heterodimer Binding Mode, Avidity, and Tumor Specificity
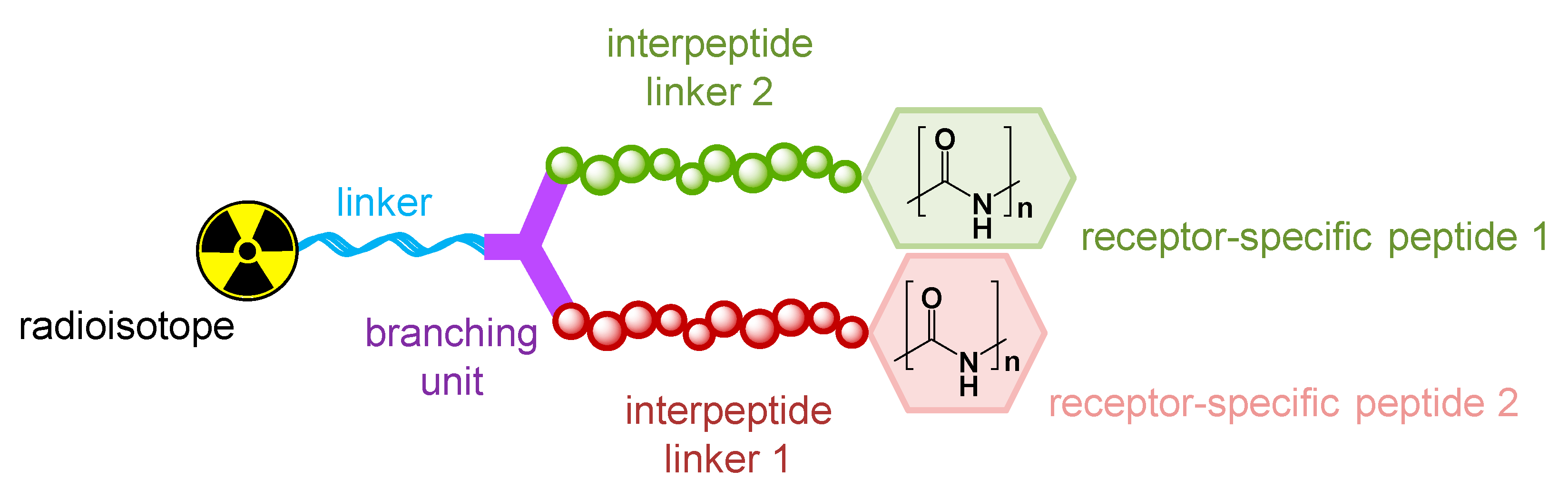
1.3. Influence of Molecular Design on Target Interaction
2. Radiolabeled Peptidic Heterodimers Developed for Improved Tumor Targeting
2.1. Heterobivalent Agents Binding the GRPR and PSMA for Improved Prostate Carcinoma Targeting
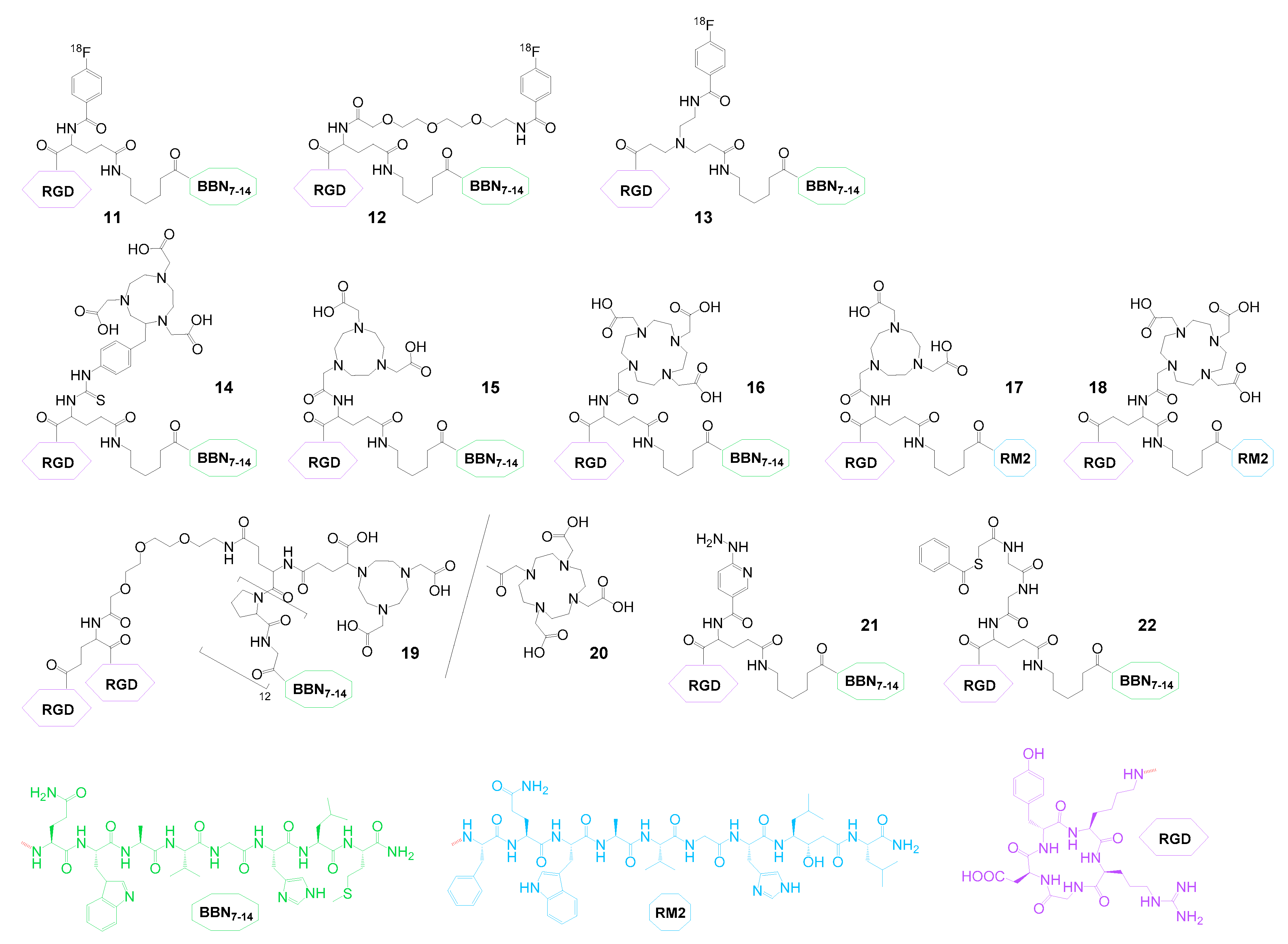
2.2. Heterobivalent Agents Targeting the GRPR and Integrin αvβ3
2.3. Further Heterobivalent Agents for PCa Imaging or Therapy
2.4. Heterobivalent Agents Developed for Breast Cancer Imaging or Therapy
2.5. Heterobivalent Agents Comprising an αvβ3 Integrin-Binding Peptide for Imaging and Therapy of Different Malignancies
3. Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Ambrosini, V.; Fani, M.; Fanti, S.; Forrer, F.; Maecke, H.R. Radiopeptide Imaging and Therapy in Europe. J. Nucl. Med. 2011, 52, 42s–55s. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, M.M.; Menda, Y. Radiopeptide Imaging and Therapy in the United States. J. Nucl. Med. 2011, 52, 56s–63s. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reubi, J.C.; Maecke, H.R. Peptide-based probes for cancer imaging. J. Nucl. Med. 2008, 49, 1735–1738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schottelius, M.; Wester, H.J. Molecular imaging targeting peptide receptors. Methods 2009, 48, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Tweedle, M.F. Peptide-Targeted Diagnostics and Radiotherapeutics. Acc. Chem. Res. 2009, 42, 958–968. [Google Scholar] [CrossRef]
- Opalinska, M.; Hubalewska-Dydejczyk, A.; Sowa-Staszczak, A. Radiolabeled peptides: Current and new perspectives. Q. J. Nucl. Med. Mol. Imaging 2017, 61, 153–167. [Google Scholar] [CrossRef]
- Rezazadeh, F.; Sadeghzadeh, N. Tumor targeting with (99m) Tc radiolabeled peptides: Clinical application and recent development. Chem. Biol. Drug Des. 2019, 93, 205–221. [Google Scholar] [CrossRef]
- Tornesello, A.L.; Buonaguro, L.; Tornesello, M.L.; Buonaguro, F.M. New Insights in the Design of Bioactive Peptides and Chelating Agents for Imaging and Therapy in Oncology. Molecules 2017, 22, 1282. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.Y.; Liu, S.; Hou, Y.P.; Tohme, M.; Park, R.; Bading, J.R.; Conti, P.S. MicroPET imaging of breast cancer alpha(v)-integrin expression with Cu-64-labeled dimeric RGD peptides. Mol. Imaging Biol. 2004, 6, 350–359. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, X.Z.; Xiong, Z.M.; Cheng, Z.; Fisher, D.R.; Liu, S.; Gambhir, S.S.; Chen, X.Y. microPET imaging of glioma integrin α(v)β(3) expression using Cu-64-labeled tetrameric RGD peptide. J. Nucl. Med. 2005, 46, 1707–1718. [Google Scholar]
- Summer, D.; Rangger, C.; Klingler, M.; Laverman, P.; Franssen, G.M.; Lechner, B.E.; Orasch, T.; Haas, H.; von Guggenberg, E.; Decristoforo, C. Exploiting the Concept of Multivalency with Ga-68- and Zr-89-Labelled Fusarinine C-Minigastrin Bioconjugates for Targeting CCK2R Expression. Contrast Media Mol. Imaging 2018, 3171794. [Google Scholar] [CrossRef] [Green Version]
- Summer, D.; Kroess, A.; Woerndle, R.; Rangger, C.; Klingler, M.; Haas, H.; Kremser, L.; Lindner, H.H.; von Guggenberg, E.; Decristoforo, C. Multimerization results in formation of re-bindable metabolites: A proof of concept study with FSC-based minigastrin imaging probes targeting CCK2R expression. PLoS ONE 2018, 13, e0201224. [Google Scholar] [CrossRef] [PubMed]
- Dijkgraaf, I.; Kruijtzer, J.A.W.; Liu, S.; Soede, A.C.; Oyen, W.J.G.; Corstens, F.H.M.; Liskamp, R.M.J.; Boerman, O.C. Improved targeting of the α(v)β(3) integrin by multimerisation of RGD peptides. Eur. J. Nucl. Med. Mol. Imaging 2007, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Dijkgraaf, I.; Yim, C.B.; Franssen, G.M.; Schuit, R.C.; Luurtsema, G.; Liu, S.A.; Oyen, W.J.G.; Boerman, O.C. PET imaging of α(v)β(3) integrin expression in tumours with Ga-68-labelled mono-, di- and tetrameric RGD peptides. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 128–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S. Radiolabeled Cyclic RGD Peptides as Integrin α(v)β(3)-Targeted Radiotracers: Maximizing Binding Affinity via Bivalency. Bioconjug. Chem. 2009, 20, 2199–2213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Liu, Z.; Chen, K.; Yan, Y.; Watzlowik, P.; Wester, H.J.; Chin, F.T.; Chen, X. 18F-labeled galacto and PEGylated RGD dimers for PET imaging of alphavbeta3 integrin expression. Mol. Imaging Biol. 2010, 12, 530–538. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.H.; Li, Z.B.; Chen, K.; Cai, W.B.; He, L.; Chin, F.T.; Li, F.; Chen, X.Y. MicroPET of tumor integrin α(v)β(3) expression using F-18-Labeled PEGylated tetrameric RGD peptide (F-18-FPRGD4). J. Nucl. Med. 2007, 48, 1536–1544. [Google Scholar] [CrossRef]
- Vauquelin, G.; Charlton, S.J. Exploring avidity: Understanding the potential gains in functional affinity and target residence time of bivalent and heterobivalent ligands. Brit. J. Pharmacol. 2013, 168, 1771–1785. [Google Scholar] [CrossRef] [Green Version]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.F.; Niu, G.; Shi, J.Y.; Liu, S.L.; Wang, F.; Liu, S.; Chen, X.Y. Ga-68-labeled cyclic RGD dimers with Gly(3) and PEG(4) linkers: Promising agents for tumor integrin α(v)β(3) PET imaging. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 947–957. [Google Scholar] [CrossRef]
- Liu, Z.F.; Liu, S.L.; Wang, F.; Liu, S.; Chen, X.Y. Noninvasive imaging of tumor integrin expression using F-18-labeled RGD dimer peptide with PEG(4) linkers. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 1296–1307. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.B.; Chen, K.; Chen, X. (68)Ga-labeled multimeric RGD peptides for microPET imaging of integrin alpha(v)beta (3) expression. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Liu, S. Radiolabeled multimeric cyclic RGD peptides as integrin α(v)β(3) targeted radiotracers for tumor imaging. Mol. Pharmaceut. 2006, 3, 472–487. [Google Scholar] [CrossRef] [PubMed]
- Kaeopookum, P.; Petrik, M.; Summer, D.; Klinger, M.; Zhai, C.; Rangger, C.; Haubner, R.; Haas, H.; Hajduch, M.; Decristoforo, C. Comparison of Ga-68-labeled RGD mono- and multimers based on a clickable siderophore-based scaffold. Nucl. Med. Biol. 2019, 78–79, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Knetsch, P.A.; Zhai, C.Y.; Rangger, C.; Blatzer, M.; Haas, H.; Kaeopookum, P.; Haubner, R.; Decristoforo, C. [Ga-68]FSC-(RGD)(3) a trimeric RGD peptide for imaging α(v)β(3) integrin expression based on a novel siderophore derived chelating scaffold-synthesis and evaluation. Nucl. Med. Biol. 2015, 42, 115–122. [Google Scholar] [CrossRef] [Green Version]
- Lobeek, D.; Franssen, G.M.; Ma, M.T.; Wester, H.J.; Decristoforo, C.; Oyen, W.J.G.; Boerman, O.C.; Terry, S.Y.A.; Rijpkema, M. In Vivo Characterization of 4 Ga-68-Labeled Multimeric RGD Peptides to Image α(v)β(3) Integrin Expression in 2 Human Tumor Xenograft Mouse Models. J. Nucl. Med. 2018, 59, 1296–1301. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.Y.; Wang, L.J.; Kim, Y.S.; Zhai, S.Z.; Jia, B.; Wang, F.; Liu, S. (TcO)-Tc-99m(MAG(2)-3G(3)-dimer): A new integrin α(v)β(3)-targeted SPECT radiotracer with high tumor uptake and favorable pharmacokinetics. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 1874–1884. [Google Scholar] [CrossRef]
- Zheng, Y.M.; Ji, S.D.; Tomaselli, E.; Yang, Y.; Liu, S. Comparison of biological properties of In-111-labeled dimeric cyclic RGD peptides. Nucl. Med. Biol. 2015, 42, 137–145. [Google Scholar] [CrossRef]
- Abiraj, K.; Jaccard, H.; Kretzschmar, M.; Helm, L.; Maecke, H.R. Novel DOTA-based prochelator for divalent peptide vectorization: Synthesis of dimeric bombesin analogues for multimodality tumor imaging and therapy. Chem. Commun. 2008, 3248–3250. [Google Scholar] [CrossRef] [Green Version]
- Lindner, S.; Michler, C.; Wängler, B.; Bartenstein, P.; Fischer, G.; Schirrmacher, R.; Wängler, C. PESIN Multimerization Improves Receptor Avidities and in Vivo Tumor Targeting Properties to GRPR-Overexpressing Tumors. Bioconjug. Chem. 2014, 25, 489–500. [Google Scholar] [CrossRef]
- Fischer, G.; Lindner, S.; Litau, S.; Schirrmacher, R.; Wangler, B.; Wangler, C. Next Step toward Optimization of GRP Receptor Avidities: Determination of the Minimal Distance between BBN(7-14) Units in Peptide Homodimers. Bioconjug. Chem. 2015, 26, 1479–1483. [Google Scholar] [CrossRef] [PubMed]
- Liolios, C.; Buchmuller, B.; Bauder-Wust, U.; Sohafer, M.; Leotta, K.; Haberkorn, U.; Eder, M.; Kopka, K. Monomeric and Dimeric Ga-68-Labeled Bombesin Analogues for Positron Emission Tomography (PET) Imaging of Tumors Expressing Gastrin-Releasing Peptide Receptors (GRPrs). J. Med. Chem. 2018, 61, 2062–2074. [Google Scholar] [CrossRef] [PubMed]
- Yim, C.B.; Boerman, O.C.; de Visser, M.; de Jong, M.; Dechesne, A.C.; Rijkers, D.T.S.; Liskamp, R.M.J. Versatile Conjugation of Octreotide to Dendrimers by Cycloaddition (“Click”) Chemistry to Yield High-Affinity Multivalent Cyclic Peptide Dendrimers. Bioconjug. Chem. 2009, 20, 1323–1331. [Google Scholar] [CrossRef] [PubMed]
- Yim, C.B.; Dijkgraaf, I.; Merkx, R.; Versluis, C.; Eek, A.; Mulder, G.E.; Rijkers, D.T.S.; Boerman, O.C.; Liskamp, R.M.J. Synthesis of DOTA-Conjugated Multimeric [Tyr(3)]Octreotide Peptides via a Combination of Cu(I)-Catalyzed “Click” Cycloaddition and Thio Acid/Sulfonyl Azide “Sulfo-Click” Amidation and Their in Vivo Evaluation. J. Med. Chem. 2010, 53, 3944–3953. [Google Scholar] [CrossRef]
- Yim, C.B.; van der Wildt, B.; Dijkgraaf, I.; Joosten, L.; Eek, A.; Versluis, C.; Rijkers, D.T.S.; Boerman, O.C.; Liskamp, R.M.J. Spacer Effects on in vivo Properties of DOTA-Conjugated Dimeric [Tyr3] Octreotate Peptides Synthesized by a “Cu-I-Click” and “Sulfo-Click” Ligation Method. ChemBioChem 2011, 12, 750–760. [Google Scholar] [CrossRef]
- Brandenburger, Y.; Rose, K.; Bagutti, C.; Eberle, A.N. Synthesis and receptor binding analysis of thirteen oligomeric alpha-MSH analogs. J. Recept. Signal Transduct. Res. 1999, 19, 467–480. [Google Scholar] [CrossRef]
- Tiesjema, B.; Merkestein, M.; Garner, K.M.; de Krom, M.; Adan, R.A.H. Multimeric alpha-MSH has increased efficacy to activate the melanocortin MC4 receptor. Eur. J. Pharmacol. 2008, 585, 24–30. [Google Scholar] [CrossRef]
- Vagner, J.; Handl, H.L.; Gillies, R.J.; Hruby, V.J. Novel targeting strategy based on multimeric ligands for drug delivery and molecular imaging: Homooligomers of alpha-MSH. Bioorg. Med. Chem. Lett. 2004, 14, 211–215. [Google Scholar] [CrossRef]
- Brabez, N.; Lynch, R.M.; Xu, L.P.; Gillies, R.J.; Chassaing, G.; Lavielle, S.; Hruby, V.J. Design, Synthesis, and Biological Studies of Efficient Multivalent Melanotropin Ligands: Tools toward Melanoma Diagnosis and Treatment. J. Med. Chem. 2011, 54, 7375–7384. [Google Scholar] [CrossRef] [Green Version]
- Hultsch, C.; Pawelke, B.; Bergmann, R.; Wuest, F. Synthesis and evaluation of novel multimeric neurotensin(8–13) analogs. Bioorg. Med. Chem. 2006, 14, 5913–5920. [Google Scholar] [CrossRef]
- Maschauer, S.; Einsiedel, J.; Reich, D.; Hubner, H.; Gmeiner, P.; Wester, H.J.; Prante, O.; Notni, J. Theranostic Value of Multimers: Lessons Learned from Trimerization of Neurotensin Receptor Ligands and Other Targeting Vectors. Pharmaceuticals 2017, 10, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohrich, A.; Bergmann, R.; Kretzschmann, A.; Noll, S.; Steinbach, J.; Pietzsch, J.; Stephan, H. A novel tetrabranched neurotensin(8–13) cyclam derivative: Synthesis, Cu-64-labeling and biological evaluation. J. Inorg. Biochem. 2011, 105, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Russnes, H.G.; Navin, N.; Hicks, J.; Borresen-Dale, A.L. Insight into the heterogeneity of breast cancer through next-generation sequencing. J. Clin. Investig. 2011, 121, 3810–3818. [Google Scholar] [CrossRef] [Green Version]
- Mannweiler, S.; Amersdorfer, P.; Trajanoski, S.; Terrett, J.A.; King, D.; Mehes, G. Heterogeneity of Prostate-Specific Membrane Antigen (PSMA) Expression in Prostate Carcinoma with Distant Metastasis. Pathol. Oncol. Res. 2009, 15, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Bedard, P.L.; Hansen, A.R.; Ratain, M.J.; Siu, L.L. Tumour heterogeneity in the clinic. Nature 2013, 501, 355–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Katzenellenbogen, J.A. The quest for improving the management of breast cancer by functional imaging: The discovery and development of 16alpha-[(18)F]fluoroestradiol (FES), a PET radiotracer for the estrogen receptor, a historical review. Nucl. Med. Biol. 2020. [Google Scholar] [CrossRef]
- Ananias, H.J.K.; van den Heuvel, M.C.; Helfrich, W.; de Jong, I.J. Expression of the Gastrin-Releasing Peptide Receptor, the Prostate Stem Cell Antigen and the Prostate-Specific Membrane Antigen in Lymph Node and Bone Metastases of Prostate Cancer. Prostate 2009, 69, 1101–1108. [Google Scholar] [CrossRef]
- Diaz, L.A.; Williams, R.T.; Wu, J.; Kinde, I.; Hecht, J.R.; Berlin, J.; Allen, B.; Bozic, I.; Reiter, J.G.; Nowak, M.A.; et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012, 486, 537–540. [Google Scholar] [CrossRef] [Green Version]
- Sosman, J.A.; Puzanov, I.; Atkins, M.B. Opportunities and obstacles to combination targeted therapy in renal cell cancer. Clin. Cancer Res. 2007, 13, 764s–769s. [Google Scholar] [CrossRef] [Green Version]
- Farag, S.; Smith, M.J.; Fotiadis, N.; Constantinidou, A.; Jones, R.L. Revolutions in treatment options in gastrointestinal stromal tumours (GISTs): The latest updates: Revolutions in treatment options in GIST. Curr. Treat. Opt. Oncol. 2020, 21, 55. [Google Scholar] [CrossRef] [PubMed]
- Gugger, M.; Reubi, J.C. Gastrin-releasing peptide receptors in non-neoplastic and neoplastic human breast. Am. J. Pathol. 1999, 155, 2067–2076. [Google Scholar] [CrossRef] [Green Version]
- Reubi, J.C.; Gugger, M.; Waser, B.; Schaer, J.C. Y-1-mediated effect of neuropeptide Y in cancer: Breast carcinomas as targets. Cancer Res. 2001, 61, 4636–4641. [Google Scholar] [PubMed]
- Reubi, J.C.; Gugger, M.; Waser, B. Co-expressed peptide receptors in breast cancer as a molecular basis for in vivo multireceptor tumour targeting. Eur. J. Nucl. Med. Mol. Imaging 2002, 29, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Valant, C.; Lane, J.R.; Sexton, P.M.; Christopoulos, A. The Best of Both Worlds? Bitopic Orthosteric/Allosteric Ligands of G Protein-Coupled Receptors. Annu. Rev. Pharmacol. 2012, 52, 153–178. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C.; Fleischmann, A.; Waser, B.; Rehmann, R. Concomitant vascular GRP-receptor and VEGF-receptor expression in human tumors: Molecular basis for dual targeting of tumoral vasculature. Peptides 2011, 32, 1457–1462. [Google Scholar] [CrossRef]
- Reubi, J.C.; Maecke, H.R. Approaches to Multireceptor Targeting: Hybrid Radioligands, Radioligand Cocktails, and Sequential Radioligand Applications. J. Nucl. Med. 2017, 58, 10s–16s. [Google Scholar] [CrossRef] [Green Version]
- Reubi, J.C.; Waser, B. Concomitant expression of several peptide receptors in neuroendocrine tumours: Molecular basis for in vivo multireceptor tumour targeting. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 781–793. [Google Scholar] [CrossRef]
- Josan, J.S.; Handl, H.L.; Sankaranarayanan, R.; Xu, L.P.; Lynch, R.M.; Vagner, J.; Mash, E.A.; Hruby, V.J.; Gillies, R.J. Cell-Specific Targeting by Heterobivalent Ligands. Bioconjug. Chem. 2011, 22, 1270–1278. [Google Scholar] [CrossRef] [Green Version]
- Vauquelin, G. Simplified models for heterobivalent ligand binding: When are they applicable and which are the factors that affect their target residence time. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2013, 386, 949–962. [Google Scholar] [CrossRef]
- Xu, L.P.; Vagner, J.; Josan, J.; Lynch, R.M.; Morse, D.L.; Baggett, B.; Han, H.Y.; Mash, E.A.; Hruby, V.J.; Gillies, R.J. Enhanced targeting with heterobivalent ligands. Mol. Cancer Ther. 2009, 8, 2356–2365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vagner, J.; Xu, L.P.; Handl, H.L.; Josan, J.S.; Morse, D.L.; Mash, E.A.; Gillies, R.J.; Hruby, V.J. Heterobivalent Ligands crosslink multiple cell-surface receptors: The human melanocortin-4 and delta-opioid receptors. Angew. Chem. Int. Ed. 2008, 47, 1685–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Josan, J.S.; Vagner, J.; Handl, H.L.; Sankaranarayanan, R.; Gillies, R.J.; Hruby, V.J. Solid-Phase Synthesis of Heterobivalent Ligands Targeted to Melanocortin and Cholecystokinin Receptors. Int. J. Pept. Res. Ther. 2008, 14, 293–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.F.; Yan, Y.J.; Chin, F.T.; Wang, F.; Chen, X.Y. Dual Integrin and Gastrin-Releasing Peptide Receptor Targeted Tumor Imaging Using F-18-labeled PEGylated RGD-Bombesin Heterodimer F-18-FB-PEG(3)-Glu-RGD-BBN. J. Med. Chem. 2009, 52, 425–432. [Google Scholar] [CrossRef]
- Li, Z.B.; Wu, Z.H.; Chen, K.; Ryu, E.K.; Chen, X.Y. F-18-labeled BBN-RGD heterodimer for prostate cancer imaging. J. Nucl. Med. 2008, 49, 453–461. [Google Scholar] [CrossRef] [Green Version]
- Lindner, S.; Fiedler, L.; Wangler, B.; Bartenstein, P.; Schirrmacher, R.; Wangler, C. Design, synthesis and in vitro evaluation of heterobivalent peptidic radioligands targeting both GRP- and VPAC(1)-Receptors concomitantly overexpressed on various malignancies—Is the concept feasible? Eur. J. Med. Chem. 2018, 155, 84–95. [Google Scholar] [CrossRef]
- Handl, H.L.; Vagner, J.; Han, H.Y.; Mash, E.; Hruby, V.J.; Gillies, R.J. Hitting multiple targets with multimeric ligands. Expert Opin. Ther. Targets 2004, 8, 565–586. [Google Scholar] [CrossRef]
- Vall-Sagarra, A.; Litau, S.; Decristoforo, C.; Wangler, B.; Schirrmacher, R.; Fricker, G.; Wangler, C. Design, Synthesis, In Vitro, and Initial In Vivo Evaluation of Heterobivalent Peptidic Ligands Targeting Both NPY(Y-1)- and GRP-Receptors-An Improvement for Breast Cancer Imaging? Pharmaceuticals 2018, 11, 65. [Google Scholar] [CrossRef] [Green Version]
- Fischer, G.; Schirrmacher, R.; Wangler, B.; Wangler, C. Radiolabeled heterobivalent peptidic ligands: An approach with high future potential for in vivo imaging and therapy of malignant diseases. ChemMedChem 2013, 8, 883–890. [Google Scholar] [CrossRef]
- Sun, X.L.; Li, Y.S.; Liu, T.; Li, Z.J.; Zhang, X.Z.; Chen, X.Y. Peptide-based imaging agents for cancer detection. Adv. Drug Deliv. Rev. 2017, 110, 38–51. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.M.; Hong, H.; Yang, S.P.; Cai, W.B. Design and Applications of Bispecific Heterodimers: Molecular Imaging and beyond. Mol. Pharmaceut. 2014, 11, 1750–1761. [Google Scholar] [CrossRef] [PubMed]
- Liolios, C.; Sachpekidis, C.; Schafer, M.; Kopka, K. Bispecific radioligands targeting prostate-specific membrane antigen and gastrin-releasing peptide receptors on the surface of prostate cancer cells. J. Labelled Comp. Radiopharm. 2019, 62, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.J.; Chen, X.Y. Peptide heterodimers for molecular imaging. Amino Acids 2011, 41, 1081–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandari, R.P.; Jiang, Z.R.; Reynolds, T.S.; Bernskoetter, N.E.; Szczodroski, A.F.; Bassuner, K.J.; Kirkpatrick, D.L.; Rold, T.L.; Sieckman, G.L.; Hoffman, T.J.; et al. Synthesis and biological evaluation of copper-64 radiolabeled [DUPA-6-Ahx-(NODAGA)-5-Ava-BBN(7-14)NH2], a novel bivalent targeting vector having affinity for two distinct biomarkers (GRPr/PSMA) of prostate cancer. Nucl. Med. Biol. 2014, 41, 355–363. [Google Scholar] [CrossRef] [Green Version]
- Eder, M.; Schafer, M.; Bauder-Wust, U.; Haberkorn, U.; Eisenhut, M.; Kopka, K. Preclinical evaluation of a bispecific low-molecular heterodimer targeting both PSMA and GRPR for improved PET imaging and therapy of prostate cancer. Prostate 2014, 74, 659–668. [Google Scholar] [CrossRef]
- Liolios, C.; Schafer, M.; Haberkorn, U.; Eder, M.; Kopka, K. Novel Bispecific PSMA/GRPr Targeting Radioligands with Optimized Pharmacokinetics for Improved PET Imaging of Prostate Cancer. Bioconjug. Chem. 2016, 27, 737–751. [Google Scholar] [CrossRef]
- Mendoza-Figueroa, M.J.; Escudero-Castellanos, A.; Ramirez-Nava, G.J.; Ocampo-Garcia, B.E.; Santos-Cuevas, C.L.; Ferro-Flores, G.; Pedraza-Lopez, M.; Avila-Rodriguez, M.A. Preparation and preclinical evaluation of Ga-68-iPSMA-BN as a potential heterodimeric radiotracer for PET-imaging of prostate cancer. J. Radioanal. Nucl. Chem. 2018, 318, 2097–2105. [Google Scholar] [CrossRef]
- Escudero-Castellanos, A.; Ocampo-Garcia, B.; Ferro-Flores, G.; Santos-Cuevas, C.; Morales-Avila, E.; Luna-Gutierrez, M.; Isaac-Olive, K. Synthesis and preclinical evaluation of the Lu-177-DOTA-PSMA(inhibitor)-Lys(3)-bombesin heterodimer designed as a radiotheranostic probe for prostate cancer. Nucl. Med. Commun. 2019, 40, 278–286. [Google Scholar] [CrossRef]
- Mitran, B.; Varasteh, Z.; Abouzayed, A.; Rinne, S.S.; Puuvuori, E.; De Rosa, M.; Larhed, M.; Tolmachev, V.; Orlova, A.; Rosenstrom, U. Bispecific GRPR-Antagonistic Anti-PSMA/GRPR Heterodimer for PET and SPECT Diagnostic Imaging of Prostate Cancer. Cancers 2019, 11, 1371. [Google Scholar] [CrossRef] [Green Version]
- Abouzayed, A.; Yim, C.B.; Mitran, B.; Rinne, S.S.; Tolmachev, V.; Larhed, M.; Rosenstrom, U.; Orlova, A. Synthesis and Preclinical Evaluation of Radio-Iodinated GRPR/PSMA Bispecific Heterodimers for the Theranostics Application in Prostate Cancer. Pharmaceutics 2019, 11, 358. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.J.; Chen, K.; Yang, M.; Sun, X.L.; Liu, S.L.; Chen, X.Y. A new F-18-labeled BBN-RGD peptide heterodimer with a symmetric linker for prostate cancer imaging. Amino Acids 2011, 41, 439–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Yan, Y.; Liu, S.; Wang, F.; Chen, X. (18)F, (64)Cu, and (68)Ga labeled RGD-bombesin heterodimeric peptides for PET imaging of breast cancer. Bioconjug. Chem. 2009, 20, 1016–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.F.; Niu, G.; Wang, F.; Chen, X.Y. Ga-68-labeled NOTA-RGD-BBN peptide for dual integrin and GRPR-targeted tumor imaging. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 1483–1494. [Google Scholar] [CrossRef]
- Zhang, J.J.; Niu, G.; Lang, L.X.; Li, F.; Fan, X.R.; Yang, X.F.; Yao, S.B.; Yan, W.G.; Huo, L.; Chen, L.B.; et al. Clinical Translation of a Dual Integrin α(v)β(3)- and Gastrin-Releasing Peptide Receptor-Targeting PET Radiotracer, Ga-68-BBN-RGD. J. Nucl. Med. 2017, 58, 228–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, A.B.; Nanda, P.K.; Rold, T.L.; Sieckman, G.L.; Szczodroski, A.F.; Hoffman, T.J.; Chen, X.Y.; Smith, C.J. Cu-64-NO2A-RGD-Glu-6-Ahx-BBN(7-14)NH2: A heterodimeric targeting vector for positron emission tomography imaging of prostate cancer. Nucl. Med. Biol. 2012, 39, 377–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.F.; Li, Z.B.; Cao, Q.Z.; Liu, S.L.; Wang, F.; Chen, X.Y. Small-Animal PET of Tumors with (64Cu)-Labeled RGD-Bombesin Heterodimer. J. Nucl. Med. 2009, 50, 1168–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Miao, Z.; Liu, H.G.; Ren, G.; Bao, A.D.; Cutler, C.S.; Shi, H.C.; Cheng, Z. Lu-177-labeled RGD-BBN heterodimeric peptide for targeting prostate carcinoma. Nucl. Med. Commun. 2013, 34, 909–914. [Google Scholar] [CrossRef]
- Durkan, K.; Jiang, Z.R.; Rold, T.L.; Sieckman, G.L.; Hoffman, T.J.; Bandari, R.P.; Szczodroski, A.F.; Liu, L.Q.; Miao, Y.B.; Reynolds, T.S.; et al. A heterodimeric [RGD-Glu-[Cu-64-NO2A]-6-Ahx-RM2] α(v)β(3)/GRPr-targeting antagonist radiotracer for PET imaging of prostate tumors. Nucl. Med. Biol. 2014, 41, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Bandara, N.; Reynolds, T.J.S.; Schehr, R.; Bandari, R.P.; Diebolder, P.J.; Krieger, S.; Xu, J.L.; Miao, Y.B.; Rogers, B.E.; Smith, C.J. Matched-pair, Y-86/Y-90-labeled, bivalent RGD/bombesin antagonist, [RGD-Glu-[DO3A]-6-Ahx-RM2], as a potential theranostic agent for prostate cancer. Nucl. Med. Biol. 2018, 62–63, 71–77. [Google Scholar] [CrossRef]
- Reynolds, T.J.S.; Schehr, R.; Liu, D.J.; Xu, J.L.; Miao, Y.B.; Hoffman, T.J.; Rold, T.L.; Lewis, M.R.; Smith, C.J. Characterization and evaluation of DOTA-conjugated Bombesin/RGD-antagonists for prostate cancer tumor imaging and therapy. Nucl. Med. Biol. 2015, 42, 99–108. [Google Scholar] [CrossRef]
- Lucente, E.; Liu, H.G.; Liu, Y.; Hu, X.; Lacivita, E.; Leopoldo, M.; Cheng, Z. Novel Cu-64 Labeled RGD(2)-BBN Heterotrimers for PET Imaging of Prostate Cancer. Bioconjug. Chem. 2018, 29, 1595–1604. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.F.; Huang, J.M.; Dong, C.Y.; Cui, L.Y.; Jin, X.N.; Jia, B.; Zhu, Z.H.; Li, F.; Wang, F. Tc-99m-Labeled RGD-BBN Peptide for Small-Animal SPECT/CT of Lung Carcinoma. Mol. Pharmaceut. 2012, 9, 1409–1417. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.Y.; Liu, Z.F.; Jin, X.N.; Jia, B.; Li, F.; Wang, F. Evaluation of Re-188-MAG(2)-RGD-bombesin for potential prostate cancer therapy. Nucl. Med. Biol. 2013, 40, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Santos-Cuevas, C.L.; Ferro-Flores, G.; de Murphy, C.A.; Ramirez, F.D.; Luna-Gutierrez, M.A.; Pedraza-Lopez, M.; Garcia-Becerra, R.; Ordaz-Rosado, D. Design, preparation, in vitro and in vivo evaluation of Tc-99m-N2S2-Tat(49-57)-bombesin: A target-specific hybrid radiopharmaceutical. Int. J. Pharmaceut. 2009, 375, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Santos-Cuevas, C.L.; Ferro-Flores, G.; Rojas-Calderon, E.L.; Garcia-Becerra, R.; Ordaz-Rosado, D.; de Murphy, C.A.; Pedraza-Lopez, M. Tc-99m-N2S2-Tat (49-57)-bombesin internalized in nuclei of prostate and breast cancer cells: Kinetics, dosimetry and effect on cellular proliferation. Nucl. Med. Commun. 2011, 32, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Escudero-Castellanos, A.; Ocampo-Garcia, B.E.; Ferro-Flores, G.; Isaac-Olive, K.; Santos-Cuevas, C.L.; Olmos-Ortiz, A.; Garcia-Quiroz, J.; Garcia-Becerra, R.; Diaz, L. Preparation and in vitro evaluation of Lu-177-iPSMA-RGD as a new heterobivalent radiopharmaceutical. J. Radioanal. Nucl. Chem. 2017, 314, 2201–2207. [Google Scholar] [CrossRef]
- Shrivastava, A.; Wang, S.H.; Raju, N.; Gierach, I.; Ding, H.M.; Tweedle, M.F. Heterobivalent dual-target probe for targeting GRP and Y1 receptors on tumor cells. Bioorg. Med. Chem. Lett. 2013, 23, 687–692. [Google Scholar] [CrossRef] [Green Version]
- Aranda-Lara, L.; Ferro-Flores, G.; Ramirez, F.D.; Ocampo-Garcia, B.; Santos-Cuevas, C.; Diaz-Nieto, L.; Isaac-Olive, K. Improved radiopharmaceutical based on Tc-99m-Bombesin-folate for breast tumour imaging. Nucl. Med. Commun. 2016, 37, 377–386. [Google Scholar] [CrossRef]
- Aranda-Lara, L.; Ferro-Flores, G.; Azorin-Vega, E.; Ramirez, F.D.; Jimenez-Mancilla, N.; Ocampo-Garcia, B.; Santos-Cuevas, C.; Isaac-Olive, K. Synthesis and evaluation of Lys(1)(alpha,gamma-Folate)Lys(3)(Lu-177-DOTA)-Bombesin(1-14) as a potential theranostic radiopharmaceutical for breast cancer. Appl. Radiat. Isot. 2016, 107, 214–219. [Google Scholar] [CrossRef]
- Capello, A.; Krenning, E.P.; Bernard, B.F.; Breeman, W.A.P.; van Hagen, M.P.; de Jong, M. Increased cell death after therapy with an Arg-Gly-Asp-linked somatostatin analog. J. Nucl. Med. 2004, 45, 1716–1720. [Google Scholar]
- Bernard, B.; Capello, A.; van Hagen, M.; Breeman, W.; Srinivasan, A.; Schmidt, M.; Erion, J.; van Gameren, A.; Krenning, E.; de Jong, M. Radiolabeled RGD-DTPA-Tyr(3)-octreotate for receptor-targeted radionuclide therapy. Cancer Biother. Radiopham. 2004, 19, 173–180. [Google Scholar] [CrossRef]
- Capello, A.; Krenning, E.P.; Bernard, B.F.; Breeman, W.A.P.; Erion, J.L.; de Jong, M. Anticancer activity of targeted proapoptotic peptides. J. Nucl. Med. 2006, 47, 122–129. [Google Scholar] [PubMed]
- Wu, H.; Chen, H.J.; Pan, D.F.; Ma, Y.F.; Liang, S.; Wan, Y.; Fang, Y. Imaging Integrin α(v)β(3) and NRP-1 Positive Gliomas with a Novel Fluorine-18 Labeled RGD-ATWLPPR Heterodimeric Peptide Probe. Mol. Imaging Biol. 2014, 16, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.F.; Liang, S.; Guo, J.; Guo, R.; Wang, H. F-18 labeled RGD-A7R peptide for dual integrin and VEGF-targeted tumor imaging in mice bearing U87MG tumors. J. Labelled Comp. Radiopharm. 2014, 57, 627–631. [Google Scholar] [CrossRef]
- Liang, S.; Ma, Y.F.; Guo, J.; Guo, R.; Wang, H. F-18-radiolabeled analogs of peptide RGD-A7R for simultaneous PET imaging of both alpha v beta 3 and VEGF in tumors. J. Radioanal. Nucl. Chem. 2015, 303, 1891–1896. [Google Scholar] [CrossRef]
- Yang, J.Q.; Guo, H.X.; Gallazzi, F.; Berwick, M.; Padilla, R.S.; Miao, Y.B. Evaluation of a Novel Arg-Gly-Asp-Conjugated α-Melanocyte Stimulating Hormone Hybrid Peptide for Potential Melanoma Therapy. Bioconjug. Chem. 2009, 20, 1634–1642. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.M.; Jeong, M.H.; Kim, D.W.; Jeong, H.J.; Lim, S.T.; Sohn, M.H. Iodine 125-labeled mesenchymal-epithelial transition factor binding peptide-click-cRGDyk heterodimer for glioma imaging. Cancer Sci. 2011, 102, 1516–1521. [Google Scholar] [CrossRef]
- Holland, J.P.; Williamson, M.J.; Lewis, J.S. Unconventional Nuclides for Radiopharmaceuticals. Mol. Imaging 2010, 9, 1–20. [Google Scholar] [CrossRef]
- Jodal, L. Beta emitters and radiation protection. Acta Oncol. 2009, 48, 308–313. [Google Scholar] [CrossRef] [Green Version]
- Ku, A.; Facca, V.J.; Cai, Z.; Reilly, R.M. Auger electrons for cancer therapy—A review. EJNMMI Radiopharm. Chem. 2019, 4, 27. [Google Scholar] [CrossRef] [Green Version]
- Markwalder, R.; Reubi, J.C. Gastrin-releasing peptide receptors in the human prostate: Relation to neoplastic transformation. Cancer Res. 1999, 59, 1152–1159. [Google Scholar] [PubMed]
- Wright, G.L.; Grob, B.M.; Haley, C.; Grossman, K.; Newhall, K.; Petrylak, D.; Troyer, J.; Konchuba, A.; Schellhammer, P.F.; Moriarty, R. Upregulation of prostate-specific membrane antigen after androgen-deprivation therapy. Urology 1996, 48, 326–334. [Google Scholar] [CrossRef]
- Wright, G.L., Jr.; Haley, C.; Beckett, M.L.; Schellhammer, P.F. Expression of prostate-specific membrane antigen in normal, benign, and malignant prostate tissues. Urol. Oncol. 1995, 1, 18–28. [Google Scholar] [CrossRef]
- Perner, S.; Hofer, M.D.; Kim, R.; Shah, R.B.; Li, H.J.; Moller, P.; Hautmann, R.E.; Gschwend, J.E.; Kuefer, R.; Rubin, M.A. Prostate-specific membrane antigen expression as a predictor of prostate cancer progression. Hum. Pathol. 2007, 38, 696–701. [Google Scholar] [CrossRef]
- Israeli, R.S.; Powell, C.T.; Corr, J.G.; Fair, W.R.; Heston, W.D.W. Expression of the Prostate-Specific Membrane Antigen. Cancer Res. 1994, 54, 1807–1811. [Google Scholar]
- von Eyben, F.E.; Picchio, M.; von Eyben, R.; Rhee, H.; Bauman, G. Ga-68-Labeled Prostate-specific Membrane Antigen Ligand Positron Emission Tomography/Computed Tomography for Prostate Cancer: A Systematic Review and Meta-analysis. Eur. Urol. Focus 2018, 4, 686–693. [Google Scholar] [CrossRef] [Green Version]
- Iagaru, A. Will GRPR Compete with PSMA as a Target in Prostate Cancer? J. Nucl. Med. 2017, 58, 1883–1884. [Google Scholar] [CrossRef] [Green Version]
- Eder, M.; Lohr, T.; Bauder-Wust, U.; Reber, M.; Mier, W.; Schafer, M.; Haberkorn, U.; Eisenhut, M. Pharmacokinetic Properties of Peptidic Radiopharmaceuticals: Reduced Uptake of (EH)(3)-Conjugates in Important Organs. J. Nucl. Med. 2013, 54, 1327–1330. [Google Scholar] [CrossRef] [Green Version]
- Pooja, D.; Gunukula, A.; Gupta, N.; Adams, D.J.; Kulhari, H. Bombesin receptors as potential targets for anticancer drug delivery and imaging. Int. J. Biochem. Cell Biol. 2019, 114. [Google Scholar] [CrossRef]
- Debordeaux, F.; Chansel-Debordeaux, L.; Pinaquy, J.B.; Fernandez, P.; Schulz, J. What about α(v)β(3) integrins in molecular imaging in oncology? Nucl. Med. Biol. 2018, 62–63, 31–46. [Google Scholar] [CrossRef]
- Nieberler, M.; Reuning, U.; Reichart, F.; Notni, J.; Wester, H.J.; Schwaiger, M.; Weinmuller, M.; Rader, A.; Steiger, K.; Kessler, H. Exploring the Role of RGD-Recognizing Integrins in Cancer. Cancers 2017, 9, 116. [Google Scholar] [CrossRef] [PubMed]
- Litau, S.; Seibold, U.; Vall-Sagarra, A.; Fricker, G.; Wangler, B.; Wangler, C. Comparative Assessment of Complex Stabilities of Radiocopper Chelating Agents by a Combination of Complex Challenge and in vivo Experiments. ChemMedChem 2015, 10, 1200–1208. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Mao, F.; Niu, G.; Peng, L.; Lang, L.; Li, F.; Ying, H.; Wu, H.; Pan, B.; Zhu, Z.; et al. (68)Ga-BBN-RGD PET/CT for GRPR and Integrin alphavbeta3 Imaging in Patients with Breast Cancer. Theranostics 2018, 8, 1121–1130. [Google Scholar] [CrossRef] [PubMed]
- Mansi, R.; Fleischmann, A.; Macke, H.R.; Reubi, J.C. Targeting GRPR in urological cancers -from basic research to clinical application. Nat. Rev. Urol. 2013, 10, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C. In-Vitro Identification of Vasoactive-Intestinal-Peptide Receptors in Human Tumors—Implications for Tumor Imaging. J. Nucl. Med. 1995, 36, 1846–1853. [Google Scholar]
- Reubi, J.C.; Laderach, U.; Waser, B.; Gebbers, J.O.; Robberecht, P.; Laissue, J.A. Vasoactive intestinal peptide/pituitary adenylate cyclase-activating peptide receptor subtypes in human tumors and their tissues of origin. Cancer Res. 2000, 60, 3105–3112. [Google Scholar]
- Valdehita, A.; Bajo, A.M.; Schally, A.V.; Varga, J.L.; Carmena, M.J.; Prieto, J.C. Vasoactive intestinal peptide (VIP) induces transactivation of EGFR and HER2 in human breast cancer cells. Mol. Cell Endocrinol. 2009, 302, 41–48. [Google Scholar] [CrossRef]
- Valdehita, A.; Carmena, M.J.; Collado, B.; Prieto, J.C.; Bajo, A.M. Vasoactive intestinal peptide (VIP) increases vascular endothelial growth factor (VEGF) expression and secretion in human breast cancer cells. Regul. Peptides 2007, 144, 101–108. [Google Scholar] [CrossRef]
- Ghosh, A.; Raju, N.; Tweedle, M.; Kumar, K. In Vitro Mouse and Human Serum Stability of a Heterobivalent Dual-Target Probe That Has Strong Affinity to Gastrin-Releasing Peptide and Neuropeptide Y1 Receptors on Tumor Cells. Cancer Biother. Radiopharm. 2017, 32, 24–32. [Google Scholar] [CrossRef] [Green Version]
- Scaranti, M.; Cojocaru, E.; Banerjee, S.; Banerji, U. Exploiting the folate receptor alpha in oncology. Nat. Rev. Clin. Oncol. 2020, 17, 349–359. [Google Scholar] [CrossRef]
- Jubb, A.M.; Strickland, L.A.; Liu, S.D.; Mak, J.; Schmidt, M.; Koeppen, H. Neuropilin-1 expression in cancer and development. J. Pathol. 2012, 226, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Perret, G.Y.; Starzec, A.; Hauet, N.; Vergote, J.; Le Pecheur, M.; Vassy, R.; Leger, G.; Verbeke, K.A.; Bormans, G.; Nicolas, P.; et al. In vitro evaluation and biodistribution of a Tc-99m-labeled anti-VEGF peptide targeting neuropilin-1. Nucl. Med. Biol. 2004, 31, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Park, K.C.; Richardson, D.R. The c-MET oncoprotein: Function, mechanisms of degradation and its targeting by novel anti-cancer agents. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129650. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
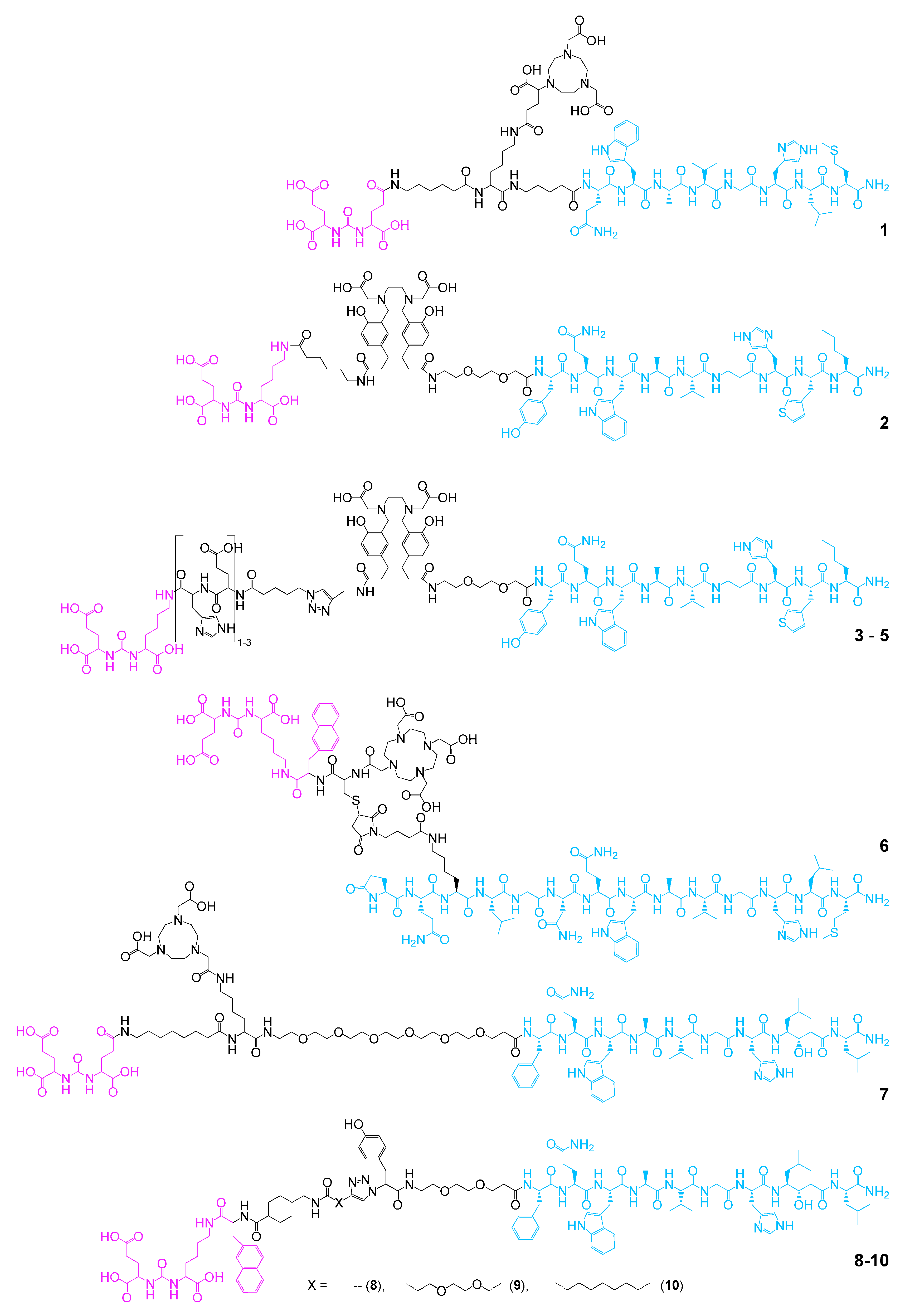{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Receptors | Compound Number | Radionuclide Used | Intended Application | In Vitro Affinity Data | Model Used for In Vivo Evaluation | Reference |
|---|---|---|---|---|---|---|
| GRPR and PSMA | 1 | 64Cu | PET imaging | IC50(GRPR): 11.1 ± 0.5 nM, IC50(PSMA): 1.2 ± 1.4 nM | PC3/AR42J and LNCaP mice | [74] |
| 2 | 68Ga | PET imaging | IC50(GRPR): 9.0 ± 1.8 nM, IC50(PSMA): 25.0 ± 5.4 nM | PC3/AR42J and LNCaP mice | [75] | |
| 3–5 | 68Ga | PET imaging | 3: IC50(GRPR): 7.3 nM, IC50(PSMA): 17.4 nM 4: IC50(GRPR): 4.4 nM, IC50(PSMA): 25.2 nM 5: IC50(GRPR): 7.1 nM, IC50(PSMA): 42.4 nM | PC3 and LNCaP mice | [76] | |
| 6 | 68Ga and 177Lu | PET imaging and therapy | [68Ga]Ga-6: Kd(GRPR): 43.7 ± 3.8 nM, Kd(PSMA): 4.4 ± 2.3 nM [177Lu]Lu-6: IC50(GRPR): 3.5 ± 0.4 nM, IC50(PSMA): 5.6 ± 1.5 nM | PC3 and LNCaP mice | [77,78] | |
| 7 | 68Ga and 111In | PET and SPECT imaging | IC50(GRPR): 4 ± 1 nM, IC50(PSMA): 824 ± 230 nM | PC3-PIP mice | [79] | |
| 8–10 | 125I | SPECT imaging and therapy | 8: IC50(GRPR): 6 ± 2 nM, IC50(PSMA): 80 ± 7 nM 9: IC50(GRPR): 13 ± 3 nM, IC50(PSMA): 98 ± 13 nM 10: IC50(GRPR): 20 ± 2 nM, IC50(PSMA): 100 ± 10 nM | PC3 and LNCaP mice | [80] | |
| GRPR and αvβ3 | 11 | 18F | PET imaging | IC50(GRPR): 32.0 ± 1.9 nM, IC50(αvβ3): 282 ± 34 nM | PC3 mice | [65] |
| 12 | 18F | PET imaging | IC50(GRPR): 73.3 ± 1.6 nM, IC50(αvβ3): 13.8 ± 1.8 nM | PC3 mice | [64] | |
| 13 | 18F | PET imaging | IC50(GRPR): 167 ± 1 nM, IC50(αvβ3): 553 ± 1 nM | PC3 mice | [81] | |
| 14 | 68Ga and 64Cu | PET imaging | IC50(GRPR): 92.8 ± 3.5 nM, IC50(αvβ3): 16.2 ± 2.8 nM | T47D and MDA-MB-435 mice | [82] | |
| 68Ga | PET imaging | IC50(GRPR): 55.9 ± 4.2 nM, IC50(αvβ3): 22.6 ± 6.7 nM | PC3 mice | [83] | ||
| 68Ga | PET imaging | - | PCa patients | [84] | ||
| 15 | 64Cu | PET imaging | IC50(GRPR): 4.0 ± 0.4 nM, IC50(αvβ3): no affinity | PC3 mice | [85] | |
| 16 | 64Cu | PET imaging | IC50(GRPR): 85.8 ± 2.1 nM, IC50(αvβ3): 21.6 ± 2.2 nM | PC3 mice | [86] | |
| 177Lu | PET imaging | - | PC3 mice | [87] | ||
| 17 | 64Cu | PET imaging | IC50(GRPR): 3.1 ± 0.3 nM, IC50(αvβ3): 518 ± 38 nM | PC3 mice | [88] | |
| 18 | 86Y and 90Y | PET imaging and therapy | IC50(GRPR): 5.7 ± 0 nM, IC50(αvβ3): 346 ± 5 nM | PC3 mice | [89] | |
| 111In and 177Lu | SPECT imaging and therapy | natIn-IC50(GRPR): 5.4 ± 1.4 nM, natIn-IC50(αvβ3): 372 ± 23 nM; natLu-IC50(GRPR): 5.8 ± 3.2 nM, natLu-IC50(αvβ3): 346 ± 53 nM | PC3 mice | [90] | ||
| 19 | 64Cu | PET imaging | IC50(GRPR): 24.3 ± 10.9 nM, IC50(αvβ3): 165.3 ± 105.4 nM | PC3 mice | [91] | |
| 20 | 64Cu | PET imaging | IC50(GRPR): 100.4 ± 73.2 nM, IC50(αvβ3): 101.2 ± 57.4 nM | PC3 mice | [91] | |
| 21 | 99mTc | SPECT imaging | IC50(GRPR): 104.7 ± 5.8 nM, IC50(αvβ3): 18.8 ± 3.7 nM | LLC mice | [92] | |
| 22 | 99mTc and 122Re | SPECT imaging and therapy | IC50(GRPR): 63.3 ± 2.9 nM, IC50(αvβ3): 13.4 ± 2.5 nM | PC3 mice | [93] | |
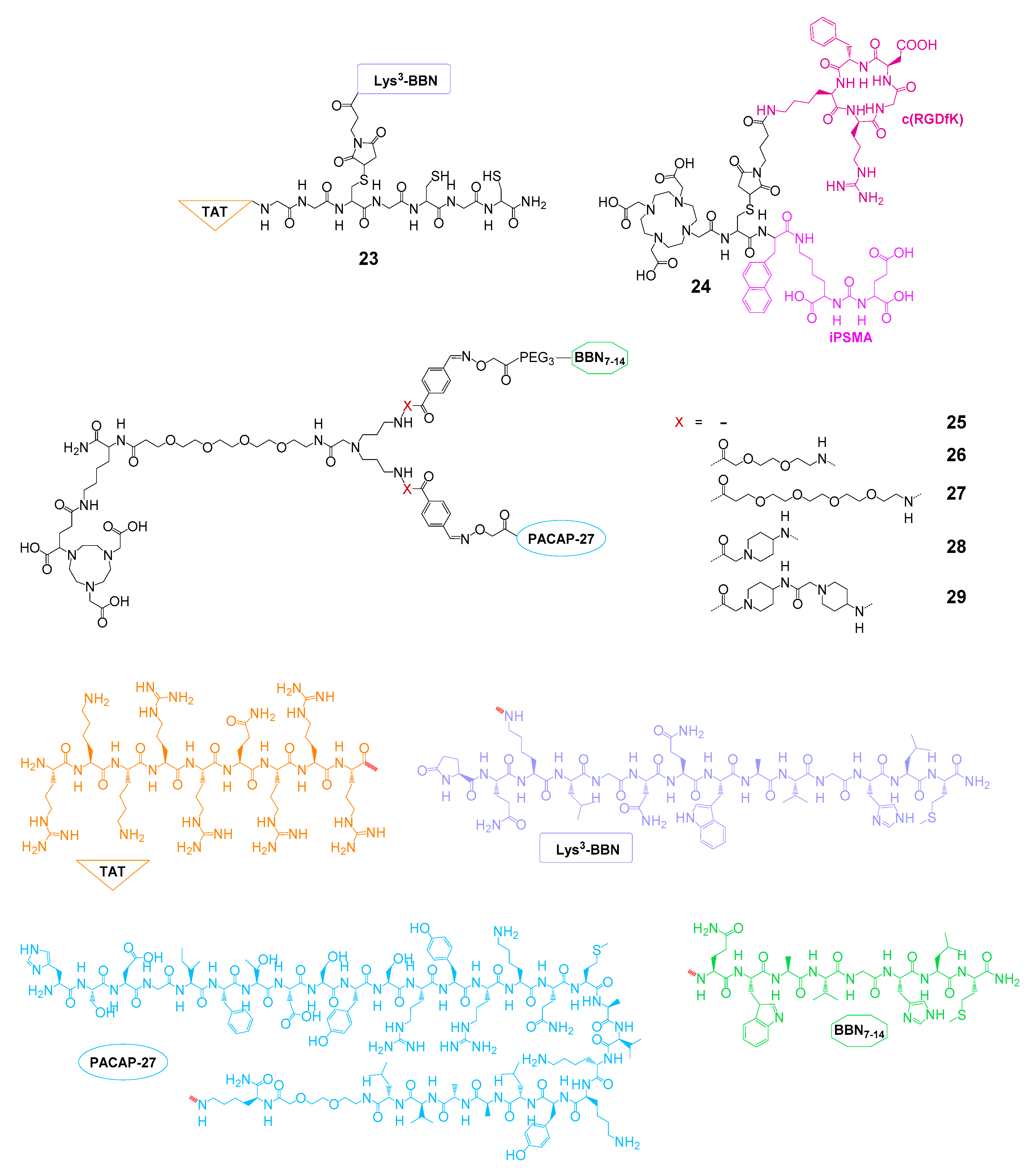
| GRPR and TAT | 23 | 99mTc | SPECT imaging and therapy | cell uptake studies, no affinities | PC3 mice | [94,95] |
| PSMA and αvβ3 | 24 | 177Lu | therapy | IC50(PSMA): 1.69 nM, IC50(αvβ3): 1.05 nM | - | [96] |
| GRPR and VPAC1R | 25–29 | 68Ga | PET imaging | cell uptake studies, no affinities | - | [66] |
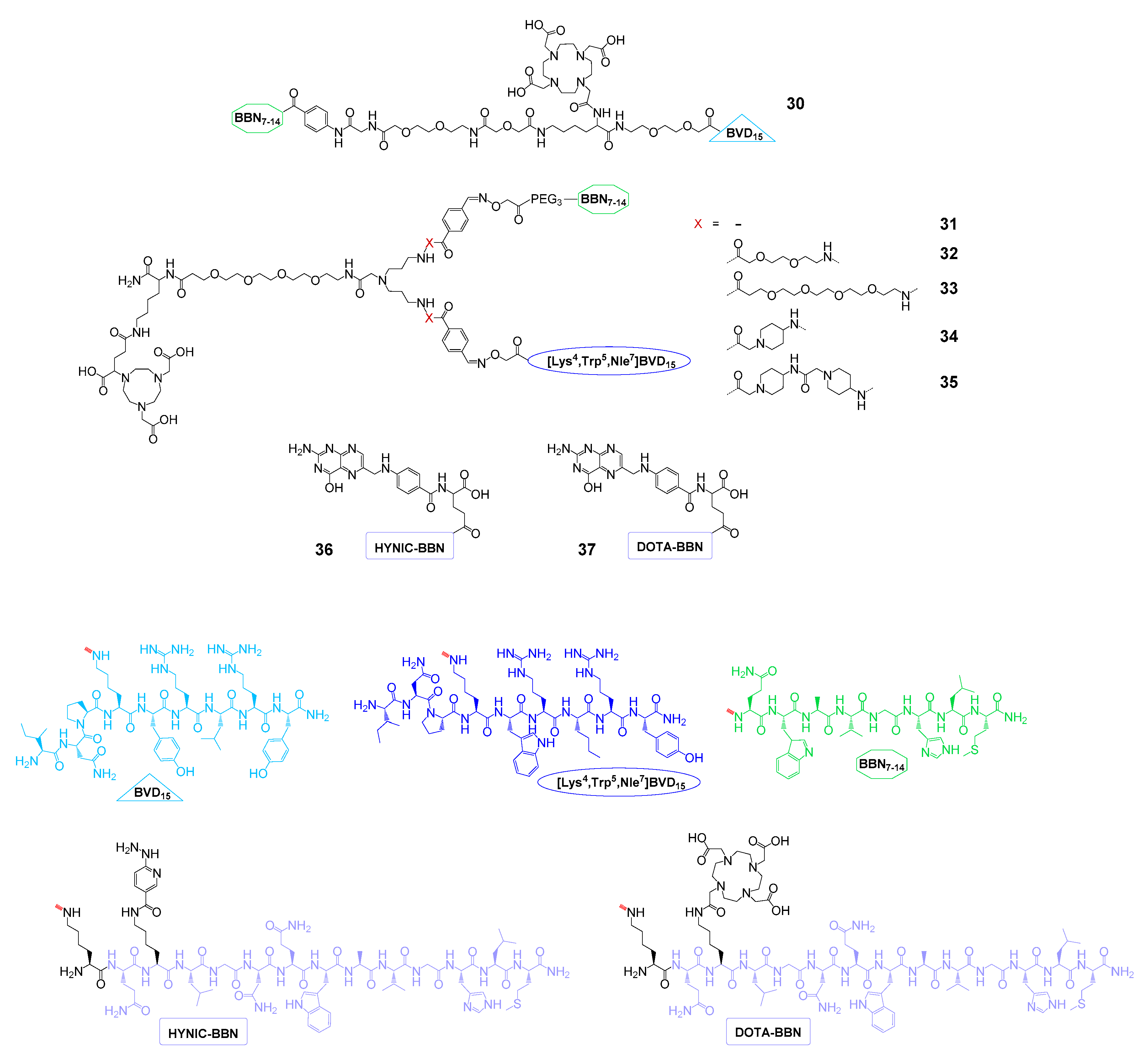
| GRPR and NPY(Y1)R | 30 | 153Gd | - | IC50(GRPR): 18.0 ± 0.7 nM, IC50(NPY(Y1)R): 80 ± 11 nM | - | [97] |
| 31–35 | 68Ga | PET imaging | - | T47D mice | [68] | |
| GRPR and FRα | 36 | 99mTc | SPECT imaging | IC50(GRPR): 3.2 ± 1.0 nM, IC50(FRα): 6.3 ± 1.5 nM | T47D mice | [98] |
| 37 | 177Lu | SPECT imaging and therapy | IC50(GRPR): 4.8 ± 0.9 nM, IC50(FRα): 9.1 ± 1.5 nM | T47D mice | [99] | |
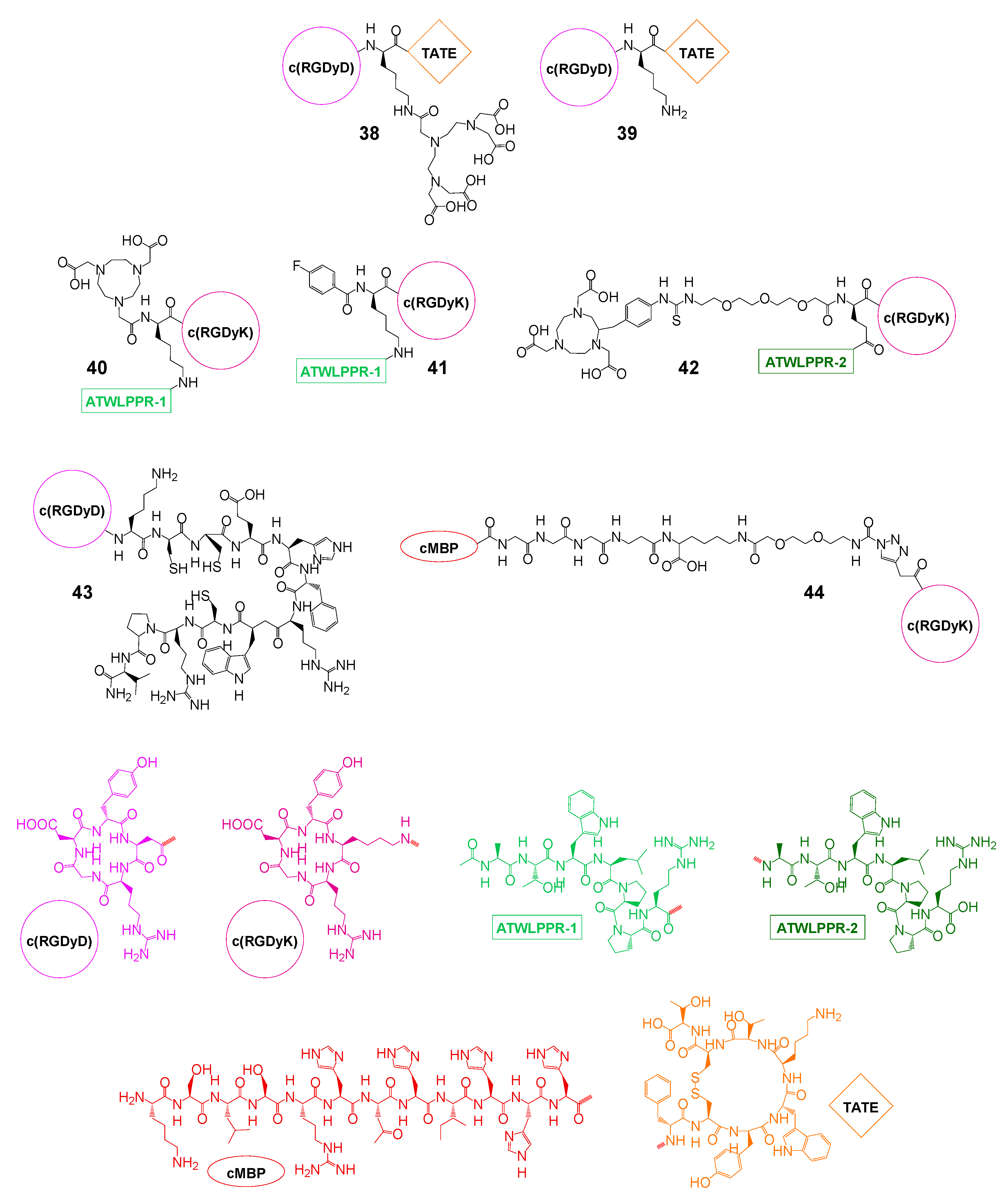
| αvβ3 and SSTR | 38 | 111In | (SPECT imaging) therapy | IC50(SSTR): 94 nM, IC50(αvβ3): n.d. | CA20948 rats | [100,101,102] |
| 39 | 125I | therapy | IC50(SSTR): 14 nM, IC50(αvβ3): n.d. | CA20948 rats | [102] | |
| αvβ3 and NRP-1 | 40 | 18F | PET imaging | IC50(NRP-1): 60.1 ± 6.5 nM, IC50(αvβ3): 43.8 ± 4.8 | U87MG mice | [103] |
| 41 | 18F | PET imaging | IC50(NRP-1): 23.7 nM, IC50(αvβ3): 21.7 nM | U87MG mice | [104] | |
| 42 | 18F | PET imaging | IC50(U87MG): 44.2 nM | U87MG mice | [105] | |
| αvβ3 and MC1R | 43 | 99mTc | SPECT imaging and therapy | IC50(B16-F1): 2.1 nM | B16-F1 mice | [106] |
| αvβ3 and c-Met | 44 | 125I | SPECT imaging | IC50(c-Met): 3.84 µM, IC50(αvβ3): 3.42 µM | U87MG mice | [107] |
| Radionuclide | Decay Mode | Half-Life | Application | Mean β-/γ-Energy |
|---|---|---|---|---|
| 18F | β+ (100%) | 109.77 min | PET imaging | 249.8 keV (β+) |
| 64Cu | β+ (17.6%) β− (38.5%) | 12.70 h | PET imaging | 278.2 keV (β+) |
| 68Ga | β+ (89.1%) | 67.71 min | PET imaging | 836.0 keV (β+) |
| 86Y | β+ (31.9%) | 14.74 h | PET imaging | 535 keV (β+) |
| 90Y | β− (100%) | 2.67 days | therapy | 933 keV (β−) |
| 99mTc | γ (100%) | 6.01 h | SPECT imaging | 140.51 keV (γ) |
| 111In | EC (100%) | 2.80 days | SPECT imaging | 171.28 keV (γ) 245.35 keV (γ) |
| 125I | EC (100%) | 60 days | therapy | 35.5 keV (γ) |
| 177Lu | β− (100%) | 6.71 days | therapy | 133 keV (β−) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Judmann, B.; Braun, D.; Wängler, B.; Schirrmacher, R.; Fricker, G.; Wängler, C. Current State of Radiolabeled Heterobivalent Peptidic Ligands in Tumor Imaging and Therapy. Pharmaceuticals 2020, 13, 173. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13080173
Judmann B, Braun D, Wängler B, Schirrmacher R, Fricker G, Wängler C. Current State of Radiolabeled Heterobivalent Peptidic Ligands in Tumor Imaging and Therapy. Pharmaceuticals. 2020; 13(8):173. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13080173
Chicago/Turabian StyleJudmann, Benedikt, Diana Braun, Björn Wängler, Ralf Schirrmacher, Gert Fricker, and Carmen Wängler. 2020. "Current State of Radiolabeled Heterobivalent Peptidic Ligands in Tumor Imaging and Therapy" Pharmaceuticals 13, no. 8: 173. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13080173