Resistance to Acetylsalicylic Acid in Patients with Coronary Heart Disease Is the Result of Metabolic Activity of Platelets

 ,
,
Abstract
:1. Introduction
2. Results
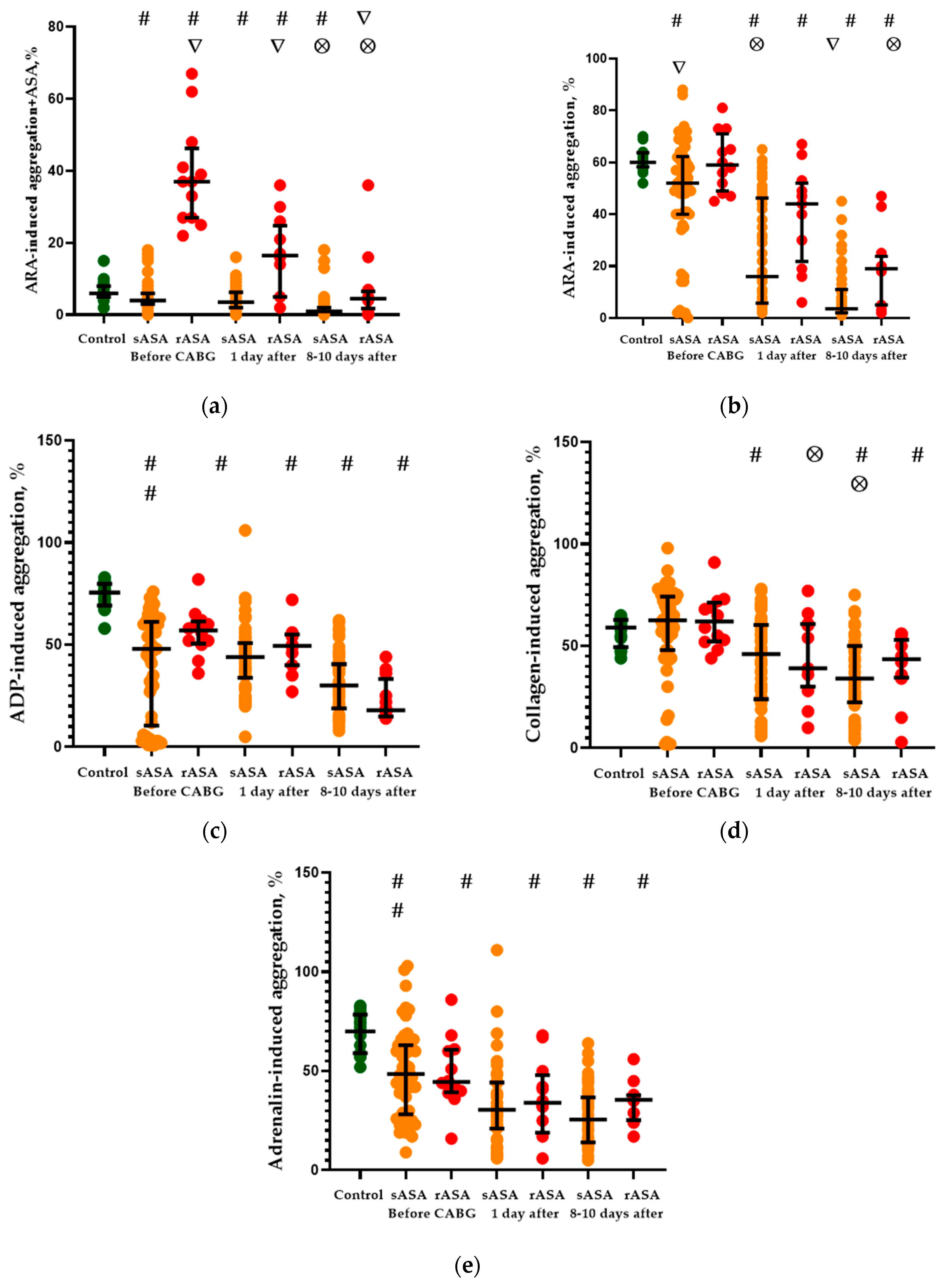
2.1. Platelet Aggregation Activity
2.2. Chemiluminescent Platelet Activity
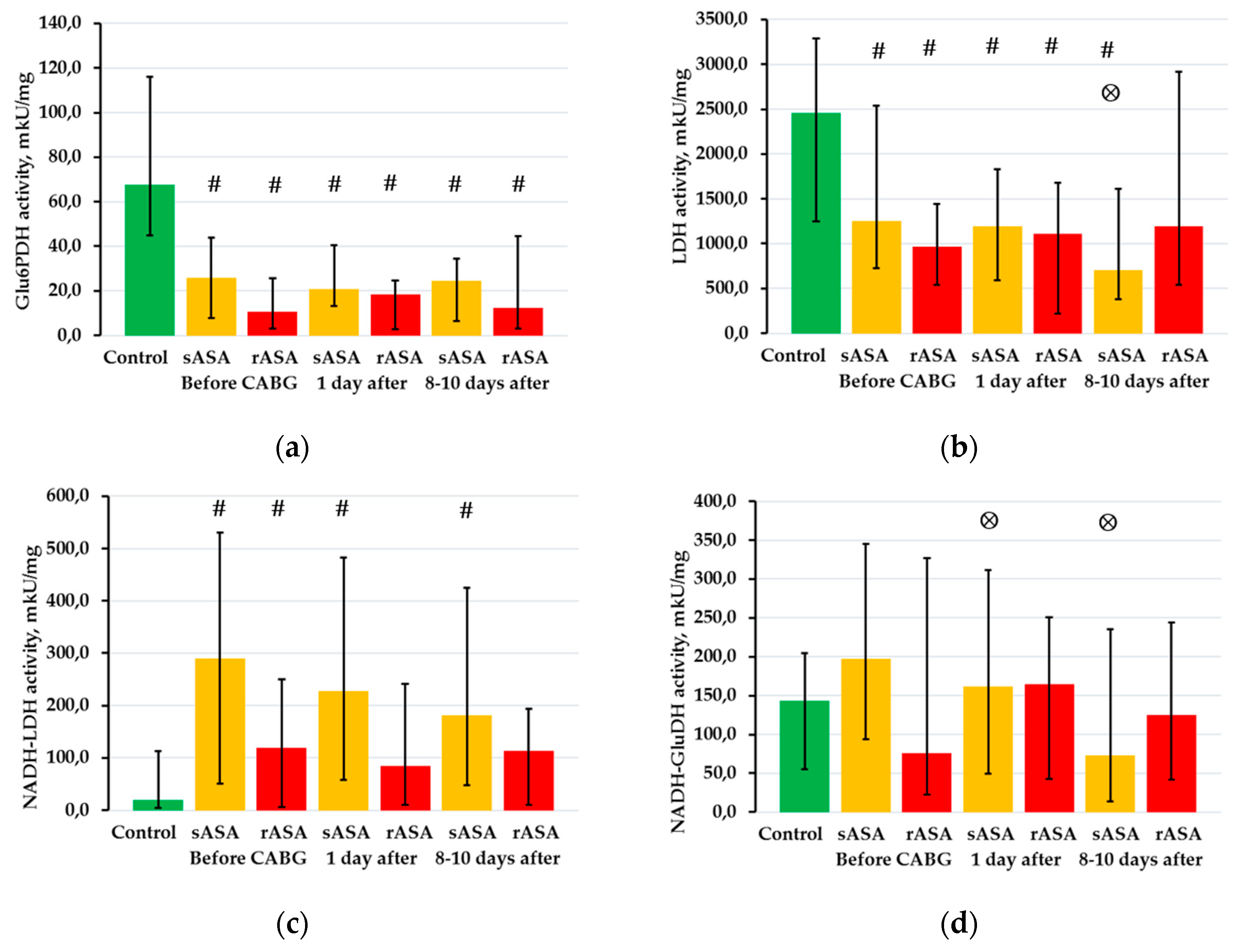
2.3. The Activity of NAD- and NADP-Dependent Dehydrogenases in Platelets
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Determination of ASA Resistance
4.3. Platelet Aggregometry Assay
4.4. Platelet Isolation
4.5. Chemiluminescent Analysis
4.6. Bioluminescent Analysis
- −
- glucose-6-phosphate dehydrogenase (Glu6PDH);
- −
- glycerol-3-phosphate dehydrogenase (Gly3PDH);
- −
- NADP-dependent malate dehydrogenase decarboxylated (NADP-MDH);
- −
- lactate dehydrogenase—according to NAD- and NADH-dependent reactions (respectively, LDH and NADH-LDH);
- −
- malate dehydrogenase—according to NAD- and NADH-dependent reactions (respectively, MDH and NADH-MDH);
- −
- NADP-dependent glutamate dehydrogenase—according to NADP- and NADPH-dependent reactions (respectively, NADP-GluDH and NADPH-GluDH);
- −
- NAD-dependent glutamate dehydrogenase—according to NAD- and NADH-dependent reactions (respectively, NAD-GluDH and NADH-GluDH);
- −
- NADP-dependent isocitrate dehydrogenase (NADP-ICDH);
- −
- NAD-dependent isocitrate dehydrogenase (NAD-ICDH);
- −
- glutathione reductase (GR).
4.7. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Della Corte, A.; Bancone, C.; Spadafora, A.; Borrelli, M.; Galdieri, N.; Quintiliano, S.N.; Bifulco, O.; De Feo, M. Postoperative bleeding in coronary artery bypass patients on double antiplatelet therapy: Predictive value of preoperative aggregometry. Eur. J. Cardiothorac. Surg. 2017, 52, 901–908. [Google Scholar] [CrossRef] [Green Version]
- DeStephan, C.M.; Schneider, D.J. Antiplatelet therapy for patients undergoing coronary artery bypass surgery. Kardiol. Pol. 2018, 76, 945–952. [Google Scholar] [CrossRef] [Green Version]
- Hybiak, J.; Broniarek, I.; Kiryczyński, G.; Los, L.D.; Rosik, J.; Machaj, F.; Sławiński, H.; Jankowska, K.; Urasińska, E. Aspirin and its pleiotropic application. Eur. J. Pharmacol. 2020, 866, 172762. [Google Scholar] [CrossRef]
- Tantry, U.S.; Navarese, E.P.; Bliden, K.P.; Gurbel, P.A. Acetylsalicylic acid and clopidogrel hyporesponsiveness following acute coronary syndromes. Kardiol. Pol. 2018, 76, 1312–1319. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, R.; Pirmohamed, M. Aspirin resistance: Effect of clinical, biochemical and genetic factors. Pharmacol. Ther. 2011, 130, 213–225. [Google Scholar] [CrossRef]
- Floyd, C.N.; Ferro, A. Mechanisms of aspirin resistance. Pharmacol. Ther. 2014, 141, 69–78. [Google Scholar] [CrossRef]
- Du, G.; Lin, Q.; Wang, J. A brief review on the mechanisms of aspirin resistance. Int. J. Cardiol. 2016, 220, 21–26. [Google Scholar] [CrossRef]
- Grinshtein, Y.I.; Kosinova, A.A.; Grinshtein, I.Y.; Subbotina, T.N.; Savchenko, A.A. The prognostic value of combinations of genetic polymorphisms in the ITGB3, ITGA2, and CYP2C19∗2 genes in predicting cardiovascular outcomes after coronary bypass grafting. Genet. Test. Mol. Biomark. 2018, 22, 259–265. [Google Scholar] [CrossRef] [Green Version]
- Fuentes, E.; Gibbins, J.M.; Holbrook, L.M.; Palomo, I. NADPH oxidase 2 (NOX2): A key target of oxidative stress-mediated platelet activation and thrombosis. Trends Cardiovasc. Med. 2018, 28, 429–434. [Google Scholar] [CrossRef]
- Qiao, J.; Arthur, J.F.; Gardiner, E.E.; Andrews, R.K.; Zeng, L.; Xu, K. Regulation of platelet activation and thrombus formation by reactive oxygen species. Redox Biol. 2018, 14, 126–130. [Google Scholar] [CrossRef]
- Meyskens, F.L., Jr.; Liu-Smith, F. Redox-Redux and NADPH Oxidase (NOX): Even More Complicated than We Thought it Might Be. J. Investig. Dermatol. 2017, 137, 1208–1210. [Google Scholar] [CrossRef]
- Pastori, D.; Pignatelli, P.; Carnevale, R.; Violi, F. Nox-2 up-regulation and platelet activation: Novel insights. Prostaglandins Other Lipid Mediat. 2015, 120, 50–55. [Google Scholar] [CrossRef]
- Violi, F.; Pignatelli, P. Platelet NOX, a novel target for anti-thrombotic treatment. Thromb. Haemost. 2014, 111, 817–823. [Google Scholar] [CrossRef]
- Paul, M.; Hemshekhar, M.; Kemparaju, K.; Girish, K.S. Berberine mitigates high glucose-potentiated platelet aggregation and apoptosis by modulating aldose reductase and NADPH oxidase activity. Free Radic. Biol. Med. 2019, 130, 196–205. [Google Scholar] [CrossRef]
- Fidler, T.P.; Rowley, J.W.; Araujo, C.; Boudreau, L.H.; Marti, A.; Souvenir, R.; Dale, K.; Boilard, E.; Weyrich, A.S.; Abel, E.D. Superoxide Dismutase 2 is dispensable for platelet function. Thromb. Haemost. 2017, 117, 1859–1867. [Google Scholar] [CrossRef]
- Lewandowski, Ł.; Kepinska, M.; Milnerowicz, H. The copper-zinc superoxide dismutase activity in selected diseases. Eur. J. Clin. Investig. 2019, 49, e13036. [Google Scholar] [CrossRef] [Green Version]
- Younus, H. Therapeutic potentials of superoxide dismutase. Int. J. Health Sci. 2018, 12, 88–93. [Google Scholar]
- Coenen, D.M.; Mastenbroek, T.G.; Cosemans, J.M.E.M. Platelet interaction with activated endothelium: Mechanistic insights from microfluidics. Blood J. Am. Soc. Hematol. 2017, 130, 2819–2828. [Google Scholar] [CrossRef]
- Holley, A.S.; Miller, J.H.; Larsen, P.D.; Harding, S.A. Relationship between Glutathione Peroxidase, Platelet Reactivity, and Reactive Oxygen Species in an Acute Coronary Syndrome Population. Ann. Clin. Lab. Sci. 2016, 46, 639–644. [Google Scholar]
- Soares, M.S.P.; da Silveira de Mattos, B.; Ávila, A.A.; Spohr, L.; Pedra, N.S.; Teixeira, F.C.; Bona, N.P.; Oliveira, P.S.; Stefanello, F.M.; Spanevello, R.M. High levels of methionine and methionine sulfoxide: Impact on adenine nucleotide hydrolysis and redox status in platelets and serum of young rats. J. Cell. Biochem. 2019, 120, 2289–2303. [Google Scholar] [CrossRef]
- Chiang, J.Y.; Lee, S.H.; Chen, Y.C.; Wu, C.K.; Chuang, J.Y.; Lo, S.C.; Yeh, H.M.; Yeh, S.S.; Hsu, C.A.; Lin, B.B.; et al. Metabolomic Analysis of Platelets of Patients With Aspirin Non-Response. Front. Pharmacol. 2019, 10, 1107. [Google Scholar] [CrossRef] [PubMed]
- Petrus, A.T.; Lighezan, D.L.; Danila, M.D.; Duicu, O.M.; Sturza, A.; Muntean, D.M.; Ionita, I. Assessment of platelet respiration as emerging biomarker of disease. Physiol. Res. 2019, 68, 347–363. [Google Scholar] [CrossRef] [PubMed]
- Aibibula, M.; Naseem, K.M.; Sturmey, R.G. Glucose metabolism and metabolic flexibility in blood platelets. J. Thromb. Haemost. 2018, 16, 2300–2314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Májek, P.; Reicheltová, Z.; Stikarová, J.; Suttnar, J.; Sobotková, A.; Dyr, J.E. Proteome changes in platelets activated by arachidonic acid, collagen, and thrombin. Proteome Sci. 2010, 8, 56. [Google Scholar] [CrossRef] [Green Version]
- Razi, M.T.; Javed, I.; Choudry, M.Z.; Khan, M.T.; Mukhtar, N. Effect of ketoprofen on lactic dehydrogenase from human platelets. Adv. Clin. Exp. Med. 2014, 23, 377–380. [Google Scholar] [CrossRef] [Green Version]
- Drohomirecka, A.; Iwaszko, A.; Walski, T.; Pliszczak-Król, A.; Wąż, G.; Graczyk, S.; Gałecka, K.; Czerski, A.; Bujok, J.; Komorowska, M. Low-level light therapy reduces platelet destruction during extracorporeal circulation. Sci. Rep. 2018, 8, 16963. [Google Scholar] [CrossRef]
- Kobzar, G.; Mardla, V.; Rätsep, I.; Samel, N. Platelet activity before and after coronary artery bypass grafting. Platelets 2006, 17, 289–291. [Google Scholar] [CrossRef]
- Sobhani, V.; Taghizadeh, M.; Moshkani-Farahani, M. Platelet indices and function response to two types of high intensity interval exercise and comparison with moderate intensity continuous exercise among men after coronary artery bypass graft: A randomized trial. ARYA Atheroscler. 2018, 14, 188–195. [Google Scholar] [CrossRef]
- Braidy, N.; Berg, J.; Clement, J.; Khorshidi, F.; Poljak, A.; Jayasena, T.; Grant, R.; Sachdev, P. Role of Nicotinamide Adenine Dinucleotide and Related Precursors as Therapeutic Targets for Age-Related Degenerative Diseases: Rationale, Biochemistry, Pharmacokinetics, and Outcomes. Antioxid. Redox Signal. 2019, 30, 251–294. [Google Scholar] [CrossRef]
- Elhassan, Y.S.; Philp, A.A.; Lavery, G.G. Targeting NAD+ in Metabolic Disease: New Insights into an Old Molecule. J. Endocr. Soc. 2017, 1, 816–835. [Google Scholar] [CrossRef]
- Moosavi, B.; Zhu, X.L.; Yang, W.C.; Yang, G.F. Genetic, epigenetic and biochemical regulation of succinate dehydrogenase function. Biol. Chem. 2020, 401, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Sellés Vidal, L.; Kelly, C.L.; Mordaka, P.M.; Heap, J.T. Review of NAD(P)H-dependent oxidoreductases: Properties, engineering and application. Biochim. Biophys. Acta Proteins Proteom. 2018, 1866, 327–347. [Google Scholar] [CrossRef] [PubMed]
- Shah, V.P.; Chegini, H.A.; Vishneski, S.R.; Weatherman, R.V.; Blackmore, P.F.; Dobrydneva, Y. Tamoxifen promotes superoxide production in platelets by activation of PI3-kinase and NADPH oxidase pathways. Thromb. Res. 2012, 129, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Fu, Y.F.; Liu, X.; Feng, G.; Xiong, D.; Mu, G.F.; Chen, F.P. ROS Promote Ox-LDL-Induced Platelet Activation by Up-Regulating Autophagy Through the Inhibition of the PI3K/AKT/mTOR Pathway. Cell. Physiol. Biochem. 2018, 50, 1779–1793. [Google Scholar] [CrossRef] [PubMed]
- Gabbasov, Z.; Ivanova, O.; Kogan-Yasny, V.; Vasilieva, E. Aspirin can stimulate luminol-enhanced chemiluminescence of activated platelets. Platelets 2010, 21, 486–489. [Google Scholar] [CrossRef]
- Khan, P.; Idrees, D.; Moxley, M.A.; Corbett, J.A.; Ahmad, F.; von Figura, G.; Sly, W.S.; Waheed, A.; Hassan, M.I. Luminol-based chemiluminescent signals: Clinical and non-clinical application and future uses. Appl. Biochem. Biotechnol. 2014, 173, 333–355. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Lu, J.; Khaidakov, M.; Mitra, S.; Ding, Z.; Raina, S.; Goyal, T.; Mehta, J.L. Aspirin suppresses cardiac fibroblast proliferation and collagen formation through downregulation of angiotensin type 1 receptor transcription. Toxicol. Appl. Pharmacol. 2012, 259, 346–354. [Google Scholar] [CrossRef]
- Michno, A.; Grużewska, K.; Bielarczyk, H.; Zyśk, M.; Szutowicz, A. Inhibition of pyruvate dehydrogenase complex activity by 3-bromopyruvate affects blood platelets responses in type 2 diabetes. Pharmacol. Rep. 2020, 72, 225–237. [Google Scholar] [CrossRef]
- Feghaly, J.; Al Hout, A.R.; Mercieca Balbi, M. Aspirin safety in glucose-6-phosphate dehydrogenase deficiency patients with acute coronary syndrome undergoing percutaneous coronary intervention. BMJ Case Rep. 2017, 2017, bcr2017220483. [Google Scholar] [CrossRef]
- Li, M.; Li, C.; Allen, A.; Stanley, C.A.; Smith, T.J. Glutamate dehydrogenase: Structure, allosteric regulation, and role in insulin homeostasis. Neurochem. Res. 2014, 39, 433–445. [Google Scholar] [CrossRef]
- Plaitakis, A.; Kalef-Ezra, E.; Kotzamani, D.; Zaganas, I.; Spanaki, C. The Glutamate Dehydrogenase Pathway and Its Roles in Cell and Tissue Biology in Health and Disease. Biology 2017, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateos-Cáceres, P.J.; Macaya, C.; Azcona, L.; Modrego, J.; Mahillo, E.; Bernardo, E.; Fernandez-Ortiz, A.; López-Farré, A.J. Different expression of proteins in platelets from aspirin-resistant and aspirin-sensitive patients. Thromb. Haemost. 2010, 103, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Stitham, J.; Gleim, S.; Di Febbo, C.; Porreca, E.; Fava, C.; Tacconelli, S.; Capone, M.; Evangelista, V.; Levantesi, G.; et al. Glucose and collagen regulate human platelet activity through aldose reductase induction of thromboxane. J. Clin. Investig. 2011, 121, 4462–4476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, A.; Rahmanian, S.; Bordbar, A.; Palsson, B.Ø.; Jamshidi, N. Network reconstruction of platelet metabolism identifies metabolic signature for aspirin resistance. Sci. Rep. 2014, 4, 3925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hino, S.; Nagaoka, K.; Nakao, M. Metabolism-epigenome crosstalk in physiology and diseases. J. Hum. Genet. 2013, 58, 410–415. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Thompson, C.B. Metabolic regulation of epigenetics. Cell Metab. 2012, 16, 9–17. [Google Scholar] [CrossRef] [Green Version]
- Kaur, R.; Kaur, M.; Singh, J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: Molecular insights and therapeutic strategies. Cardiovasc. Diabetol. 2018, 17, 121. [Google Scholar] [CrossRef]
- Pretorius, L.; Thomson, G.J.A.; Adams, R.C.M.; Nell, T.A.; Laubscher, W.A.; Pretorius, E. Platelet activity and hypercoagulation in type 2 diabetes. Cardiovasc. Diabetol. 2018, 17, 141. [Google Scholar] [CrossRef] [Green Version]
- Walinjkar, R.S.; Khadse, S.; Kumar, S.; Bawankule, S.; Acharya, S. Platelet Indices as a Predictor of Microvascular Complications in Type 2 Diabetes. Indian J. Endocrinol. Metab. 2019, 23, 206–210. [Google Scholar] [CrossRef]
- El Haouari, M. Platelet Oxidative Stress and its Relationship with Cardiovascular Diseases in Type 2 Diabetes Mellitus Patients. Curr. Med. Chem. 2019, 26, 4145–4165. [Google Scholar] [CrossRef]
- Harima, K.; Honda, S.; Mikami, K.; Kitajima, M.; Urushizaka, M.; Tomisawa, T.; Hagii, J.; Metoki, H.; Yasujima, M.; Osanai, T. Collagen-Induced Platelet Aggregates, Diabetes, and Aspirin Therapy Predict Clinical Outcomes in Acute Ischemic Stroke. J. Stroke Cerebrovasc. Dis. 2019, 28, 2302–2310. [Google Scholar] [CrossRef] [PubMed]
- Verdoia, M.; Schaffer, A.; Barbieri, L.; Cassetti, E.; Nardin, M.; Bellomo, G.; Marino, P.; Sinigaglia, F.; De Luca, G. Novara Atherosclerosis Study (NAS) group. Diabetes, glucose control and mean platelet volume: A single-centre cohort study. Diabetes Res. Clin. Pract. 2014, 104, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Al-Sofiani, M.E.; Derenbecker, R.; Quartuccio, M.; Kalyani, R.R. Aspirin for Primary Prevention of Cardiovascular Disease in Diabetes: A Review of the Evidence. Curr. Diabetes Rep. 2019, 19, 107. [Google Scholar] [CrossRef] [PubMed]
- Al-Sofiani, M.E.; Yanek, L.R.; Faraday, N.; Kral, B.G.; Mathias, R.; Becker, L.C.; Becker, D.M.; Vaidya, D.; Kalyani, R.R. Diabetes and Platelet Response to Low-Dose Aspirin. J. Clin. Endocrinol. Metab. 2018, 103, 4599–4608. [Google Scholar] [CrossRef] [PubMed]
- Kapłon-Cieślicka, A.; Rosiak, M.; Postuła, M.; Serafin, A.; Kondracka, A.; Opolski, G.; Filipiak, K.J. Predictors of high platelet reactivity during aspirin treatment in patients with type 2 diabetes. Kardiol. Pol. 2013, 71, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, J.; Gulati, G.; Uppal, G.; Gong, J. Assessment of the Reliability of the Sysmex XE-5000 Analyzer to Detect Platelet Clumps. Lab. Med. 2016, 47, 189–194. [Google Scholar] [CrossRef] [Green Version]
- Savchenko, A.A. Evaluation of NAD(P)-dependent dehydrogenase activities in neutrophilic granulocytes by the bioluminescent method. Bull. Exp. Biol. Med. 2015, 159, 692–695. [Google Scholar] [CrossRef]
- Esimbekova, E.N.; Torgashina, I.G.; Kratasyuk, V.A. Comparative study of immobilized and soluble NADH:FMN-oxidoreductase-luciferase coupled enzyme system. Biochemistry 2009, 74, 695–700. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Parameters | Control 1 | Before CABG | 1 Day After CABG | 8–10 days after CABG | |||
|---|---|---|---|---|---|---|---|
| sASA Patients 2 | rASA Patients 3 | sASA Patients 4 | rASA Patients 5 | sASA Patients 6 | rASA Patients 7 | ||
| Spontaneous lucigenin-enhanced chemiluminescence | |||||||
| Tmax, sec. | 213 (80–450) | 813 (88–2841) # | 185 (35–249) ∇ | 789 (283–2043) # | 66 (41–115) ∇ | 938 (565–1908) # | 88 (65–420) ∇ |
| Imax, r.u. | 80 (73–93) | 117 (78–566) # | 80 (72–92) ∇ | 105 (87–331) # | 94 (90–103) # | 173 (100–351) # | 106 (78–118) #,∇,⊗ |
| S, r.u.× sec. × 102 | 2.38 (1.76–2.75) | 3.01 (1.79–7.74) # | 2.25 (1.95–3.13) | 4.05 (2.44–8.17) # | 2.38 (1.97–3.58) ∇ | 4.24 (2.75–6.58) # | 2.89 (2.22–3.66) |
| ADP-induced lucigenin-enhanced chemiluminescence | |||||||
| Tmax, sec. | 96 (49–608) | 1036 (346–3743) # | 577 (342–1166) | 745 (355–1008) #,⊗ | 285 (217–341) ∇,⊗ | 1266 (621–2198) # | 495 (263–1099) |
| Imax, r.u. | 80 (76–127) | 127 (84–498) # | 81 (73–109) ∇ | 360 (91–595) # | 98 (82–106) ∇ | 238 (119–455) # | 102 (81–129) ∇ |
| S, r.u. × sec. × 102 | 2,75 (1.87–3.65) | 4.15 (2.51–10.89) # | 2.33 (2.06–3.32) ∇ | 4.86 (2.20–9.50) # | 3.05 (2.54–3.48) | 4.49 (3.35–7.31) # | 2.99 (2.49–3.80) ∇ |
| AI | 1.01 (0.86–1.87) | 1.12 (0.90–1.59) | 1.06 (0.92–1.40) | 1.11 (0.82–1.40) | 1.22 (0.91–1.37) | 1.22 (0.99–1.54) | 1.09 (1.02–1.17) |
| Spontaneous luminol-enhanced chemiluminescence | |||||||
| Tmax, sec. | 71 (0–464) | 230 (45–1748) | 71 (69–81) | 336 (71–998) | 852 (26–2394) ⊗ | 269 (71–848) # | 54 (4–445) ∇ |
| Imax, r.u. | 80 (77–110) | 122 (80–611) # | 84 (80–381) | 205 (95–490) # | 137 (90–167) # | 561 (127–1116) #,⊗ | 165 (129–388) # |
| S, r.u. × sec. × 102 | 2.62 (2.22–3.13) | 2.96 (2.10–8.99) | 3.01 (2.31–3.41) | 3.87 (2.71–9.70) # | 3.94 (3.20–4.75) # | 5.60 (3.96–12.35) #,⊗ | 3.63 (3.14–5.08) #,⊗ |
| ADP-induced luminol-enhanced chemiluminescence | |||||||
| Tmax, sec. | 154 (0–471) | 455 (45–2197) | 68 (13–743) # | 634 (89–1567) # | 97 (69–241) ∇ | 631 (264–1483) # | 117 (28–530) ∇ |
| Imax, r.u. | 77 (71–100) | 113 (79–519) # | 90 (75–342) | 294 (96–785) # | 145 (88–188) # | 690 (156–1346) #,⊗ | 156 (120–398) #,⊗ |
| S, r.u. × sec. × 102 | 2.35 (2.07–3.34) | 3.06 (2.24–8.35) | 3,27 (2.48–3.58) | 5.55 (2.37–11.51) # | 3.62 (3.20–5.25) # | 6.87 (3.87–20.11) #,⊗ | 4.74 (2.60–5.92) |
| AI | 0.99 (0.71–1.26) | 1.04 (0.73–1.45) | 1.10 (1.04–1.39) | 1.09 (0.84–1.32) | 1.00 (0.69–1.13) | 1.16 (0.85–1.37) | 1.06 (0.78–1.28) |
| Parameters | All Patients | sASA Patients | rASA Patients | p |
|---|---|---|---|---|
| Gender, n (%) Female/male | 17 (25.7%)/ 49 (74.3%) | 3 (26.0%)/ 9 (74.0%) | 14 (25.0%)/ 40 (75.0%) | 0.661 |
| Age (years), Me (C25—C75) | 62.0 (60.5–62.0) | 62.5 (56.0–65.0) | 63.0 (55.0–65.3) | 0.716 |
| Smokers (current), % | 36.4 | 35.8 | 38.4 | 0.575 |
| Total cholesterol, mmol/L | 4.49 (3.8–5.86) | 4.7 (3.8–5.9) | 4.2 (3.8–5.8) | 0.912 |
| Leukocytes, 109/L | 7.66 (6.66–7.66) | 9.5 (7.7–12.2) | 9.7 (7.8–10.6) | 0.605 |
| Platelets, 109/L | 228 (209–228) | 233 (189.5–296.5) | 258.5 (218.5–341.3) | 0.151 |
| Erythrocyte, 1012/L | 5.03 (4.74–5.03) | 4.05 (3.57–4.91) | 3.85 (3.4–4.6) | 0.103 |
| Hemoglobin, g/L | 143 (134.25–143) | 118 (102–138) | 111.5 (98.8–133) | 0.264 |
| Creatinine, mmol/L | 110 (96.25–110) | 106 (93–122) | 115 (98–137.8) | 0.282 |
| Stable angina grade II, % | 65.2 | 64.7 | 65.3 | 0.590 |
| Stable angina grade III, % | 34.8 | 35.3 | 34.7 | 0.590 |
| Diabetes mellitus, % | 25.7 | 22.4 | 29.4 | 0.633 |
| Myocardial infarction in anamnesis, % | 63.6 | 61.2 | 70.6 | 0.608 |
| Obesity, % | 34.8 | 26.5 | 52.9 | 0.599 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grinshtein, Y.I.; Savchenko, A.A.; Kosinova, A.A.; Goncharov, M.D. Resistance to Acetylsalicylic Acid in Patients with Coronary Heart Disease Is the Result of Metabolic Activity of Platelets. Pharmaceuticals 2020, 13, 178. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13080178
Grinshtein YI, Savchenko AA, Kosinova AA, Goncharov MD. Resistance to Acetylsalicylic Acid in Patients with Coronary Heart Disease Is the Result of Metabolic Activity of Platelets. Pharmaceuticals. 2020; 13(8):178. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13080178
Chicago/Turabian StyleGrinshtein, Yuriy I., Andrei A. Savchenko, Aleksandra A. Kosinova, and Maxim D. Goncharov. 2020. "Resistance to Acetylsalicylic Acid in Patients with Coronary Heart Disease Is the Result of Metabolic Activity of Platelets" Pharmaceuticals 13, no. 8: 178. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13080178