The Interaction of Fluorinated Glycomimetics with DC-SIGN: Multiple Binding Modes Disentangled by the Combination of NMR Methods and MD Simulations

 , and
, and 
Abstract
:
1. Introduction
2. Results and Discussion
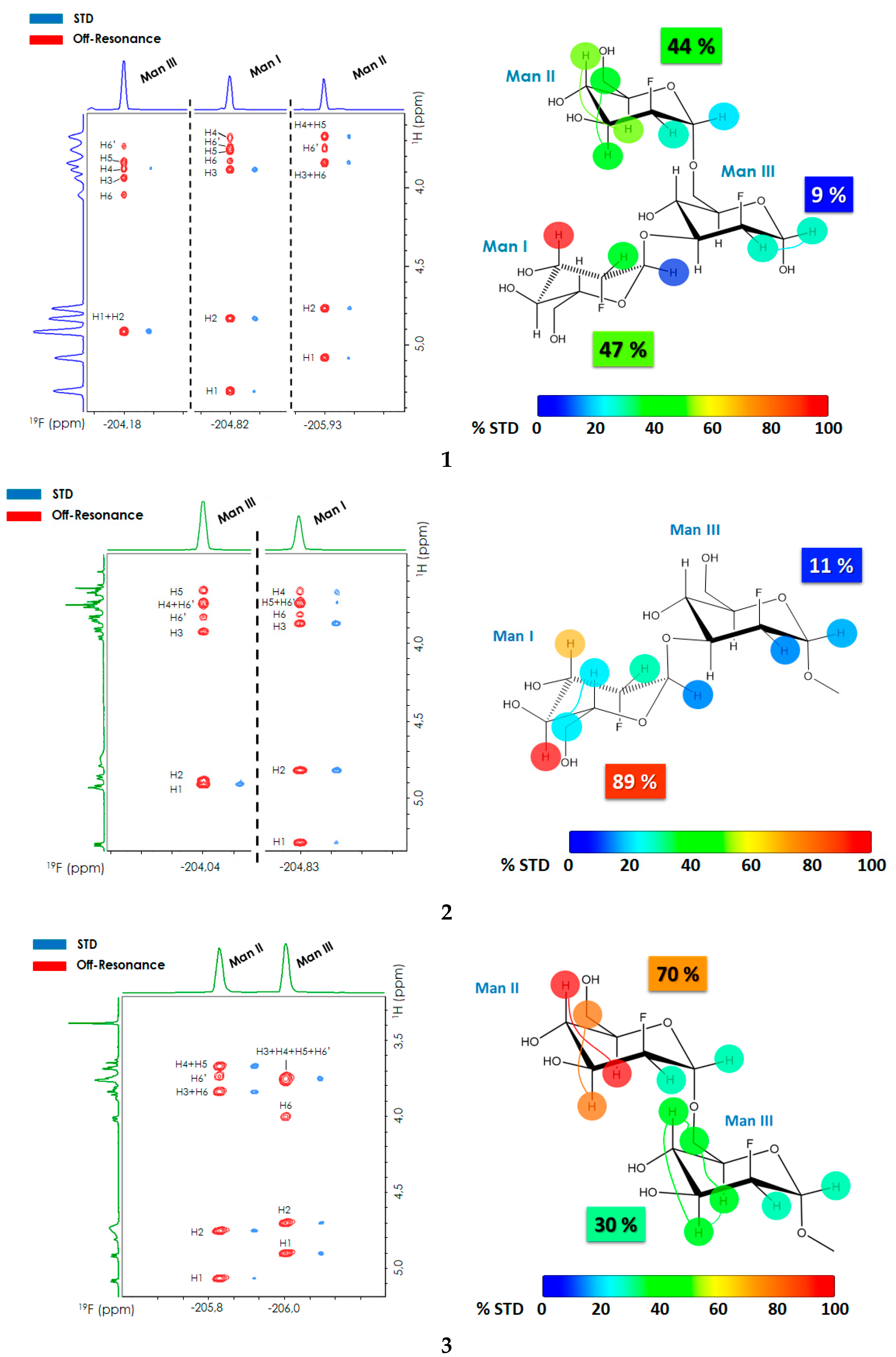
2.1. 2D-STD-TOCSYreF Experiments and Affinity Measurements
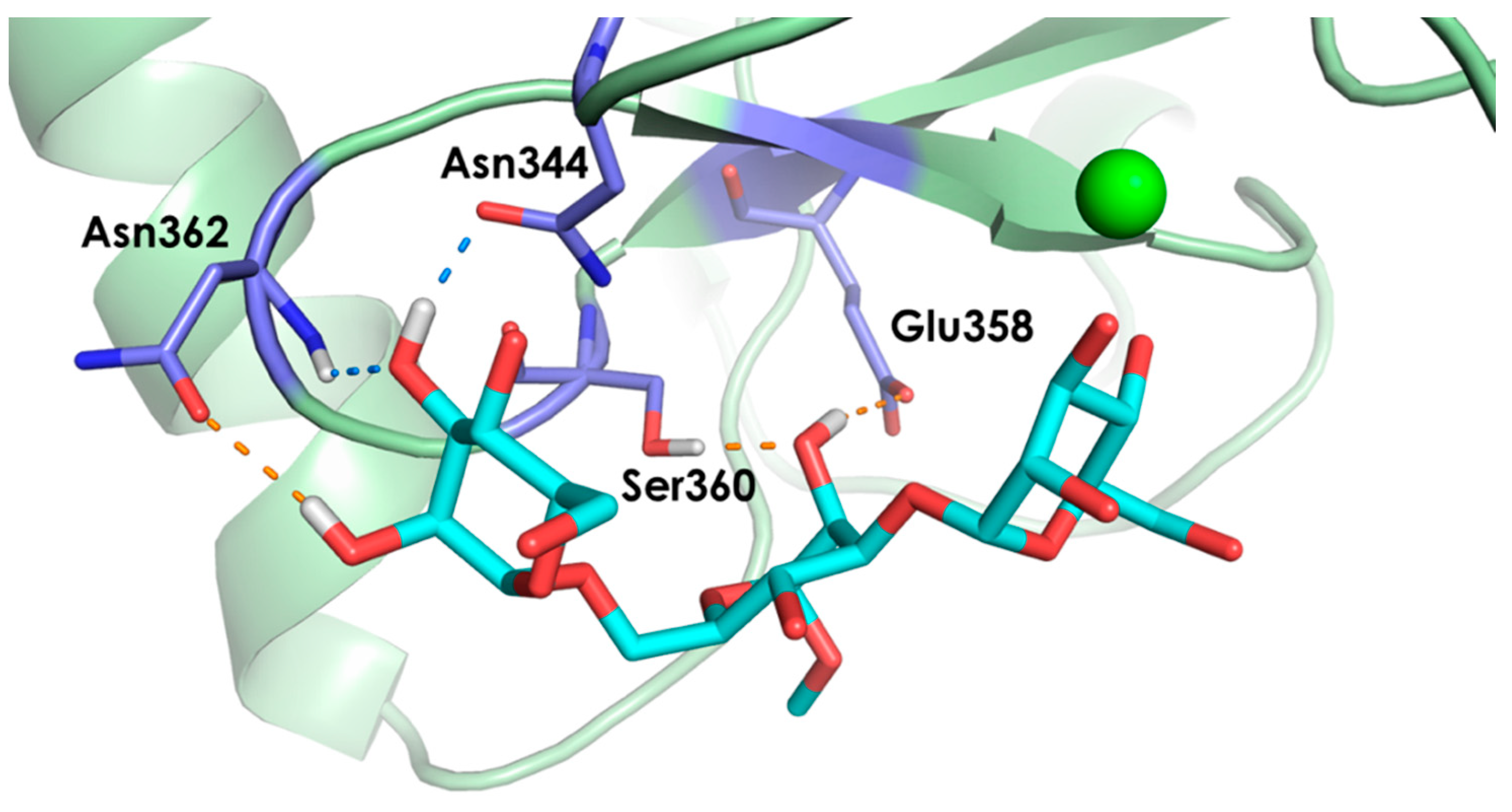
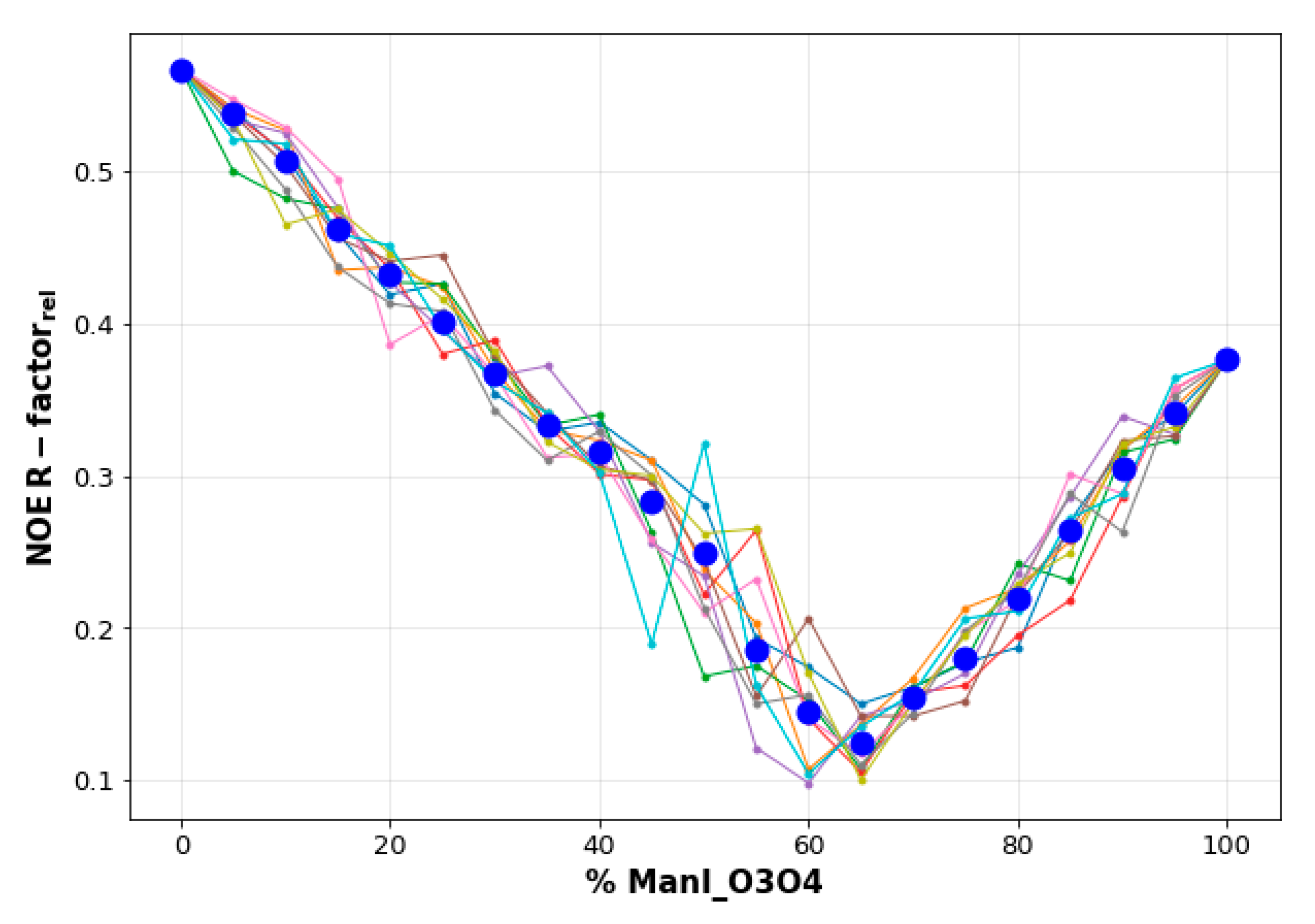
2.2. Molecular Dynamics Simulations
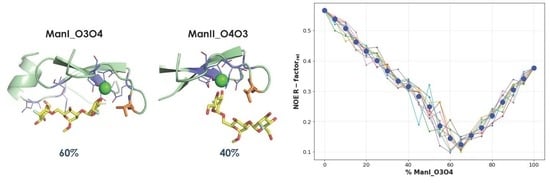
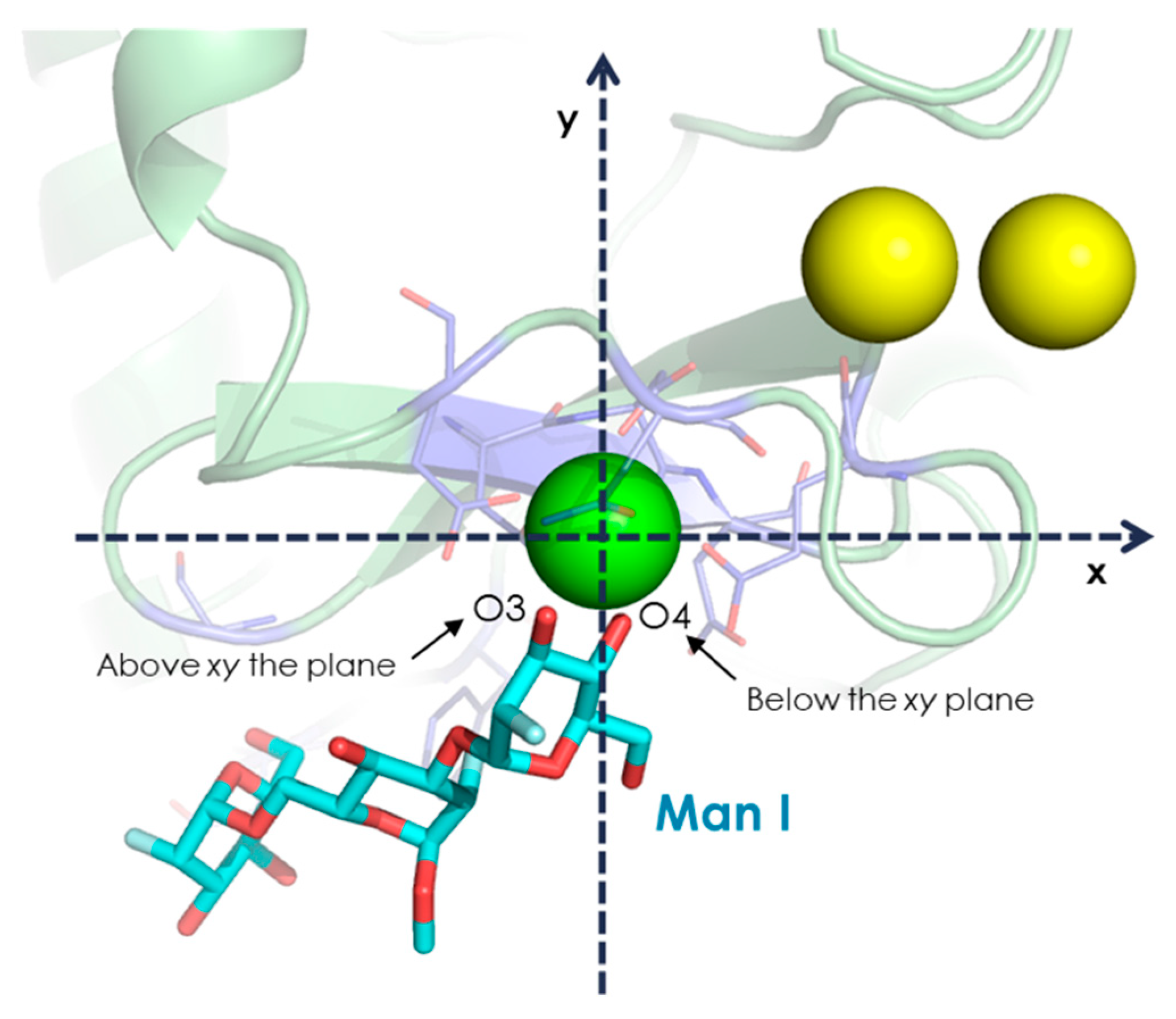
- The vicinal O3 and O4 atoms of any Man residue coordinate the key Ca2+.
- The corresponding Man moiety was superimposed with the equivalent Man unit deposited on PDB 2IT5 [45]. This crystallographic structure contains two alternative O3-O4 (major) and O4-O3 (minor) poses, respectively.
2.3. CORCEMA-ST and Best-Model STD Fitting
3. Materials and Methods
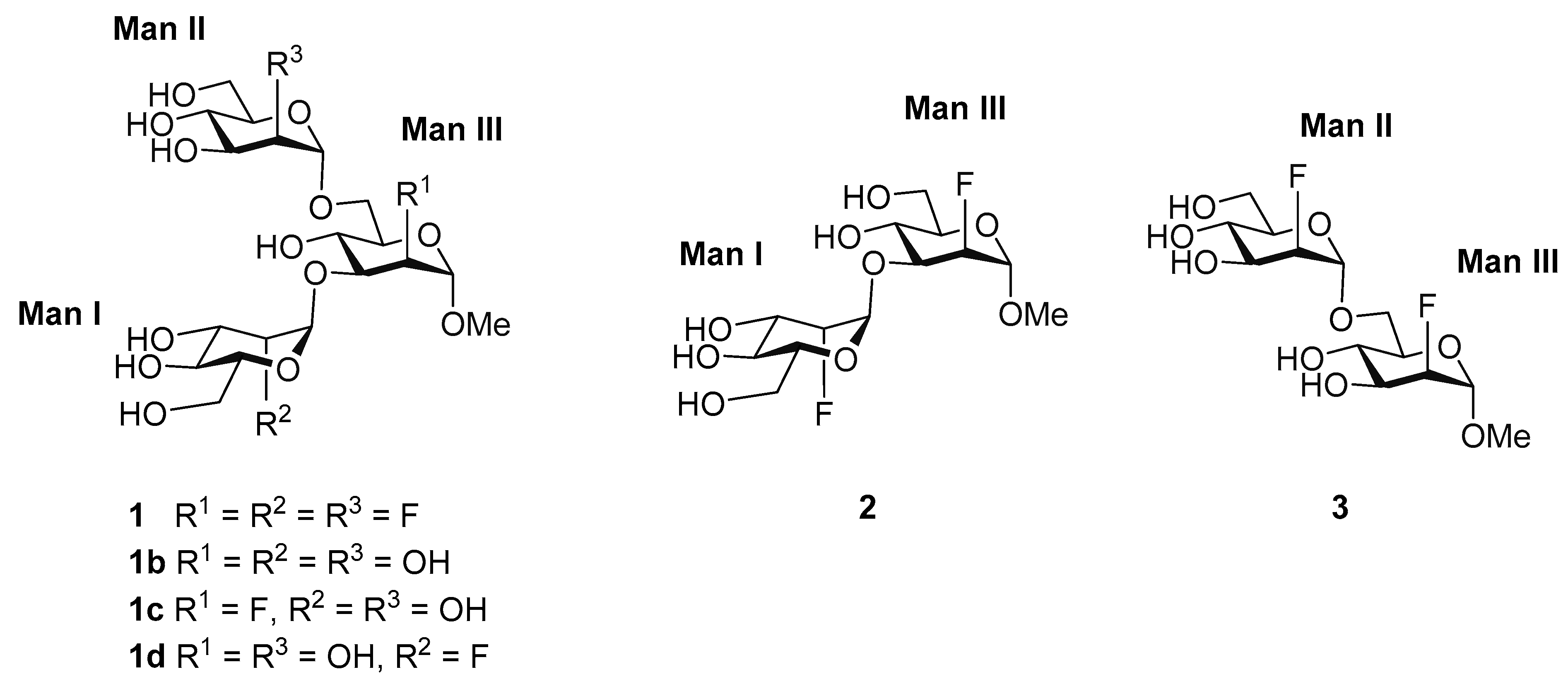
3.1. Man-Based Ligands
3.2. DC-SIGN ECD Preparation
3.3. NMR Experiments
3.4. Molecular Dynamics Simulations
3.5. CORCEMA-ST and Best-Model STD Fitting
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef] [PubMed]
- André, S.; Kaltner, H.; Manning, J.C.; Murphy, P.V.; Gabius, H.J. Lectins: Getting familiar with translators of the sugar code. Molecules 2015, 20, 1788–1823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, E.; Schetters, S.T.T.; Van Kooyk, Y. The tumour glyco-code as a novel immune checkpoint for immunotherapy. Nat. Rev. Immunol. 2018, 18, 204–211. [Google Scholar] [CrossRef] [PubMed]
- van Kooyk, Y.; Rabinovich, G.A. Protein-glycan interactions in the control of innate and adaptive immune responses. Nat. Immunol. 2008, 9, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Kleene, R.; Schachner, M. Glycans and neural cell interactions. Nat. Rev. Neurosci. 2004, 5, 195–208. [Google Scholar] [CrossRef]
- Rahfeld, P.; Sim, L.; Moon, H.; Constantinescu, I.; Morgan-Lang, C.; Hallam, S.J.; Kizhakkedathu, J.N.; Withers, S.G. An enzymatic pathway in the human gut microbiome that converts A to universal O type blood. Nat. Microbiol. 2019, 4, 1475–1485. [Google Scholar] [CrossRef]
- Naresh, K.; Schumacher, F.; Hahm, H.S.; Seeberger, P.H. Pushing the limits of automated glycan assembly: Synthesis of a 50mer polymannoside. Chem. Commun. (Camb). 2017, 53, 9085–9088. [Google Scholar] [CrossRef] [Green Version]
- Schönemann, W.; Cramer, J.; Mühlethaler, T.; Fiege, B.; Silbermann, M.; Rabbani, S.; Dätwyler, P.; Zihlmann, P.; Jakob, R.P.; Sager, C.P.; et al. Improvement of Aglycone π-Stacking Yields Nanomolar to Sub-nanomolar FimH Antagonists. ChemMedChem 2019, 14, 749–757. [Google Scholar] [CrossRef]
- Fernández de Toro, B.; Peng, W.; Thompson, A.J.; Domínguez, G.; Cañada, F.J.; Pérez-Castells, J.; Paulson, J.C.; Jiménez-Barbero, J.; Canales, A. Avenues to Characterize the Interactions of Extended N-Glycans with Proteins by NMR Spectroscopy: The Influenza Hemagglutinin Case. Angew. Chem. Int. Ed. Engl. 2018, 57, 15051–15055. [Google Scholar] [CrossRef]
- Parsons, T.B.; Struwe, W.B.; Gault, J.; Yamamoto, K.; Taylor, T.A.; Raj, R.; Wals, K.; Mohammed, S.; Robinson, C.V.; Benesch, J.L.P.; et al. Optimal Synthetic Glycosylation of a Therapeutic Antibody. Angew. Chem., Int. Ed. 2016, 55, 2361–2367. [Google Scholar] [CrossRef]
- Klukowski, P.; Schubert, M. Chemical shift-based identification of monosaccharide spin-systems with NMR spectroscopy to complement untargeted glycomics. Bioinformatics 2019, 35, 293–300. [Google Scholar] [CrossRef]
- Tamburrini, A.; Colombo, C.; Bernardi, A. Design and synthesis of glycomimetics: Recent advances. Med. Res. Rev. 2020, 40, 495–531. [Google Scholar] [CrossRef] [PubMed]
- Franconetti, A.; López, O.; Fernandez-Bolanos, J.G. Carbohydrates: Potential Sweet Tools against Cancer. Curr. Med. Chem. 2020, 27, 1206–1242. [Google Scholar] [CrossRef] [PubMed]
- Egger, J.; Weckerle, C.; Cutting, B.; Schwardt, O.; Rabbani, S.; Lemme, K.; Ernst, B. Nanomolar E-Selectin Antagonists with Prolonged Half-Lives by a Fragment-Based Approach. J. Am. Chem. Soc. 2013, 135, 9820–9828. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Paulson, J.C. CD22 Ligands on a Natural N-Glycan Scaffold Efficiently Deliver Toxins to B-Lymphoma Cells. J. Am. Chem. Soc. 2017, 139, 12450–12458. [Google Scholar] [CrossRef] [PubMed]
- Peterson, K.; Kumar, R.; Stenström, O.; Verma, P.; Verma, P.R.; Hakansson, M.; Kahl-Knutsson, B.; Zetterberg, F.; Leffler, H.; Akke, M.; et al. Systematic Tuning of Fluoro-galectin-3 Interactions Provides Thiodigalactoside Derivatives With Single-Digit nM Affinity and High Selectivity. J. Med. Chem. 2018, 61, 1164–1175. [Google Scholar]
- Hevey, R. Strategies for the development of glycomimetic drug candidates. Pharmaceuticals 2019, 12, 55. [Google Scholar] [CrossRef] [Green Version]
- Rillahan, C.D.; Antonopoulos, A.; Lefort, C.T.; Sonon, R.; Azadi, P.; Ley, K.; Dell, A.; Haslam, S.M.; Paulson, J.C. Global metabolic inhibitors of sialyl- and fucosyltransferases remodel the glycome. Nat. Chem. Biol. 2012, 8, 661–668. [Google Scholar] [CrossRef] [Green Version]
- Mei, H.; Han, J.; Fustero, S.; Medio-Simon, M.; Sedgwick, D.M.; Santi, C.; Ruzziconi, R.; Soloshonok, V.A. Fluorine-containing drugs approved by the FDA in 2018. Chemistry 2019, 25, 11797–11819. [Google Scholar] [CrossRef]
- Mei, H.; Remete, A.M.; Zou, Y.; Moriwaki, H.; Fustero, S.; Kiss, L.; Soloshonok, V.A.; Han, J. Fluorine-containing drugs approved by the FDA in 2019. Chin. Chem. Lett. 2020. [Google Scholar] [CrossRef]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in Medicinal Chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Linclau, B.; Ardá, A.; Reichardt, N.C.; Sollogoub, M.; Unione, L.; Vincent, S.P.; Jiménez-Barbero, J. Fluorinated carbohydrates as chemical probes for molecular recognition studies. Current status and perspectives. Chem. Soc. Rev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Schulze, J.; Baukmann, H.; Wawrzinek, R.; Fuchsberger, F.F.; Specker, E.; Aretz, J.; Nazaré, M.; Rademacher, C. CellFy: A Cell-Based Fragment Screen against C-Type Lectins. ACS Chem. Biol. 2018, 13, 3229–3235. [Google Scholar] [CrossRef] [PubMed]
- Wamhoff, E.C.; Hanske, J.; Schnirch, L.; Aretz, J.; Grube, M.; Silva, D.V.; Rademacher, C. 19F NMR-Guided Design of Glycomimetic Langerin Ligands. ACS Chem. Biol. 2016, 11, 2407–2413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalvit, C.; Vulpetti, A. Ligand-Based Fluorine NMR Screening: Principles and Applications in Drug Discovery Projects. J. Med. Chem. 2019, 62, 2218–2244. [Google Scholar]
- Valverde, P.; Delgado, S.; Martínez, J.D.; Vendeville, J.-B.; Malassis, J.; Linclau, B.; Reichardt, N.-C.; Cañada, F.J.; Jiménez-Barbero, J.; Ardá, A. Molecular insights into DC-SIGN binding to self-antigens: The interaction with the blood group A/B antigens. ACS Chem. Biol. 2019, 14, 1660–1671. [Google Scholar] [CrossRef]
- Figdor, C.G.; van Kooyk, Y.; Adema, G.J. C-type Lectin Receptors on Dendritic Cells and Langerhans Cells. Nat. Rev. Immunol. 2002, 2, 77–84. [Google Scholar] [CrossRef]
- van Vliet, S.J.; García-Vallejo, J.J.; van Kooyk, Y. Dendritic cells and C-type lectin receptors: Coupling innate to adaptive immune responses. Cell Biol. 2008, 86, 580–587. [Google Scholar] [CrossRef]
- Valverde, P.; Martinez, J.D.; Cañada, F.J.; Arda, A.; Jimenez-Barbero, J. Molecular recognition in C-type lectins: The cases of DC-SIGN, Langerin, MGL, and L-sectin. ChemBioChem 2020. [Google Scholar] [CrossRef]
- Nagae, M.; Yamaguchi, Y. Structural Aspects of Carbohydrate Recognition Mechanisms of C-Type Lectins. Curr. Top. Microbiol. Immunol. 2019, 1–30. [Google Scholar] [CrossRef]
- Cummings, R.D.; McEver, R.P. Essentials of Glycobiology, 3rd ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor: Suffolk County, NY, USA, 2005; Volume 17, Chapter 34. [Google Scholar]
- Geijtenbeek, T.B.; Gringhuis, S.I. C-type lectin receptors in the control of T helper cell differentiation. Nat. Rev. Immunol. 2016, 16, 433–448. [Google Scholar] [CrossRef] [PubMed]
- Mason, C.P.; Tarr, A.W. Human Lectins and Their Roles in Viral Infections. Molecules 2015, 20, 2229–2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, D.S.; Gregorio, G.; Bitton, N.; Hendrickson, W.A.; Littman, D.R. DC-SIGN-Mediated Internalization of HIV Is Required for Trans-Enhancement of T Cell Infection. Immunity 2002, 16, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Hodges, A.; Sharrocks, K.; Edelmann, M.; Baban, D.; Moris, A.; Schwartz, O.; Drakesmith, H.; Davies, K.; Kessler, B.; McMichael, A.; et al. Activation of the lectin DC-SIGN induces an immature dendritic cell phenotype triggering Rho-GTPase activity required for HIV-1 replication. Nat. Immunol. 2007, 8, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, R.; Mitra, D.; Chakrabarti, S. HIV-1 gp120 protein downregulates Nef induced IL-6 release in immature dentritic cells through interplay of DC-SIGN. PLoS ONE 2013, 8, e59073. [Google Scholar] [CrossRef]
- Simmons, G.; Reeves, J.D.; Grogan, C.C.; Vandenberghe, L.H.; Baribaud, F.; Whitbeck, J.C.; Burke, E.; Buchmeier, M.J.; Soilleux, E.J.; Riley, J.L.; et al. DC-SIGN and DC-SIGNR bind ebola glycoproteins and enhance infection of macrophages and endothelial cells. Virology 2003, 305, 115–123. [Google Scholar] [CrossRef] [Green Version]
- Tassaneetrithep, B.; Burgess, T.H.; Granelli-Piperno, A.; Trumpfheller, C.; Finke, J.; Sun, W.; Eller, M.A.; Pattanapanyasat, K.; Sarasombath, S.; Birx, D.L.; et al. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J. Exp. Med. 2003, 197, 823–829. [Google Scholar] [CrossRef] [Green Version]
- Alen, M.M.; Dallmeier, K.; Balzarini, J.; Neyts, J.; Schols, D. Crucial role of the N-glycans on the viral E-envelope glycoprotein in DC-SIGN-mediated dengue virus infection. Antiviral Res. 2012, 96, 280–287. [Google Scholar] [CrossRef]
- Liu, P.; Ridilla, M.; Patel, P.; Betts, L.; Gallichotte, E.; Shahidi, L.; Thompson, N.L.; Jacobson, K. Beyond attachment: Roles of DC-SIGN in dengue virus infection. Traffic 2017, 18, 218–231. [Google Scholar] [CrossRef] [Green Version]
- van Liempt, E.; Bank, C.M.C.; Mehta, P.; García-Vallejo, J.J.; Kawar, Z.S.; Geyer, R.; Alvarez, R.A.; Cummings, R.D.; van Kooyk, Y.; van Die, I. Specificity of DC-SIGN for mannose- and fucose-containing glycans. FEBS Lett. 2006, 580, 6123–6131. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, D.A.; Fadden, A.J.; Drickamer, K. A novel mechanism of carbohydrate recognition by the C-type lectins DC-SIGN and DC-SIGNR subunit organization and binding to multivalent ligands. J. Biol. Chem. 2001, 276, 28939–28945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Feinberg, H.; Conroy, E.; Mitchell, D.A.; Alvarez, R.; Blixt, O.; Taylor, M.E.; Weis, W.I.; Drickamer, K. Structural basis for distinct ligand-binding and targeting properties of the receptors DC-SIGN and DC-SIGNR. Nat. Struct. Mol. Biol. 2004, 11, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Diercks, T.; Infantino, A.S.; Unione, L.; Jiménez-Barbero, J.; Oscarson, S.; Gabius, H.-J. Fluorinated Carbohydrates as Lectin Ligands: Synthesis of OH/F-Substituted N-Glycan Core Trimannoside and Epitope Mapping by 2D STD-TOCSYreF NMR spectroscopy. Chem. Eur. J. 2018, 24, 15761–15765. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, H.; Castelli, R.; Drickamer, K.; Seeberger, P.H.; Weis, W.I. Multiple modes of binding enhance the affinity of DC-SIGN for high mannose N-linked glycans found on viral glycoproteins. J. Biol. Chem. 2007, 282, 4202–4209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angulo, J.; Diaz, I.; Reina, J.J.; Tabarani, G.; Fieschi, F.; Rojo, J.; Nieto, P.M. Saturation Transfer Difference (STD) NMR Spectroscopy Characterization of Dual Binding Mode of a Mannose Disaccharide to DC-SIGN. ChemBioChem 2008, 9, 2225–2227. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, H.; Mitchell, D.A.; Drickamer, K.; Weis, W.I. Structural Basis for Selective Recognition of Oligosaccharides by DC-SIGN and DC-SIGNR. Science 2001, 294, 2163–2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez, J.D.; Valverde, P.; Delgado, S.; Romanò, C.; Linclau, B.; Reichardt, N.C.; Oscarson, S.; Ardá, A.; Jiménez-Barbero, J.; Cañada, F.J. Unraveling Sugar Binding Modes to DC-SIGN by Employing Fluorinated Carbohydrates. Molecules 2019, 24, 2337. [Google Scholar] [CrossRef] [Green Version]
- Thépaut, M.; Guzzi, C.; Sutkeviciute, I.; Sattin, S.; Ribeiro-Viana, R.; Varga, N.; Chabrol, E.; Rojo, J.; Bernardi, A.; Angulo, J.; et al. Structure of a glycomimetic ligand in the carbohydrate recognition domain of C-type lectin DC-SIGN. Structural requirements for selectivity and ligand design. J. Am. Chem. Soc. 2013, 135, 2518–2529. [Google Scholar] [CrossRef]
- Guzzi, C.; Alfarano, P.; Sutkeviciute, I.; Sattin, S.; Ribeiro-Viana, R.; Fieschi, F.; Bernardi, A.; Weiser, J.; Rojo, J.; Angulo, J.; et al. Detection and quantitative analysis of two independent binding modes of a small ligand responsible for DC-SIGN clustering. Org. Biomol. Chem. 2016, 14, 335–344. [Google Scholar] [CrossRef] [Green Version]
- Jayalakshmi, V.; Krishna, N.R. Complete relaxation and conformational exchange matrix (CORCEMA) analysis of intermolecular saturation transfer effects in reversibly forming ligand-receptor complexes. J. Magn. Reson. 2002, 155, 106–118. [Google Scholar] [CrossRef]
- Jayalakshmi, V.; Krishna, N.R. CORCEMA refinement of the bound ligand conformation within the protein binding pocket in reversibly forming weak complexes using STD-NMR intensities. J. Magn. Reson. 2004, 168, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Marcelo, F.; Garcia-Martin, F.; Matsushita, T.; Sardinha, J.; Coelho, H.; Oude-Vrielink, A.; Koller, C.; André, S.; Cabrita, E.J.; Gabius, H.J.; et al. Delineating Binding Modes of Gal/GalNAc and Structural Elements of the Molecular Recognition of Tumor-Associated Mucin Glycopeptides by the Human Macrophage Galactose-Type Lectin. Chemistry 2014, 20, 16147–16155. [Google Scholar] [CrossRef]
- Dalvit, C. Theoretical analysis of the competition ligand-based NMR experiments and selected applications to fragment screening and binding constant measurements. Concepts Magn. Reson. 2008, 32A, 341–372. [Google Scholar] [CrossRef]
- Doerr, A.J.; Case, M.A.; Pelczer, I.; McLendon, G.L. Design of a functional protein for molecular recognition: Specificity of ligand binding in a metal-assembled protein cavity probed by 19F NMR. J. Am. Chem. Soc. 2004, 126, 4192–4198. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, K.N.; Yongye, A.B.; Tschampel, S.M.; González-Outeiriño, J.; Daniels, C.R.; Foley, B.L.; Woods, R.J. GLYCAM06: A generalizable biomolecular force field. Carbohydrates. J. Comput. Chem. 2008, 29, 622–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Antechamber, An Accessory Software Package For Molecular Mechanical Calculations. J. Comput. Chem. 2005, 25, 1157–1174. [Google Scholar] [CrossRef]
- Li, P.; Song, L.F.; Merz, K.M., Jr. Systematic Parameterization of Monovalent Ions Employing the Nonbonded Model. J. Chem. Theory Comput. 2015, 11, 1645–1657. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99S. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. Inc. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jayalakshmi, V.; Krishna, N.R. Determination of the conformation of trimethoprim in the binding pocket of bovine dihydrofolate reductase from a STD-NMR intensity-restrained CORCEMA-ST optimization. J. Am. Chem. Soc. 2005, 127, 14080–14084. [Google Scholar] [CrossRef]
- Gimeno, A.; Reichardt, N.C.; Cañada, F.J.; Perkams, L.; Unverzagt, C.; Jiménez-Barbero, J.; Ardá, A. NMR and molecular recognition of N-Glycans: Remote modifications of the saccharide chain modulate binding features. ACS Chem. Biol. 2017, 12, 1104–1112. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | H | % STD |
|---|---|---|
| 1 | ManI-1 | 16 |
| ManI-2 | 42 | |
| ManI-3 | 100 | |
| ManIII-1/2 | 31 | |
| ManII-1 | 19 | |
| ManII-2 | 31 | |
| ManII-3/6 | 47 | |
| ManII-4/5 | 52 | |
| 2 | ManI-1 | 15 |
| ManI-2 | 33 | |
| ManI-3 | 74 | |
| ManI-4 | 100 | |
| ManI-5 | 22 | |
| ManIII-1 | 17 | |
| ManIII-2 | 14 | |
| 3 | ManIII-1 | 33 |
| ManIII-2 | 31 | |
| ManIII-3/4/5/6 | 47 | |
| ManII-1 | 31 | |
| ManII-2 | 45 | |
| ManII-3/6 | 86 | |
| ManII-4/5 | 100 |
| Structure | a KI (mM) | SD (mM) | b r2 |
|---|---|---|---|
| 1 | 2.5 | 0.7 | 0.91 |
| 1b | 1.1 | 0.2 | 0.97 |
| 1c | 1.0 | 0.1 | 0.99 |
| 1d | 1.1 | 0.2 | 0.98 |
| 2 | 9.6 | 3.5 | 0.77 |
| 3 | 4.0 | 0.6 | 0.98 |
| Ligand | Binding Poses | Complex Stability (ns) 1 |
|---|---|---|
| 1 | ManI_O3-O4 | 321 |
| ManI_O4-O3 | 9 | |
| ManII_O3-O4 | 27 | |
| ManII_O4-O3 | 22 | |
| 2 | ManI_O3-O4 | 77 |
| ManI_O4-O3 | 18 | |
| 3 | ManII_O3-O4 | 400 |
| ManII_O4-O3 | 22 | |
| ManIII_O3-O4 | 19 | |
| ManIII_O4-O3 | 1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez, J.D.; Infantino, A.S.; Valverde, P.; Diercks, T.; Delgado, S.; Reichardt, N.-C.; Ardá, A.; Cañada, F.J.; Oscarson, S.; Jiménez-Barbero, J. The Interaction of Fluorinated Glycomimetics with DC-SIGN: Multiple Binding Modes Disentangled by the Combination of NMR Methods and MD Simulations. Pharmaceuticals 2020, 13, 179. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13080179
Martínez JD, Infantino AS, Valverde P, Diercks T, Delgado S, Reichardt N-C, Ardá A, Cañada FJ, Oscarson S, Jiménez-Barbero J. The Interaction of Fluorinated Glycomimetics with DC-SIGN: Multiple Binding Modes Disentangled by the Combination of NMR Methods and MD Simulations. Pharmaceuticals. 2020; 13(8):179. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13080179
Chicago/Turabian StyleMartínez, J. Daniel, Angela S. Infantino, Pablo Valverde, Tammo Diercks, Sandra Delgado, Niels-Christian Reichardt, Ana Ardá, Francisco Javier Cañada, Stefan Oscarson, and Jesús Jiménez-Barbero. 2020. "The Interaction of Fluorinated Glycomimetics with DC-SIGN: Multiple Binding Modes Disentangled by the Combination of NMR Methods and MD Simulations" Pharmaceuticals 13, no. 8: 179. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13080179