Tumor Immunotherapy Using A2A Adenosine Receptor Antagonists




1
iHuman Institute, ShanghaiTech University, Shanghai 201210, China
2
School of Life Science and Technology, ShanghaiTech University, Shanghai 201210, China
3
Department of Integrated Structural and Computational Biology, Scripps Research, La Jolla, CA 92037, USA
*
Author to whom correspondence should be addressed.
Pharmaceuticals 2020, 13(9), 237; https://0-doi-org.brum.beds.ac.uk/10.3390/ph13090237
Submission received: 14 August 2020
/
Revised: 31 August 2020
/
Accepted: 2 September 2020
/
Published: 8 September 2020
(This article belongs to the Special Issue GPCRs: Ligands and beyond 2022)
Abstract
:The A2A adenosine receptor (A2AAR) plays critical roles in human physiology and pathophysiology, which makes it an important drug target. Previous drug-discovery efforts targeting the A2AAR have been focused on the use of A2AAR antagonists for the treatment of Parkinson’s disease. More recently, the A2AAR has attracted additional attention for its roles in immuno-oncology, and a number of A2AAR antagonists are currently used as lead compounds for antitumor drugs in both preclinical models and clinical trials. This review surveys recent advances in the development of A2AAR antagonists for cancer immunotherapy. The therapeutic potential of representative A2AAR antagonists is discussed based on both animal efficacy studies and clinical data.
1. Introduction
The A2A adenosine receptor (A2AAR) is a family A G protein-coupled receptor (GPCR) [1]. There are four subtypes of adenosine receptors (A1, A2A, A2B, and A3), and all of them can be activated by extracellular adenosine [1]. The A1 and A2A receptors are widely distributed in both the central nervous system (CNS) and the periphery, while the density of A2B and A3 in the brain is very low [2]. The A1, A2A, and A2B adenosine receptors are conserved throughout evolution and are highly homologous across different species, whereas A3 varies substantially [3]. Sequence homology is about 49% between A1 and A3, and 59% between the A2A and A2B receptors. In terms of endogenous ligand binding, the A1, A2A, and A3 receptors have high affinity, whereas A2B shows low binding affinity for adenosine [4]. Upon adenosine binding and the resulting activation, the A1 and A3 receptors couple primarily to the Gi protein, leading to a decrease in the intracellular concentration of cAMP, whereas A2A and A2B couple preferentially to the Gs protein, which leads to an increase in intracellular cAMP levels [4]. These diverse downstream effects of the different adenosine receptor subtypes require high selectivity of synthetic ligands to be used as A2AAR antagonists [5].
Over the past decades, drug-discovery efforts have led to numerous A2AAR-targeting ligands, including both agonists and antagonists [6]. A2AAR agonists have been examined as anti-inflammatory agents and as coronary vasodilators [7]. Regadenoson, a selective A2AAR agonist, has been approved as a pharmacological stress agent in myocardial perfusion imaging (MPI) [8]. Early studies of A2AAR antagonists have been focused on their use to treat Parkinson’s disease (PD) [9,10]. This is based on the fact that A2AAR is prone to forming heterodimers with the dopamine D2 receptor in the CNS, and that activation of A2AAR inhibits the activation of the D2 receptor [2]. A2AAR antagonists can prevent this inhibition and enhance dopaminergic activity, leading to the desired therapeutic effects in PD patients. A number of A2AAR antagonists have shown encouraging anti-Parkinson effects in animal models of PD, and a number of clinical trials have been conducted to evaluate their therapeutic potential [11]. These include the compounds istradefylline (also known as KW-6002, Kyowa Hakko Kirin), PBF-509 (PaloBiofarma), V81444 (Vernalis), ST1535 and ST4206 (Sigma-Tau), V2006 (Biogen), SCH-420814 (Merck Sharp & Dohme), and SYN115 (Biotie Therapies) [9]. Although most of these compounds failed to show statistically significant therapeutic effects in the clinic, istradefylline showed efficacy as an adjunctive treatment to levodopa/carbidopa in PD patients by reducing the “off” episodes. It was approved in Japan in 2013 [12] and was also approved by the US FDA in August 2019.
The role of adenosine as an immunosuppressive factor was first reported in 1975, when it was demonstrated that adenosine inhibits lymphocyte-mediated cytolysis via increasing intracellular cAMP levels [13]. It was later reported that the concentration of adenosine is significantly enhanced compared to adjacent tissues in the tumor microenvironment (TME) [14], creating an “adenosine protective ring” that helps the tumor to fight off attacks from the immune system. Regarding the immunosuppressive role of adenosine, it has been demonstrated that among the four adenosine receptor subtypes, A2AAR is the dominant receptor for extracellular adenosine, leading to an increase in intracellular cAMP and consequently the functional inhibition of immune cells. Genetic knockout of the A2AAR suppressed the immunosuppression by adenosine, and small molecule A2AAR antagonists have similar effects [15,16]. Overall, studies with different A2AAR antagonists have thus shown that A2AAR is a promising target for the development of novel immunotherapies of cancer [17,18,19].
When A2AAR gained interest as an immuno-oncology drug target, a small library of A2AAR antagonists had already been reported, mainly as a result of earlier efforts in the development of A2AAR antagonists as anti-PD agents. This greatly facilitated the mechanism-of-action and proof-of-concept studies of A2AAR antagonists as novel anticancer agents. Furthermore, the A2AAR is one of the most extensively studied GPCRs in structural biology, being one of the GPCRs for which both the antagonist-bound and agonist-bound structures have been reported [20,21]. The structure of an agonist-A2AAR-Gs ternary complex has also been reported [22]. Conformational dynamics of the A2AAR have been demonstrated recently, using solution NMR methods [23,24]. These structural biology data greatly facilitate the design of novel compounds with high binding affinity and target selectivity. The promising role of A2AAR in immuno-oncology and the potential of combination therapies using A2AAR antagonists together with other checkpoint inhibitors, encourage continuous efforts in this area. Therefore, a new wave of drug-discovery efforts targeting the A2AAR has been emerging.
2. Mechanism of Action of A2AAR Antagonists in Immuno-Oncology
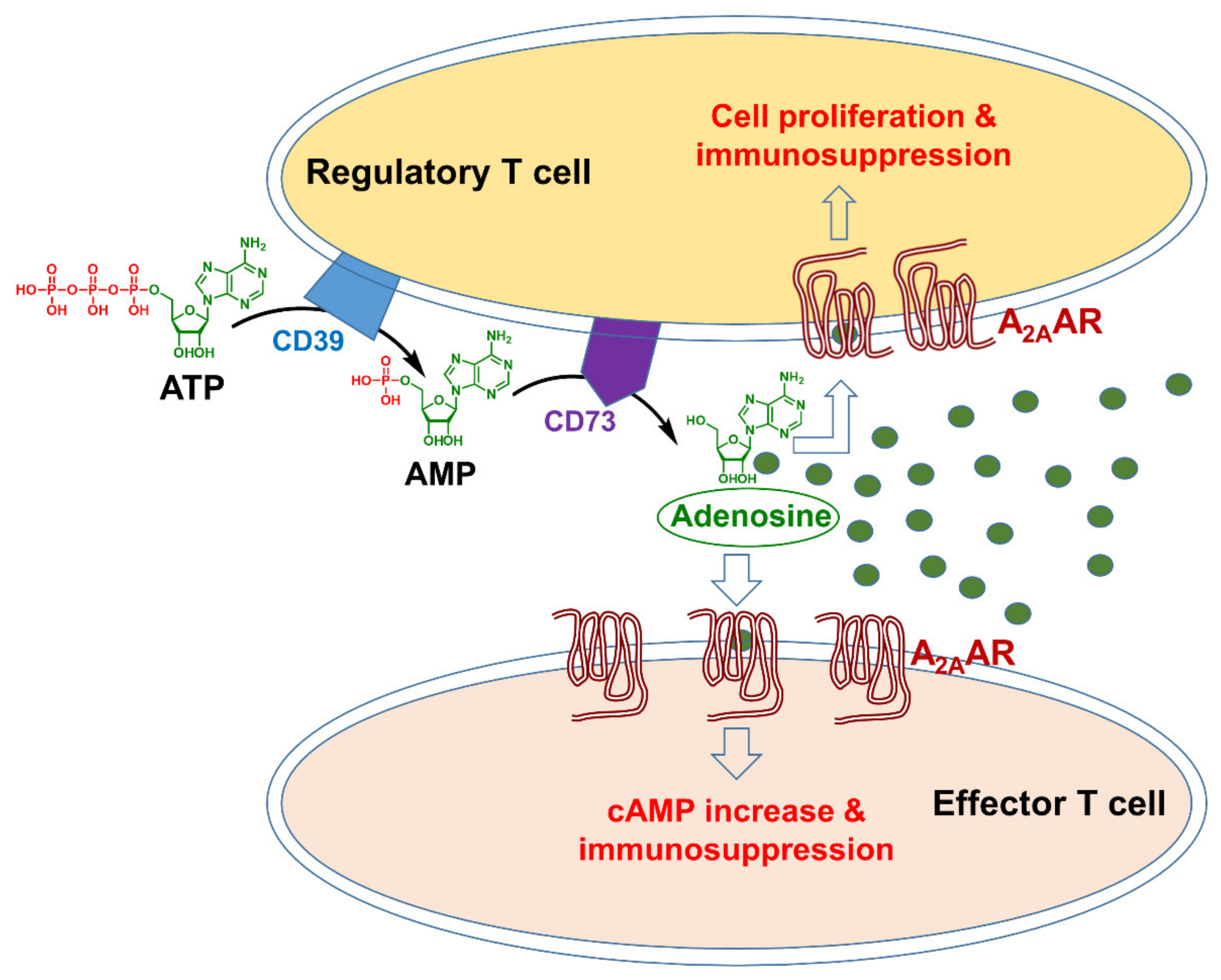
A line of evidence has highlighted the importance of adenosine as a critical immunosuppressive factor that accumulates in the TME [4,14,25]. The concentration of this nucleoside, which is present at low nanomolar levels in the interstitial fluids of unstressed tissues, can rapidly increase in response to pathophysiological conditions, such as hypoxia, ischemia, inflammation, or tissue injury [25]. Many factors can lead to extracellular aggregation of adenosine triphosphate (ATP) in the TME, for example, hypoxia, increased metabolism, and apoptosis. Two nucleotidases expressed on the cell surface, CD39 and CD73, catalyze the conversion of ATP to AMP (adenosine monophosphate) and of AMP to adenosine, respectively (Figure 1). CD39 is expressed by regulatory T and B cells [26], and CD73 is expressed on the regulatory T (Treg) cells and various stromal cells in the bone marrow, such as mesenchymal stem cells, fibroblasts, and endothelial cells [27]. Preclinical studies have shown that with the impact of these agents, adenosine concentrations in the TME can increase more than 10-fold [14], leading to immunosuppression by tumor tissues.
Besides its roles in the regulation of the immune system, adenosine is also involved in angiogenesis and tumor cell proliferation [4]. Different tumor types actually show altered purine metabolism, which facilitates the production of adenosine and/or reduces its degradation, thereby resulting in a “protective adenosine halo” that contributes to cancer progression [18,28].
It was reported almost two decades ago that A2AAR is the dominant receptor for adenosine in its immunosuppressive roles, so that blocking A2AAR signaling could downscale the immunosuppression by adenosine [15]. It has also been reported that A2AAR protects tumors from the cytotoxic effects of functional T cells [16]. Hypoxia-driven accumulation of extracellular adenosine in the TME triggers the suppression of activated immune cells via A2AARs on their cell surfaces. This molecular pathway is of critical importance for immunosuppression in the TME [29]. The main immune cells that A2AAR acts on are CD8+ T cells and the natural killer (NK) cells. Blocking A2AAR with the antagonist SCH-58261 enhanced interferon gamma levels and the cytotoxic CD8+ T cell response, leading to tumor suppression [30]. The stimulation of the A2AAR also suppresses the maturation of NK cells and their cytotoxic effects in vitro and promoted tumor metastasis in mice by decreasing the expression of the cytotoxic protein granzyme B by NK cells [30].
Adenosine binding to A2AAR also suppresses antitumor immunity through its action on the Treg cells, which play a key role in regulating or suppressing the function of effector T cells in the immune system (Figure 1). In the TME, suppressed immunity by Treg cells supports the uncontrolled growth of cancer cells. An increased number of Treg cells in the TME thus represents a barrier to successful immunotherapies [31], therefore blocking the A2AAR prevents immunosuppression by tissue-produced adenosine as well as by Treg cells.
Adenosine also promotes angiogenesis of solid tumors [4]. Although A2BAR has been shown to be the main receptor that mediates the proangiogenic role of adenosine, through inducing the secretion of the vascular endothelial growth factor (VEGF) [32], A2AAR has also been demonstrated to play a role [4]. In the early stages of human lung cancer, the expression of A2AAR on endothelial cells is increased, which may indicate that these receptors are involved in promoting the growth of blood vessels and thereby support early tumor growth and proliferation [33]. Alternatively, A2AAR knockout led to reduced tumor angiogenesis and concomitant tumor growth suppression [34].
Many preclinical studies have shown that A2AAR antagonists can block the immunosuppressive effects of adenosine, making A2AAR antagonists a novel class of potential therapeutic agents in immuno-oncology [35]. For example, both gene knockout of A2AAR and its inactivation by small molecule A2AAR antagonists could liberate tumor-reactive CD8+ T cells from tumor-induced immunosuppression [34]. The combination of A2AAR antagonists with immune checkpoint inhibitors, such as anti-PD-1/PD-L1 or CTLA4 antibodies, led to significantly enhanced antitumor effects [19,36]. In mouse experiments, the combination of A2AAR antagonists with adoptive cellular immunotherapies (ACT) also showed enhanced antitumor effects [37].
Overall, the available literature emphasizes the complexity of tumor immunity, and understanding of the roles of adenosine-A2AAR signaling in immuno-oncology is still evolving. The important roles of A2BAR in the immunosuppressive roles of adenosine are worth noting [38], and simultaneous inhibition of A2AAR, A2BAR and the upstream nucleotidases CD39 and CD73 may be necessary for optimal therapeutic efficacy [4]. Preclinical studies have convincingly demonstrated the anticancer efficacy of A2AAR antagonists [18,30,39,40,41]. A2AAR antagonists have also been combined with other approaches of immunotherapy to potentiate additive effects on tumor control and markedly enhance antitumor immunity in mouse models [19,36,40,42,43]. Considering the promise of the work surveyed in this review, we can look forward to more and more A2AAR antagonists emerging as potential drugs in the field of immuno-oncology.
3. A2AAR Antagonists in Preclinical and Clinical Studies
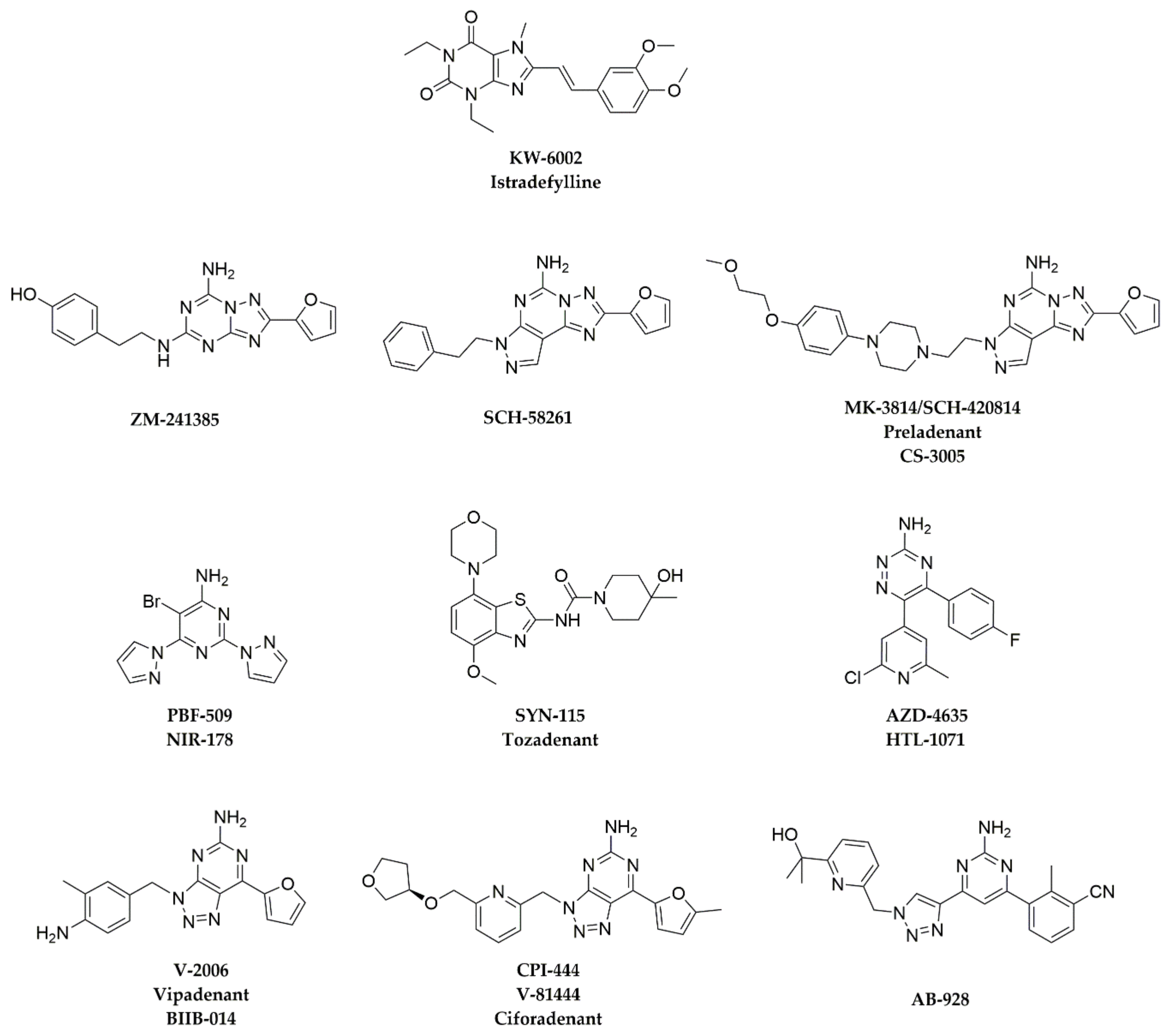
Representative A2AAR antagonists are reviewed in this section for which preclinical antitumor effects have been reported and/or clinical trials have been initiated. Chemical structures of these A2AAR antagonists are shown in Figure 2, and their binding affinities to A2AAR and other adenosine receptors are summarized in Table 1.
3.1. ZM-241385
The compound ZM-241385 was reported by Zeneca Pharmaceuticals (now AstraZeneca) as one of the first A2AAR antagonists. It shows a binding affinity of 0.8 nM for A2AAR, with 318-, 62-, and >1000-fold selectivity against A1AR, A2BAR, and A3AR, respectively (Table 1) [45,46]. In functional assays, it showed an IC50 of 54 nM in a G protein-mediated cAMP assay [52]. ZM-241385 has been widely used in biological studies of A2AAR [53,54], but there have been few in vivo studies in tumor models. ZM-241385 was tested both alone and in combination with an anti-CTLA4 mAb in a B16F10 mouse melanoma model, and it was found that melanoma-bearing mice treated with ZM-241365 alone showed a marked tumor growth inhibition compared with controls, and the combination therapy showed significant tumor growth delay compared with either controls or each agent alone [55]. The results showed that when an A2AAR antagonist is combined with an anti-CTLA4 mAb, the therapeutic effects of inhibiting tumor growth and antitumor immune responses can be enhanced [55]. However, ZM-241385 has poor pharmacokinetic properties and low bioavailability [56]. Therefore, no clinical trials have been initiated. The bicyclic non-xanthine scaffold of ZM-241385, with the furan and –NH2 substitutions, has served as a pharmacophore for the design of novel A2AAR antagonists having high selectivity (Figure 2).
3.2. SCH-58261
Compound SCH-58261 is a potent and selective A2AAR antagonist developed by Schering-Plough. It shows a binding affinity of 0.6 nM for A2AAR, with 478-, 8351-, and >10,000-fold selectivity against A1AR, A2BAR, and A3AR, respectively (Table 1) [47]. In functional assays, it showed an IC50 of 17 nM in the G protein-mediated cAMP assay [57]. SCH-58261 has also been widely used for characterizing A2AAR [58]. In CD73+ mouse tumor models, SCH-58261 was found to enhance tumor immunotherapy and suppress metastases [30,43]. In melanoma and breast cancer mouse models, SCH-58261 prolonged survival and reduced metastatic burden when used in combination with an anti-mouse CD73 mAb [59]. However, SCH-58261 has poor physicochemical and pharmacokinetic properties [58], and no clinical studies have been initiated.
3.3. MK-3814
MK-3814 (or SCH-420814, preladenant, CS-3005) is a structural derivative of SCH-58261. To improve A2AAR selectivity and physicochemical and pharmacokinetic properties, hydrophilic groups were added to the side chain of SCH-58261 to give SCH-420814/MK-3814 [48]. MK-3814 exhibits higher affinity for human A2AAR, with a Ki value of 1.1 nM, and high selectivity of over 1000-fold for human A2AAR compared with the other adenosine receptors (Table 1) [48]. MK-3814 is orally active and shows good pharmacokinetic properties [60]. A phase Ib/II study of MK-3814 used alone and in combination with the anti-PD-1 drug pembrolizumab (Keytruda) in patients with advanced solid tumors was initiated in June 2017 (NCT03099161). Unfortunately, the trial was terminated early because the data did not support the study endpoints.
3.4. PBF-509
PBF-509 (or NIR-178) is a potent and selective A2AAR antagonist discovered by Palobiofarma, which was licensed to Novartis through an agreement in 2015. It has a high affinity for A2AAR (Ki = 12 nM), with 208-, 83-, and 416-fold selectivity against A1AR, A2BAR, and A3AR, respectively (Table 1) [18]. It showed a KB value of 72.8 nM in the agonist-mediated cAMP accumulation assay in A2AARSNAP-expressing HEK cells [61]. PBF-509 has been tested in a mouse model of B16F10 melanoma, which was gene-modified to express CD73, as well as an MCA205 model, which endogenously expresses high levels of CD73. Oral administration of PBF-509 (15 or 30 mg/kg/day) significantly reduced the tumor burden of mice in both models [18]. A phase I/II study of PBF-509 both alone and in combination with anti-PD-1 mAb PDR001 in non-small cell lung cancer patients was started in October 2015 (NCT02403193); the results showed that the compound was well tolerated, and clinical benefit was observed in immunotherapy-exposed and -naive patients, irrespective of the PD-L1 status [62].
3.5. SYN-115
Compound SYN-115 (tozadenant) was first reported by Roche [49], which was later licensed to Synosia Therapeutics. SYN-115 has a benzothiophene scaffold that is structurally not related to xanthine or adenine, and it shows a binding affinity of 5 nM for A2AAR, with 270-, 140-, and 314-fold selectivity against A1AR, A2BAR, and A3AR, respectively (Table 1) [49]. SYN-115 was generally well-tolerated in phase II trials for PD and was advanced to phase III [63], but eventually the clinical trials for PD were discontinued. In a mouse model of CD73-expressing AT-3ovadim breast carcinoma, SYN-115 had no single-agent activity but significantly enhanced the antitumor efficacy of the anti-PD-1 mAb RMP1-14 by promoting antitumor T-cell responses [36]. No subsequent clinical cancer studies have been registered as yet.
3.6. AZD-4635
AZD-4635 (HTL-1071) is an orally available A2AAR antagonist. It was discovered by Heptares Therapeutics (now a wholly owned subsidiary of Sosei Heptares), and AstraZeneca licensed exclusive global rights to the compound in 2015. AZD-4635 binds to human A2AAR with a Ki of 1.7 nM and with 94-, 37-, and >5000-fold selectivity against A1AR, A2BAR, and A3AR, respectively (Table 1) [50]. AZD-4635 was reported to inhibit adenosine-mediated cAMP accumulation in both human and mouse A2AAR-expressing cells [50]. For the human A2AAR, it inhibited the cAMP increase induced by 1 μM adenosine with an IC50 of 10 nM [50]. In an MC-38 syngeneic mouse colorectal tumor model, treatment with AZD-4635 at 50 mg/kg BID led to a reduction in tumor growth [50]. When used in combination with an anti-PD-L1 mAb, the tumor suppressive effects were further enhanced. AZD-4635 increased expression of the genes associated with immune activation and increased expression of co-stimulatory markers on antigen-presenting cells (APCs) [50].
AZD-4635 is currently in phase II clinical trials for the treatment of solid tumors. In June 2016, phase I studies of continuous oral monotherapy with AZD-4635 alone or in combination with various other drugs (durvalumab, abiraterone acetate, enzalutamide, oleclumab, and docetaxel) were initiated in patients with advanced solid malignancies [NCT02740985]. In May 2018, MedImmune LLC. started a clinical study of AZD-4635 in combination with the anti-CD73 antibody oleclumab (MEDI9447) to investigate the safety, tolerability, and antitumor activity of novel combination therapies administered in subjects with advanced non-small cell lung cancer (NSCLC) [NCT03381274]. In August 2019, a phase II study of AZD-4635 in patients with prostate cancer in combination with the anti-PD-L1 antibody durvalumab and anti-CD73 oleclumab was also initiated [NCT04089553]. One now looks forward to seeing if these studies will demonstrate efficacy of AZD-4635 either as a single agent and/or in combination with other anticancer therapeutics.
3.7. CPI-444
CPI-444 (ciforadenant, previously known as V-81444) is an orally available A2AAR antagonist created by Vernalis [64]. CPI-444 was designed to address the chemical structural liabilities that may have led to toxicity concerns for compound V-2006 (vipadenant, previously known as BIIB014, a non-xanthine, selective A2AAR antagonist [51]). It showed a binding affinity of 3.54 nM for A2AAR, with 54-, 431-, and 693-fold selectivity against A1AR, A2BAR, and A3AR, respectively (Table 1) [19]. In functional assays, CPI-444 showed an IC50 of 70 nM in the G protein-mediated cAMP assay [19]. In multiple murine tumor models, including the MC-38 and CT-26 colon tumors, the B16F10 melanoma and the RENCA renal cell cancer model, CPI-444 induced antitumor immune responses, and suppressed tumor growth as a single agent; it also augmented the efficacy of anti-PD-1/PD-L1 and anti-CTLA-4 agents [19]. CPI-444 was also reported to decrease the expression of multiple checkpoint pathways and to improve T cell infiltration and effector functions in MC-38, CT-26, and B16OVA tumor models [37]. In February 2015, CPI-444 was licensed by Corvus Pharmaceuticals for further development of therapeutic applications.
A clinical study of CPI-444 to evaluate its pharmacokinetics has been completed (NCT03237988), and a phase I/Ib study evaluating the safety and clinical activity of CPI-444 alone and in combination with anti-PD-1 atezolizumab in patients with advanced solid tumors was initiated in 2016 (NCT02655822). In April 2018, Corvus sponsored a study of CPI-006, an anti-CD73 mAb, alone and in combination with CPI-444 and pembrolizumab for patients with advanced cancers (NCT03454451). A phase Ib study of CPI-444 as a single agent and in combination with the anti-CD38 antibody daratumumab in relapsed or refractory multiple myeloma was initiated in February 2020 (NCT04280328). Results from these trials should be available in the near future.
3.8. AB-928
AB-928 is an A2AAR and A2BAR dual antagonist discovered by Arcus Biosciences, which inhibits A2AAR and A2BAR with similar potencies (Ki = 1.5 and 2.0 nM, respectively) and with 42- and 326-fold selectivity against A1AR and A3AR, respectively (Table 1) [42]. Given that both A2AAR and A2BAR play important roles in the immunosuppression by adenosine, dual antagonists may have better anticancer effects [4]. In a preclinical model of AT-3-OVA-bearing mice, AB-928 combined with chemotherapy by doxorubicin or oxaliplatin resulted in a significant reduction in the tumor growth rate when compared to chemotherapy alone [65]. In healthy volunteers, AB-928 was well tolerated up to the highest dose tested (200 mg once daily) and did not affect any physiologic parameters that are potentially sensitive to adenosine inhibition [66]. Researchers found that plasma levels of over 1 μM of the compound were associated with over 90% adenosine receptor inhibition [66]. These data resulted in further clinical development of oral AB-928 in cancer patients [66].
A number of clinical trials have been initiated to study the anticancer effects of AB-928 in combination with other agents. For example, a phase I study to evaluate the safety and tolerability of AB-928 in combination with the anti-PD-1 antibody zimberelimab in patients with advanced malignancies was started in August 2018 (NCT03629756), and a phase I study to evaluate the clinical efficacy of AB-928 combined with the drug combination “mFOLFOX” in participants with advanced metastatic gastroesophageal cancer was started in October 2018 (NCT03720678). In January 2020, a phase II study to evaluate AB-928 in combination with the anti-PD-1 zimberelimab or with AB-154 (a humanized mAb targeting human T cell immune-receptor with Ig and ITIM domains (TIGIT)) in participants with PD-L1-positive NSCLC, was started (NCT04262856). In June 2020, a phase Ib/II study to evaluate the antitumor activity and safety of an AB-928-based combination therapy in participants with metastatic castrate resistant prostate cancer in which AB-928 was combined with zimberelimab, enzalutamide, docetaxel, and the anti-CD73 antibody AB-680 was started (NCT04381832). Data from these trials should reveal the efficacy of A2AAR and A2BAR dual antagonists in cancer immunotherapy.
3.9. EOS-100850
EOS-100850 is an A2AAR antagonist reported by iTeos Therapeutics for which the chemical structure has not been disclosed as yet. Based on a patent publication by iTeos [67], EOS-100850 may contain a novel core structure (5-aminothiazolo[5,4-e][1,2,4]triazolo[1,5-c]pyrimidin-2(3H)-one), which would be similar to the tricyclic structure of A2AAR antagonists, such as SCH-58261 and MK-3814. Little has been published regarding the preclinical profiling of this compound. A phase I/Ib first-in-human study of EOS-100850 in patients with advanced solid tumors was started in January 2019 (NCT03873883). Clinical studies of EOS-100850 in combination with other agents in patients with melanoma, prostate cancer, and triple-negative breast cancer have also been planned [68].
4. Binding Modes of Antagonists in Complexes with A2AAR
For four of the antagonists presented in Figure 2 and Table 1, crystal structures of complexes with A2AAR are available. These structures greatly contribute to the understanding of the binding modes of A2AAR ligands and, therefore, help with the design of novel lead compounds for development of drugs that target A2AAR. This section surveys the available structures.
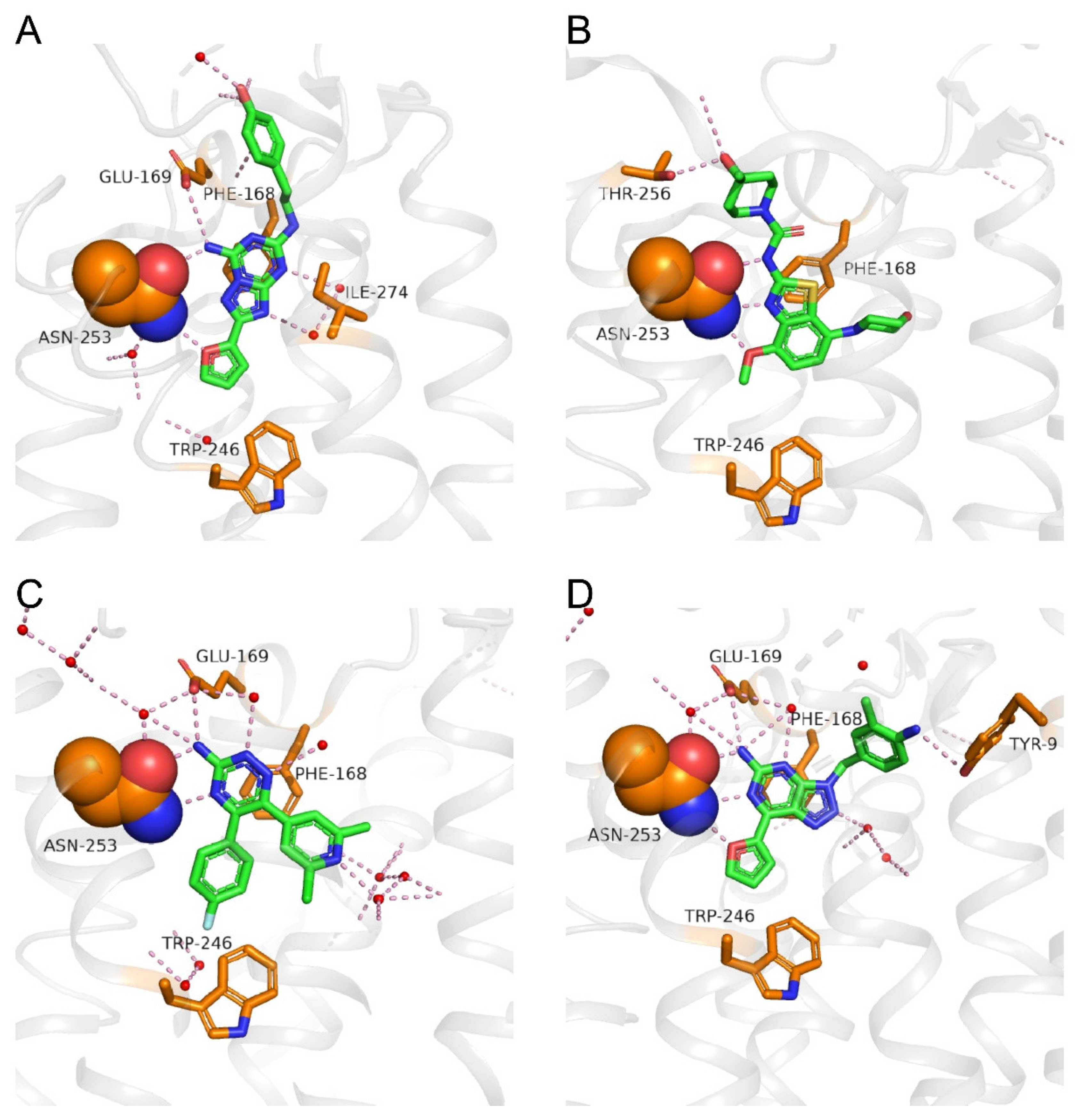
A crystal structure of ZM-241385 bound to A2AAR solved in 2008 (PDB: 3EML) [20] showed the inactive conformation of the receptor (Figure 3A). ZM-241385 binds to the receptor in an extended conformation, with the furan head inserting deep into the binding pocket and the phenol tail located at the entry of the orthosteric binding site [20]. The ligand-binding pocket is bottomed by Trp2466.48, and ZM-241385 is anchored by an aromatic stacking interaction with Phe1685.29. The furan ring forms a hydrogen bond with the side chain NH2 group of Asn2536.55, and the NH2 group on the bicyclic triazolotriazine core of ZM-241385 forms hydrogen bonds with the side chain C=O groups of Asn2536.55 and Glu1695.30. The tail phenol group of ZM-241385 forms a hydrogen bond with a water molecule at the pocket entry. In a different ZM-241385-bound A2AAR structure solved in 2011, a salt bridge between His264ECL3 and Glu169 is broken (not shown in Figure 3A), and the tail phenol group points to a side pocket instead of the pocket entry (PDB: 3PWH) [69].
A crystal structure of SYN-115 bound to A2AAR reported in 2018 (PDB: 5OLO) [70] shows that SYN-115 fits into a similar pocket to that occupied by ZM-241385 (Figure 3A,B). There is an aromatic stacking interaction between the benzothiazole core of SYN-115 and residue Phe1685.29. Three hydrogen bonds could be seen between the ligand and the side chain of Asn2536.55 in which the oxygen of the methoxy group and the nitrogen of the thiazole ring interact with the side chain NH2 group of Asn2536.55 and act as hydrogen bond acceptors, while the NH of SYN-115 forms a hydrogen bond with the side chain C=O group of Asn2536.55. Similar to ZM-241385, the hydroxyl tail of SYN-115 points toward the pocket entry, where it forms a hydrogen bond with Thr2566.58.
Crystal structures of AZD-4635 analogs (3-amino-1,2,4-triazine compounds) bound to the A2AAR reported in 2012 [71], and a more recently presented higher resolution structure of AZD-4635-bound A2AAR (PDB: 6GT3) show that AZD-4635 lies deep in the binding pocket and occupies a region next to a cluster of “unhappy” waters (Figure 3C). Unlike ZM-241385 or SYN-115, AZD-4635 does not have a “tail” group that would reach out to the pocket entry. Hydrogen bonds are observed between the triazine nitrogen at position 4 and the side chain NH2 group of Asn2536.55 and between the NH2 group on the triazine ring and the side chain C=O groups of Asn2536.55 and Glu1695.30.
A crystal structure of compound V-2006 bound to A2AAR (PDB: 5OLH) [70] shows that the bicyclic core and the furan substituent of V-2006 share very similar interactions to those of ZM-241385, namely the aromatic stacking with Phe1685.29 and the hydrogen bonds with Asn2536.55 and Glu1695.30 (Figure 3D). Unlike ZM-241385, the 2-methylaniline group of V-2006 does not point to the pocket entry but fits into a side pocket, where it interacts with Tyr91.35.
Overall, although the chemical structures of the four A2AAR antagonists (Figure 2) are quite different, they are buried in the orthosteric pocket of A2AAR, where they show many common interactions with key residues in the pocket (Figure 3). The basal region of the orthosteric site is delimited by Trp2466.48, which engages in Van der Waals contacts to aromatic substituents, such as furan (ZM-241385 and V-2006) and benzene (AZD-4635). The aromatic stacking interaction with Phe1685.29 and the hydrogen bonds with Asn2536.55 are common interactions for all four A2AAR antagonists. When a –NH2 group is attached to the bicyclic or tricyclic core of A2AAR antagonists (Figure 2), a hydrogen bond with Glu1695.30 is often seen. Additional studies of crystal structures of A2AAR complexes with different ligands and their binding kinetics [23,52,72,73,74] also contribute to the foundation for the discovery of new chemotypes of A2AAR antagonists, either through virtual screening approaches or structure-based drug design.
5. Conclusions and Future Perspectives
In the tumor microenvironment, adenosine suppresses the antitumor activity of effector T cells and other cytotoxic immune cells mainly through its action on A2AAR. Blocking A2AAR thus has the potential to markedly enhance antitumor immunity. Preclinical in vivo animal studies have demonstrated efficacy of A2AAR antagonists in tumor immunotherapy, providing incentives to develop A2AAR antagonists for use in immuno-oncology.
The complex pathogenesis of all cancerous growths makes it challenging to achieve efficacious and lasting therapeutic effects for single-drug therapies, mainly because of imminent drug resistance. To obtain more effective antitumor effects, drug combinations are commonly applied. This is also true for A2AAR antagonists. In most preclinical models, A2AAR antagonists show only mild tumor-suppressing effects as stand-alone agents, while significantly better results could be achieved when A2AAR antagonists are used in combination with other antitumor agents. In clinical trials, A2AAR antagonists are most often tested in combination with other check-point inhibitors (in most cases anti-PD-1/PD-L1 or anti-CTLA-4 antibodies) as well as with chemotherapies. Ongoing studies will reveal novel combination regimens, improved dosing and timing of interventions in different tumor types and patient subpopulations.
Based on the prominent role of A2AAR in immuno-oncology, new antagonist chemotypes are being developed based on structural information on ligand binding modes as well as reported structure–activity relationships. Because of the high adenosine concentrations in the TME, high binding affinity is necessary for A2AAR antagonist drug candidates. Target selectivity is of course also very important, especially versus A1AR and A3AR because of their opposite downstream effects on the regulation of cAMP levels. Selectivity versus A2BAR is a quite different issue, as illustrated by the fact that dual A2AAR and A2BAR antagonists are also being developed. Finally, considering that otherwise potentially promising A2AAR antagonists had to be abandoned in drug-discovery projects because of poor physicochemical and pharmacokinetic properties, multi-parametric optimization of lead compounds is yet another avenue to be further explored in the development of clinically useful A2AAR antagonists.
Author Contributions
Conceptualization, J.C.; writing—original draft preparation, J.Z.; writing—review and editing, J.Z., W.Y., W.D., K.W., J.C. All authors have read and agreed to the published version of the manuscript.
Funding
The APC was funded by the Shanghai Municipal Government and ShanghaiTech University.
Acknowledgments
The authors would like to thank the Shanghai Municipal Government and ShanghaiTech University for financial support. K.W. is the Cecil H. and Ida M. Green Professor at Scripps Research.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Linden, J.; Müller, C.E. International union of basic and clinical pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—An update. Pharm. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Shook, B.C.; Jackson, P.F. Adenosine A2A receptor antagonists and Parkinson’s disease. ACS Chem. Neurosci. 2011, 2, 555–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klinger, M.; Freissmuth, M.; Nanoff, C. Adenosine receptors: G protein-mediated signalling and the role of accessory proteins. Cell Signal. 2002, 14, 99–108. [Google Scholar] [CrossRef]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709–724. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Müller, C.E. Medicinal chemistry of adenosine, P2Y and P2X receptors. Neuropharmacology 2016, 104, 31–49. [Google Scholar] [CrossRef] [Green Version]
- Waarde, A.; Dierckx, R.; Zhou, X.; Khanapur, S.; Tsukada, H.; Ishiwata, K.; Luurtsema, G.; de Vries, E.F.J.; Elsinga, P.H. Potential therapeutic applications of adenosine A2A receptor ligands and opportunities for A2A receptor imaging. Med. Res. Rev. 2018, 38, 5–56. [Google Scholar] [CrossRef]
- Garnock-Jones, K.P.; Curran, M.P. Regadenoson. Am. J. Cardiovasc. Drugs 2011, 10, 65–71. [Google Scholar] [CrossRef]
- Hage, F.G.; Ghimire, G.; Lester, D.; McKay, J.; Bleich, S.; El-Hajj, S.; Iskandrian, A.E. The prognostic value of regadenoson myocardial perfusion imaging. J. Nucl. Cardiol. 2015, 22, 1214–1221. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.Y.; Zhang, X.H.; Zhen, X.C. Development of adenosine A2A receptor antagonists for the treatment of Parkinson’s disease: A recent update and challenge. ACS Chem. Neurosci. 2019, 10, 783–791. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Gao, Z.G.; Matricon, P.; Eddy, M.T.; Carlsson, J. Adenosine A2A receptor antagonists: From caffeine to selective non-xanthines. Br. J. Pharmacol. 2020, in press. [Google Scholar] [CrossRef]
- Pinna, A. Adenosine A2A receptor antagonists in Parkinson’s disease: Progress in clinical trials from the newly approved istradefylline to drugs in early development and those already discontinued. CNS Drugs 2014, 28, 455–474. [Google Scholar] [CrossRef] [PubMed]
- Dungo, R.; Deeks, E.D. Istradefylline: First global approval. Drugs 2013, 73, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Wolberg, G.; Zimmerman, T.P.; Hiemstra, K.; Winston, M.; Chu, L. Adenosine inhibition of lymphocyte-mediated cytolysis: Possible role of cyclic adenosine monophosphate. Science 1975, 187, 957–959. [Google Scholar] [CrossRef] [PubMed]
- Blay, J.; White, T.D.; Hoskin, D.W. The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res. 1997, 57, 2602–2605. [Google Scholar] [PubMed]
- Ohta, A.; Sitkovsky, M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 2001, 414, 916–919. [Google Scholar] [CrossRef]
- Ohta, A.; Elieser Gorelik, E.; Prasad, S.J.; Ronchese, F.; Lukashev, D.; Wong, M.K.K.; Huang, X.; Caldwell, S.; Liu, K.; Smith, P.; et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13132–13137. [Google Scholar] [CrossRef] [Green Version]
- Hatfield, S.M.; Sitkovsky, M. A2A adenosine receptor antagonists to weaken the hypoxia-HIF-1alpha driven immunosuppression and improve immunotherapies of cancer. Curr. Opin. Pharmacol. 2016, 29, 90–96. [Google Scholar] [CrossRef] [Green Version]
- Mediavilla-Varela, M.; Castro, J.; Chiappori, A.; Noyes, D.; Hernandez, D.C.; Allard, B.; Stagg, J.; Antonia, S.J. A novel antagonist of the immune checkpoint protein adenosine A2A receptor restores tumor-infiltrating lymphocyte activity in the context of the tumor microenvironment. Neoplasia 2017, 19, 530–536. [Google Scholar] [CrossRef]
- Willingham, S.B.; Ho, P.Y.; Hotson, A.; Hill, C.; Piccione, E.C.; Hsieh, J.; Liu, L.; Buggy, J.J.; McCaffery, I.; Miller, R.A. A2AR antagonism with CPI-444 induces antitumor responses and augments efficacy to anti-PD-L1 and anti-CTLA-4 in preclinical models. Cancer Immunol Res. 2018, 6, 1136–1149. [Google Scholar] [CrossRef] [Green Version]
- Jaakola, V.; Griffith, M.T.; Hanson, M.A.; Cherezov, V.; Chien, E.Y.T.; Lane, J.R.; IJzerman, A.P.; Stevens, R.C. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 2008, 322, 1211–1216. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Wu, H.; Katritch, V.; Han, G.W.; Jacobson, K.A.; Gao, Z.G.; Cherezov, V.; Stevens, R.C. Structure of an agonist-bound human A2A adenosine receptor. Science 2011, 332, 322–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, B.; Nehme, R.; Warne, T.; Leslie, A.G.; Tate, C.G. Structure of the adenosine A2A receptor bound to an engineered G protein. Nature 2016, 536, 104–107. [Google Scholar] [CrossRef]
- Susac, L.; Eddy, M.T.; Didenko, T.; Stevens, R.C.; Wüthrich, K. A2A adenosine receptor functional states characterized by 19F-NMR. Proc. Natl. Acad. Sci. USA 2018, 115, 12733–12738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eddy, M.T.; Lee, M.Y.; Gao, Z.G.; White, K.L.; Didenko, T.; Horst, R.; Audet, M.; Stanczak, P.; McClary, K.M.; Han, G.W.; et al. Allosteric coupling of drug binding and intracellular signaling in the A2A adenosine receptor. Cell 2018, 172, 68–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasko, G.; Linden, J.; Cronstein, B.; Pacher, P. Adenosine receptors: Therapeutic aspects for inflammatory and immune diseases. Nat. Rev. Drug Discov. 2008, 7, 759–770. [Google Scholar] [CrossRef]
- Moesta, A.K.; Li, X.Y.; Smyth, M.J. Targeting CD39 in cancer. Nat. Rev. Immunol. 2020. [Google Scholar] [CrossRef]
- Colgan, S.P.; Eltzschig, H.K.; Eckle, T.; Thompson, L.F. Physiological roles for ecto-5’-nucleotidase (CD73). Purinergic. Signal. 2006, 2, 351–360. [Google Scholar] [CrossRef] [Green Version]
- Young, A.; Ngiow, S.F.; Madore, J.; Reinhardt, J.; Landsberg, J.; Chitsazan, A.; Rautela, J.; Bald, T.; Barkauskas, D.S.; Ahern, E.; et al. Targeting adenosine in BRAF-mutant melanoma reduces tumor growth and metastasis. Cancer Res. 2017, 77, 4684–4696. [Google Scholar] [CrossRef] [Green Version]
- Kazemi, M.H.; Mohseni, S.R.; Hojjat-Farsangi, M.; Anvari, E.; Ghalamfarsa, G.; Mohammadi, H.; Jadidi-Niaragh, F. Adenosine and adenosine receptors in the immunopathogenesis and treatment of cancer. J. Cell Physiol. 2018, 233, 2032–2057. [Google Scholar] [CrossRef] [Green Version]
- Beavis, P.A.; Divisekera, U.; Paget, C.; Chow, M.T.; John, L.B.; Devaud, C.; Dwyer, K.; Stagg, J.; Smyth, M.J.; Darcy, P.K. Blockade of A2A receptors potently suppresses the metastasis of CD73+ tumors. Proc. Natl. Acad. Sci. USA 2013, 110, 14711–14716. [Google Scholar] [CrossRef] [Green Version]
- Ohta, A.; Kini, R.; Ohta, A.; Subramanian, M.; Madasu, M.; Sitkovsky, M. The development and immunosuppressive functions of CD4+ CD25+ FoxP3+ regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front. Immunol. 2012, 3, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorrentino, C.; Miele, L.; Porta, A.; Pinto, A.; Morello, S. Myeloid-derived suppressor cells contribute to A2B adenosine receptor-induced VEGF production and angiogenesis in a mouse melanoma model. Oncotarget 2015, 6, 27478–27489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmada, A.; Ahmada, S.; Gloverb, L.; Millera, S.M.; Shannonc, J.M.; Guoa, X.; Franklind, W.A.; Bridgesc, J.P.; Schaacke, J.B.; Colganb, S.P.; et al. Adenosine A2A receptor is a unique angiogenic target of HIF-2α in pulmonary endothelial cells. Proc. Natl. Acad. Sci. USA 2009, 106, 10684–10689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kjaergaard, J.; Hatfield, S.; Jones, G.; Ohta, A.; Sitkovsky, M. A2A adenosine receptor gene deletion or synthetic A2A antagonist liberate tumor-reactive CD8+ T Cells from tumor-induced immunosuppression. J. Immunol. 2018, 201, 782–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Congreve, M.; Brown, G.A.; Borodovsky, A.; Lamb, M.L. Targeting adenosine A2A receptor antagonism for treatment of cancer. Expert Opin. Drug Discov. 2018, 13, 997–1003. [Google Scholar] [CrossRef]
- Beavis, P.A.; Milenkovski, N.; Henderson, M.A.; John, L.B.; Allard, B.; Loi, S.; Kershaw, M.H.; Stagg, J.; Darcy, P.K. Adenosine receptor 2A blockade increases the efficacy of anti-PD-1 through enhanced antitumor T-cell responses. Cancer Immunol. Res. 2015, 3, 506–517. [Google Scholar] [CrossRef] [Green Version]
- Leone, R.D.; Sun, I.M.; Oh, M.H.; Sun, I.H.; Wen, J.; Englert, J.; Powell, J.D. Inhibition of the adenosine A2A receptor modulates expression of T cell coinhibitory receptors and improves effector function for enhanced checkpoint blockade and ACT in murine cancer models. Cancer Immunol. Immunother. 2018, 67, 1271–1284. [Google Scholar] [CrossRef]
- Gao, Z.G.; Jacobson, K.A. A2B adenosine receptor and cancer. Int. J. Mol. Sci. 2019, 20, 5139. [Google Scholar] [CrossRef] [Green Version]
- Leone, R.D.; Lo, Y.C.; Powell, J.D. A2AR antagonists: Next generation checkpoint blockade for cancer immunotherapy. Comput. Struct. Biotechnol. J. 2015, 13, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Cekic, C.; Linden, J. Adenosine A2A receptors intrinsically regulate CD8+ T cells in the tumor microenvironment. Cancer Res. 2014, 74, 7239–7249. [Google Scholar] [CrossRef] [Green Version]
- Sepulveda, C.; Palomo, I.; Fuentes, E. Role of adenosine A2B receptor overexpression in tumor progression. Life Sci. 2016, 166, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Walters, M.J.; Tan, J.B.; Becker, A.; Yi, F.F.; Park, T.; Leleti, M.R.; Rosen, B.; Sharif, E.; Debien, L.; Young, S.; et al. Characterization of the potent and selective A2AR antagonist AB928 for the treatment of cancer. Cancer Res. 2017, 77, AM2017–AM4572. [Google Scholar]
- Mittal, D.; Young, A.; Stannard, K.; Yong, M.; Teng, M.W.; Allard, B.; Stagg, J.; Smyth, M.J. Antimetastatic effects of blocking PD-1 and the adenosine A2A receptor. Cancer Res. 2014, 74, 3652–3658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoyama, S.; Ichimura, M.; Ikeda, K.; Ishii, A.; Kanda, T.; Koga, K.; Koike, N.; Kurokawa, M.; Kuwana, Y.; Mori, A.; et al. Progress in pursuit of therapeutic A2A antagonists: The adenosine A2A receptor selective antagonist KW-6002: Research and development toward a novel nondopaminergic therapy for Parkinson’s disease. Neurology 2003, 61, S97–S100. [Google Scholar] [CrossRef]
- Poucher, S.M.; Keddie, J.R.; Singh, P.; Stoggall, S.M.; Caulkett, P.W.R.; Jones, G.; Collis, M.G. The in vitro pharmacology of ZM-241385, a potent, non-xanthine, A2A selective adenosine receptor antagonist. Br. J. Pharmacol. 1995, 115, 1096–1102. [Google Scholar] [CrossRef] [Green Version]
- De Zwart, M.; Vollinga, R.C.; Beukers, M.W.; Sleegers, D.F.; von Frijtag Drabbe Künze, J.K.; de Groote, M.; IJzerman, A.P. Potent antagonists for the human adenosine A2B receptor. Derivatives of the triazolotriazine adenosine receptor antagonist ZM-241385 with high affinity. Drug Develop. Res. 1999, 48, 95–103. [Google Scholar] [CrossRef]
- Ongini, E.; Dionisotti, S.; Irenius, S.G.E.; Fredholm, B.B. Comparison of CGS-5943, ZM-241385 and SCH-58261 as antagonists at human adenosine receptors. Naunyn Schmiedeberg’s Arch. Pharmacol. 1999, 359, 7–10. [Google Scholar] [CrossRef]
- Neustadt, B.R.; Hao, J.; Lindo, N.; Greenlee, W.J.; Stamford, A.W.; Tulshian, D.; Ongini, E.; Hunter, J.; Monopoli, A.; Bertorelli, R.; et al. Potent, selective, and orally active adenosine A2A receptor antagonists: Arylpiperazine derivatives of pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidines. Bioorg. Med. Chem. Lett. 2007, 17, 1376–1380. [Google Scholar] [CrossRef]
- Flohr, A.; Moreau, J.; Poli, S.M.; Riemer, C.; Steward, L. 4-Hydroxy-4-methyl-piperidine-1-carboxylic acid (4-methoxy-7-morpholin-4-yl-benzothiazol-2-yl)-amide. US Patent 20,050,261,289, 24 November 2005. [Google Scholar]
- Borodovsky, A.; Wang, Y.; Ye, M.; Shaw, J.C.; Sachsenmeier, K.F.; Deng, N.; DelSignore, K.J.; Fretland, A.J.; Clarke, J.D.; Goodwin, R.J.; et al. Abstract 5580: Preclinical pharmacodynamics and antitumor activity of AZD-4635, a novel adenosine 2A receptor inhibitor that reverses adenosine mediated T cell suppression. AACR Annu. Meet. 2017. [Google Scholar] [CrossRef]
- Gillespie, R.J.; Bamford, S.J.; Botting, R.; Comer, M.; Denny, S.; Gaur, S.; Griffin, M.; Jordan, A.M.; Knight, A.R.; Lerpiniere, J.; et al. Antagonists of the human A2A adenosine receptor. 4. Design, synthesis, and preclinical evaluation of 7-aryltriazolo[4,5-d]pyrimidines. J. Med. Chem. 2009, 52, 33–47. [Google Scholar] [CrossRef]
- Uustare, A.; Vonk, A.; Terasmaa, A.; Fuxe, K.; Rinken, A. Kinetic and functional properties of [3H] ZM-241385, a high affinity antagonist for adenosine A2A receptors. Life Sci. 2005, 76, 1513–1526. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.J.; Rhee, S.Y.; Ryu, J.H.; Cheong, J.H.; Kwon, K.; Yang, S.I.; Park, S.H.; Lee, J.; Kim, H.Y.; Han, S.H.; et al. Activation of adenosine A2A receptor up-regulates BDNF expression in rat primary cortical neurons. Neurochem. Res. 2011, 36, 2259–2269. [Google Scholar] [CrossRef]
- Da Rocha Lapa, F.; da Silva, M.D.; de Almeida Cabrini, D.; Santos, A.R. Anti-inflammatory effects of purine nucleosides, adenosine and inosine, in a mouse model of pleurisy: Evidence for the role of adenosine A2 receptors. Purinergic Signal. 2012, 8, 693–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannone, R.; Miele, L.; Maiolino, P.; Pinto, A.; Morello, S. Adenosine limits the therapeutic effectiveness of anti-CTLA4 mAb in a mouse melanoma model. Am. J. Cancer. Res. 2014, 4, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Keddie, J.R.; Poucher, S.M.; Shaw, G.R.; Brooks, R.; Collis, M.G. In vivo characterisation of ZM-241385, a selective adenosine A2A receptor antagonist. Eur. J. Pharmacol. 1996, 301, 107–113. [Google Scholar] [CrossRef]
- Yang, M.; Soohoo, D.; Soelaiman, S.; Kalla, R.; Zablocki, J.; Chu, N.; Leung, K.; Yao, L.; Diamond, I.; Belardinelli, L.; et al. Characterization of the potency, selectivity, and pharmacokinetic profile for six adenosine A2A receptor antagonists. Naunyn Schmiedebergs Arch. Pharmacol. 2007, 375, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Kiselgof, E.; Tulshian, D.B.; Arik, L.; Zhang, H.; Fawzi, A. 6-(2-Furanyl)-9H-purin-2-amine derivatives as A2A adenosine antagonists. Bioorg. Med. Chem. Lett. 2005, 15, 2119–2122. [Google Scholar] [CrossRef]
- Loi, S.; Pommey, S.; Haibe-Kains, B.; Beavis, P.A.; Darcy, P.K.; Smyth, M.J.; Stagg, J. CD73 promotes anthracycline resistance and poor prognosis in triple negative breast cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 11091–11096. [Google Scholar] [CrossRef] [Green Version]
- Hodgson, R.A.; Bertorelli, R.; Varty, G.B.; Lachowicz, J.E.; Forlani, A.; Fredduzzi, S.; Cohen-Williams, M.E.; Higgins, G.A.; Impagnatiello, F.; Nicolussi, E.; et al. Characterization of the potent and highly selective A2A receptor antagonists preladenant and SCH-412348 [7-[2-[4-2,4-difluorophenyl]-1-piperazinyl]ethyl]-2-(2-furanyl)-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-amine] in rodent models of movement disorders and depression. J. Pharmacol. Exp. Ther. 2009, 330, 294–303. [Google Scholar] [CrossRef] [Green Version]
- Nunez, F.; Taura, J.; Camacho, J.; Lopez-Cano, M.; Fernandez-Duenas, V.; Castro, N.; Castro, J.; Ciruela, F. PBF509, an adenosine A2A receptor antagonist with efficacy in rodent models of movement disorders. Front. Pharmacol. 2018, 9, 1200. [Google Scholar] [CrossRef] [Green Version]
- Chiappori, A.; Williams, C.; Creelan, B.; Tanvetyanon, T.; Gray, J.; Haura, E.; Chen, D.T.; Thapa, R.; Beg, A.; Boyle, T.; et al. Phase I/II study of the A2AR antagonist NIR178 (PBF-509), an oral immunotherapy, in patients (pts) with advanced NSCLC. J. Thorac. Oncol. 2018, 13. [Google Scholar] [CrossRef] [Green Version]
- Hauser, R.A.; Olanow, C.W.; Kieburtz, K.D.; Pourcher, E.; Docu-Axelerad, A.; Lew, M.; Kozyolkin, O.; Neale, A.; Resburg, C.; Meya, U.; et al. Tozadenant (SYN115) in patients with Parkinson’s disease who have motor fluctuations on levodopa: A phase 2b, double-blind, randomised trial. Lancet. Neurol. 2014, 13, 767–776. [Google Scholar] [CrossRef]
- Bamford, S.J.; Gillespie, R.J.; Todd, R.S. Triazolo[4,5-d]pyramidine derivatives and their use as purine receptor antagonists. WO2,009,156,737, 30 December 2009. [Google Scholar]
- Schindler, U.; Seitz, L.; Ashok, D.; Piovesan, D.; Tan, J.; DiRenzo, D.; Yin, F.; Leleti, M.; Rosen, B.; Miles, D.; et al. AB928, a dual antagonist of the A2AR and A2BR adenosine receptors, leads to greater immune activation and reduced tumor growth when combined with chemotherapy. Eur. J. Cancer 2018, 92, S14–S15. [Google Scholar] [CrossRef]
- Seitz, L.; Jin, L.; Leleti, M.; Ashok, D.; Jeffrey, J.; Rieger, A.; Tiessen, R.G.; Arold, G.; Tan, J.B.L.; Powers, J.P.; et al. Safety, tolerability, and pharmacology of AB928, a novel dual adenosine receptor antagonist, in a randomized, phase 1 study in healthy volunteers. Investig. New Drugs 2019, 37, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Crosignani, S.; Dickinson, P.; de Matas, M.; Houthuys, E.J.K.H.; Marillier, R.G.; Martinoli, C.; de Henau, O.; Deriessens, G. Thiocarbamate derivatives as A2A inhibitors, pharmaceutical composition thereof and combinations with anticancer agents. WO2,020,053,263, 19 March 2020. [Google Scholar]
- iTeos Therapeutics. Available online: https://www.iteostherapeutics.com/ (accessed on 14 August 2020).
- Dore, A.S.; Robertson, N.; Errey, J.C.; Ng, I.; Hollenstein, K.; Tehan, B.; Hurrell, E.; Bennett, K.; Congreve, M.; Magnani, F.; et al. Structure of the adenosine A2A receptor in complex with ZM-241385 and the xanthines XAC and caffeine. Structure 2011, 19, 1283–1293. [Google Scholar] [CrossRef] [Green Version]
- Rucktooa, P.; Cheng, R.K.Y.; Segala, E.; Geng, T.; Errey, J.C.; Brown, G.A.; Cooke, R.M.; Marshall, F.H.; Dore, A.S. Towards high throughput GPCR crystallography: In meso soaking of adenosine A2A receptor crystals. Sci. Rep. 2018, 8, 41. [Google Scholar] [CrossRef] [Green Version]
- Congreve, M.; Andrews, S.P.; Dore, A.S.; Hollenstein, K.; Hurrell, E.; Langmead, C.J.; Mason, J.S.; Ng, I.W.; Tehan, B.; Zhukov, A.; et al. Discovery of 1,2,4-triazine derivatives as adenosine A2A antagonists using structure based drug design. J. Med. Chem. 2012, 55, 1898–1903. [Google Scholar] [CrossRef]
- Guo, D.; Xia, L.; van Veldhoven, J.P.; Hazeu, M.; Mocking, T.; Brussee, J.; Ijzerman, A.P.; Heitman, L.H. Binding kinetics of ZM-241385 derivatives at the human adenosine A2A receptor. Chem. Med. Chem. 2014, 9, 752–761. [Google Scholar] [CrossRef]
- Sun, B.; Bachhawat, P.; Chu, M.L.; Wood, M.; Ceska, T.; Sands, Z.A.; Mercier, J.; Lebon, F.; Kobilka, T.S.; Kobilka, B.K. Crystal structure of the adenosine A2A receptor bound to an antagonist reveals a potential allosteric pocket. Proc. Natl. Acad. Sci. USA 2017, 114, 2066–2071. [Google Scholar] [CrossRef] [Green Version]
- Igonet, S.; Raingeval, C.; Cecon, E.; Pucic-Bakovic, M.; Lauc, G.; Cala, O.; Baranowski, M.; Perez, J.; Jockers, R.; Krimm, I.; et al. Enabling STD-NMR fragment screening using stabilized native GPCR: A case study of adenosine receptor. Sci. Rep. 2018, 8, 8142. [Google Scholar] [CrossRef]
Figure 1.
Adenosine-A2AAR signaling in the tumor microenvironment (the green circles represent adenosine molecules). ATP = adenosine triphosphate; AMP = adenosine monophosphate.
Figure 1.
Adenosine-A2AAR signaling in the tumor microenvironment (the green circles represent adenosine molecules). ATP = adenosine triphosphate; AMP = adenosine monophosphate.

Figure 2.
Chemical structures of A2AAR antagonists discussed in this chapter.

Figure 3.
Binding modes of A2AAR with different antagonists. (A) ZM-241385; the key amino acid residues: Asn2536.55, Glu1695.30, Phe1685.29, Trp2466.48, and Ile2747.39 are identified (PDB ID: 3EML). (B) SYN-115; the key amino acid residues: Asn2536.55, Thr2566.58, Phe1685.29, and Trp2466.48 are identified (PDB ID: 5OLO). (C) AZD-4635; the key amino acid residues: Asn2536.55, Glu1695.30, Phe1685.29, and Trp2466.48 are identified (PDB ID: 6GT3). (D) V-2006; the key amino acid residues: Asn2536.55, Glu1695.30, Tyr91.35, Phe1685.29, and Trp2466.48 are identified (PDB ID: 5OLH). The A2AAR back bone is colored gray, and the amino acid side chains that interact with the ligands are shown as sticks and colored by element (carbon, yellow; nitrogen, blue; oxygen, red; sulfur, yellow; the side chain of Asn2536.55 is shown in a space filling presentation). The antagonists are shown as sticks and colored by element (carbon, green; nitrogen, blue; oxygen, red; sulfur, yellow). Polar contacts are presented as dashed lines, and water molecules are shown as red spheres.
Figure 3.
Binding modes of A2AAR with different antagonists. (A) ZM-241385; the key amino acid residues: Asn2536.55, Glu1695.30, Phe1685.29, Trp2466.48, and Ile2747.39 are identified (PDB ID: 3EML). (B) SYN-115; the key amino acid residues: Asn2536.55, Thr2566.58, Phe1685.29, and Trp2466.48 are identified (PDB ID: 5OLO). (C) AZD-4635; the key amino acid residues: Asn2536.55, Glu1695.30, Phe1685.29, and Trp2466.48 are identified (PDB ID: 6GT3). (D) V-2006; the key amino acid residues: Asn2536.55, Glu1695.30, Tyr91.35, Phe1685.29, and Trp2466.48 are identified (PDB ID: 5OLH). The A2AAR back bone is colored gray, and the amino acid side chains that interact with the ligands are shown as sticks and colored by element (carbon, yellow; nitrogen, blue; oxygen, red; sulfur, yellow; the side chain of Asn2536.55 is shown in a space filling presentation). The antagonists are shown as sticks and colored by element (carbon, green; nitrogen, blue; oxygen, red; sulfur, yellow). Polar contacts are presented as dashed lines, and water molecules are shown as red spheres.


{kind=link}
{kind=link}
{kind=link}
Table 1.
A2AAR antagonists and their affinity for adenosine receptors.
| Compound | Ki (nM) | References | |||
|---|---|---|---|---|---|
| A1AR | A2AAR | A2BAR | A3AR | ||
| KW-6002 | 9600 | 12 | 1800 | >3000 | [44] |
| ZM-241385 | 255 | 0.8 | 50 | >10,000 | [45,46] |
| SCH-58261 | 287 | 0.6 | 5011 | >10,000 | [47] |
| MK-3814 | >1000 | 1.1 | >1700 | >1000 | [48] |
| PBF-509 | 2500 | 12 | 1000 | 5000 | [18] |
| SYN-115 | 1350 | 5 | 700 | 1570 | [49] |
| AZD-4635 | 160 | 1.7 | 64 | >10,000 | [50] |
| V-2006 | 68 | 1.3 | 63 | 1005 | [51] |
| CPI-444 | 192 | 3.54 | 1528 | 2455 | [19] |
| AB-928 | 64 | 1.5 | 2.0 | 489 | [42] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhang, J.; Yan, W.; Duan, W.; Wüthrich, K.; Cheng, J. Tumor Immunotherapy Using A2A Adenosine Receptor Antagonists. Pharmaceuticals 2020, 13, 237. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13090237
AMA Style
Zhang J, Yan W, Duan W, Wüthrich K, Cheng J. Tumor Immunotherapy Using A2A Adenosine Receptor Antagonists. Pharmaceuticals. 2020; 13(9):237. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13090237
Chicago/Turabian StyleZhang, Jinfeng, Wenzhong Yan, Wenwen Duan, Kurt Wüthrich, and Jianjun Cheng. 2020. "Tumor Immunotherapy Using A2A Adenosine Receptor Antagonists" Pharmaceuticals 13, no. 9: 237. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13090237
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.