Vascular α1A Adrenergic Receptors as a Potential Therapeutic Target for IPAD in Alzheimer’s Disease


 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Post-Mortem Delay Does Not Adversely Affect AR Staining
2.2. The Overall Pattern of Immunocytochemistry for α1A-AR on the Cerebral Vessels Is Not Affected by Age
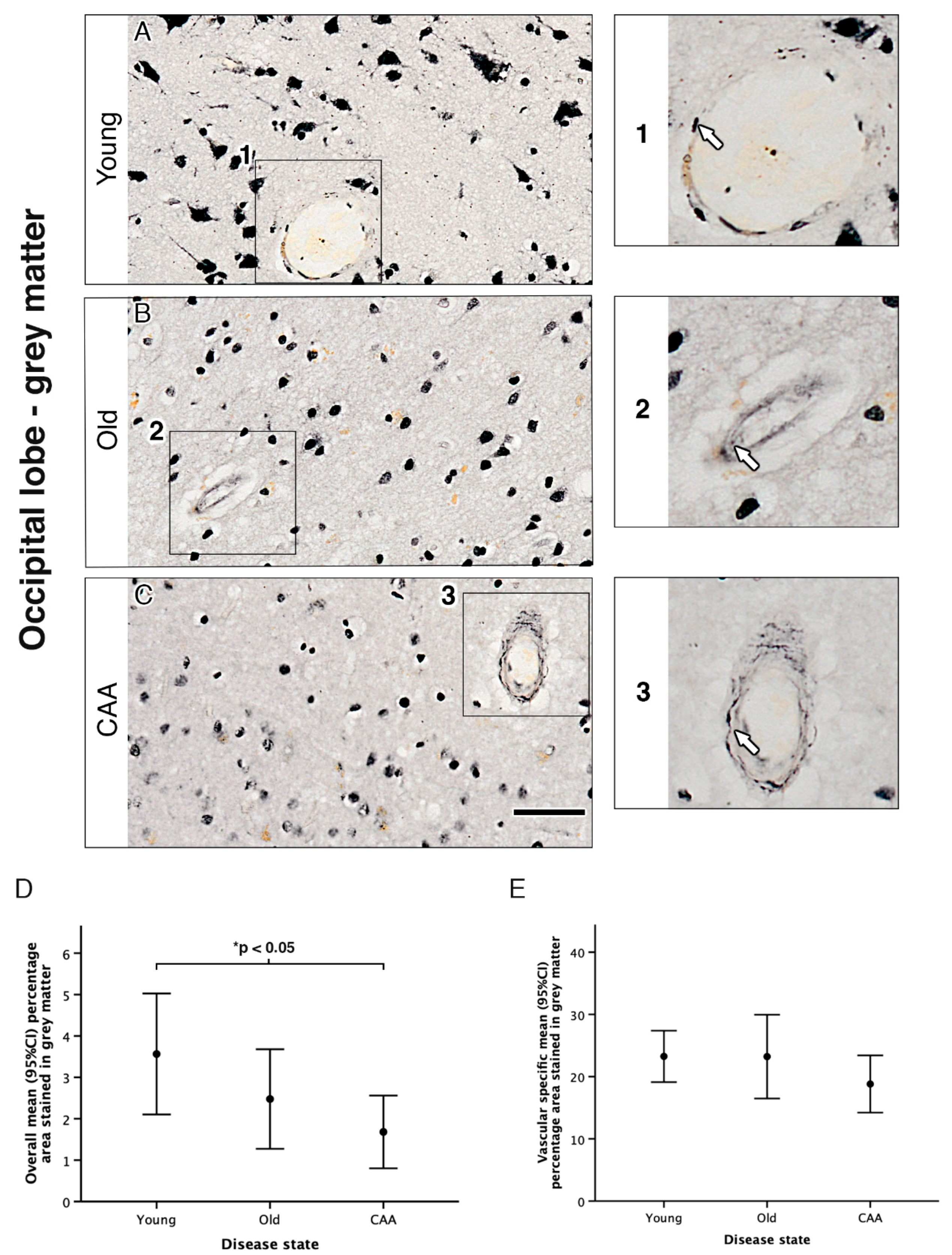
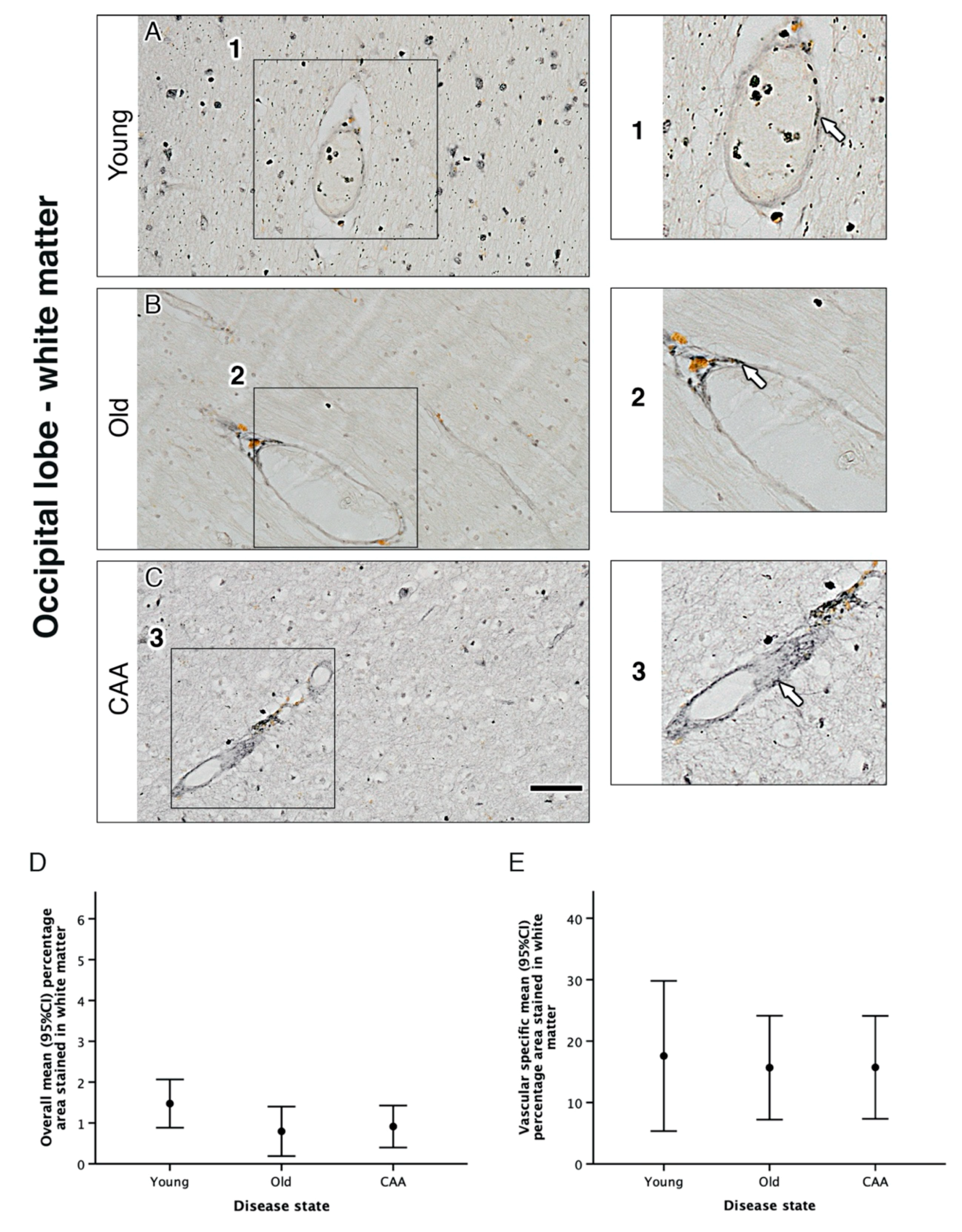
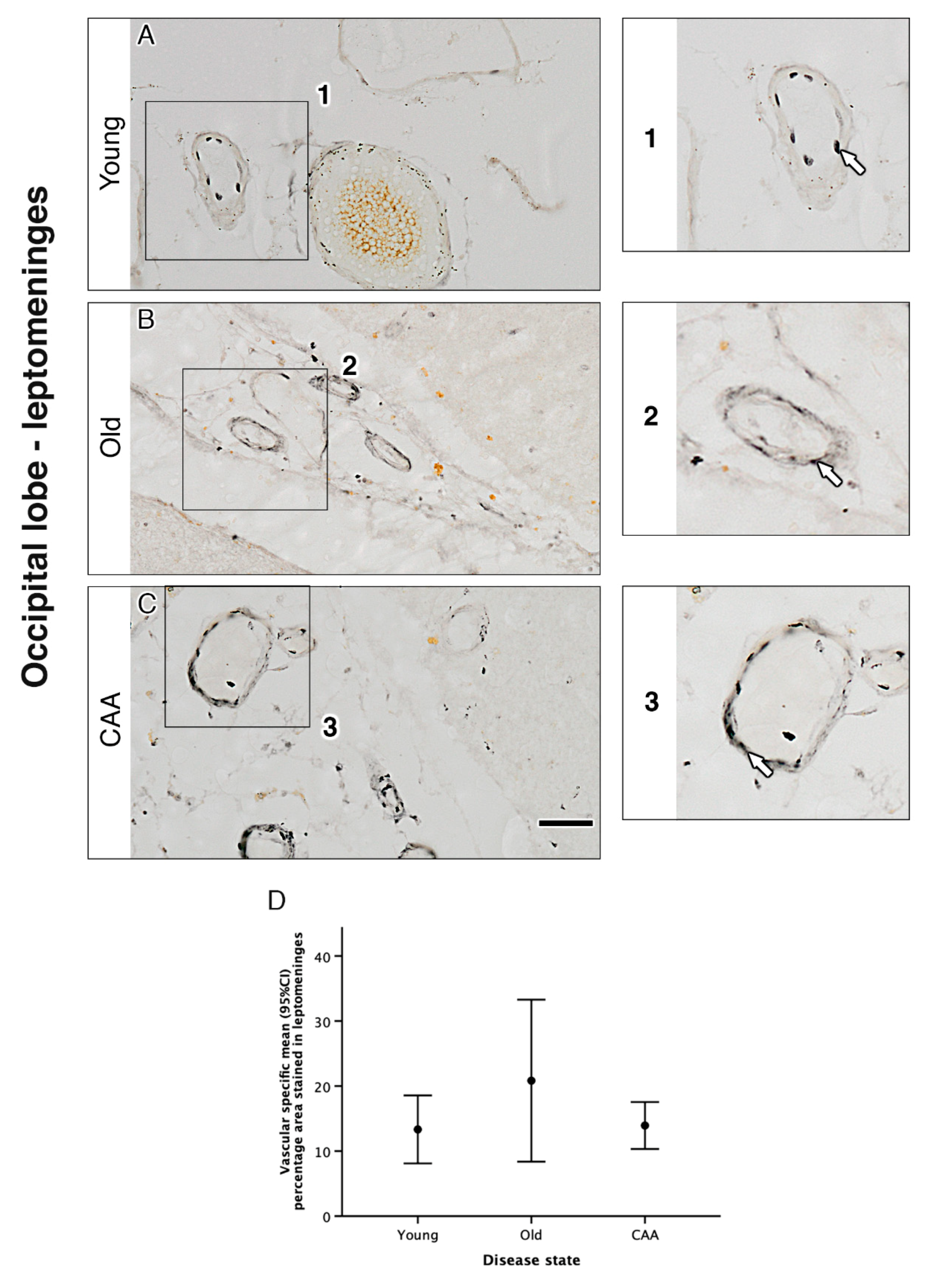
2.3. Vascular α1A-AR Expression Is Unaltered with Age and Disease in the Occipital Lobe
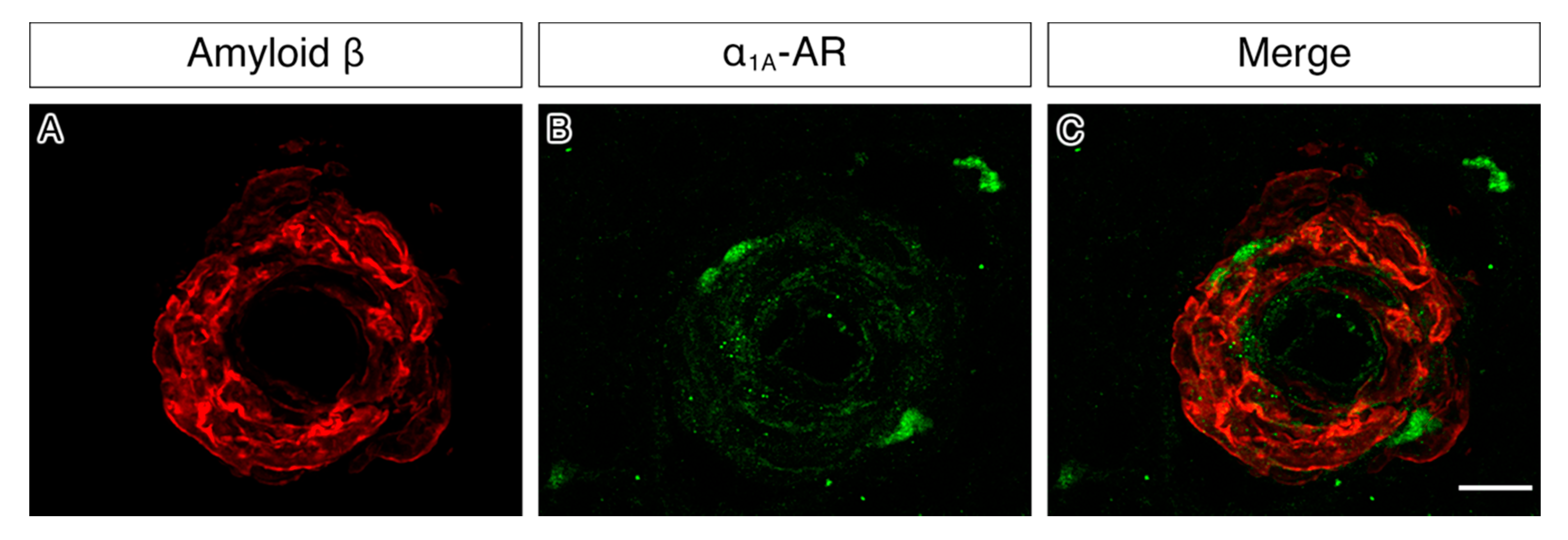
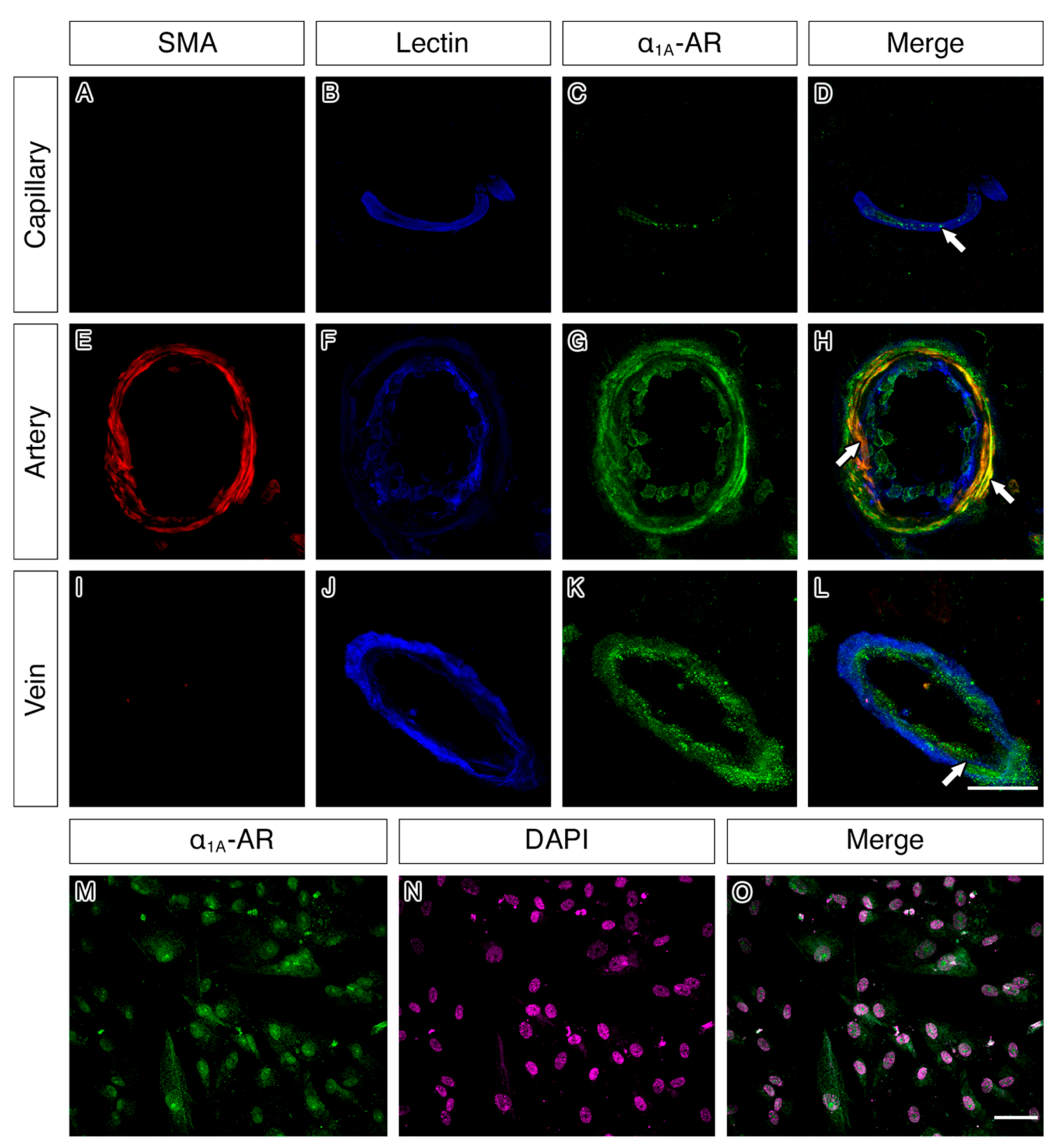
2.4. α1A-AR Co-Localises with Endothelial and Smooth Muscle Cell Markers in Capillaries, Arteries and Veins and with Cultured Vascular Smooth Muscle Cells
3. Discussion
4. Materials and Methods
4.1. Brain Tissue Cohort
4.2. Cell Culture
4.3. Immunohistochemistry on Human Tissue
4.4. Immunofluorescence on Human Tissue
4.5. Immunofluorescence on Cell Cultures
4.6. Imaging
4.7. Image Analysis and Statistics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Attems, J.; Jellinger, K.; Thal, D.R.; Van Nostrand, W. Review: Sporadic cerebral amyloid angiopathy. Neuropathol. Appl. Neurobiol. 2011, 37, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Taylor, X.; Cisternas, P.; You, Y.; You, Y.; Xiang, S.; Marambio, Y.; Zhang, J.; Vidal, R.; Lasagna-Reeves, C.A. A1 reactive astrocytes and a loss of TREM2 are associated with an early stage of pathology in a mouse model of cerebral amyloid angiopathy. J. Neuroinflamm. 2020, 17, 223. [Google Scholar] [CrossRef]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revesz, T.; Ghiso, J.; Lashley, T.; Plant, G.; Rostagno, A.; Frangione, B.; Holton, J.L. Cerebral Amyloid Angiopathies: A Pathologic, Biochemical, and Genetic View. J. Neuropathol. Exp. Neurol. 2003, 62, 885–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain—Implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, M.D.; Montagne, A.; Sagare, A.P.; Nation, D.A.; Schneider, L.S.; Chui, H.C.; Harrington, M.G.; Pa, J.; Law, M.; Wang, D.J.J.; et al. Vascular dysfunction-The disregarded partner of Alzheimer’s disease. Alzheimer’s Dement. 2019, 15, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Carare, R.O.; Bernardes-Silva, M.; Newman, T.A.; Page, A.M.; Nicoll, J.A.R.; Perry, V.H.; Weller, R.O. Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: Significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol. Appl. Neurobiol. 2008, 34, 131–144. [Google Scholar] [CrossRef]
- Morris, A.W.J.; Sharp, M.M.; Albargothy, N.J.; Fernandes, R.; Hawkes, C.A.; Verma, A.; Weller, R.O.; Carare, R.O. Vascular basement membranes as pathways for the passage of fluid into and out of the brain. Acta Neuropathol. 2016, 131, 725–736. [Google Scholar] [CrossRef] [Green Version]
- Albargothy, N.J.; Johnston, D.A.; Sharp, M.M.; Weller, R.O.; Verma, A.; Hawkes, C.A.; Carare, R.O. Convective influx/glymphatic system: Tracers injected into the CSF enter and leave the brain along separate periarterial basement membrane pathways. Acta Neuropathol. 2018, 136, 139–152. [Google Scholar] [CrossRef] [Green Version]
- Hawkes, C.A.; Härtig, W.; Kacza, J.; Schliebs, R.; Weller, R.O.; Nicoll, J.A.; Carare, R.O. Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathol. 2011, 121, 431–443. [Google Scholar] [CrossRef]
- Hawkes, C.A.; Gatherer, M.; Sharp, M.M.; Dorr, A.; Yuen, H.M.; Kalaria, R.; Weller, R.O.; Carare, R.O. Regional differences in the morphological and functional effects of aging on cerebral basement membranes and perivascular drainage of amyloid-β from the mouse brain. Aging Cell 2013, 12, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C.A.; Sullivan, P.M.; Hands, S.; Weller, R.O.; Nicoll, J.A.R.; Carare, R.O. Disruption of Arterial Perivascular Drainage of Amyloid-β from the Brains of Mice Expressing the Human APOE ε4 Allele. PLoS ONE 2012, 7, e41636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldea, R.; Weller, R.O.; Wilcock, D.M.; Carare, R.O.; Richardson, G. Cerebrovascular Smooth Muscle Cells as the Drivers of Intramural Periarterial Drainage of the Brain. Front. Aging Neurosci. 2019, 11, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Veluw, S.J.; Hou, S.S.; Calvo-Rodriguez, M.; Arbel-Ornath, M.; Snyder, A.C.; Frosch, M.P.; Greenberg, S.M.; Bacskai, B.J. Vasomotion as a Driving Force for Paravascular Clearance in the Awake Mouse Brain. Neuron 2020, 105, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Carare, R.O.; Aldea, R.; Bulters, D.; Alzetani, A.; Birch, A.A.; Richardson, G.; Weller, R.O. Vasomotion Drives Periarterial Drainage of Aβ from the Brain. Neuron 2020, 105, 400–401. [Google Scholar] [CrossRef]
- Bell, R.D.; Deane, R.; Chow, N.; Long, X.; Sagare, A.; Singh, I.; Streb, J.W.; Guo, H.; Rubio, A.; Van Nostrand, W.; et al. SRF and myocardin regulate LRP-mediated amyloid-beta clearance in brain vascular cells. Nat. Cell Biol. 2009, 11, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Bell, R.D.; Zlokovic, B.V. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Harik, S.I.; Sromek, S.M.; Kalaria, R.N. Alpha- and beta-adrenergic receptors of the rat cerebral cortex and cerebral microvessels in aging, and their response to denervation. Neurobiol. Aging 1991, 12, 567–573. [Google Scholar] [CrossRef]
- Andrés-Benito, P.; Fernández-Dueñas, V.; Carmona, M.; Escobar, L.A.; Torrejón-Escribano, B.; Aso, E.; Ciruela, F.; Ferrer, I. Locus coeruleus at asymptomatic early and middle Braak stages of neurofibrillary tangle pathology. Neuropathol. Appl. Neurobiol. 2017, 43, 373–392. [Google Scholar] [CrossRef] [Green Version]
- Betts, M.J.; Cardenas-Blanco, A.; Kanowski, M.; Spottke, A.; Teipel, S.J.; Kilimann, I.; Jessen, F.; Düzel, E. Locus coeruleus MRI contrast is reduced in Alzheimer’s disease dementia and correlates with CSF Aβ levels. Alzheimer’s Dement. 2019, 11, 281–285. [Google Scholar] [CrossRef]
- Betts, M.J.; Kirilina, E.; Otaduy, M.C.G.; Ivanov, D.; Acosta-Cabronero, J.; Callaghan, M.F.; Lambert, C.; Cardenas-Blanco, A.; Pine, K.; Passamonti, L.; et al. Locus coeruleus imaging as a biomarker for noradrenergic dysfunction in neurodegenerative diseases. Brain 2019, 142, 2558–2571. [Google Scholar] [CrossRef] [PubMed]
- Haase, N.; Herse, F.; Spallek, B.; Haase, H.; Morano, I.; Qadri, F.; Szijártó, I.A.; Rohm, I.; Yilmaz, A.; Warrington, J.P.; et al. Amyloid- Peptides Activate 1-Adrenergic Cardiovascular Receptors. Hypertension 2013, 62, 966–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szot, P.; White, S.S.; Greenup, J.L.; Leverenz, J.B.; Peskind, E.R.; Raskind, M.A. Changes in adrenoreceptors in the prefrontal cortex of subjects with dementia: Evidence of compensatory changes. Neuroscience 2007, 146, 471–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsouri, L.; Vizcaychipi, M.P.; McArthur, S.; Harrison, I.F.; Suárez-Calvet, M.; Lleo, A.; Lloyd, D.G.; Ma, D.; Sastre, M. Prazosin, an α1-adrenoceptor antagonist, prevents memory deterioration in the APP23 transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1105–1115. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.Y.; Shofer, J.B.; Rohde, K.; Hart, K.L.; Hoff, D.J.; McFall, Y.H.; Raskind, M.A.; Peskind, E.R. Prazosin for the Treatment of Behavioral Symptoms in Patients With Alzheimer Disease With Agitation and Aggression. Am. J. Geriatr. Psychiatry 2009, 17, 744–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagano, K.; Kwon, C.; Ishida, J.; Hashimoto, T.; Kim, J.-D.; Kishikawa, N.; Murao, M.; Kimura, K.; Kasuya, Y.; Kimura, S.; et al. Cooperative action of APJ and α1A-adrenergic receptor in vascular smooth muscle cells induces vasoconstriction. J. Biochem. 2019, 166, 383–392. [Google Scholar] [CrossRef]
- Chen, Y.; Peng, Y.; Che, P.; Gannon, M.; Liu, Y.; Li, L.; Bu, G.; Van Groen, T.; Jiao, K.; Wang, Q. α2A adrenergic receptor promotes amyloidogenesis through disrupting APP-SorLA interaction. Proc. Natl. Acad. Sci. USA 2014, 111, 17296–17301. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Gannon, M.; Chen, Y.; Yan, S.; Zhang, S.; Feng, W.; Tao, J.; Sha, B.; Liu, Z.; Saito, T.; et al. β-amyloid redirects norepinephrine signaling to activate the pathogenic GSK3β/tau cascade. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Michel, M.C.; Wieland, T.; Tsujimoto, G. How reliable are G-protein-coupled receptor antibodies? Naunyn Schmiedebergs Arch. Pharmacol. 2009, 379, 385–388. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a | Percentage Area Stained for Overall α1A Adrenergic Receptor in… | ||||||||||||
| Grey Matter | White Matter | ||||||||||||
| Disease state | N | Mean (SD) | (Min, Max) | N | Mean (SD) | (Min, Max) | |||||||
| Young (Y) | 5 | 3.56 (1.18) | (2.68, 5.62) | 5 | 1.47 (0.47) | (1.11, 2.14) | |||||||
| Old Non-demented (O) | 5 | 2.48 (0.97) | (1.48, 3.97) | 5 | 0.79 (0.49) | (0.38, 1.48) | |||||||
| CAA (C) | 5 | 1.68 (0.71) | (0.59, 2.56) | 5 | 0.91 (0.41) | (0.39, 1.53) | |||||||
| b | Grey Matter | White Matter | |||||||||||
| Multiple Comparisons between Disease States | N | Mean Difference (95% CI) | p1 | N | Mean Difference (95% CI) | p1 | |||||||
| O–Y | 10 | −1.09 (−2.79, 0.62) | 0.305 | 10 | −0.68 (−1.49, 0.13) | 0.113 | |||||||
| C–Y | 10 | −1.88 (−3.59, −0.18) | 0.029 | 10 | −0.56 (−1.37, 0.25) | 0.233 | |||||||
| C–O | 10 | −0.79 (−2.50, 0.91) | 0.658 | 10 | 0.12 (−0.69, 0.93) | 1.000 | |||||||
| c | Percentage Area Stained for Vascular Specific α1A Adrenergic Receptor in… | ||||||||||||
| Grey Matter | White Matter | Leptomeninges | |||||||||||
| Disease State | N | Mean (SD) | (Min, Max) | N | Mean (SD) | (Min, Max) | N | Mean (SD) | (Min, Max) | ||||
| Young (Y) | 5 | 23.25 (3.33) | (17.56, 25.78) | 5 | 17.58 (9.84) | (8.84, 34.09) | 5 | 13.33 (4.21) | (6.81, 17.99) | ||||
| Old non-demented (O) | 5 | 23.22 (5.42) | (15.40, 29.40) | 5 | 15.67 (6.81) | (7.21, 21.89) | 5 | 20.82 (10.03) | (10.67, 34.60) | ||||
| CAA (C) | 5 | 18.81 (3.71) | (15.54, 24.99) | 5 | 15.72 (6.75) | (7.02, 23.12) | 5 | 13.93 (2.91) | (10.71, 18.22) | ||||
| d | Grey Matter | White Matter | Leptomeninges | ||||||||||
| Multiple Comparisons between Disease States | N | Mean Difference (95% CI) | p1 | N | Mean Difference (95% CI) | p1 | N | Mean Difference (95% CI) | p1 | ||||
| O–Y | 10 | −0.03 (−7.51, 7.44) | 1.000 | 10 | −1.91 (−15.86, 12.05) | 1.000 | 10 | 7.50 (−3.93, 18.93) | 0.280 | ||||
| C–Y | 10 | −4.44 (−11.92, 3.03) | 0.373 | 10 | −1.86 (−15.81, 12.09) | 1.000 | 10 | 0.61 (−10.82, 12.04) | 1.000 | ||||
| C–O | 10 | −4.41 (−11.88, 3.06) | 0.381 | 10 | 0.05 (−13.90, 14.00) | 1.000 | 10 | −6.89 (−18.32, 4.54) | 0.359 | ||||
| Source | Age | Sex | pm Delay/Hrs | Category |
|---|---|---|---|---|
| Edinburgh | 51 | M | 78 | Young |
| Edinburgh | 41 | F | 50 | Young |
| Edinburgh | 60 | M | 52 | Young |
| Edinburgh | 59 | F | 53 | Young |
| Edinburgh | 33 | M | 47 | Young |
| Newcastle | 73 | M | 25 | Old non-demented |
| Newcastle | 90 | M | 18 | Old non-demented |
| Newcastle | 95 | F | 66 | Old non-demented |
| Newcastle | 95 | M | 21 | Old non-demented |
| Newcastle | 89 | F | 98 | Old non-demented |
| Newcastle | 67 | M | 46 | CAA |
| Newcastle | 86 | F | 51 | CAA |
| Newcastle | 73 | M | 7 | CAA |
| Newcastle | 74 | F | 49 | CAA |
| Newcastle | 87 | F | 54 | CAA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frost, M.; Keable, A.; Baseley, D.; Sealy, A.; Andreea Zbarcea, D.; Gatherer, M.; Yuen, H.M.; Sharp, M.M.; Weller, R.O.; Attems, J.; et al. Vascular α1A Adrenergic Receptors as a Potential Therapeutic Target for IPAD in Alzheimer’s Disease. Pharmaceuticals 2020, 13, 261. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13090261
Frost M, Keable A, Baseley D, Sealy A, Andreea Zbarcea D, Gatherer M, Yuen HM, Sharp MM, Weller RO, Attems J, et al. Vascular α1A Adrenergic Receptors as a Potential Therapeutic Target for IPAD in Alzheimer’s Disease. Pharmaceuticals. 2020; 13(9):261. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13090261
Chicago/Turabian StyleFrost, Miles, Abby Keable, Dan Baseley, Amber Sealy, Diana Andreea Zbarcea, Maureen Gatherer, Ho Ming Yuen, Matt MacGregor Sharp, Roy O. Weller, Johannes Attems, and et al. 2020. "Vascular α1A Adrenergic Receptors as a Potential Therapeutic Target for IPAD in Alzheimer’s Disease" Pharmaceuticals 13, no. 9: 261. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13090261