Journey on Naphthoquinone and Anthraquinone Derivatives: New Insights in Alzheimer’s Disease



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
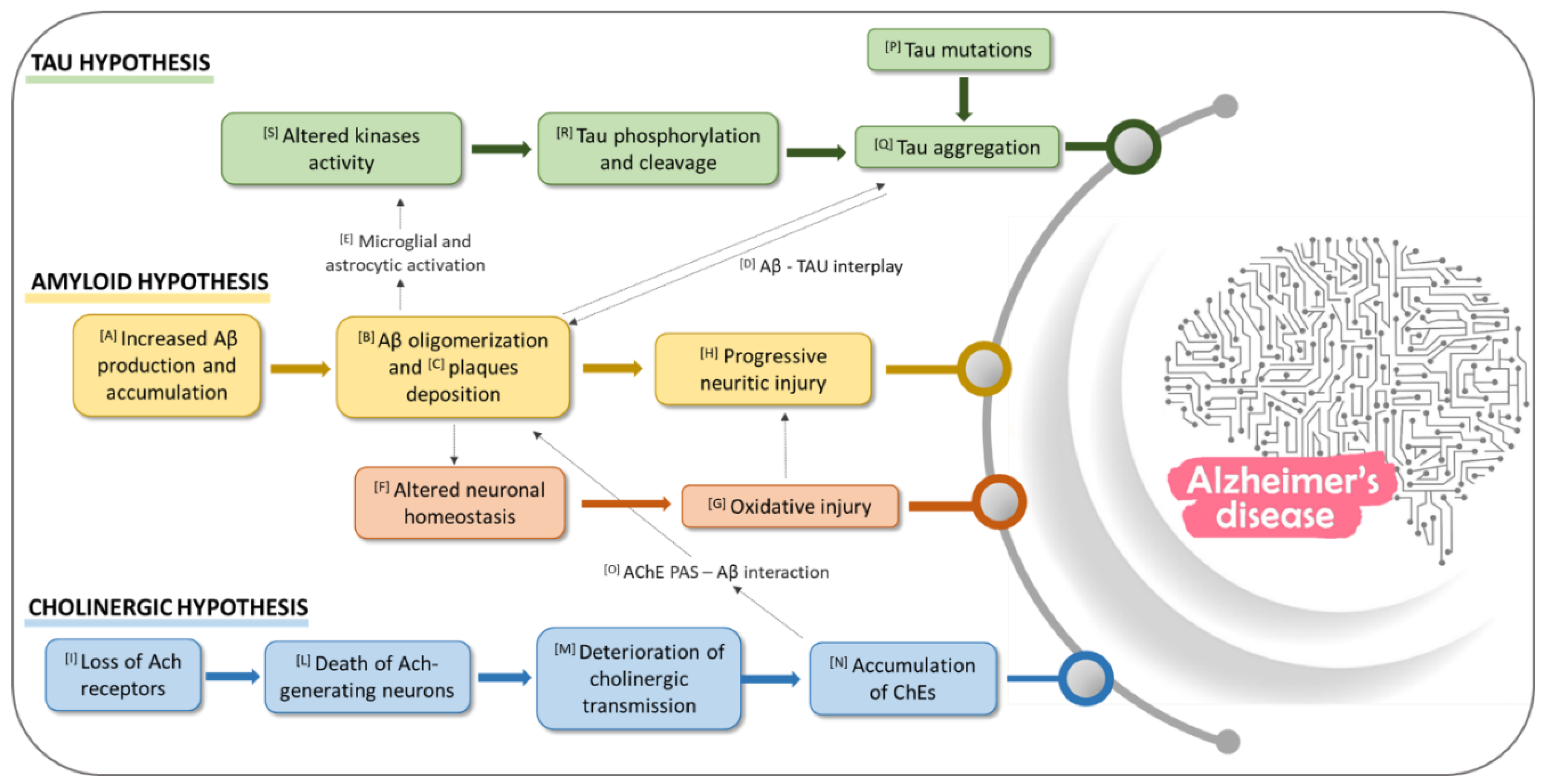
1.1. Role of Aβ in AD
1.2. Role of Cholinesterase Enzymes (ChEs) in AD
1.3. Role of Tau Protein in AD
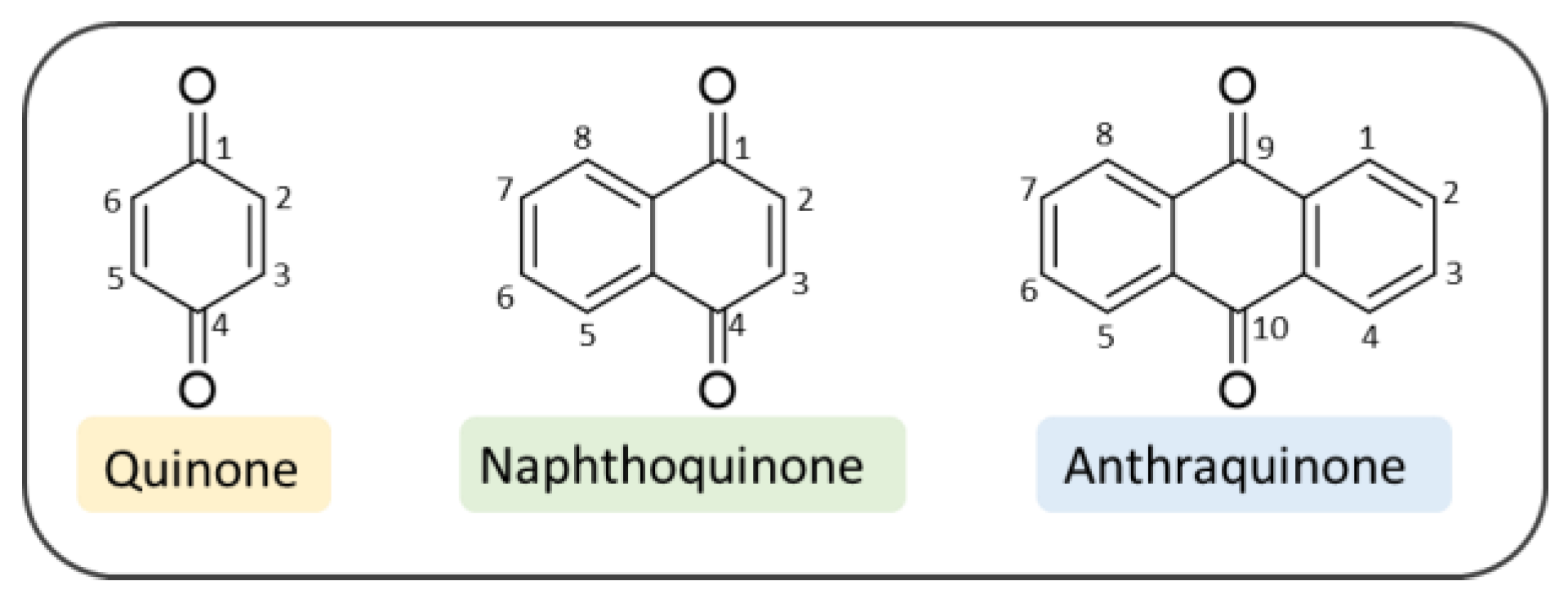
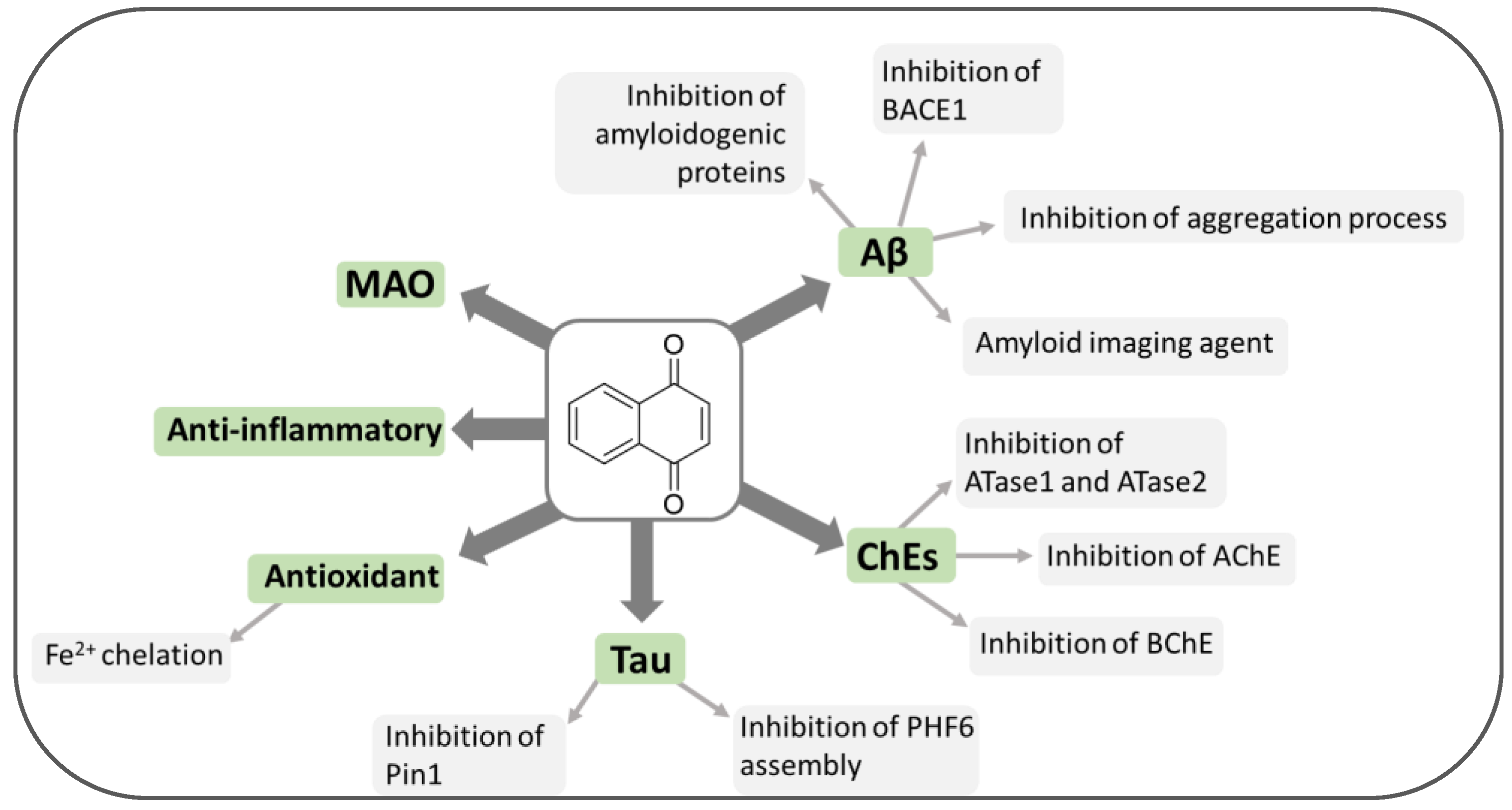
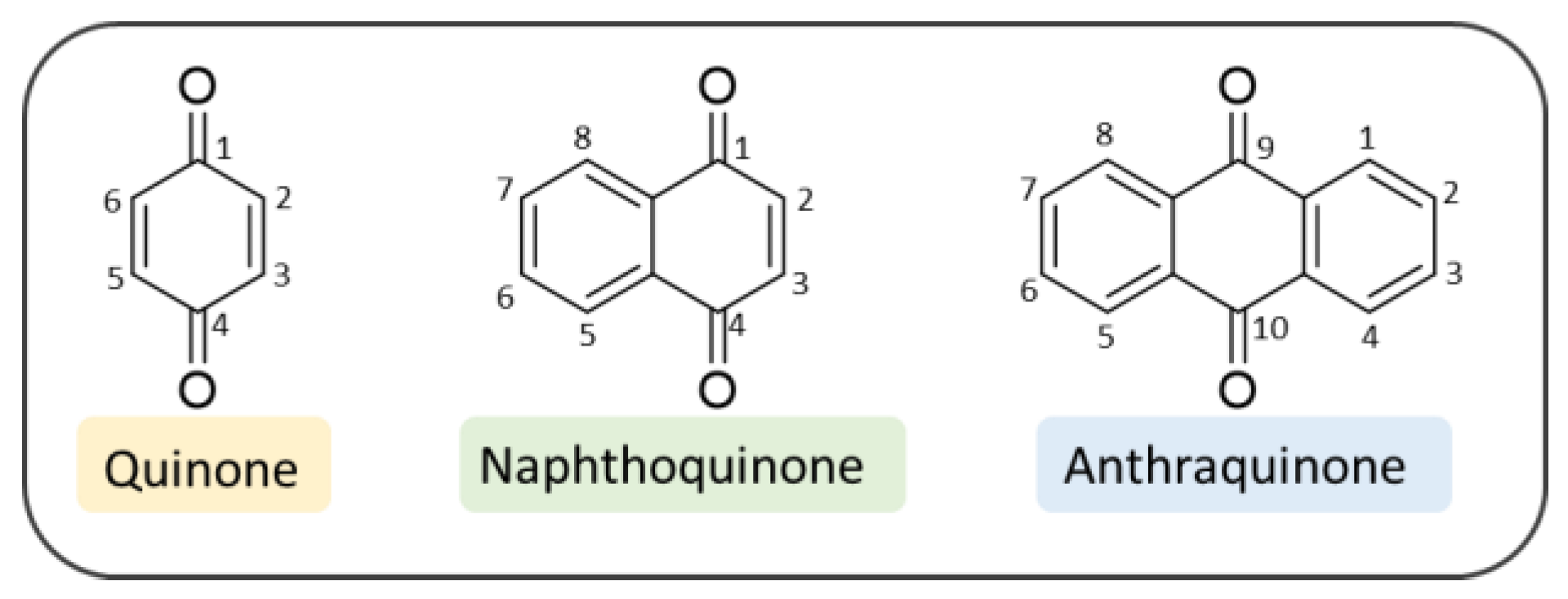
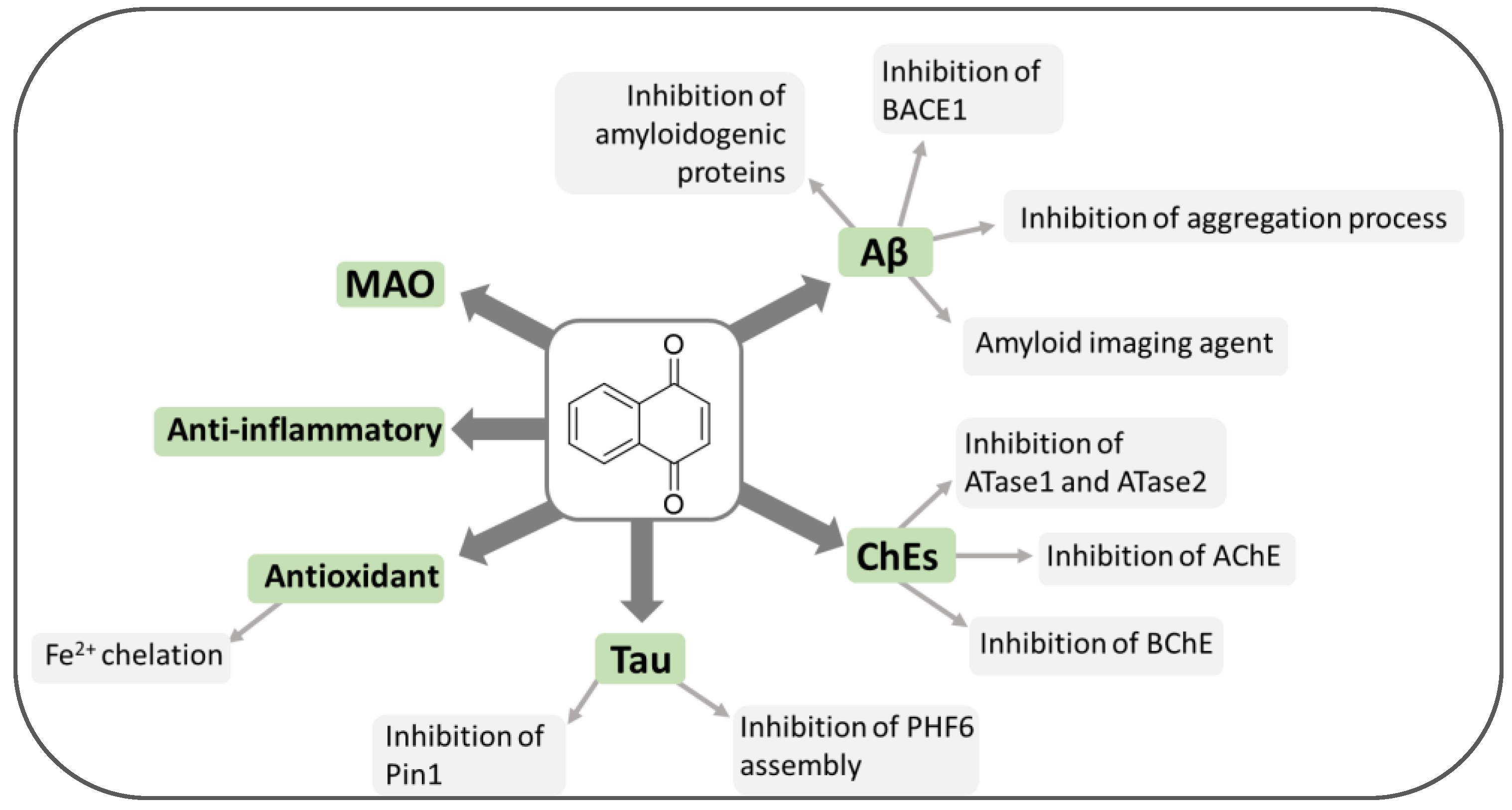
2. Quinone-Based Scaffolds for the Development of Novel Agents against AD
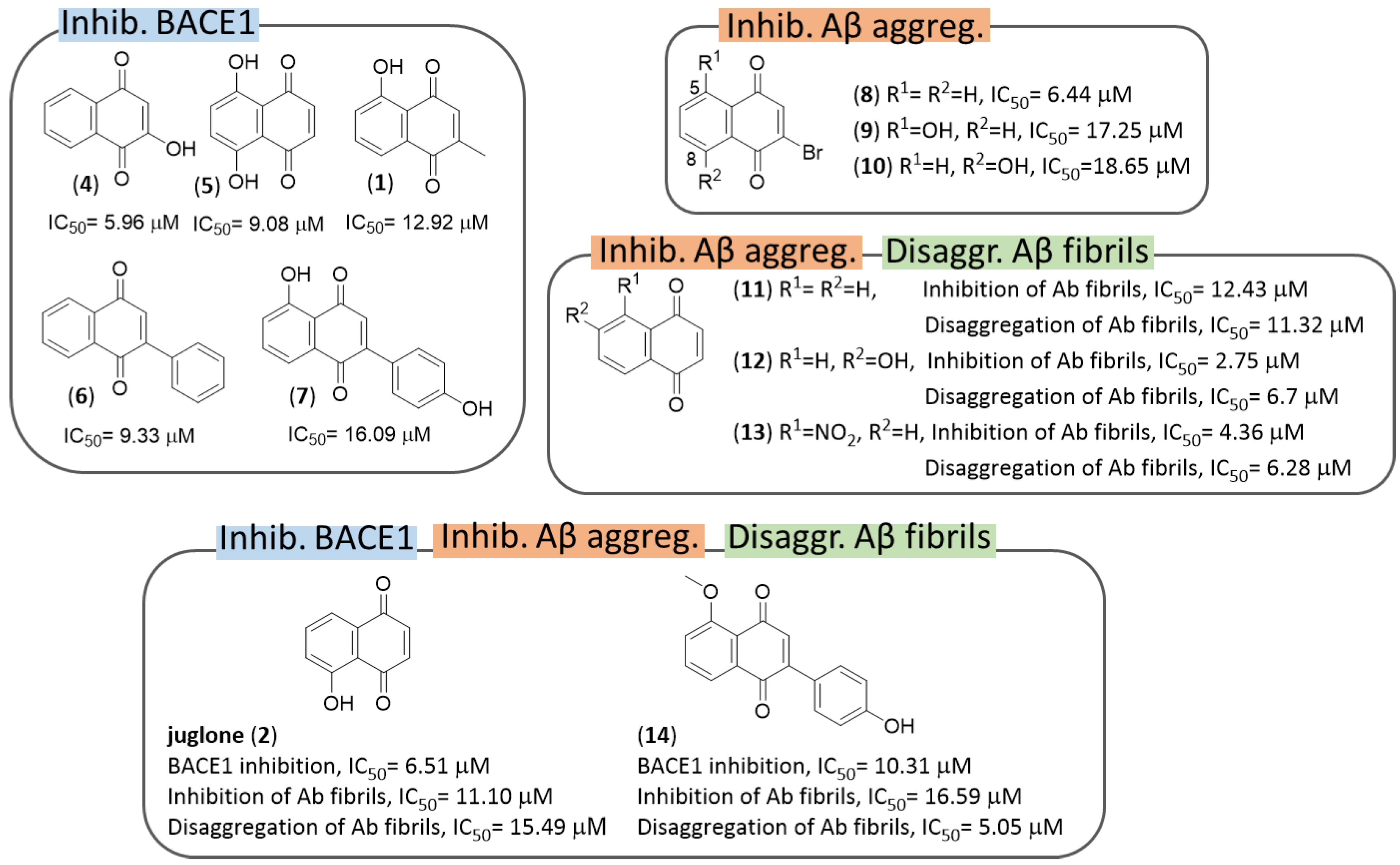
3. Naphthoquinones
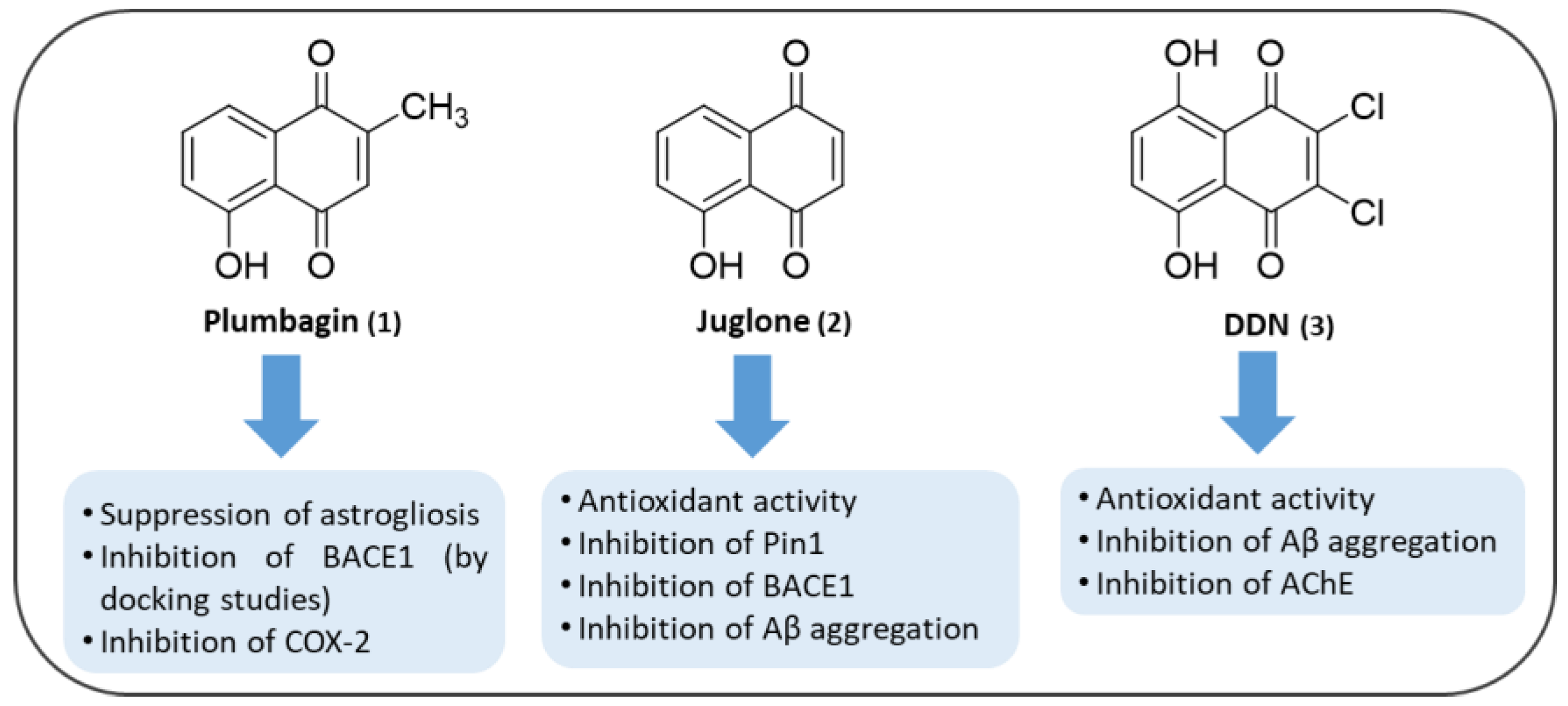
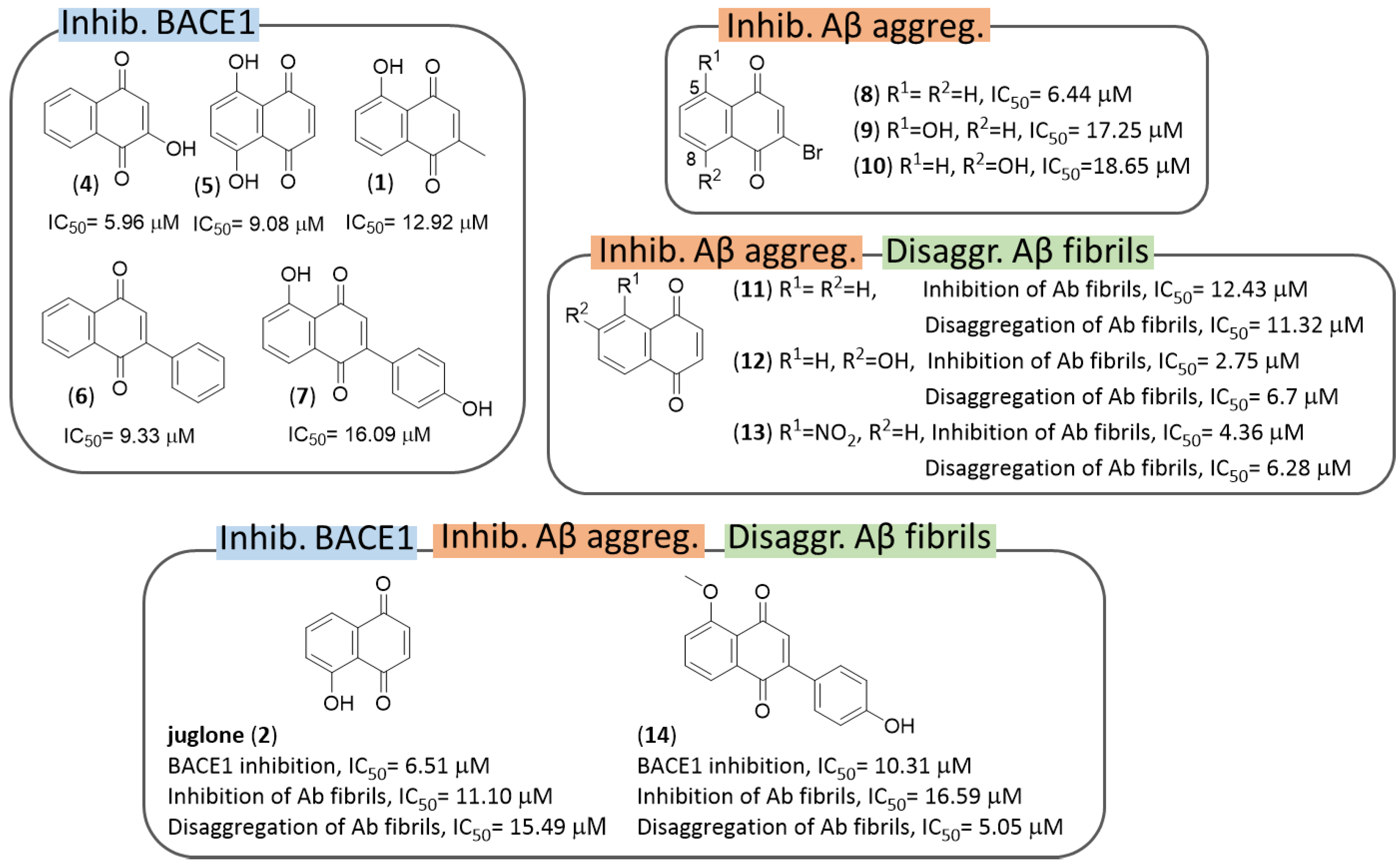
3.1. NQs from Natural Sources
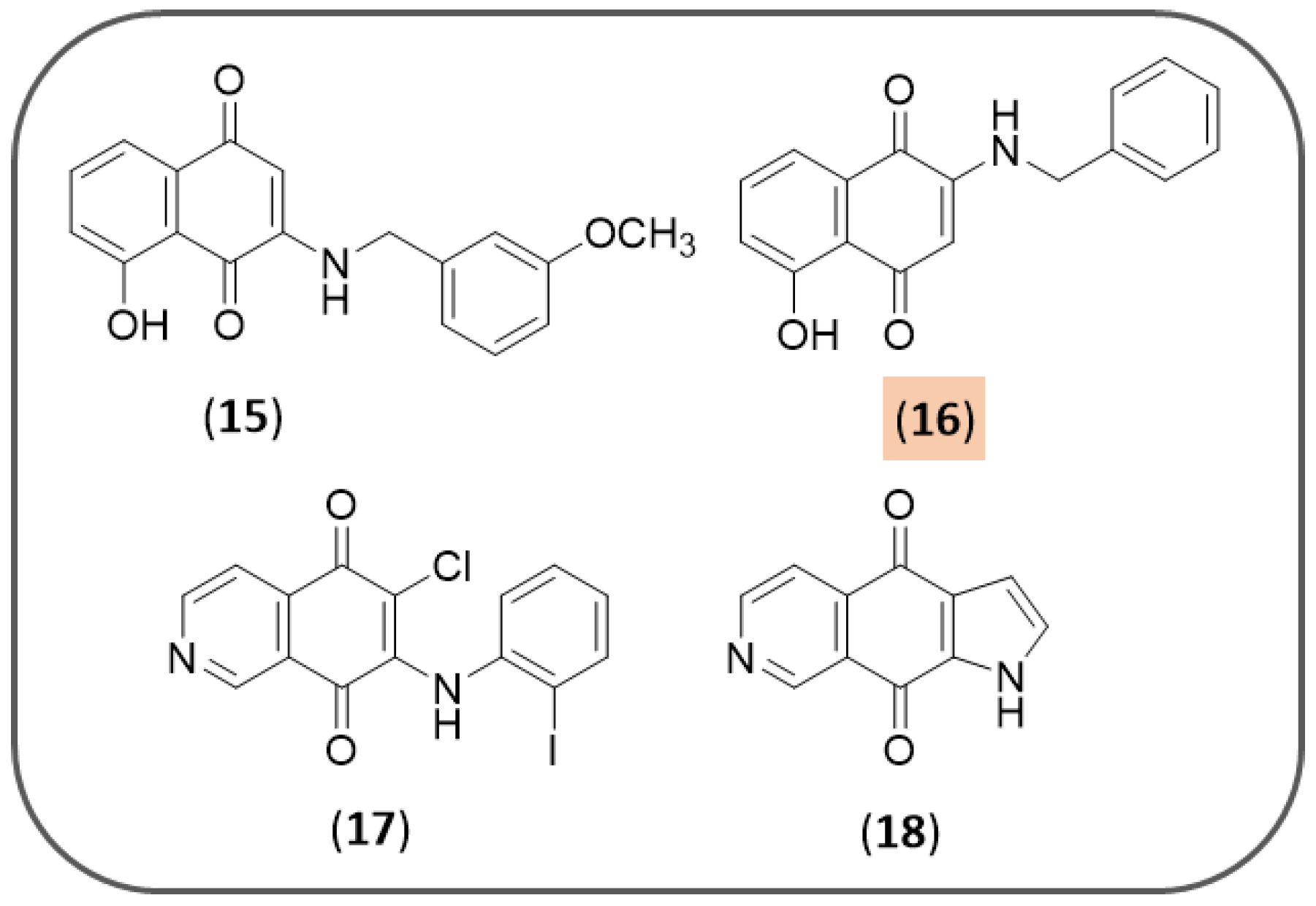
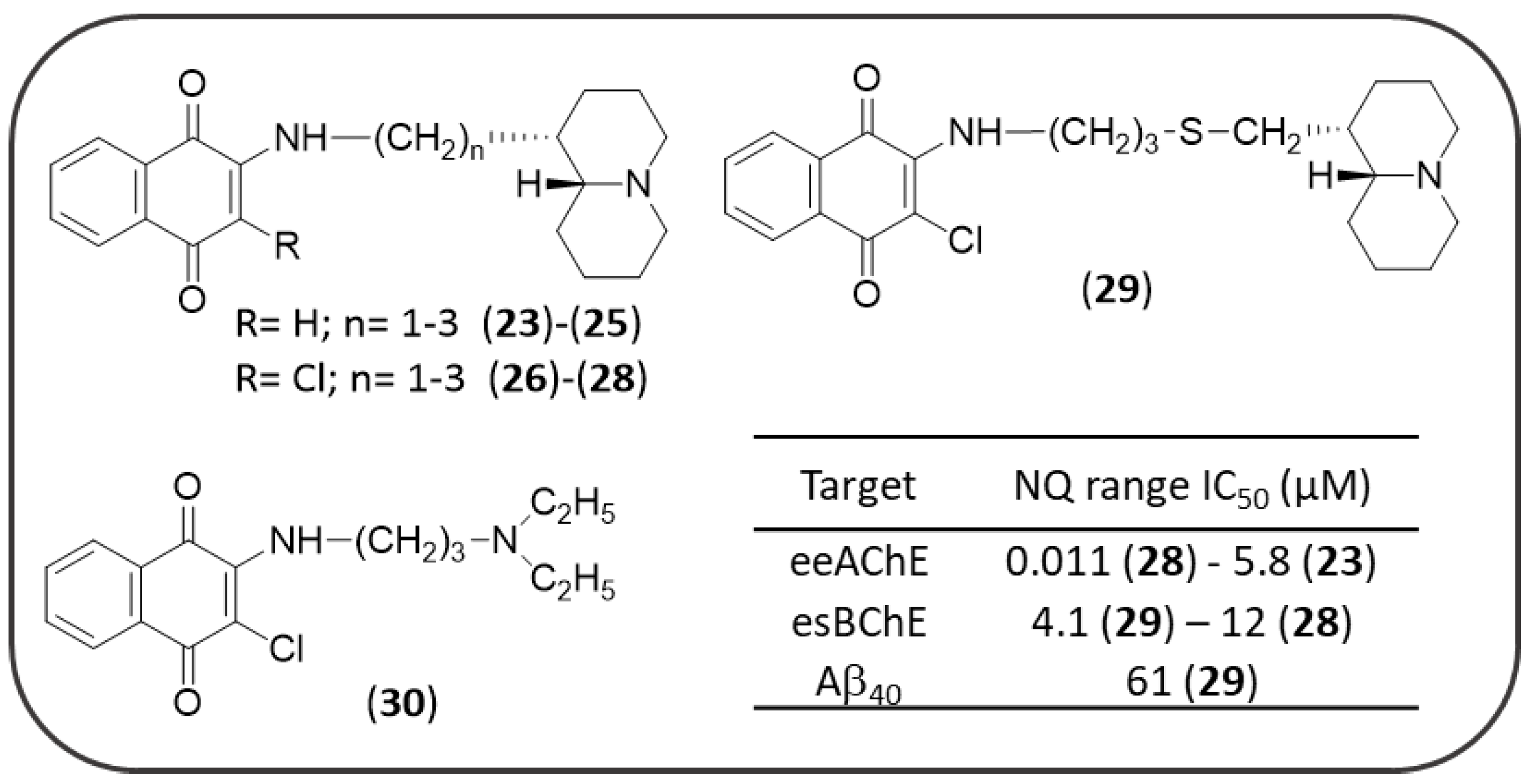
3.2. Synthetic NQ Derivatives
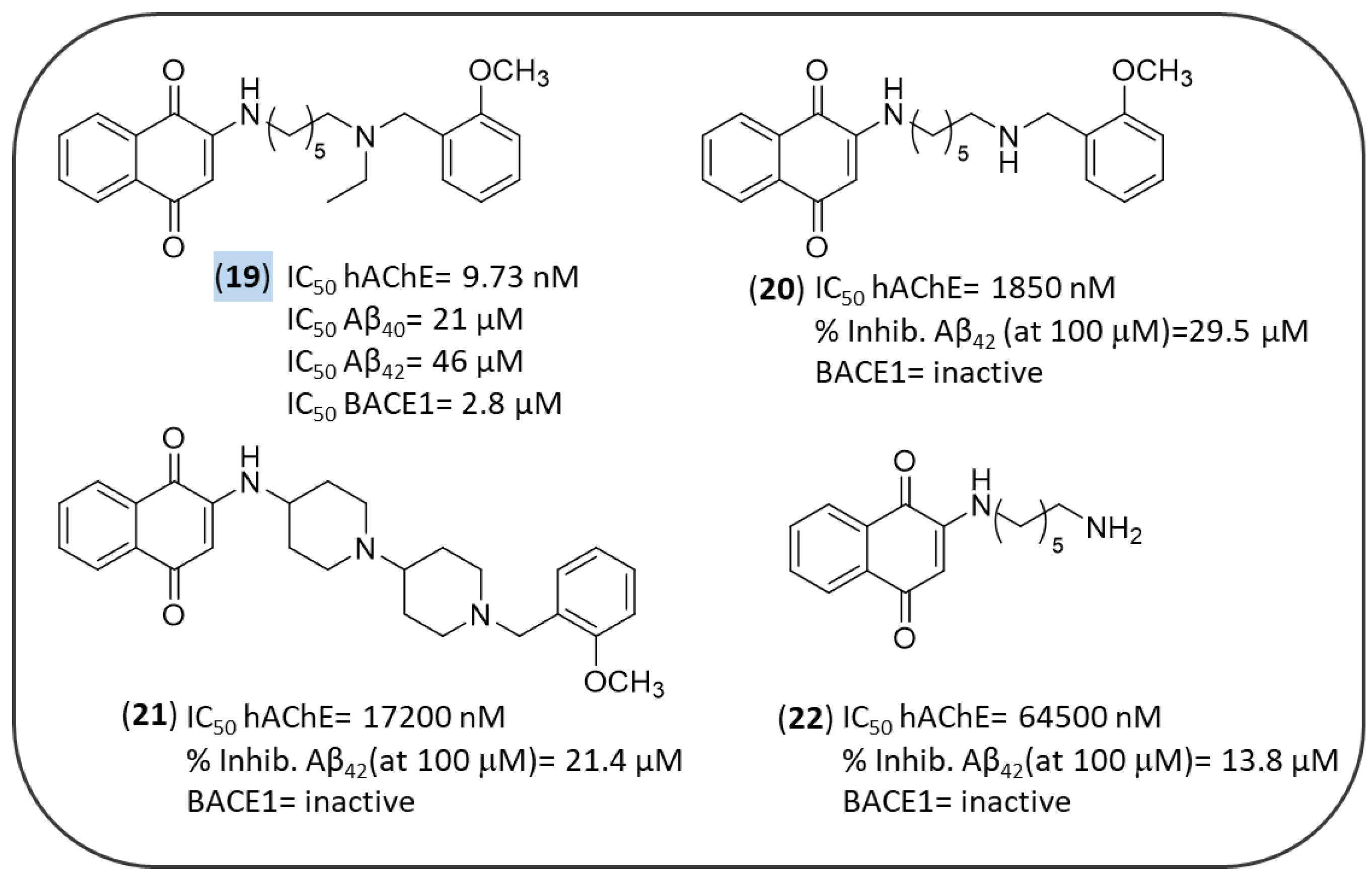
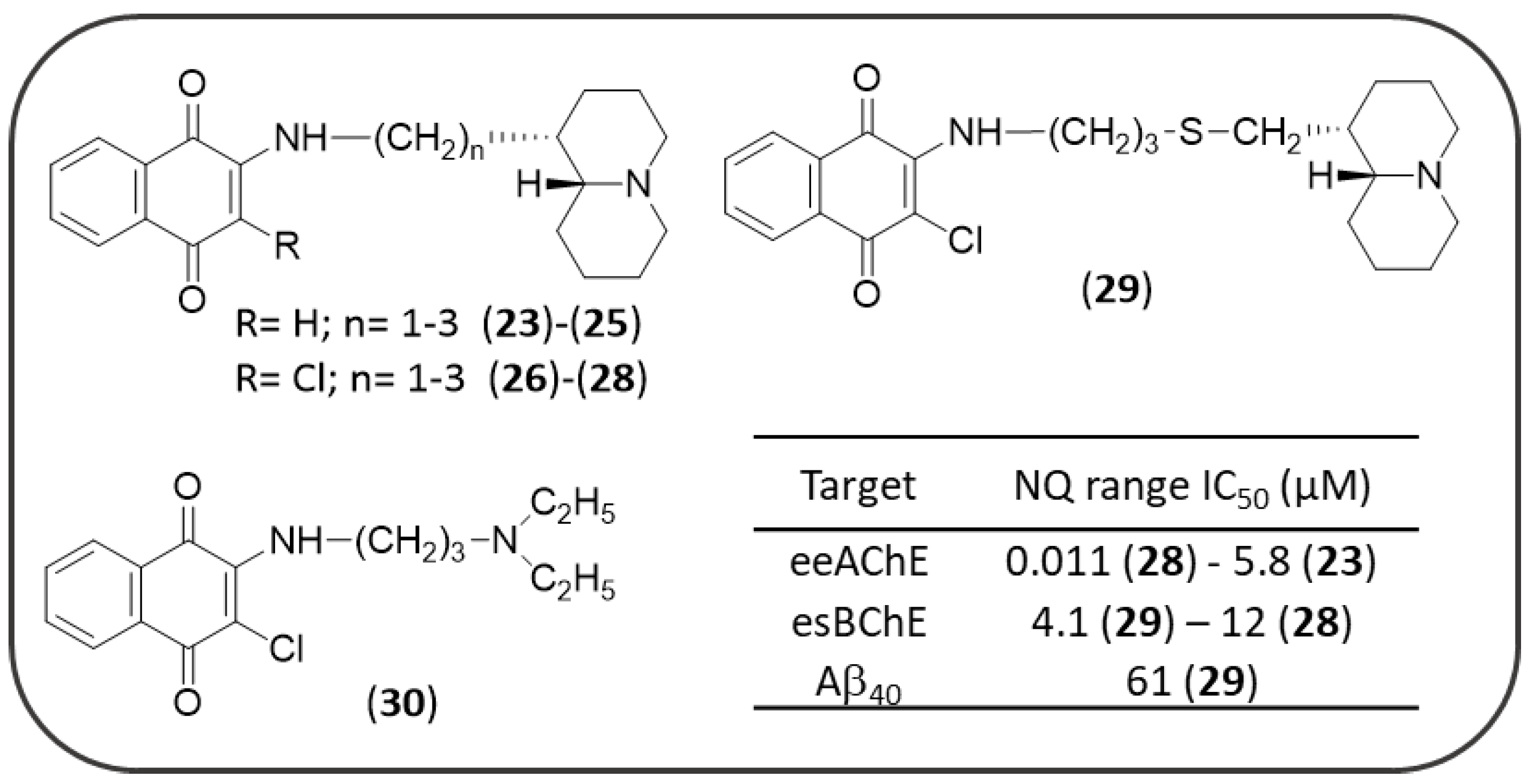
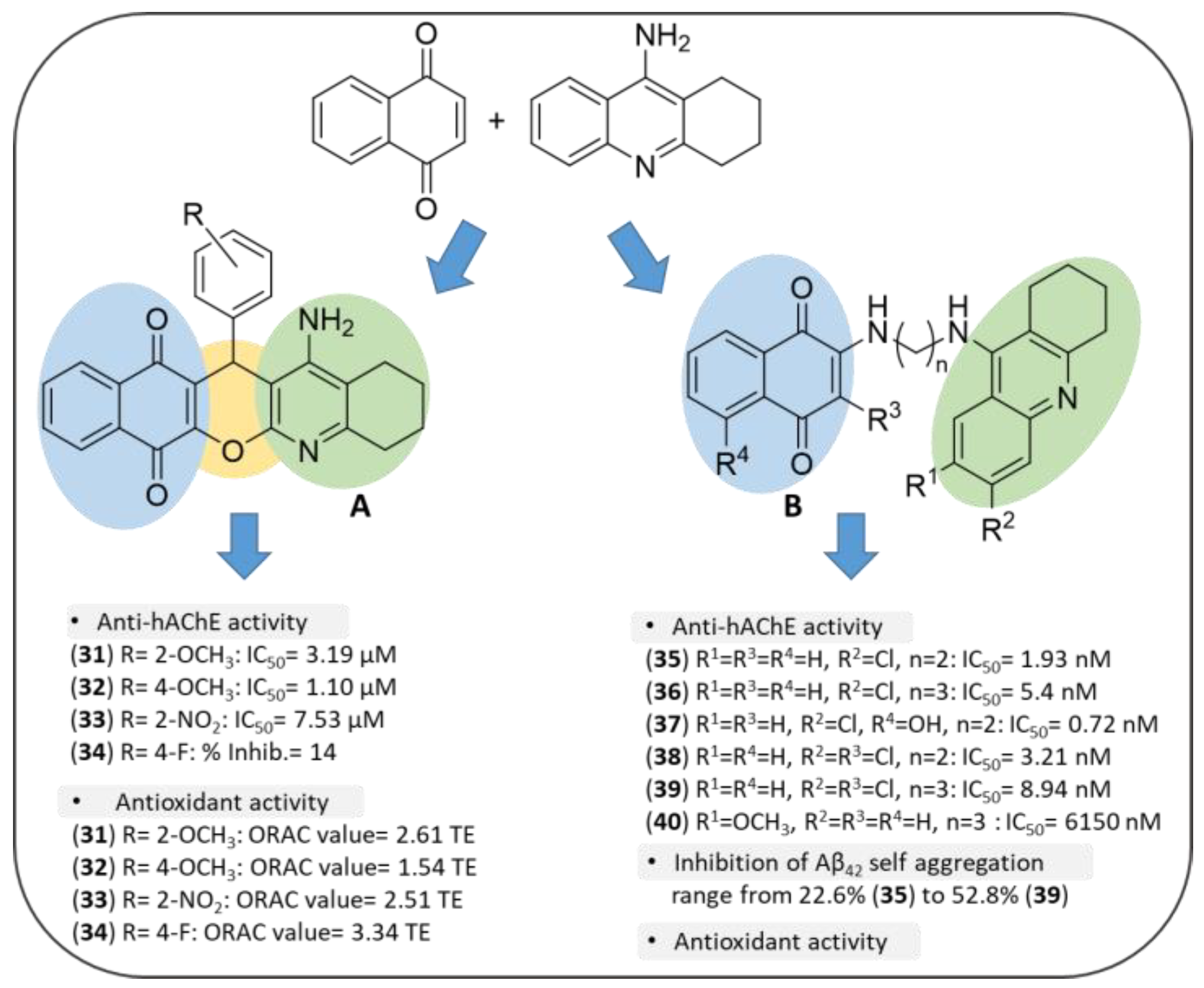
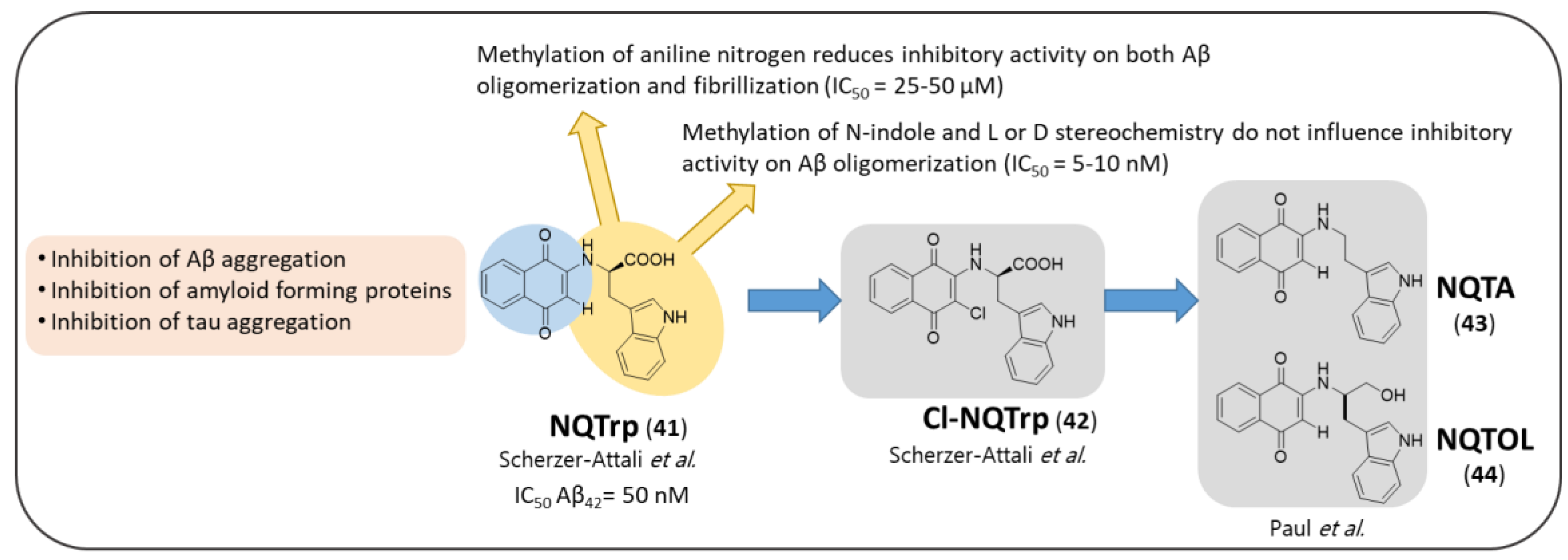
3.3. NQ-Based Hybrids
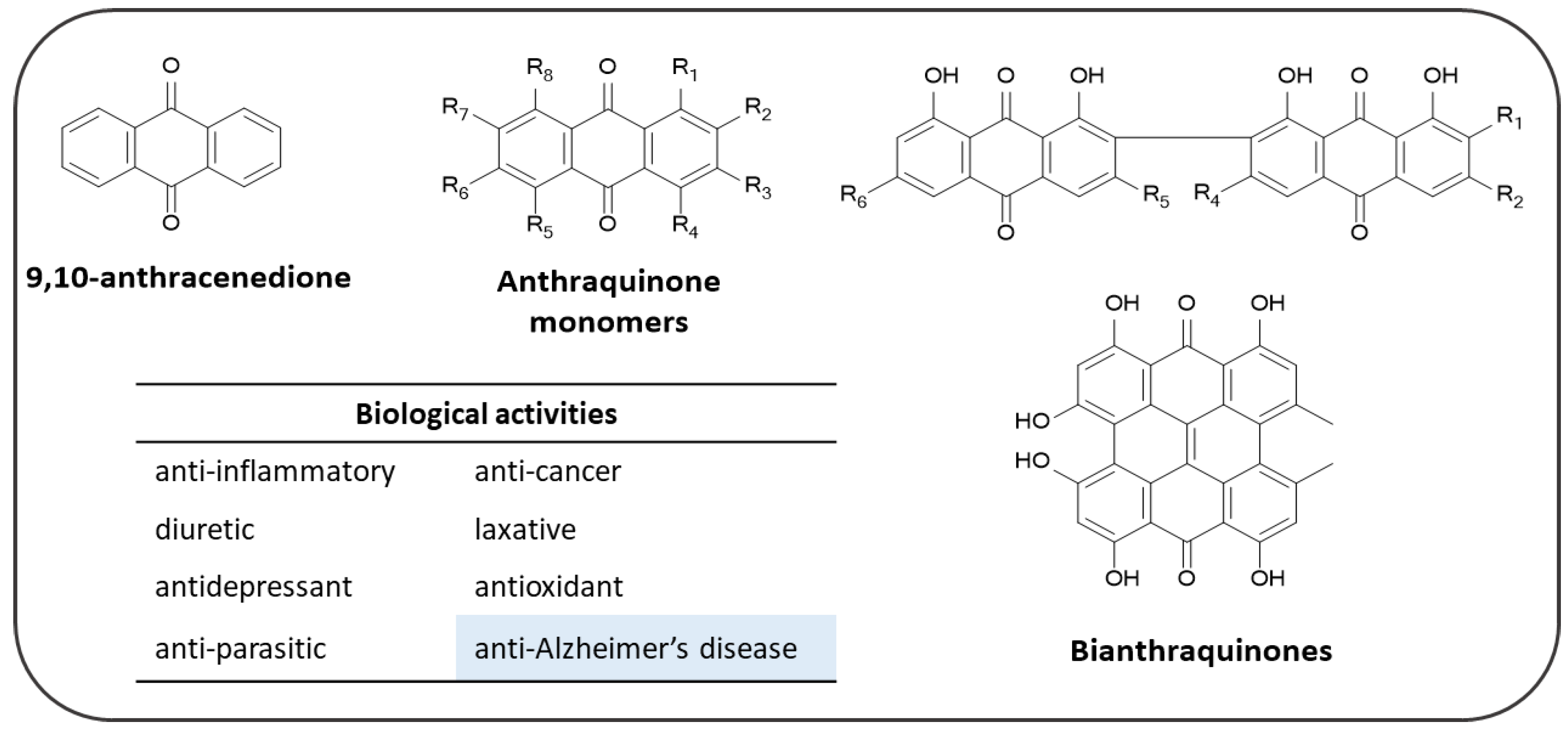
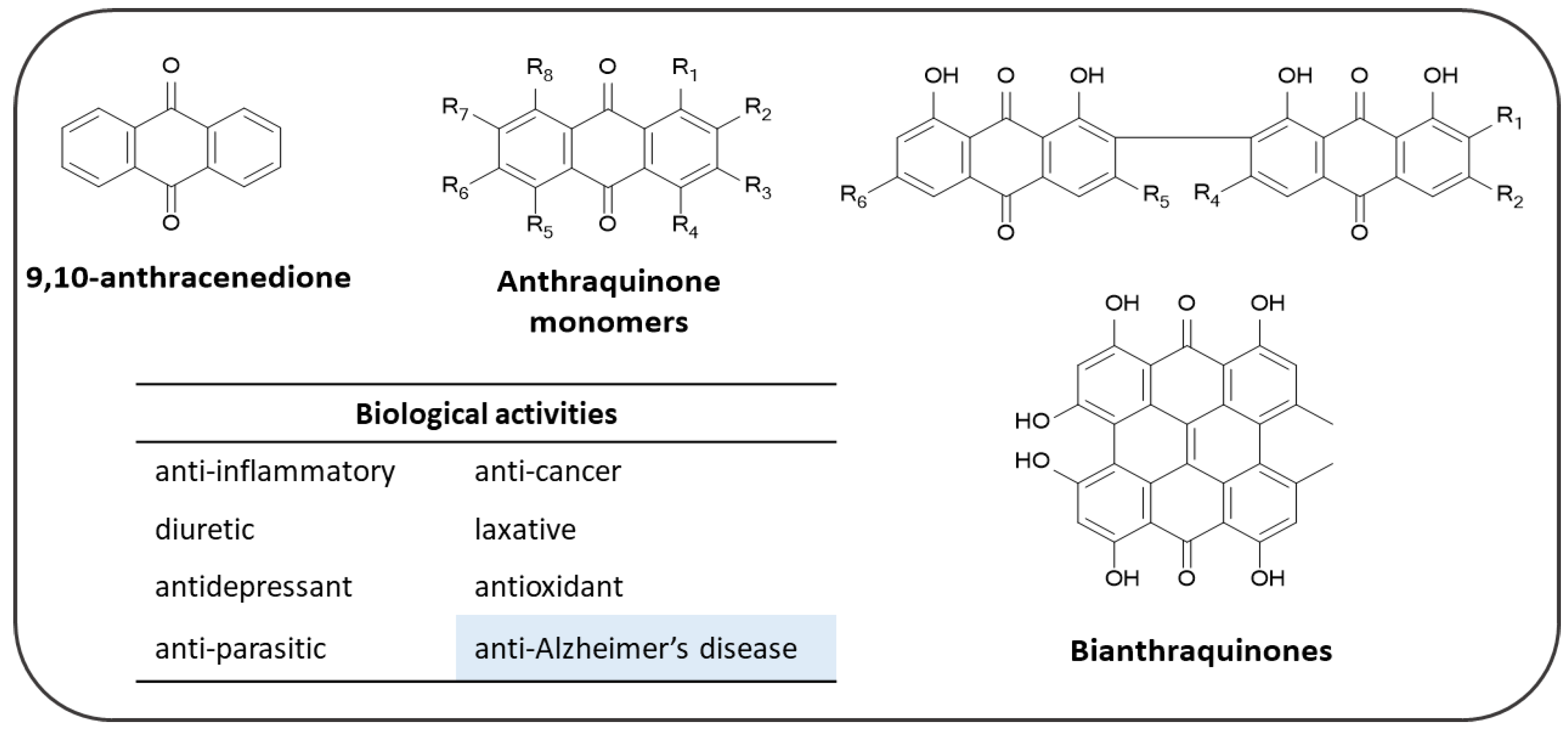
4. Anthraquinones
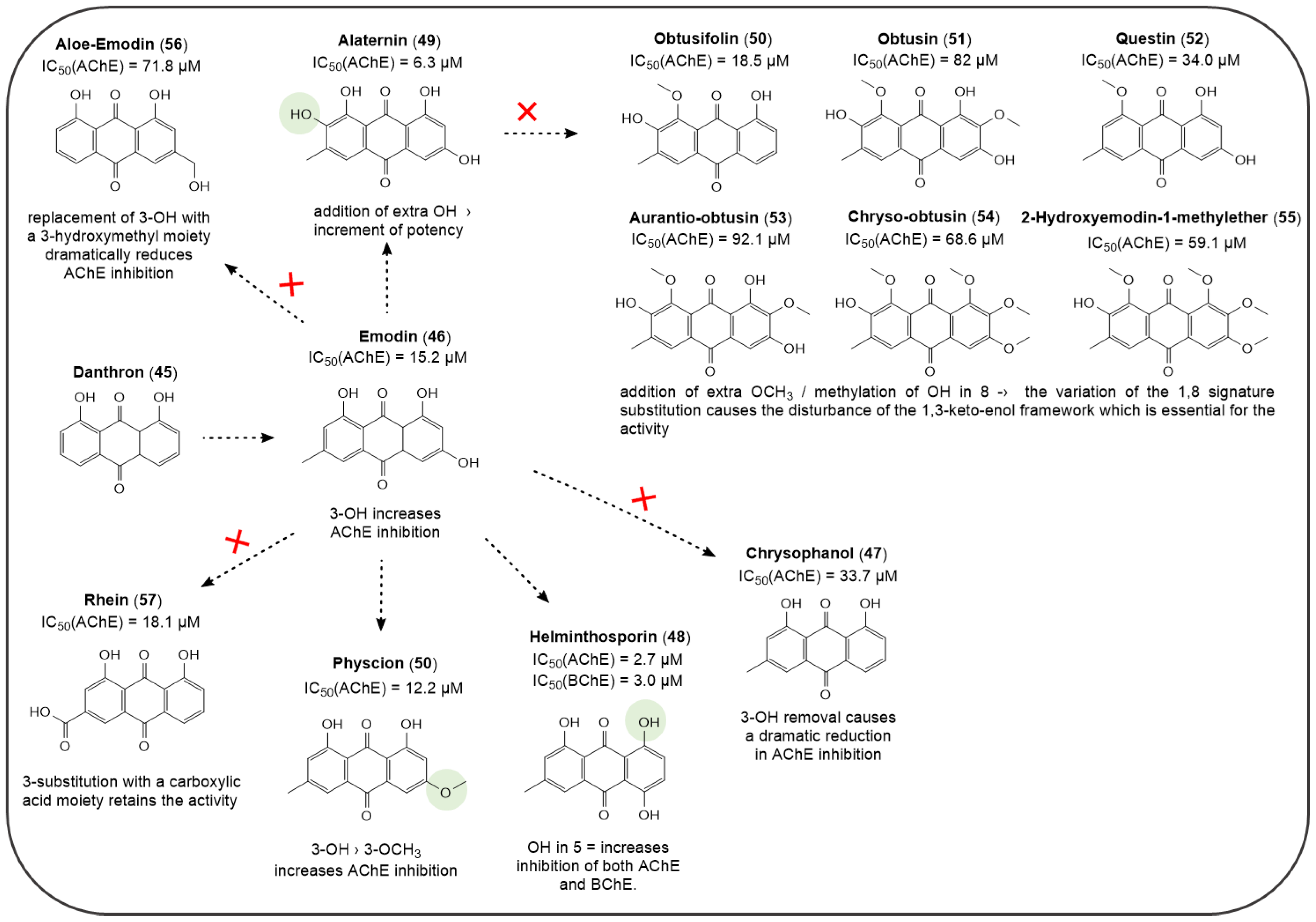
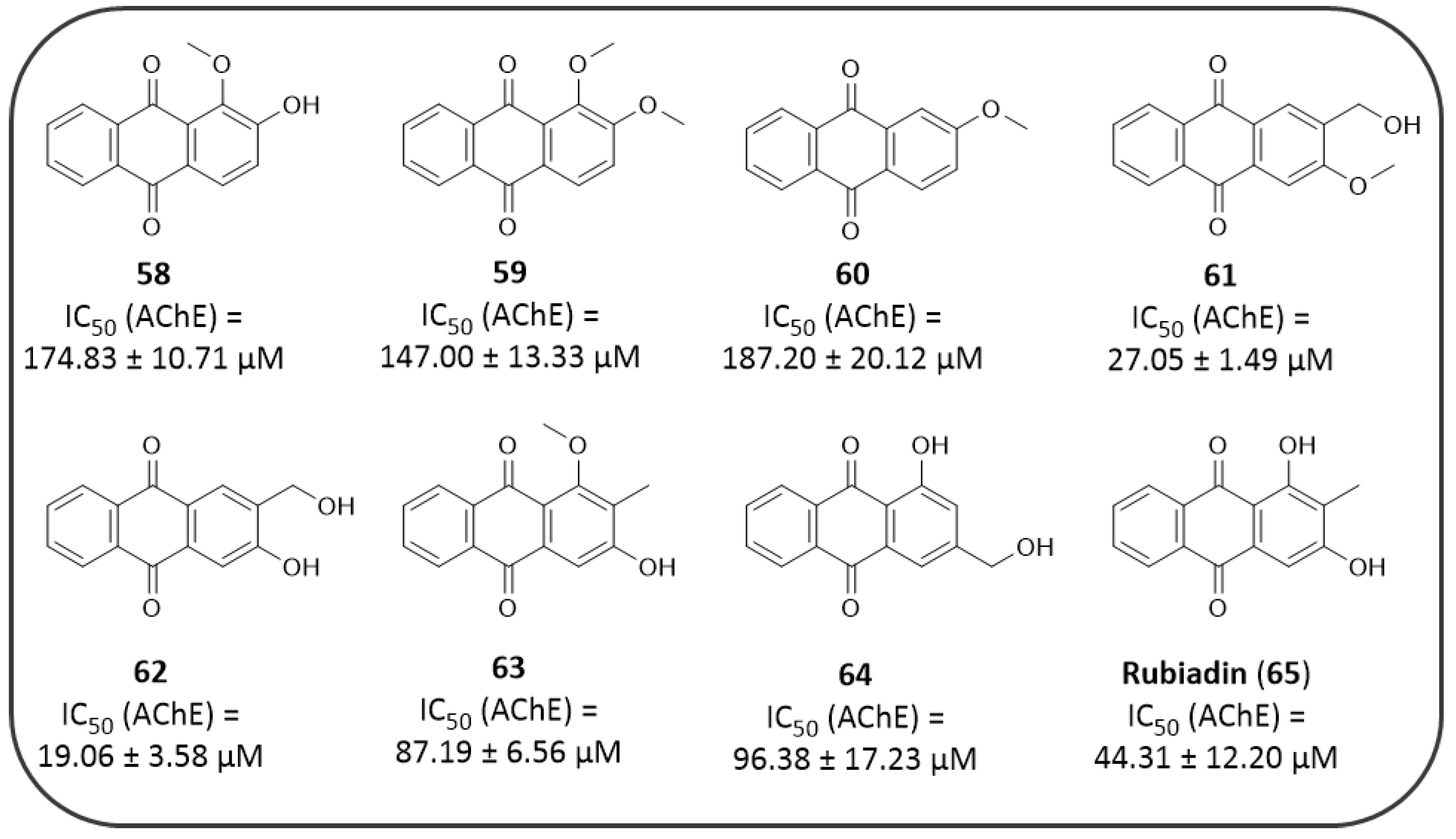
4.1. AQs from Natural Sources: Cholinesterase Inhibitors
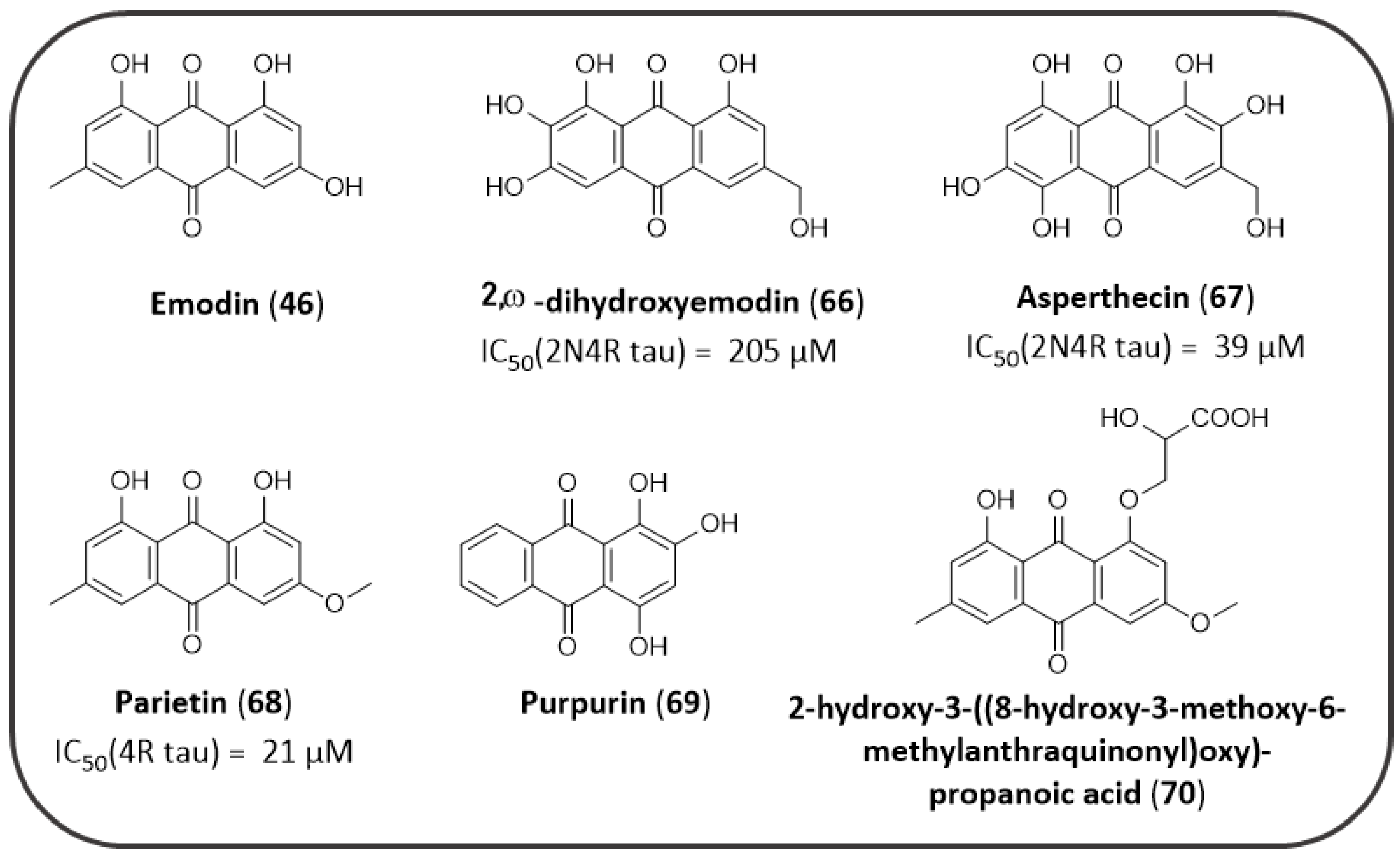
4.2. AQs from Natural Sources: Tau Aggregation Inhibitors
4.3. AQs from Natural Sources: BACE1 Inhibitors and Antioxidants
4.4. Synthetic AQ Derivatives
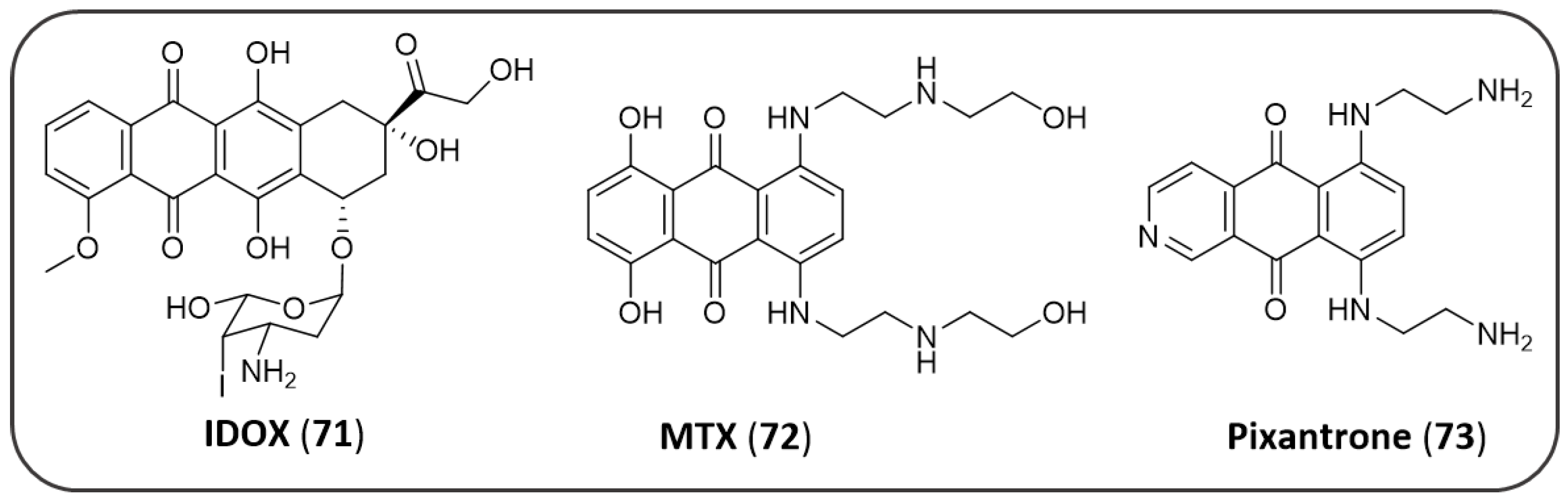
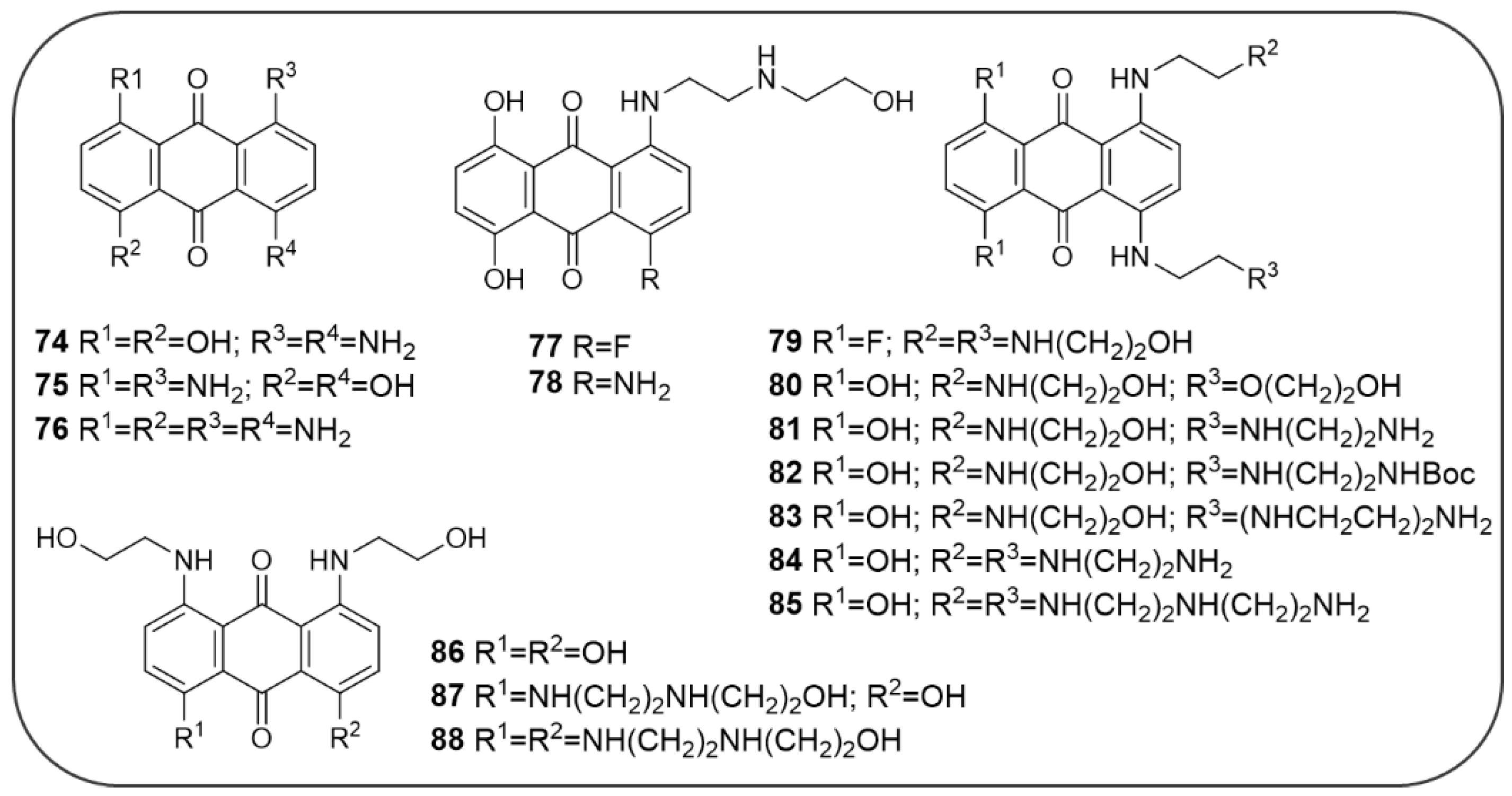
4.5. AQ-Based Hybrids
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Adelina, C. The costs of dementia: Advocacy, media and stigma. In World Alzheimer Report 2019: Attitudes to Dementia; Alzheimer’s Disease International: London, UK, 2019; Chapter 3; pp. 100–101. Available online: https://www.alzint.org/u/WorldAlzheimerReport2019.pdf (accessed on 20 September 2019).
- Glenner, G.G.; Murphy, M.A. Amyloidosis of the nervous system. J. Neurol. Sci. 1989, 94, 1–28. [Google Scholar] [CrossRef]
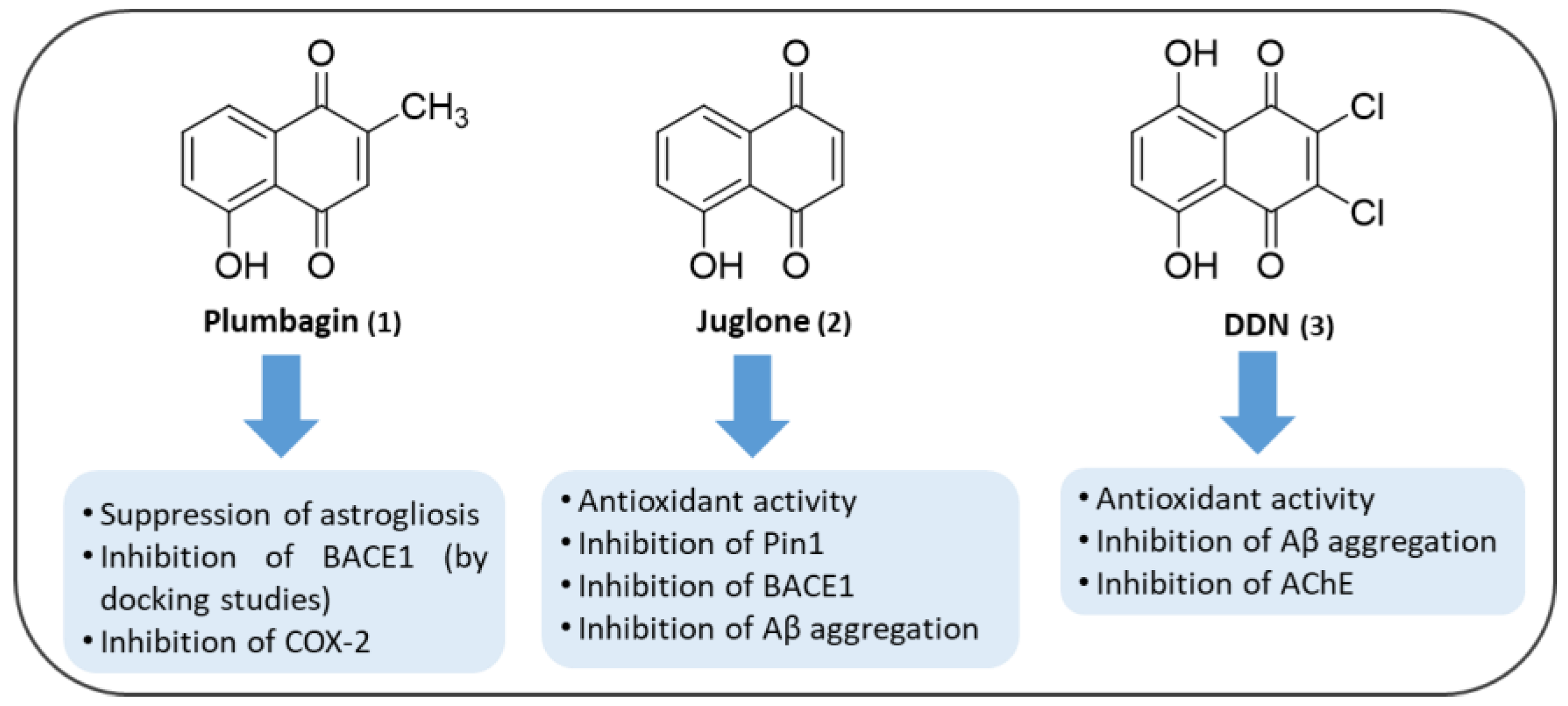
- Nakhate, K.T.; Bharne, A.P.; Verma, V.S.; Aru, D.N.; Kokare, D.M. Plumbagin ameliorates memory dysfunction in streptozotocin induced Alzheimer’s disease via activation of Nrf2/ARE pathway and inhibition of β-secretase. Biomed. Pharmacother. 2018, 101, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terry, R.D.; Gonatas, N.K.; Weiss, M. Ultrastructural studies in Alzheimer’s presenile dementia. Am. J. Pathol. 1964, 44, 269–297. [Google Scholar]
- Coyle, J.; Puttfarcken, P. Oxidative stress, glutamate, and neurodegenerative disorders. Science 1993, 262, 689–695. [Google Scholar] [CrossRef]
- Huang, X.; Moir, R.D.; Tanzi, R.E.; Bush, A.I.; Rogers, J.T. Redox-active metals, oxidative stress, and Alzheimer’s disease pathology. Ann. N. Y. Acad. Sci. 2004, 1012, 153–163. [Google Scholar] [CrossRef]
- Alam, J.; Sharma, L. Potential Enzymatic Targets in Alzheimer’s: A Comprehensive Review. Curr. Drug Targets 2018, 20, 316–339. [Google Scholar] [CrossRef]
- Blaikie, L.; Kay, G.; Lin, P.K.T. Current and emerging therapeutic targets of alzheimer’s disease for the design of multi-target directed ligands. Med. Chem. Comm. 2019, 10, 2052–2072. [Google Scholar] [CrossRef]
- Guo, J.; Wang, Z.; Liu, R.; Huang, Y.; Zhang, N.; Zhang, R. Memantine, Donepezil, or Combination Therapy—What is the best therapy for Alzheimer’s Disease? A Network Meta-Analysis. Brain Behav. 2020, 10, e01831. [Google Scholar] [CrossRef]
- Selkoe, D. Normal and Abnormal Biology of the β-Amyloid Precursor Protein. Annu. Rev. Neurosci. 1994, 17, 489–517. [Google Scholar] [CrossRef]
- Nhan, H.S.; Chiang, K.; Koo, E.H. The multifaceted nature of amyloid precursor protein and its proteolytic fragments: Friends and foes. Acta Neuropathol. 2015, 129, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Multhaup, G.; Huber, O.; Buée, L.; Galas, M.C. Amyloid Precursor Protein (APP) Metabolites APP Intracellular Fragment (AICD), Aβ42, and Tau in Nuclear Roles. J. Biol. Chem. 2015, 290, 23515–23522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goate, A.; Chartier-Harlin, M.C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef] [PubMed]
- De Jonghe, C. Pathogenic APP mutations near the gamma-secretase cleavage site differentially affect Aβ secretion and APP C-terminal fragment stability. Hum. Mol. Genet. 2001, 10, 1665–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, O.S.; Vasenina, E.E. 25 Years of the Amyloid Hypothesis of the Origin of Alzheimer’s Disease: Advances, Failures, and New Perspectives. Neurosci. Behav. Physiol. 2017, 47, 1065–1070. [Google Scholar] [CrossRef]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Agrawal, N.; Skelton, A.A. Structure and Function of Alzheimer’s Amyloid βeta Proteins from Monomer to Fibrils: A Mini Review. Protein J. 2019, 38, 425–434. [Google Scholar] [CrossRef]
- Yoshiike, Y.; Chui, D.-H.; Akagi, T.; Tanaka, N.; Takashima, A. Specific Compositions of Amyloid-β Peptides as the Determinant of Toxic β-Aggregation. J. Biol. Chem. 2003, 278, 23648–23655. [Google Scholar] [CrossRef] [Green Version]
- Kirkitadze, M.D.; Condron, M.M.; Teplow, D.B. Identification and characterization of key kinetic intermediates in amyloid β-protein fibrillogenesis. J. Mol. Biol. 2001, 312, 1103–1119. [Google Scholar] [CrossRef]
- Walsh, D.M.; Lomakin, A.; Benedek, G.B.; Condron, M.M.; Teplow, D.B. Amyloid β-protein fibrillogenesis: Detection of a protofibrillar intermediate. J. Biol. Chem. 1997, 272, 22364–22372. [Google Scholar] [CrossRef] [Green Version]
- Marina, G.B.; Kirkitadze, D.; Lomakin, A.; Vollers, S.S.; Benedek, G.B.; Teplow, D.B. Amyloid β-protein (Aβ) assembly: Aβ40 and Aβ42 oligomerize through distinct pathways. Proc. Natl. Acad. Sci. USA 2003, 100, 330–335. [Google Scholar] [CrossRef] [Green Version]
- Kirkitadze, M.D.; Kowalska, A. Molecular mechanisms initiating amyloid β-fibril formation in Alzheimer’s disease. Acta Biochim. Pol. 2005, 52, 417–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, J.; Higgins, G. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Ettcheto, M.; Petrov, D.; Abad, S.; Pedrós, I.; Marin, M.; Olloquequi, J.; Camins, A. Review of the advances in treatment for Alzheimer disease: Strategies for combating β-amyloid protein. Neurol. Engl. Ed. 2018, 33, 47–58. [Google Scholar] [CrossRef]
- Hsiao, C.C.; Rombouts, F.; Gijsen, H.J.M. New evolutions in the BACE1 inhibitor field from 2014 to 2018. Bioorg. Med. Chem. Lett. 2019, 29, 761–777. [Google Scholar] [CrossRef] [PubMed]
- Moussa-Pacha, N.M.; Abdin, S.M.; Omar, H.A.; Alniss, H.; Al-Tel, T.H. BACE1 inhibitors: Current status and future directions in treating Alzheimer’s disease. Med. Res. Rev. 2020, 40, 339–384. [Google Scholar] [CrossRef]
- Nalivaeva, N.N.; Turner, A.J. Targeting amyloid clearance in Alzheimer’s disease as a therapeutic strategy. Br. J. Pharmacol. 2019, 176, 3447–3463. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Götz, J. Amyloid-β and tau—A toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 2011, 12, 67–72. [Google Scholar] [CrossRef]
- Guo, J.P.; Arai, T.; Miklossy, J.; McGeer, P.L. Aβ and tau form soluble complexes that may promote self aggregation of both into the insoluble forms in Alzheimer’s diseases. Proc. Natl. Acad. Sci. USA 2006, 103, 1953–1958. [Google Scholar] [CrossRef] [Green Version]
- Kaur, D.; Sharma, V.; Deshmukh, R. Activation of microglia and astrocytes: A roadway to neuroinflammation and Alzheimer’s disease. Inflammopharmacology 2019, 27, 663–677. [Google Scholar] [CrossRef]
- Blurton-Jones, M.; LaFerla, F. Pathways by Which Aβ Facilitates Tau Pathology. Curr. Alzheimer Res. 2006, 3, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Roher, A.E.; Baudry, J.; Chaney, M.O.; Kuo, Y.M.; Stine, W.B.; Emmerling, M.R. Oligomerization and fibril assembly of the amyloid-β protein. Biochim. Biophys. Acta 2000, 1502, 31–43. [Google Scholar] [CrossRef] [Green Version]
- Bartus, R.T.; Dean, R.L., III; Beer, B.; Lippa, A.S. The cholinergic hypothesis of geriatric memory dysfunction. Science 1982, 217, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Maloney, A.J. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976, 2, 1403. [Google Scholar] [CrossRef]
- Picciotto, M.R.; Higley, M.J.; Mineur, Y.S. Acetylcholine as a Neuromodulator: Cholinergic Signaling Shapes Nervous System Function and Behavior. Neuron 2012, 76, 116–129. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.-Q.; Mobley, W.C. Exploring the Pathogenesis of Alzheimer Disease in Basal Forebrain Cholinergic Neurons: Converging Insights from Alternative Hypotheses. Front. Neurosci. 2019, 13, 446. [Google Scholar] [CrossRef] [Green Version]
- Francis, P.T.; Palmer, A.M.; Snape, M.; Wilcock, G.K. The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J. Neurol. Neurosurg. Psychiatry 1999, 66, 137–147. [Google Scholar] [CrossRef]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [Green Version]
- David, L.; Cheah, E.; Cygler, M.; Dijkstra, B.; Frolow, F.; Sybille, M.; Harel, M.; James Remington, S.; Silman, I.; Schrag, J.; et al. The α/β hydrolase fold. Protein Eng. Des. Sel. 1992, 5, 197–211. [Google Scholar] [CrossRef] [Green Version]
- Mehta, M.; Adem, A.; Sabbagh, M. New Acetylcholinesterase Inhibitors for Alzheimer’s Disease. Int. J. Alzheimers Dis. 2012, 2012, 1–8. [Google Scholar] [CrossRef]
- Deture, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacobini, E. Cholinesterases: New roles in brain function and in Alzheimer’s disease. Neurochem. Res. 2003, 28, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Stanciu, G.D.; Luca, A.; Rusu, R.N.; Bild, V.; Chiriac, S.I.B.; Solcan, C.; Bild, W.; Ababei, D.C. Alzheimer’s disease pharmacotherapy in relation to cholinergic system involvement. Biomolecules 2020, 10, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tougu, V. Acetylcholinesterase: Mechanism of catalysis and inhibition. Curr. Med. Chem. CNS Agents 2001, 1, 155–170. [Google Scholar] [CrossRef]
- Zhang, Y.; Kua, J.; McCammon, J.A. Role of the Catalytic Triad and Oxyanion Hole in Acetylcholinesterase Catalysis: An ab initio QM/MM Study. J. Am. Chem. Soc. 2002, 124, 10572–10577. [Google Scholar] [CrossRef] [PubMed]
- Silman, I.; Sussman, J.L. Acetylcholinesterase: How is structure related to function? Chem. Biol. Interact. 2008, 175, 3–10. [Google Scholar] [CrossRef]
- Rees, T.M.; Brimijoin, S. The role of acetylcholinesterase in the pathogenesis of Alzheimer’s disease. Drugs Today 2003, 39, 75–83. [Google Scholar] [CrossRef]
- Augustin, N.; Nuthakki, V.K.; Abdullaha, M.; Hassan, Q.P.; Gandhi, S.G.; Bharate, S.B. Discovery of Helminthosporin, an Anthraquinone Isolated from Rumex abyssinicus Jacq as a Dual Cholinesterase Inhibitor. ACS Omega 2020, 5, 1616–1624. [Google Scholar] [CrossRef] [Green Version]
- Inestrosa, N.C.; Dinamarca, M.C.; Alvarez, A. Amyloid-cholinesterase interactions: Implications for Alzheimer’s disease. FEBS J. 2008, 275, 625–632. [Google Scholar] [CrossRef]
- VandeVrede, L.; Boxer, A.L.; Polydoro, M. Targeting tau: Clinical trials and novel therapeutic approaches. Neurosci. Lett. 2020, 731, 134919. [Google Scholar] [CrossRef]
- Naseri, N.N.; Wang, H.; Guo, J.; Sharma, M.; Luo, W. The complexity of tau in Alzheimer’s disease. Neurosci. Lett. 2019, 705, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Drombosky, K.; Hou, Z.; Sari, L.; Kashmer, O.; Ryder, B.; Perez, V.; Woodard, D.; Lin, M.; Diamond, M.; et al. Tau local structure shields amyloid motif and controls aggregation propensity. bioRxiv 2018, 330266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noble, W.; Hanger, D.P.; Miller, C.C.J.; Lovestone, S. The importance of tau phosphorylation for neurodegenerative diseases. Front. Neurol. 2013, 4, 1–11. [Google Scholar] [CrossRef] [Green Version]
- López López, L.I.; Nery Flores, S.D.; Silva Belmares, S.Y.; Sáenz Galindo, A. Naphthoquinones: Biological properties and synthesis of lawsone and derivatives—A structured review. Vitae 2014, 21, 248–258. [Google Scholar]
- Vukic, M.D.; Vukovic, N.L.; Djelic, G.T.; Popovic, S.L.; Zaric, M.M.; Baskic, D.D.; Krstic, G.B.; Tesevic, V.V.; Kacaniova, M.M. Antibacterial and cytotoxic activities of naphthoquinone pigments from onosma Visianii clem. EXCLI J. 2017, 16, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Tandon, V.K.; Chhor, R.B.; Singh, R.V.; Rai, S.; Yadav, D.B. Design, synthesis and evaluation of novel 1,4-naphthoquinone derivatives as antifungal and anticancer agents. Bioorganic Med. Chem. Lett. 2004, 14, 1079–1083. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.H.; Yoo, J.; Park, S.H.; Jung, J.K.; Cho, H.; Chung, Y. Synthesis and evaluation of antitumor activity of novel 1,4-naphthoquinone derivatives (IV). Arch. Pharm. Res. 2006, 29, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Alemán, J.; Jacobsen, C.B.; Frisch, K.; Overgaard, J.; Jørgensen, K.A. Organocatalytic asymmetric vinylogous addition to quinones—Formation of optically active α-aryl ketones. Chem. Commun. 2002, 8, 632–634. [Google Scholar] [CrossRef]
- Sun, J.W.; Wang, X.S.; Liu, Y. Copper(II)-catalyzed sequential C,N-difunctionalization of 1,4-naphthoquinone for the synthesis of benzo[f]indole-4,9-diones under base-free condition. J. Org. Chem. 2013, 78, 10560–10566. [Google Scholar] [CrossRef]
- Lee, J.; Panek, J.S. Synthesis of Isochromene-Type Scaffolds via Single-Flask Diels–Alder-[4 + 2]-Annulation Sequence of a Silyl-Substituted Diene with Menadione. Org. Lett. 2014, 16, 3320–3323. [Google Scholar] [CrossRef]
- Albrecht, Ł.; Gómez, C.V.; Jacobsen, C.B.; Jørgensen, K.A. 1,4-naphthoquinones in H-bond-directed trienamine-mediated strategies. Org. Lett. 2013, 15, 3010–3013. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Liu, T.; Tao, H.; Wang, C.J. A facile access to enantioenriched isoindolines via one-pot sequential Cu(I)-catalyzed asymmetric 1,3-dipolar cycloaddition/aromatization. Org. Lett. 2012, 14, 6230–6233. [Google Scholar] [CrossRef] [PubMed]
- Murphy, B.; Goodrich, P.; Hardacre, C.; Oelgemöller, M. Green photochemistry: Photo-Friedel–Crafts acylations of 1,4-naphthoquinone in room temperature ionic liquids. Green Chem. 2009, 11, 1867–1870. [Google Scholar] [CrossRef]
- Yu, J.-S. 1,4-Naphthoquinone. Synlett 2014, 25, 2377–2378. [Google Scholar] [CrossRef] [Green Version]
- Berkessel, A.; Guixà, M.; Schmidt, F.; Neudörfl, J.M.; Lex, J. Highly enantioselective epoxidation of 2-methylnaphthoquinone (vitamin K3) mediated by new cinchona alkaloid phase-transfer catalysts. Chem. A Eur. J. 2007, 13, 4483–4498. [Google Scholar] [CrossRef]
- Scherzer-Attali, R.; Pellarin, R.; Convertino, M.; Frydman-Marom, A.; Egoz-Matia, N.; Peled, S.; Levy-Sakin, M.; Shalev, D.E.; Caflisch, A.; Gazit, E.; et al. Complete phenotypic recovery of an Alzheimer’s disease model by a quinone-tryptophan hybrid aggregation inhibitor. PLoS ONE 2010, 5, e11101. [Google Scholar] [CrossRef]
- Nam, K.N.; Son, M.S.; Park, J.H.; Lee, E.H. Shikonins attenuate microglial inflammatory responses by inhibition of ERK, Akt, and NF-κB: Neuroprotective implications. Neuropharmacology 2008, 55, 819–825. [Google Scholar] [CrossRef]
- Castagnoli, K.P.; Steyn, S.J.; Petzer, J.P.; Van Der Schyf, C.J.; Castagnoli, N. Neuroprotection in the MPTP parkinsonian C57BL/6 mouse model by a compound isolated from tobacco. Chem. Res. Toxicol. 2001, 14, 523–527. [Google Scholar] [CrossRef]
- Bermejo-Bescós, P.; Martín-Aragón, S.; Jiménez-Aliaga, K.L.; Ortega, A.; Molina, M.T.; Buxaderas, E.; Orellana, G.; Csákÿ, A.G. In vitro antiamyloidogenic properties of 1,4-naphthoquinones. Biochem. Biophys. Res. Commun. 2010, 400, 169–174. [Google Scholar] [CrossRef]
- Dutra, R.C.; Campos, M.M.; Santos, A.R.S.; Calixto, J.B. Medicinal plants in Brazil: Pharmacological studies, drug discovery, challenges and perspectives. Pharmacol. Res. 2016, 112, 4–29. [Google Scholar] [CrossRef]
- Raskin, I.; Ribnicky, D.M.; Komarnytsky, S.; Ilic, N.; Poulev, A.; Borisjuk, N.; Brinker, A.; Moreno, D.A.; Ripoll, C.; Yakoby, N.; et al. Plants and human health in the twenty-first century. Trends Biotechnol. 2002, 20, 522–531. [Google Scholar] [CrossRef]
- Soejarto, D.D.; Farnsworth, N.R. Tropical rain forests: Potential source of new drugs? Perspect. Biol. Med. 1989, 32, 244–256. [Google Scholar] [CrossRef] [PubMed]
- Tshisikhawe, M.P.; van Rooyen, M.W.; Bhat, R.B. An evaluation of the extent and threat of bark harvesting of medicinal plant species in the venda region, limpopo province, south africa. Phyton (B. Aires) 2012, 81, 89–100. [Google Scholar]
- Gordaliza, M. Natural products as leads to anticancer drugs. Clin. Transl. Oncol. 2007, 9, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Coseri, S. Natural Products and their Analogues as Efficient Anticancer Drugs. Mini-Rev. Med. Chem. 2009, 9, 560–571. [Google Scholar] [CrossRef]
- de Carvalho da Silva, F.; Francisco Ferreira, V. Natural Naphthoquinones with Great Importance in Medicinal Chemistry. Curr. Org. Synth. 2016, 13, 334–371. [Google Scholar] [CrossRef]
- Thomson, R.H.; Thomson, R.H. Benzoquinones. In Naturally Occurring Quinones IV; Springer Netherlands: Heidelberg, Germany, 1997; pp. 1–111. ISBN 978-94-009-1551-0. [Google Scholar]
- Patai, S.; Rappoport, Z. The Chemistry of the Quinonoid Compounds. In The Chemistry of the Quinonoid Compounds; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1988; Volume 2, pp. 1–878. ISBN 9780470772119. [Google Scholar]
- Babula, P.; Adam, V.; Havel, L.; Kizek, R. Noteworthy secondary metabolites naphthoquinone—Their occurence, pharmacological properties and analysis. Curr. Pharm. Anal. 2009, 5, 47–68. [Google Scholar] [CrossRef]
- Van der Vijver, L.M. Distribution of plumbagin in the Plumbaginaceae. Phytochemistry 1972, 11, 3247–3248. [Google Scholar] [CrossRef]
- Hazra, B.; Das Sarma, M.; Sanyal, U. Separation methods of quinonoid constituents of plants used in Oriental traditional medicines. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2004, 812, 259–275. [Google Scholar] [CrossRef]
- Padhye, S.; Dandawate, P.; Yusufi, M.; Ahmad, A.; Sarkar, F.H. Perspectives on medicinal properties of plumbagin and its analogs. Med. Res. Rev. 2012, 32, 1131–1158. [Google Scholar] [CrossRef]
- Ahmad, A.; Syed, F.A.; Singh, S.; Hadi, S.M. Prooxidant activity of resveratrol in the presence of copper ions: Mutagenicity in plasmid DNA. Toxicol. Lett. 2005, 159, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.S.; Ahmad, A.; Hadi, S.M. Anti-oxidant, pro-oxidant properties of tannic acid and its binding to DNA. Chem. Biol. Interact. 2000, 125, 177–189. [Google Scholar] [CrossRef]
- Checker, R.; Sharma, D.; Sandur, S.K.; Khanam, S.; Poduval, T.B. Anti-inflammatory effects of plumbagin are mediated by inhibition of NF-kappaB activation in lymphocytes. Int. Immunopharmacol. 2009, 9, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J. NF-κB-regulated, proinflammatory miRNAs in Alzheimer’s disease. Alzheimer’s Res. Ther. 2012, 4, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Son, T.G.; Camandola, S.; Arumugam, T.V.; Cutler, R.G.; Telljohann, R.S.; Mughal, M.R.; Moore, T.A.; Luo, W.; Yu, Q.S.; Johnson, D.A.; et al. Plumbagin, a novel Nrf2/ARE activator, protects against cerebral ischemia. J. Neurochem. 2010, 112, 1316–1326. [Google Scholar] [CrossRef] [Green Version]
- Colaric, M.; Veberic, R.; Solar, A.; Hudina, M.; Stampar, F. Phenolic acids, syringaldehyde, and juglone in fruits of different cultivars of Juglans regia L. J. Agric. Food Chem. 2005, 53, 6390–6396. [Google Scholar] [CrossRef]
- Inbaraj, J.J.; Chignell, C.F. Cytotoxic Action of Juglone and Plumbagin: A Mechanistic Study Using HaCaT Keratinocytes. Chem. Res. Toxicol. 2004, 17, 55–62. [Google Scholar] [CrossRef]
- Ahmad, T.; Suzuki, Y.J. Juglone in Oxidative Stress and Cell Signaling. Antioxidants 2019, 8, 91. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.M.; Jurgens, T.M.; Hufford, C.D. Antimicrobial activity of juglone. Phyther. Res. 1990, 4, 11–14. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, A.; Li, Y.; Zhao, X.; Lv, S.; Zhu, W.; Jin, Y. Anticancer activity and mechanism of juglone on human cervical carcinoma hela cells. Can. J. Physiol. Pharmacol. 2012, 90, 1553–1558. [Google Scholar] [CrossRef]
- Fang, F.; Chen, S.; Ma, J.; Cui, J.; Li, Q.; Meng, G.; Wang, L. Juglone suppresses epithelial-mesenchymal transition in prostate cancer cells via the protein kinase b/glycogen synthase kinase-3β/snail signaling pathway. Oncol. Lett. 2018, 16, 2579–2584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugie, S.; Okamoto, K.; Rahman, K.M.W.; Tanaka, T.; Kawai, K.; Yamahara, J.; Mori, H. Inhibitory effects of plumbagin and juglone on azoxymethane-induced intestinal carcinogenesis in rats. Cancer Lett. 1998, 127, 177–183. [Google Scholar] [CrossRef]
- Chobot, V.; Hadacek, F. Milieu-dependent pro- and antioxidant activity of juglone may explain linear and nonlinear effects on seedling development. J. Chem. Ecol. 2009, 35, 383–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Chen, Y.; Zhang, Y.; Du, J.; Lv, Y.; Mo, S.; Liu, Y.; Ding, F.; Wu, J.; Li, J. Juglone potentiates TRAIL-induced apoptosis in human melanoma cells via activating the ROS-p38-p53 pathway. Mol. Med. Rep. 2017, 16, 9645–9651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Liu, K.; Wang, X.F.; Sun, D.J. Juglone reduces growth and migration of U251 glioblastoma cells and disrupts angiogenesis. Oncol. Rep. 2017, 38, 1959–1966. [Google Scholar] [CrossRef]
- Jin, R. A DFT study on the radical scavenging activity of juglone and its derivatives. J. Mol. Struct. Theochem. 2010, 939, 9–13. [Google Scholar] [CrossRef]
- De Castro, E.; De Castro, S.H.; Johnson, T.E. Isolation of long-lived mutants in Caenorhabditis elegans using selection for resistance to juglone. Free Radic. Biol. Med. 2004, 37, 139–145. [Google Scholar] [CrossRef]
- Tamafo Fouegue, A.D.; Ghogomu, J.N.; Bikélé Mama, D.; Nkungli, N.K.; Younang, E. Structural and Antioxidant Properties of Compounds Obtained from Fe2+ Chelation by Juglone and Two of Its Derivatives: DFT, QTAIM, and NBO Studies. Bioinorg. Chem. Appl. 2016, 2016, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Galano, A.; Mazzone, G.; Alvarez-Diduk, R.; Marino, T.; Alvarez-Idaboy, J.R.; Russo, N. Food Antioxidants: Chemical Insights at the Molecular Level. Annu. Rev. Food Sci. Technol. 2016, 7, 335–352. [Google Scholar] [CrossRef]
- Leopoldini, M.; Russo, N.; Toscano, M. The molecular basis of working mechanism of natural polyphenolic antioxidants. Food Chem. 2011, 125, 288–306. [Google Scholar] [CrossRef]
- Bentes, A.L.A.; Borges, R.S.; Monteiro, W.R.; De Macedo, L.G.M.; Alves, C.N. Structure of dihydrochalcones and related derivatives and their scavenging and antioxidant activity against oxygen and nitrogen radical species. Molecules 2011, 16, 1749–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leopoldini, M.; Marino, T.; Russo, N.; Toscano, M. Antioxidant properties of phenolic compounds: H-atom versus electron transfer mechanism. J. Phys. Chem. A 2004, 108, 4916–4922. [Google Scholar] [CrossRef]
- Gülçin, I.; Huyut, Z.; Elmastaş, M.; Aboul-Enein, H.Y. Radical scavenging and antioxidant activity of tannic acid. Arab. J. Chem. 2010, 3, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Galas, M.C.; Dourlen, P.; Bégard, S.; Ando, K.; Blum, D.; Hamdane, M.; Buée, L. The peptidylprolyl cis/trans-isomerase Pin1 modulates stress-induced dephosphorylation of Tau in neurons: Implication in a pathological mechanism related to Alzheimer disease. J. Biol. Chem. 2006, 281, 19296–19304. [Google Scholar] [CrossRef] [Green Version]
- Pandareesh, M.D.; Chauhan, V.; Chauhan, A. Walnut Supplementation in the Diet Reduces Oxidative Damage and Improves Antioxidant Status in Transgenic Mouse Model of Alzheimer’s Disease. J. Alzheimers. Dis. 2018, 64, 1295–1305. [Google Scholar] [CrossRef] [Green Version]
- Hennig, L.; Christner, C.; Kipping, M.; Schelbert, B.; Rücknagel, K.P.; Grabley, S.; Küllertz, G.; Fischer, G. Selective inactivation of parvulin-like peptidyl-prolyl cis/trans isomerases by juglone. Biochemistry 1998, 37, 5953–5960. [Google Scholar] [CrossRef]
- Jordens, J.; Janssens, V.; Longin, S.; Stevens, I.; Martens, E.; Bultynck, G.; Engelborghs, Y.; Lescrinier, E.; Waelkens, E.; Goris, J.; et al. The protein phosphatase 2A phosphatase activator is a novel peptidyl-prolyl cis/trans-isomerase. J. Biol. Chem. 2006, 281, 6349–6357. [Google Scholar] [CrossRef] [Green Version]
- Hamdane, M.; Smet, C.; Sambo, A.V.; Leroy, A.; Wieruszeski, J.M.; Delobel, P.; Maurage, C.A.; Ghestem, A.; Wintjens, R.; Bégard, S.; et al. Pin1: A therapeutic target in Alzheimer neurodegeneration. J. Mol. Neurosci. 2002, 19, 275–287. [Google Scholar] [CrossRef]
- Khelifi, I.; Tourrette, A.; Dhouafli, Z.; Bouajila, J.; Efferth, T.; Abdelfatah, S.; Ksouri, R.; Hayouni, E.A. The antioxidant 2,3-dichloro,5,8-dihydroxy,1,4-naphthoquinone inhibits acetyl-cholinesterase activity and amyloid β 42 aggregation: A dual target therapeutic candidate compound for the treatment of Alzheimer’s disease. Biotechnol. Appl. Biochem. 2020, bab.1870. [Google Scholar] [CrossRef]
- Chung, Y.C.; Chien, C.T.; Teng, K.Y.; Chou, S.T. Antioxidative and mutagenic properties of Zanthoxylum ailanthoides Sieb & zucc. Food Chem. 2006, 97, 418–425. [Google Scholar] [CrossRef]
- Woo, H.C.; Seong, S.H.; Lee, S.A.; Xiang, H.H.; Kyong, S.L.; Myung, K.L.; Bang, Y.H.; Jai, S.R. Monoamine oxidase inhibitory naphthoquinones from the roots of Lithospermum erythrorhizon. Arch. Pharm. Res. 2005, 28, 400–404. [Google Scholar] [CrossRef]
- Mostert, S.; Petzer, A.; Petzer, J.P. Evaluation of Natural and Synthetic 1,4-naphthoquinones as Inhibitors of Monoamine Oxidase. Chem. Biol. Drug Des. 2016, 87, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z. Monoamine oxidase inhibitors: Promising therapeutic agents for Alzheimer’s disease (Review). Mol. Med. Rep. 2014, 9, 1533–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabrese, V.; Cornelius, C.; Mancuso, C.; Pennisi, G.; Calafato, S.; Bellia, F.; Bates, T.E.; Giuffrida Stella, A.M.; Schapira, T.; Dinkova Kostova, A.T.; et al. Cellular stress response: A novel target for chemoprevention and nutritional neuroprotection in aging, neurodegenerative disorders and longevity. Neurochem. Res. 2008, 33, 2444–2471. [Google Scholar] [CrossRef] [PubMed]
- Neo Shin, N.; Jeon, H.; Jung, Y.; Baek, S.; Lee, S.; Yoo, H.C.; Bae, G.H.; Park, K.; Yang, S.H.; Han, J.M.; et al. Fluorescent 1,4-Naphthoquinones to Visualize Diffuse and Dense-Core Amyloid Plaques in APP/PS1 Transgenic Mouse Brains. ACS Chem. Neurosci. 2019, 10, 3031–3044. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.L.; Chiriano, G.P.; Bartolini, M.; Mancini, F.; Bottegoni, G.; Maestri, V.; Czvitkovich, S.; Windisch, M.; Cavalli, A.; Minarini, A.; et al. Synthesis of monomeric derivatives to probe memoquin’s bivalent interactions. J. Med. Chem. 2011, 54, 8299–8304. [Google Scholar] [CrossRef]
- Bolognesi, M.L.; Bartolini, M.; Tarozzi, A.; Morroni, F.; Lizzi, F.; Milelli, A.; Minarini, A.; Rosini, M.; Hrelia, P.; Andrisano, V.; et al. Multitargeted drugs discovery: Balancing anti-amyloid and anticholinesterase capacity in a single chemical entity. Bioorg. Med. Chem. Lett. 2011, 21, 2655–2658. [Google Scholar] [CrossRef]
- Bolognesi, M.L.; Banzi, R.; Bartolini, M.; Cavalli, A.; Tarozzi, A.; Andrisano, V.; Minarini, A.; Rosini, M.; Tumiatti, V.; Bergamini, C.; et al. Novel Class of Quinone-Bearing Polyamines as Multi-Target-Directed Ligands to Combat Alzheimer’s Disease. J. Med. Chem. 2007, 50, 4882–4897. [Google Scholar] [CrossRef]
- Bartolini, M.; Bertucci, C.; Cavrini, V.; Andrisano, V. β-Amyloid aggregation induced by human acetylcholinesterase: Inhibition studies. Biochem. Pharmacol. 2003, 65, 407–416. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Alvarez, A.; Pérez, C.A.; Moreno, R.D.; Vicente, M.; Linker, C.; Casanueva, O.I.; Soto, C.; Garrido, J. Acetylcholinesterase accelerates assembly of amyloid-β-peptides into Alzheimer’s fibrils: Possible role of the peripheral site of the enzyme. Neuron 1996, 16, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Czvitkovich, S.; Duller, S.; Mathiesen, E.; Lorenzoni, K.; Imbimbo, B.P.; Hutter-Paier, B.; Windisch, M.; Wronski, R. Comparison of pharmacological modulation of APP metabolism in primary chicken telencephalic neurons and in a human neuroglioma cell line. J. Mol. Neurosci. 2011, 43, 257–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonelli, M.; Catto, M.; Tasso, B.; Novelli, F.; Canu, C.; Iusco, G.; Pisani, L.; De Stradis, A.; Denora, N.; Sparatore, A.; et al. Multitarget therapeutic leads for Alzheimer’s disease: Quinolizidinyl derivatives of Bi- and tricyclic systems as dual inhibitors of cholinesterases and β-amyloid (Aβ) aggregation. ChemMedChem 2015, 10, 1040–1053. [Google Scholar] [CrossRef] [PubMed]
- Puglielli, L. Two endoplasmic reticulum (ER)/ER Golgi intermediate compartment-based lysine acetyltransferases posttranslationally regulate BACE1 levels. J. Biol. Chem. 2009, 284, 2482–2492. [Google Scholar]
- Ding, Y.; Ko, M.H.; Pehar, M.; Kotch, F.; Peters, N.R.; Luo, Y.; Salamat, S.M.; Puglielli, L. Biochemical inhibition of the acetyltransferases ATase1 and ATase2 reduces β-secretase (BACE1) levels and Aβ generation. J. Biol. Chem. 2012, 287, 8424–8433. [Google Scholar] [CrossRef] [Green Version]
- Morphy, R.; Kay, C.; Rankovic, Z. From magic bullets to designed multiple ligands. Drug Discov. Today 2004, 9, 641–651. [Google Scholar] [CrossRef]
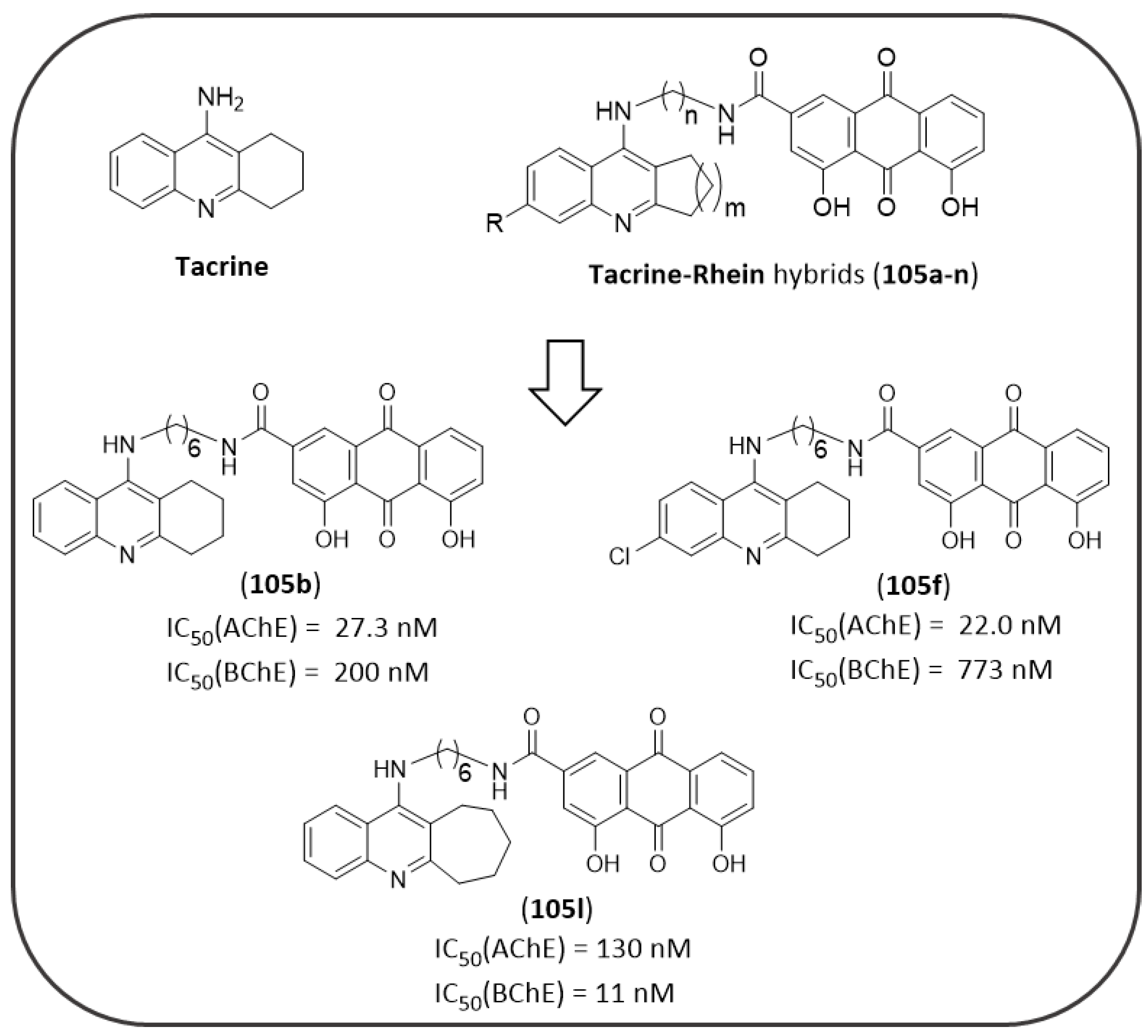
- Watkins, P.B.; Zimmerman, H.J.; Knapp, M.J.; Gracon, S.I.; Lewis, K.W. Hepatotoxic Effects of Tacrine Administration in Patients with Alzheimer’s Disease. JAMA 1994, 271, 992–998. [Google Scholar] [CrossRef]
- Romero, A.; Cacabelos, R.; Oset-Gasque, M.J.; Samadi, A.; Marco-Contelles, J. Novel tacrine-related drugs as potential candidates for the treatment of Alzheimer’s disease. Bioorganic Med. Chem. Lett. 2013, 23, 1916–1922. [Google Scholar] [CrossRef]
- Chioua, M.; Serrano, E.; Dgachi, Y.; Martin, H.; Jun, D.; Janockova, J.; Sepsova, V.; Soukup, O.; Moraleda, I.; Chabchoub, F.; et al. Synthesis, Biological Assessment and Molecular Modeling of Racemic QuinoPyranoTacrines for Alzheimer’s Disease Therapy. ChemistrySelect 2018, 3, 461–466. [Google Scholar] [CrossRef]
- Nepovimova, E.; Uliassi, E.; Korabecny, J.; Peña-Altamira, L.E.; Samez, S.; Pesaresi, A.; Garcia, G.E.; Bartolini, M.; Andrisano, V.; Bergamini, C.; et al. Multitarget drug design strategy: Quinone-tacrine hybrids designed to block amyloid-β aggregation and to exert anticholinesterase and antioxidant effects. J. Med. Chem. 2014, 57, 8576–8589. [Google Scholar] [CrossRef]
- Galdeano, C.; Viayna, E.; Sola, I.; Formosa, X.; Camps, P.; Badia, A.; Clos, M.V.; Relat, J.; Ratia, M.; Bartolini, M.; et al. Huprine-tacrine heterodimers as anti-amyloidogenic compounds of potential interest against Alzheimer’s and prion diseases. J. Med. Chem. 2012, 55, 661–669. [Google Scholar] [CrossRef]
- Soukup, O.; Jun, D.; Zdarova-Karasova, J.; Patocka, J.; Musilek, K.; Korabecny, J.; Krusek, J.; Kaniakova, M.; Sepsova, V.; Mandikova, J.; et al. A Resurrection of 7-MEOTA: A Comparison with Tacrine. Curr. Alzheimer Res. 2013, 10, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Pawar, A.P.; DuBay, K.F.; Zurdo, J.; Chiti, F.; Vendruscolo, M.; Dobson, C.M. Prediction of “aggregation-prone” and “aggregation- susceptible” regions in proteins associated with neurodegenerative diseases. J. Mol. Biol. 2005, 350, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Xu, W.; Mu, Y.; Derreumaux, P. Atomic and dynamic insights into the beneficial effect of the 1,4-naphthoquinon-2-yl- l -tryptophan inhibitor on Alzheimer’s Aβ1-42 dimer in terms of aggregation and toxicity. ACS Chem. Neurosci. 2014, 5, 148–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherzer-Attali, R.; Farfara, D.; Cooper, I.; Levin, A.; Ben-Romano, T.; Trudler, D.; Vientrov, M.; Shaltiel-Karyo, R.; Shalev, D.E.; Segev-Amzaleg, N.; et al. Naphthoquinone-tyrptophan reduces neurotoxic Aβ*56 levels and improves cognition in Alzheimer’s disease animal model. Neurobiol. Dis. 2012, 46, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Scherzer-Attali, R.; Shaltiel-Karyo, R.; Adalist, Y.H.; Segal, D.; Gazit, E. Generic inhibition of amyloidogenic proteins by two naphthoquinone-tryptophan hybrid molecules. Proteins Struct. Funct. Bioinforma. 2012, 80, 1962–1973. [Google Scholar] [CrossRef] [PubMed]
- Frenkel-Pinter, M.; Tal, S.; Scherzer-Attali, R.; Abu-Hussien, M.; Alyagor, I.; Eisenbaum, T.; Gazit, E.; Segal, D. Cl-NQTrp Alleviates Tauopathy Symptoms in a Model Organism through the Inhibition of Tau Aggregation-Engendered Toxicity. Neurodegener. Dis. 2017, 17, 73–82. [Google Scholar] [CrossRef]
- Frenkel-Pinter, M.; Tal, S.; Scherzer-Attali, R.; Abu-Hussien, M.; Alyagor, I.; Eisenbaum, T.; Gazit, E.; Segal, D. Naphthoquinone-Tryptophan Hybrid Inhibits Aggregation of the Tau-Derived Peptide PHF6 and Reduces Neurotoxicity. J. Alzheimer’s Dis. 2016, 51, 165–178. [Google Scholar] [CrossRef]
- Dammers, C.; Yolcu, D.; Kukuk, L.; Willbold, D.; Pickhardt, M.; Mandelkow, E.; Horn, A.H.C.; Sticht, H.; Malhis, M.N.; Will, N.; et al. Selection and characterization of tau binding D-enantiomeric peptides with potential for therapy of Alzheimer disease. PLoS ONE 2016, 11, e0167432. [Google Scholar] [CrossRef] [Green Version]
- Friedhoff, P.; Von Bergen, M.; Mandelkow, E.M.; Mandelkow, E. Structure of tau protein and assembly into paired helical filaments. Biochim. Biophys. Acta—Mol. Basis Dis. 2000, 1502, 122–132. [Google Scholar] [CrossRef]
- Scherzer-Attali, R.; Convertino, M.; Pellarin, R.; Gazit, E.; Segal, D.; Caflisch, A. Methylations of tryptophan-modified naphthoquinone affect its inhibitory potential toward aβ aggregation. J. Phys. Chem. B 2013, 117, 1780–1789. [Google Scholar] [CrossRef]
- Paul, A.; Viswanathan, G.K.; Mahapatra, S.; Balboni, G.; Pacifico, S.; Gazit, E.; Segal, D. Antagonistic Activity of Naphthoquinone-Based Hybrids toward Amyloids Associated with Alzheimer’s Disease and Type-2 Diabetes. ACS Chem. Neurosci. 2019, 10, 3510–3520. [Google Scholar] [CrossRef] [PubMed]
- Linse, S. Mechanism of amyloid protein aggregation and the role of inhibitors. Pure Appl. Chem. 2019, 91, 211–229. [Google Scholar] [CrossRef]
- Arosio, P.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Chemical kinetics for drug discovery to combat protein aggregation diseases. Trends Pharmacol. Sci. 2014, 35, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Gazit, E. A possible role for π-stacking in the self-assembly of amyloid fibrils. FASEB J. 2002, 16, 77–83. [Google Scholar] [CrossRef]
- Bertolani, A.; Pirrie, L.; Stefan, L.; Houbenov, N.; Haataja, J.S.; Catalano, L.; Terraneo, G.; Giancane, G.; Valli, L.; Milani, R.; et al. Supramolecular amplification of amyloid self-assembly by iodination. Nat. Commun. 2015, 6, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Makin, O.S.; Atkins, E.; Sikorski, P.; Johansson, J.; Serpell, L.C. Molecular basis for amyloid fibril formation and stability. Proc. Natl. Acad. Sci. USA 2005, 102, 315–320. [Google Scholar] [CrossRef] [Green Version]
- KrishnaKumar, V.G.; Paul, A.; Gazit, E.; Segal, D. Mechanistic insights into remodeled Tau-derived PHF6 peptide fibrils by Naphthoquinone-Tryptophan hybrids. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Convertino, M.; Pellarin, R.; Catto, M.; Carotti, A.; Caflisch, A. 9,10-Anthraquinone hinders β-aggregation: How does a small molecule interfere with Aβ-peptide amyloid fibrillation? Protein Sci. 2009, 18, 792–800. [Google Scholar] [CrossRef] [Green Version]
- Churches, Q.I.; Caine, J.; Cavanagh, K.; Epa, V.C.; Waddington, L.; Tranberg, C.E.; Meyer, A.G.; Varghese, J.N.; Streltsov, V.; Duggan, P.J. Naturally occurring polyphenolic inhibitors of amyloid β aggregation. Bioorg. Med. Chem. Lett. 2014, 24, 3108–3112. [Google Scholar] [CrossRef]
- Chaturvedi, S.K.; Zaidi, N.; Alam, P.; Khan, J.M.; Qadeer, A.; Siddique, I.A.; Asmat, S.; Zaidi, Y.; Khan, R.H. Unraveling Comparative Anti-Amyloidogenic Behavior of Pyrazinamide and D-Cycloserine: A Mechanistic Biophysical Insight. PLoS ONE 2015, 10, e0136528. [Google Scholar] [CrossRef] [Green Version]
- Tjernberg, L.O.; Näslundt, J.; Lindqvist, F.; Johansson, J.; Karlström, A.R.; Thyberg, J.; Tereniust, L.; Nordstedt, C. Arrest of β-amyloid fibril formation by a pentapeptide ligand. J. Biol. Chem. 1996, 271, 8545–8548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz-Muñoz, G.; Miranda, I.L.; Sartori, S.K.; de Rezende, D.C.; Diaz, M.A.N. Anthraquinones: An Overview. Stud. Nat. Prod. Chem. 2018, 58, 313–338. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, J.G. Health functions and structure-activity relationships of natural anthraquinones from plants. Food Funct. 2018, 9, 6063–6080. [Google Scholar] [CrossRef] [PubMed]
- Malik, E.M.; Müller, C.E. Anthraquinones As Pharmacological Tools and Drugs. Med. Res. Rev. 2016, 36, 705–748. [Google Scholar] [CrossRef]
- Jung, H.A.; Ali, M.Y.; Jung, H.J.; Jeong, H.O.; Chung, H.Y.; Choi, J.S. Inhibitory activities of major anthraquinones and other constituents from Cassia obtusifolia against β-secretase and cholinesterases. J. Ethnopharmacol. 2016, 191, 152–160. [Google Scholar] [CrossRef]
- Orhan, I.; Tosun, F.; Şener, B. Coumarin, anthroquinone and stilbene derivatives with anticholinesterase activity. Z. Naturforsch Sect. C J. Biosci. 2008, 63, 366–370. [Google Scholar] [CrossRef]
- Zhang, J.H.; Xin, H.L.; Xu, Y.M.; Shen, Y.; He, Y.Q.; Lin, B.; Song, H.T.; Yang, H.Y.; Qin, L.P.; Zhang, Q.Y.; et al. Morinda officinalis How—A comprehensive review of traditional uses, phytochemistry and pharmacology. J. Ethnopharmacol. 2018, 213, 230–255. [Google Scholar] [CrossRef]
- Lee, Y.K.; Bang, H.J.; Oh, J. Bin; Whang, W.K. Bioassay-Guided Isolated Compounds from Morinda officinalis Inhibit Alzheimer’s Disease Pathologies. Molecules 2017, 22, 1638. [Google Scholar] [CrossRef] [Green Version]
- Pickhardt, M.; Gazova, Z.; Von Bergen, M.; Khlistunova, I.; Wang, Y.; Hascher, A.; Mandelkow, E.M.; Biernat, J.; Mandelkow, E. Anthraquinones inhibit tau aggregation and dissolve Alzheimer’s paired helical filaments in vitro and in cells. J. Biol. Chem. 2005, 280, 3628–3635. [Google Scholar] [CrossRef] [Green Version]
- Paranjape, S.R.; Chiang, Y.M.; Sanchez, J.F.; Entwistle, R.; Wang, C.C.C.; Oakley, B.R.; Gamblin, T.C. Inhibition of tau aggregation by three aspergillus nidulans secondary metabolites: 2, ω -dihydroxyemodin, asperthecin, and asperbenzaldehyde. Planta Med. 2014, 80, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Viswanathan, G.K.; Shwartz, D.; Losev, Y.; Arad, E.; Shemesh, C.; Pichinuk, E.; Engel, H.; Raveh, A.; Jelinek, R.; Cooper, I.; et al. Purpurin modulates Tau-derived VQIVYK fibrillization and ameliorates Alzheimer’s disease-like symptoms in animal model. Cell. Mol. Life Sci. 2020, 77, 2795–2813. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, K.; Rank, K.B.; Evans, D.B.; Sharma, S.K. Role of cysteine-291 and cysteine-322 in the polymerization of human tau into Alzheimer-like filaments. Biochem. Biophys. Res. Commun. 2001, 285, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Areche, C.; Zapata, F.; González, M.; Díaz, E.; Montecinos, R.; Hernández, M.; Melo, F.; Cornejo, A. Anthraquinone Derivative Reduces Tau Oligomer Progression by Inhibiting Cysteine-Cysteine Interaction. ChemistryOpen 2019, 8, 554–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandiera, T.; Lansen, J.; Post, C.; Varasi, M. Inhibitors of Aβ Peptide Aggregation as Potential anti-Alzheimer Agents. Curr. Med. Chem. 1997, 4, 159–170. [Google Scholar]
- Colombo, R.; Carotti, A.; Catto, M.; Racchi, M.; Lanni, C.; Verga, L.; Caccialanza, G.; De Lorenzi, E. CE can identify small molecules that selectively target soluble oligomers of amyloid β protein and display antifibrillogenic activity. Electrophoresis 2009, 30, 1418–1429. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Peacey, E.; Dickson, J.; Donahue, C.P.; Zheng, S.; Varani, G.; Wolfe, M.S. Mitoxantrone analogues as ligands for a stem-loop structure of tau Pre-mRNA. J. Med. Chem. 2009, 52, 6523–6526. [Google Scholar] [CrossRef] [Green Version]
- Hong, C.; Luo, W.; Yao, D.; Su, Y. Bin; Zhang, X.; Tian, R.G.; Wang, C.J. Novel aromatic-polyamine conjugates as cholinesterase inhibitors with notable selectivity toward butyrylcholinesterase. Bioorg. Med. Chem. 2014, 22, 3213–3219. [Google Scholar] [CrossRef]
- Wang, J.; Li, W.; Qin, J.; Wang, L.; Wei, S.; Tang, H. Assessment of novel azaanthraquinone derivatives as potent multi-target inhibitors of inflammation and amyloid-β aggregation in Alzheimer’s disease. Bioorg. Chem. 2019, 83, 477–486. [Google Scholar] [CrossRef]
- Crowe, A.; Ballatore, C.; Hyde, E.; Trojanowski, J.Q.; Lee, V.M.Y. High throughput screening for small molecule inhibitors of heparin-induced tau fibril formation. Biochem. Biophys. Res. Commun. 2007, 358, 1–6. [Google Scholar] [CrossRef] [Green Version]
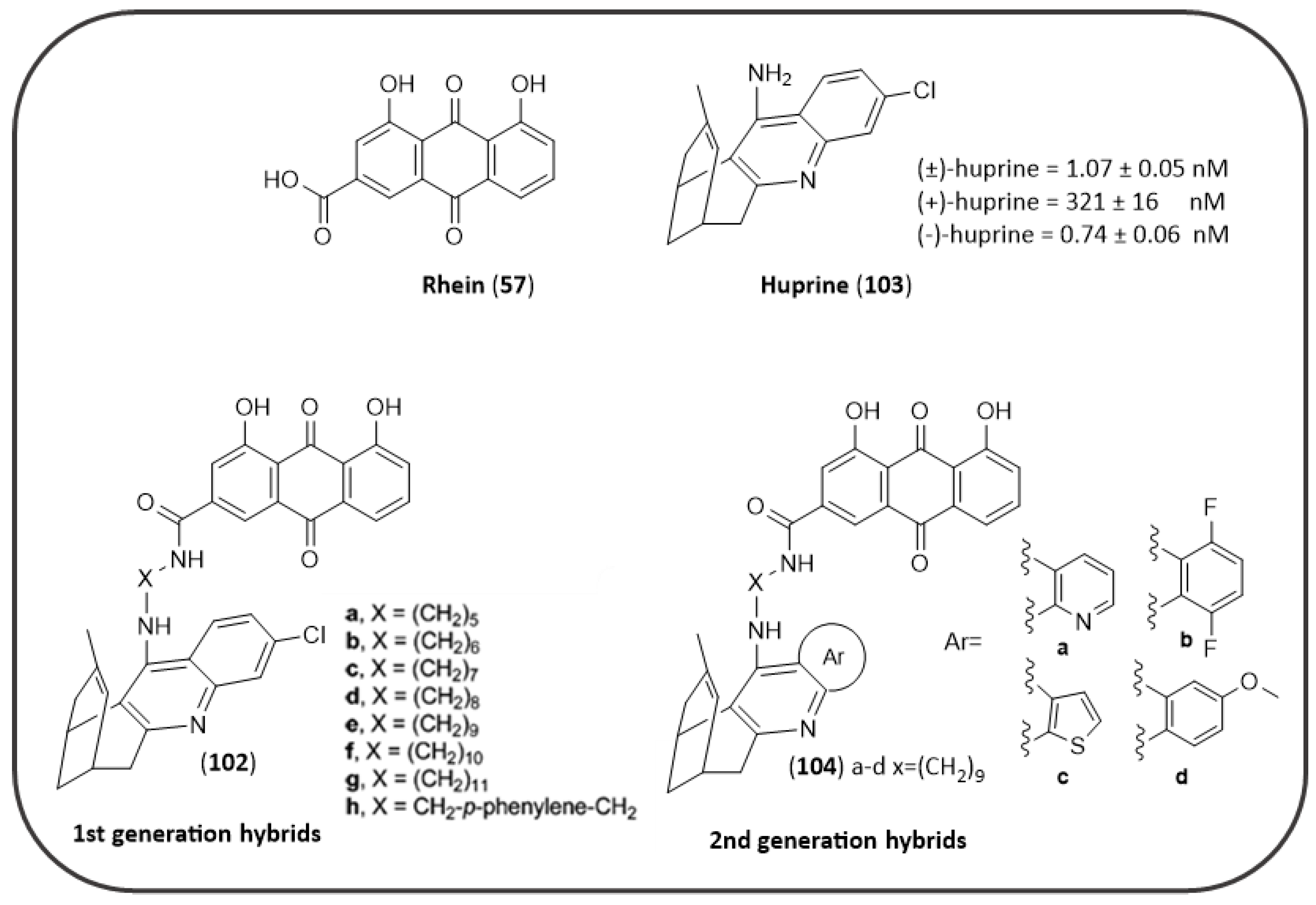
- Viayna, E.; Sola, I.; Bartolini, M.; De Simone, A.; Tapia-Rojas, C.; Serrano, F.G.; Sabaté, R.; Juárez-Jiménez, J.; Pérez, B.; Luque, F.J.; et al. Synthesis and multitarget biological profiling of a novel family of rhein derivatives as disease-modifying anti-Alzheimer agents. J. Med. Chem. 2014, 57, 2549–2567. [Google Scholar] [CrossRef] [Green Version]
- Camps, P.; Cusack, B.; Mallender, W.D.; El Achab, R.E.; Morral, J.; Muñoz-Torrero, D.; Rosenberry, T.L. Huprine X is a novel high-affinity inhibitor of acetylcholinesterase that is of interest for treatment of Alzheimer’s disease. Mol. Pharmacol. 2000, 57, 409–417. [Google Scholar] [PubMed]
- Pérez-Areales, F.J.; Betari, N.; Viayna, A.; Pont, C.; Espargaró, A.; Bartolini, M.; De Simone, A.; Rinaldi Alvarenga, J.F.; Pérez, B.; Sabate, R.; et al. Design, synthesis and multitarget biological profiling of second-generation anti-Alzheimer rhein–huprine hybrids. Future Med. Chem. 2017, 9, 965–981. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Y.; Jiang, N.; Xie, S.S.; Wang, K.D.G.; Wang, X.B.; Kong, L.Y. Design, synthesis and evaluation of novel tacrine-rhein hybrids as multifunctional agents for the treatment of Alzheimer’s disease. Org. Biomol. Chem. 2014, 12, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, F.J.; Inestrosa, N.C. Interactions of AChE with Aβ Aggregates in Alzheimer’s Brain: Therapeutic Relevance of IDN. Front. Mol. Neurosci. 2011, 4, 19. [Google Scholar] [CrossRef] [Green Version]
- Turon-Estrada, A.; López-Pousa, S.; Gelada-Batlle, E.; Garre-Olmo, J.; Lozano-Gallego, M.; Hernández-Ferràndiz, M.; Fajardo-Tibau, C.; Morante-Muñoz, V.; Vilalta-Franch, J. Tolerance and adverse events of treatment with acetylcholinesterase inhibitors in a clinical sample of patients with very slight and mild Alzheimer’s disease over a six-month period. Rev. Neurol. 2003, 36, 421–424. [Google Scholar] [CrossRef]
- Delrieu, J.; Piau, A.; Caillaud, C.; Voisin, T.; Vellas, B. Managing Cognitive Dysfunction through the Continuum of Alzheimerʼs Disease. CNS Drugs 2011, 25, 213–226. [Google Scholar] [CrossRef]
- Scheltens, P.; Blennow, K.; Breteler, M.M.B.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Prati, F.; Cavalli, A.; Bolognesi, M. Navigating the Chemical Space of Multitarget-Directed Ligands: From Hybrids to Fragments in Alzheimer’s Disease. Molecules 2016, 21, 466. [Google Scholar] [CrossRef] [Green Version]





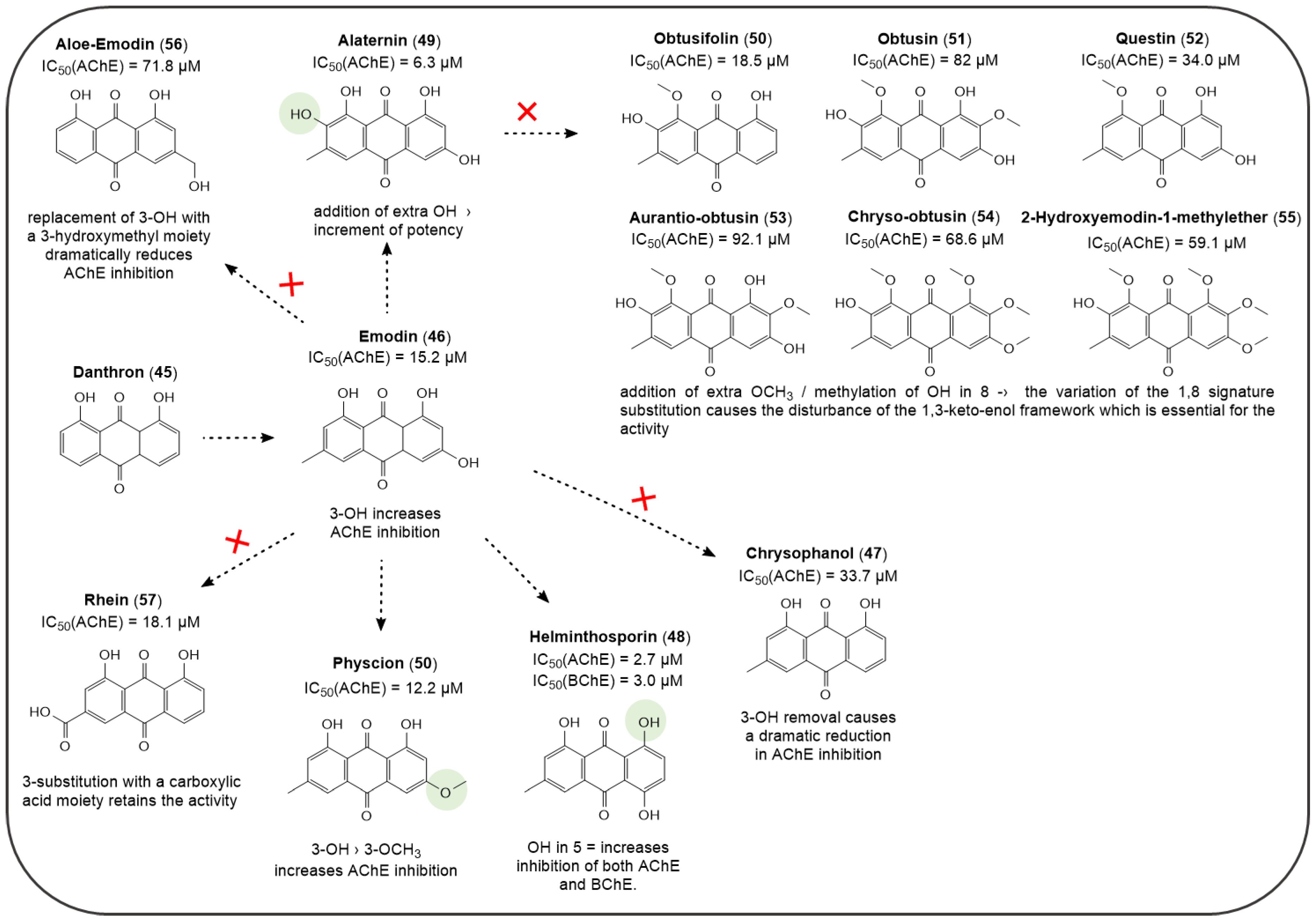


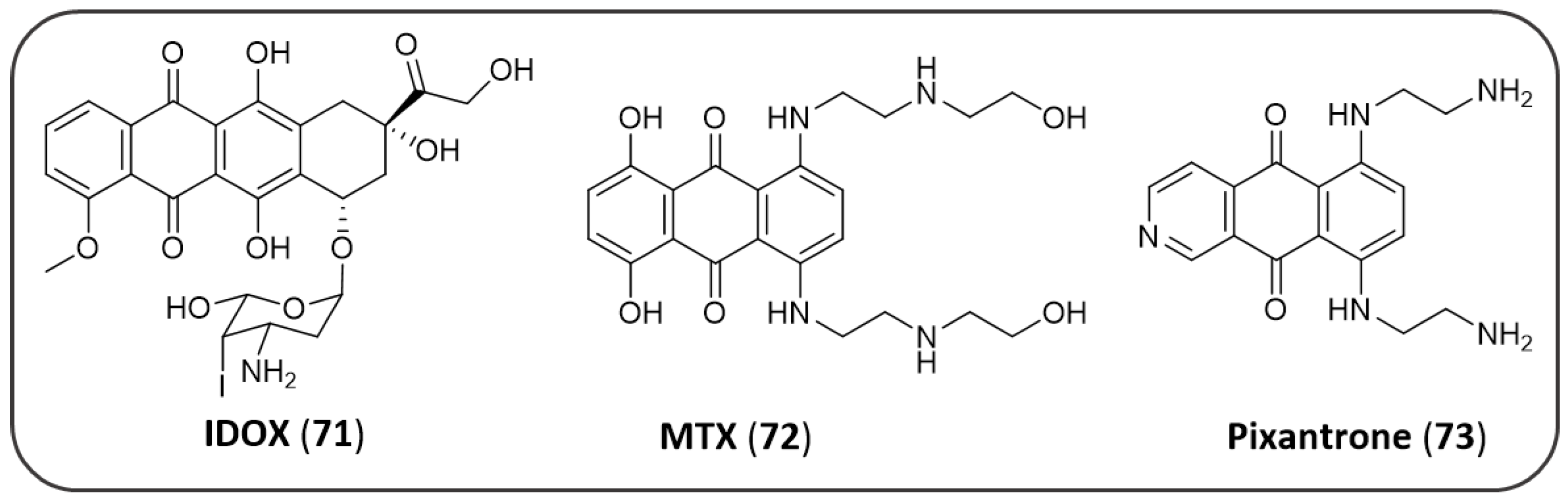
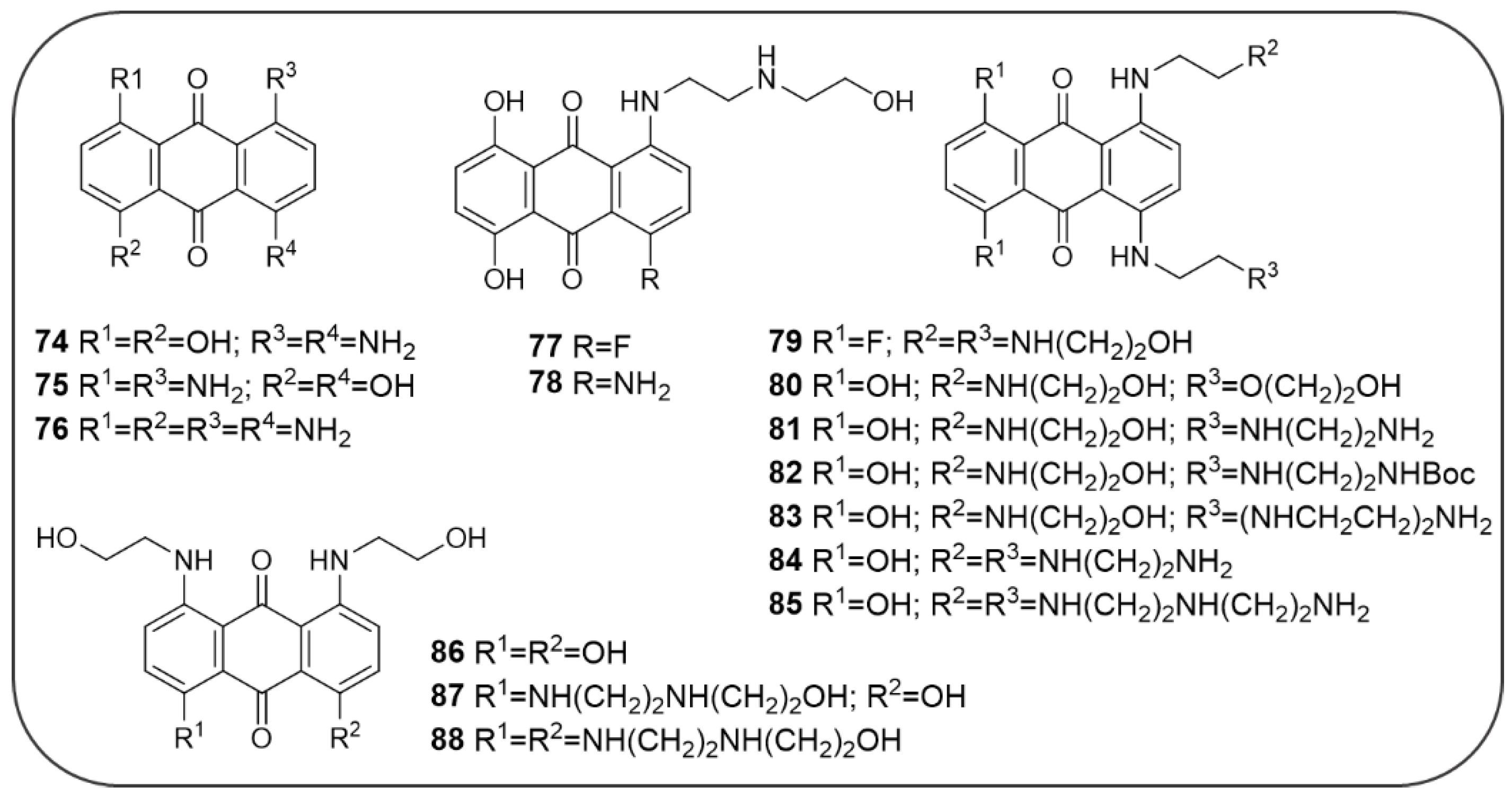

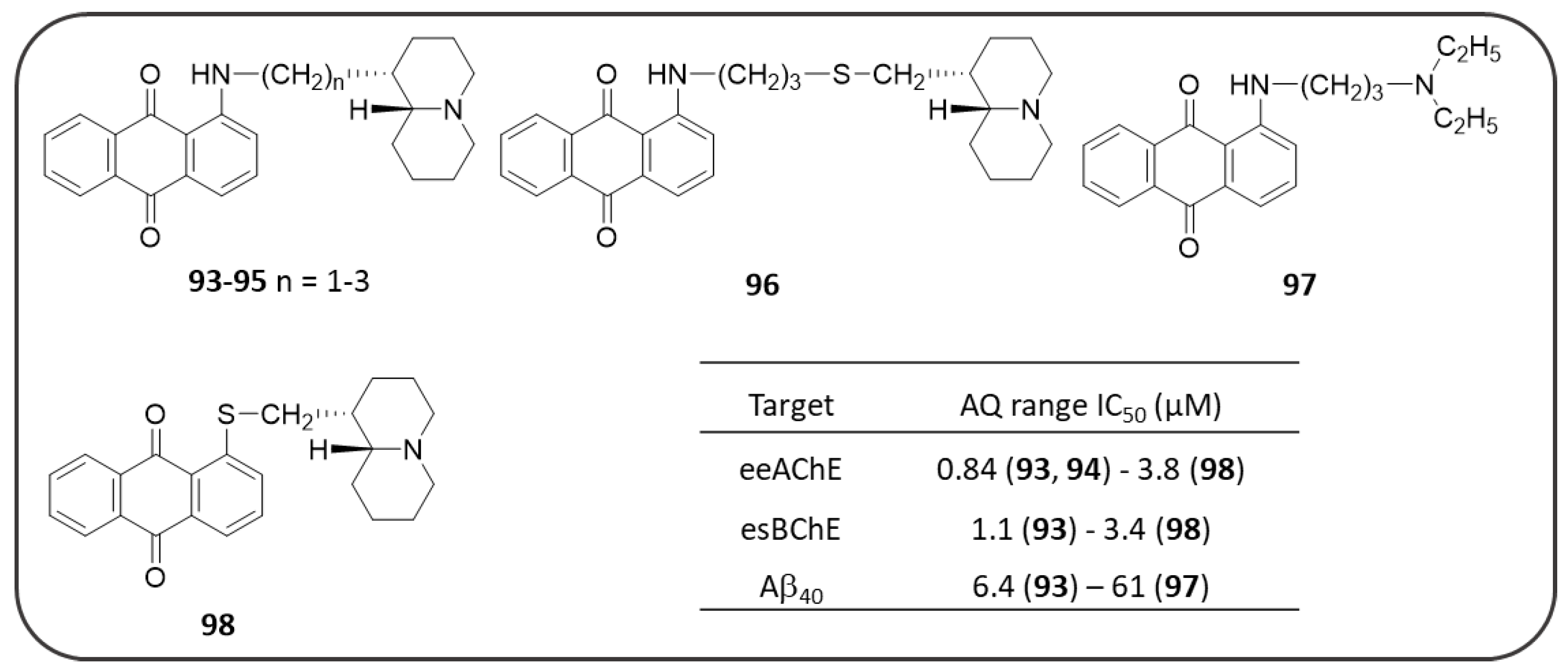



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campora, M.; Francesconi, V.; Schenone, S.; Tasso, B.; Tonelli, M. Journey on Naphthoquinone and Anthraquinone Derivatives: New Insights in Alzheimer’s Disease. Pharmaceuticals 2021, 14, 33. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14010033
Campora M, Francesconi V, Schenone S, Tasso B, Tonelli M. Journey on Naphthoquinone and Anthraquinone Derivatives: New Insights in Alzheimer’s Disease. Pharmaceuticals. 2021; 14(1):33. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14010033
Chicago/Turabian StyleCampora, Marta, Valeria Francesconi, Silvia Schenone, Bruno Tasso, and Michele Tonelli. 2021. "Journey on Naphthoquinone and Anthraquinone Derivatives: New Insights in Alzheimer’s Disease" Pharmaceuticals 14, no. 1: 33. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14010033