Modifications on the Tetrahydroquinoline Scaffold Targeting a Phenylalanine Cluster on GPER as Antiproliferative Compounds against Renal, Liver and Pancreatic Cancer Cells

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results and Discussion
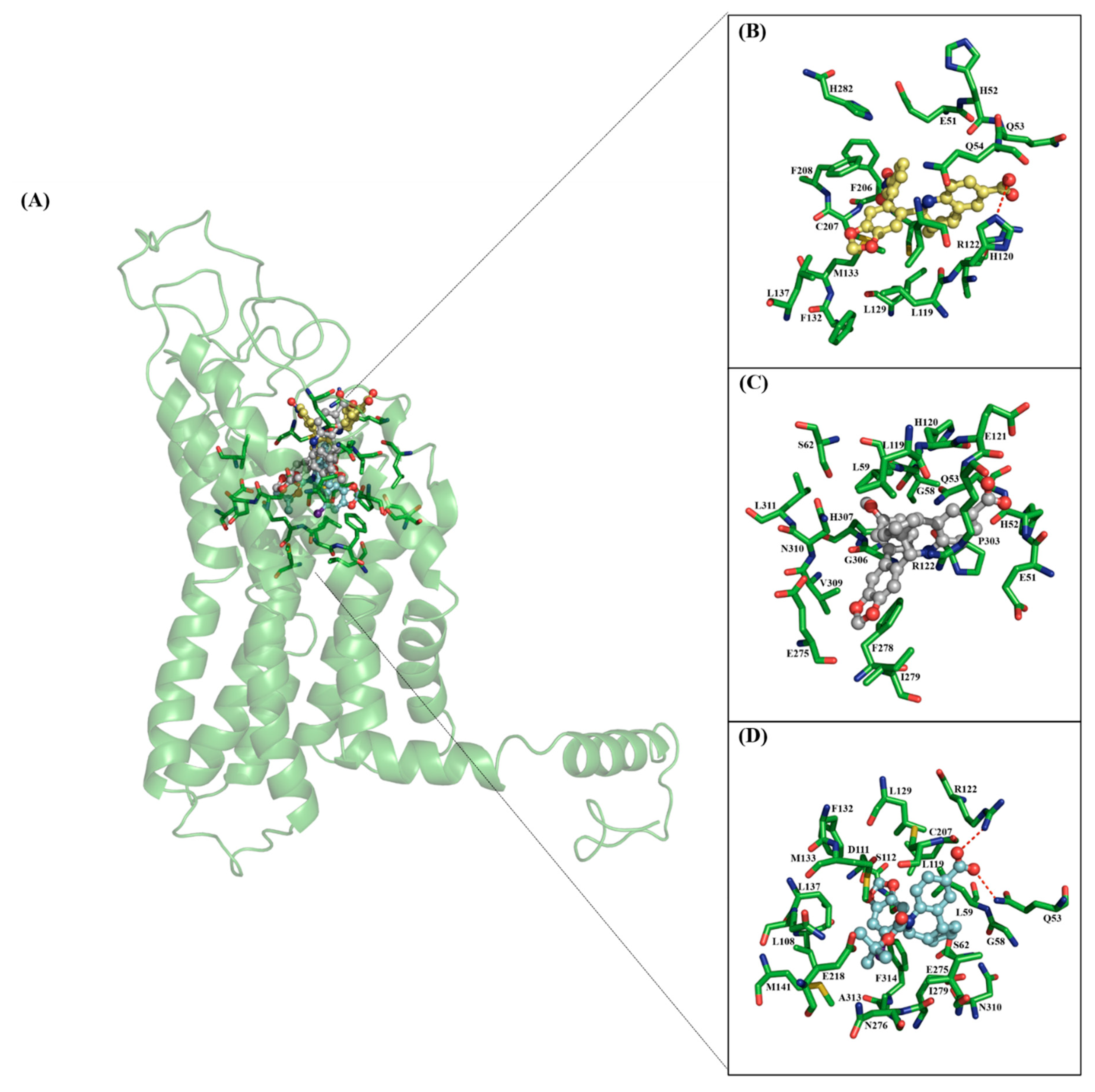
2.1. Docking Calculations
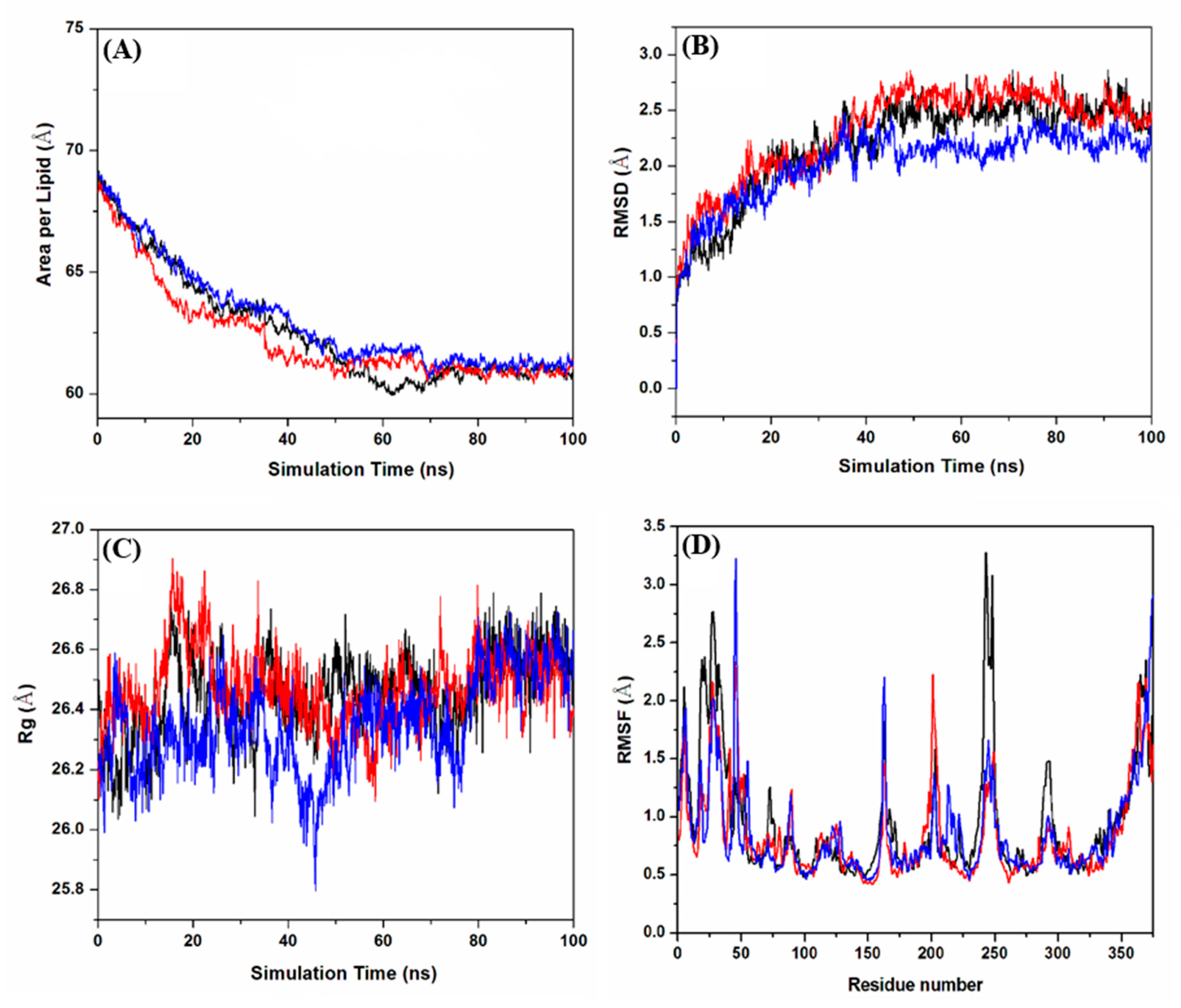
2.2. Molecular Dynamics Simulations
2.2.1. Evaluation of the Equilibrium of Membrane-Embedded MD GPER–Ligand Complexes
2.2.2. Clustering Analysis
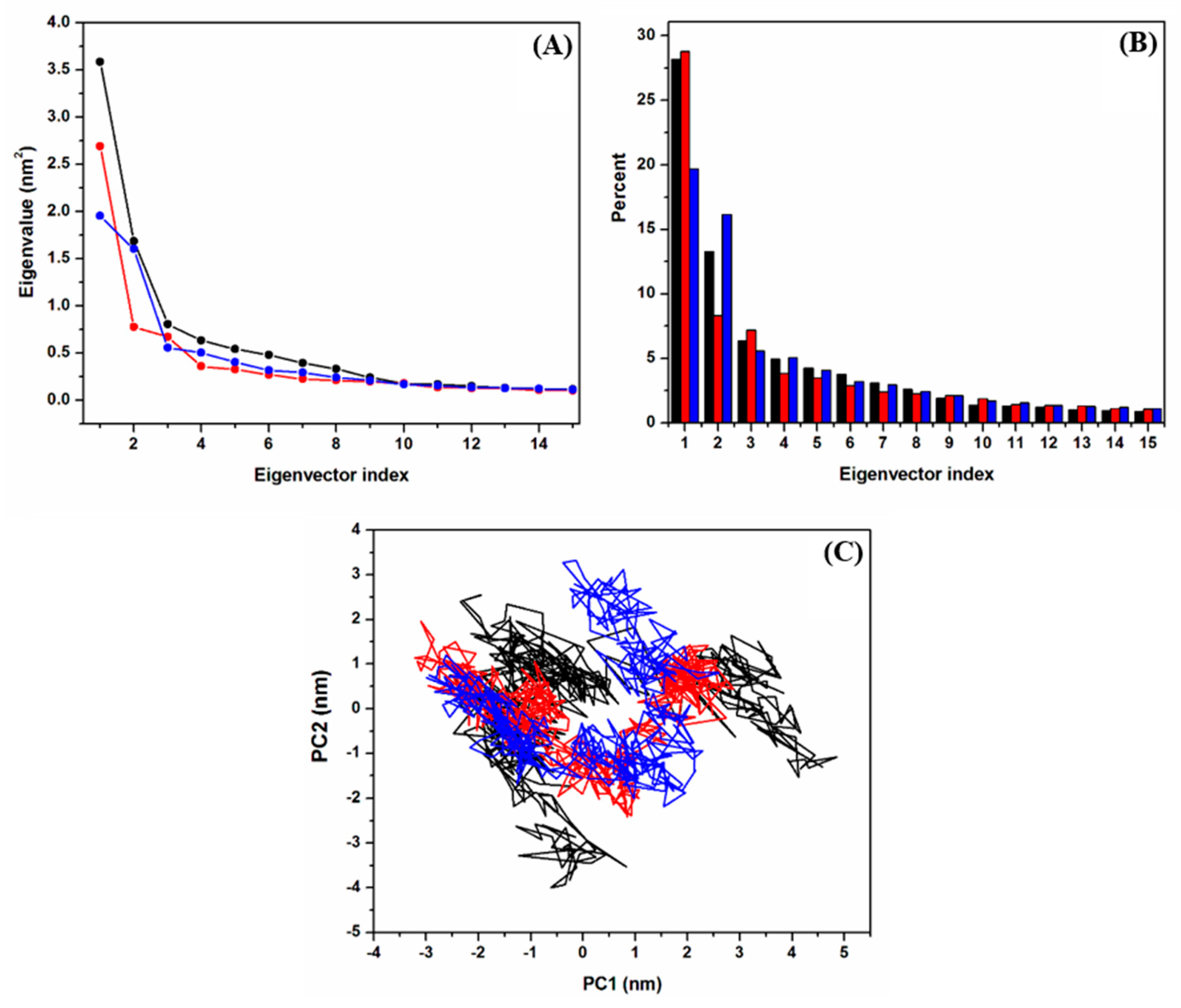
2.2.3. Principal Component Analysis (PCA)
2.2.4. Structural Analysis of the Empty and Bound GPER Systems
2.2.5. Absolute Binding Free Energy Calculations
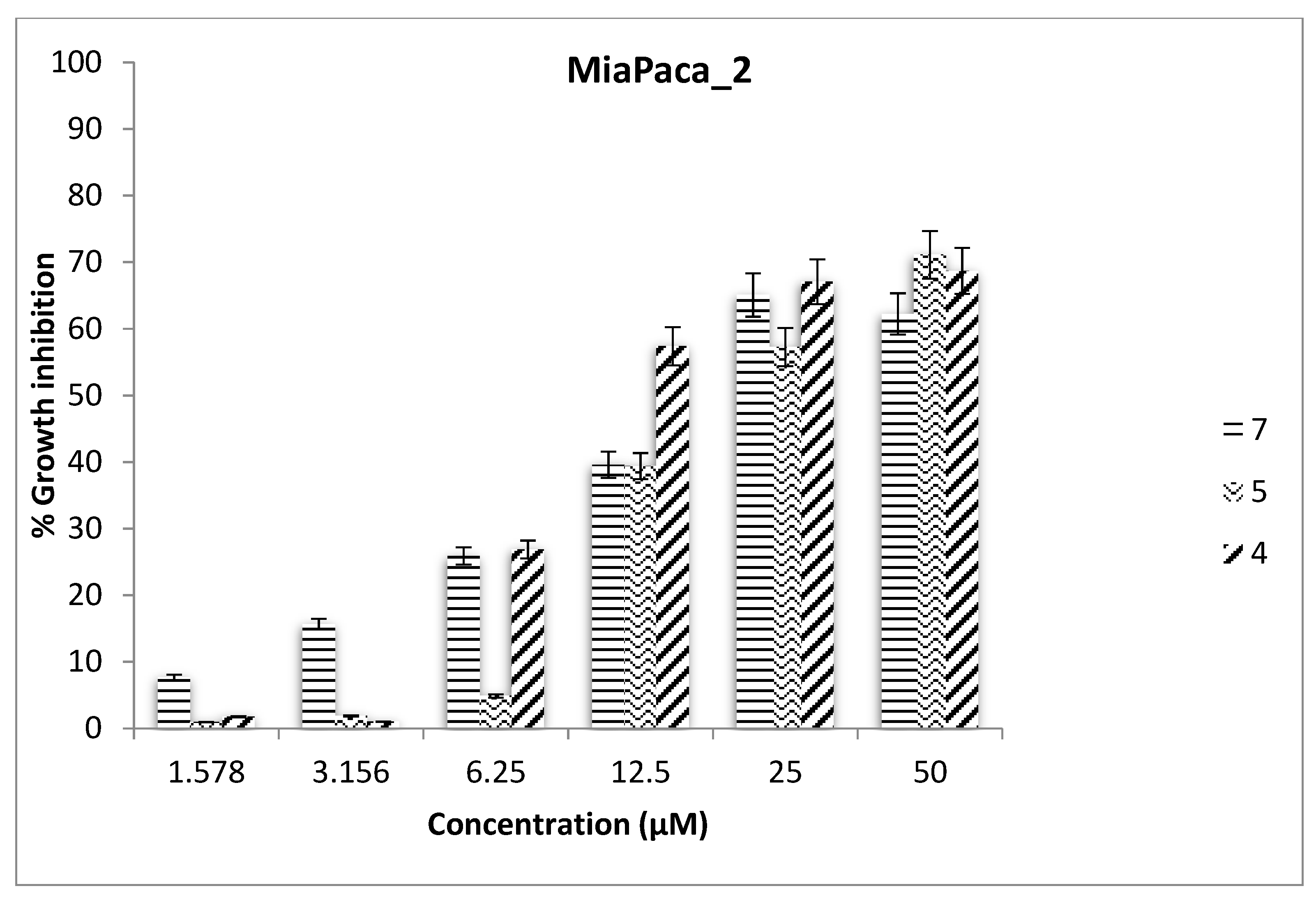
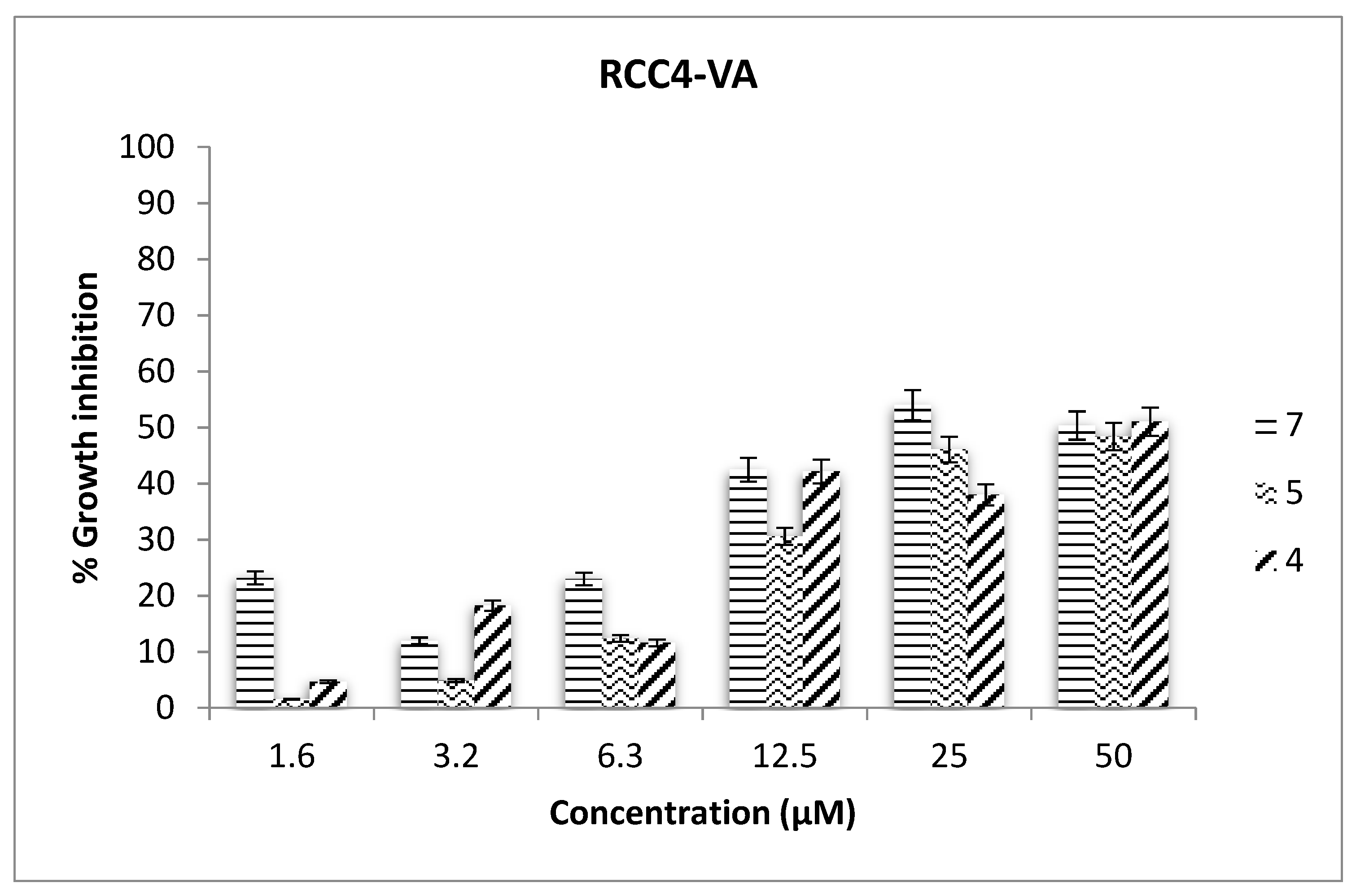
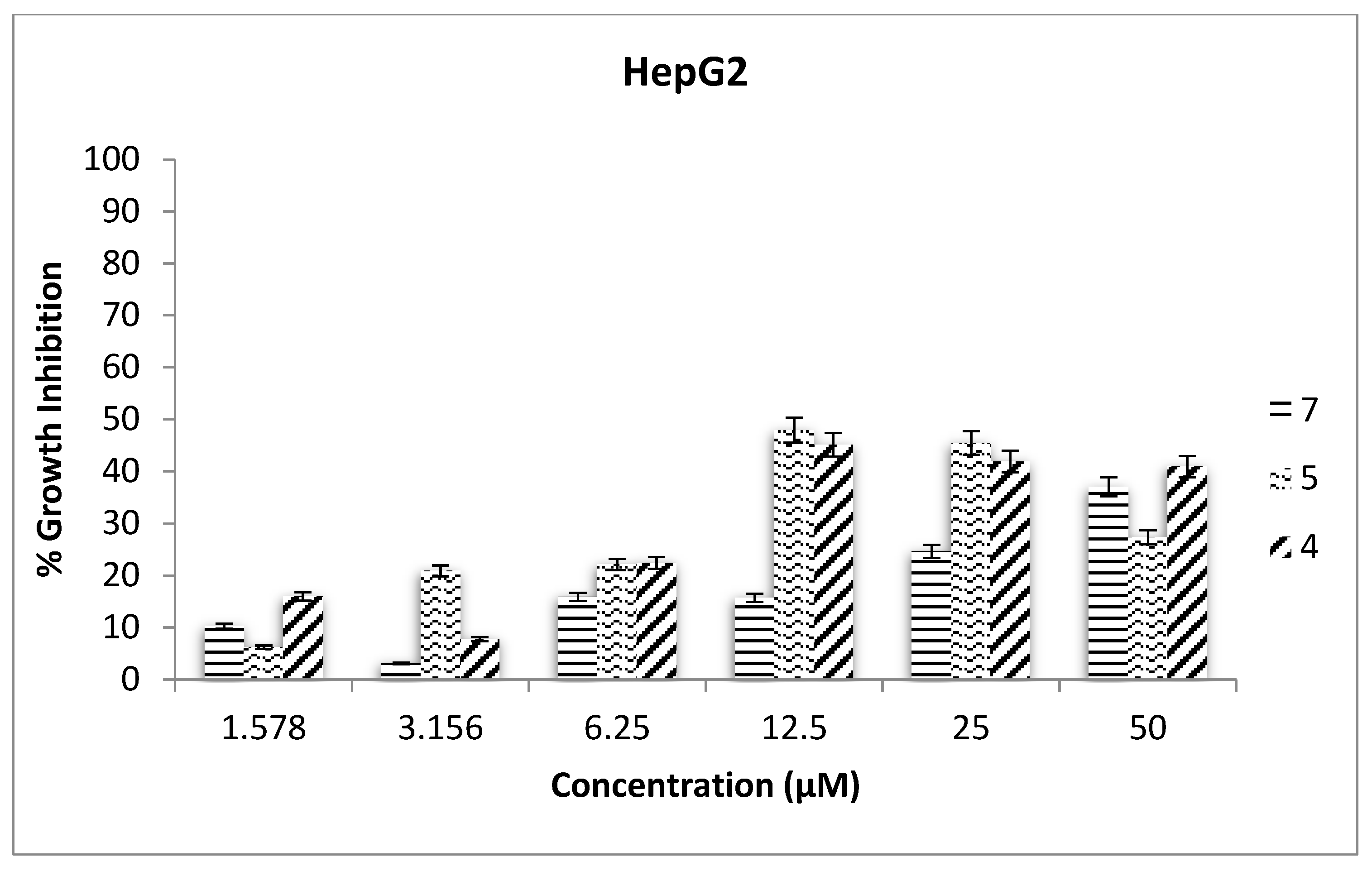
2.3. Antiproliferative Assays
3. Materials and Methods
3.1. Docking, Physicochemical and ADME-Tox Properties Analysis
3.2. Embedding of the GPER–Ligand Complexes into the Membrane
3.3. MD Simulations of the Membrane-Embedded GPER–Ligand Complexes
3.4. Analysis of MD Simulations
3.5. Principal Component Analysis
3.6. Calculation of Absolute Binding Free Energies
3.7. Calculation of Entropy Contributions
3.8. Chemical Synthesis
3.8.1. Synthesis of (3aS, 4R, 9bR)-4-(6-Bromobenzo[d][1,3]dioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-carboxylic acid, G1-PABA 1
3.8.2. Synthesis of (3aS, 4R, 9bR)-4-(6-(3-Nitrophenyl)benzo[d][1,3]dioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-carboxylic acid, 4
3.8.3. Synthesis of (3aS, 4R, 9bR)-4-(6-(3-Methoxyphenyl)benzo[d][1,3]dioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-carboxylic acid, 5
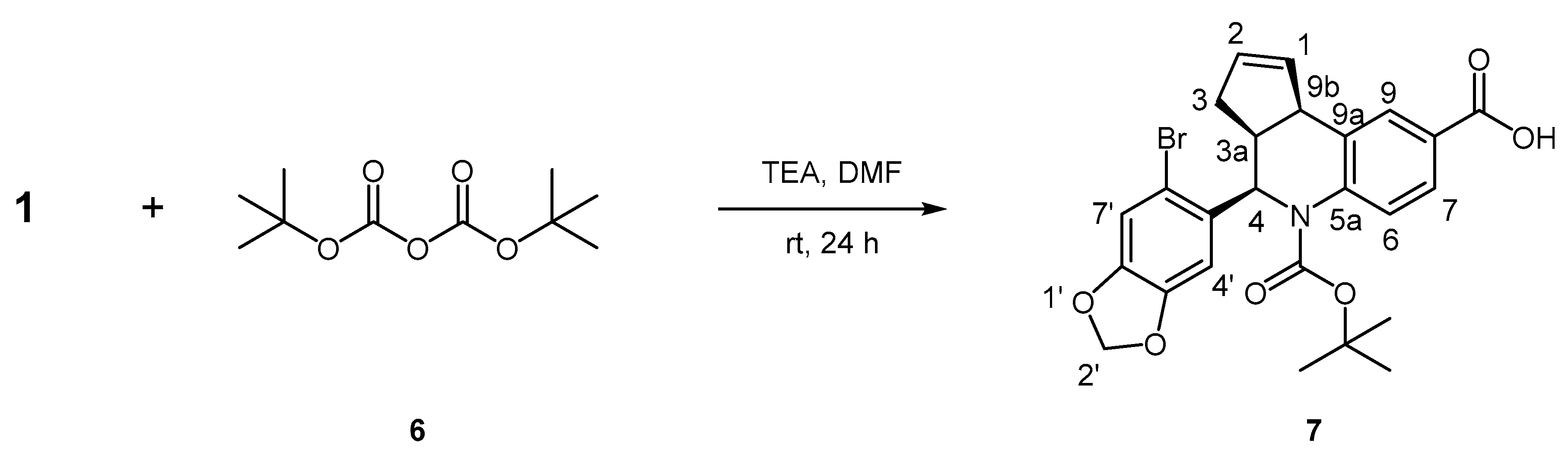
3.8.4. Synthesis of (3aS, 4R, 9bR)-4-(6-Bromobenzo[d][1,3]dioxol-5-yl)-5-(tert-butoxycarbonyl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-carboxylic acid, 7
3.9. Purity Analysis by HPLC
3.10. Cell Culture
3.11. Antiproliferative Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Allred, D.C. Issues and updates: Evaluating estrogen receptor-α, progesterone receptor, and HER2 in breast cancer. Mod. Pathol. 2010, 23, S52–S59. [Google Scholar] [CrossRef] [Green Version]
- DeRoo, B.J.; Korach, K.S. Estrogen receptors and human disease. J. Clin. Investig. 2006, 116, 561–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prossnitz, E.R.; Arterburn, J.B.; Sklar, L.A. GPR30: A G protein-coupled receptor for estrogen. Mol. Cell. Endocrinol. 2007, 265, 138–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olde, B.; Leeb-Lundberg, L.M.F. GPR30/GPER1: Searching for a role in estrogen physiology. Trends Endocrinol. Metab. 2009, 20, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Signalling: Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 2002, 3, 639–650. [Google Scholar] [CrossRef]
- Dorsam, R.T.; Gutkind, J.S. G-protein-coupled receptors and cancer. Nat. Rev. Cancer 2007, 7, 79–94. [Google Scholar] [CrossRef]
- Revankar, C.M.; Cimino, D.F.; Sklar, L.A.; Arterburn, J.B.; Prossnitz, E.R. A Transmembrane Intracellular Estrogen Receptor Mediates Rapid Cell Signaling. Science 2005, 307, 1625–1630. [Google Scholar] [CrossRef] [Green Version]
- Thomas, P.; Pang, Y.; Filardo, E.J.; Dong, J. Identity of an Estrogen Membrane Receptor Coupled to a G Protein in Human Breast Cancer Cells. Endocrinology 2005, 146, 624–632. [Google Scholar] [CrossRef] [Green Version]
- Filardo, E.J.; Quinn, J.A.; Bland, K.I.; Frackelton, A.R. Estrogen-Induced Activation of Erk-1 and Erk-2 Requires the G Protein-Coupled Receptor Homolog, GPR30, and Occurs via Trans-Activation of the Epidermal Growth Factor Receptor through Release of HB-EGF. Mol. Endocrinol. 2000, 14, 1649–1660. [Google Scholar] [CrossRef]
- Bologa, C.G.; Revankar, C.M.; Young, S.M.; Edwards, B.S.; Arterburn, J.B.; Kiselyov, A.S.; Parker, M.A.; Tkachenko, S.E.; Savchuck, N.P.; Sklar, L.A.; et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat. Chem. Biol. 2006, 2, 207–212. [Google Scholar] [CrossRef]
- Burai, R.; Ramesh, C.; Shorty, M.; Curpan, R.; Bologa, C.G.; Sklar, L.A.; Oprea, T.I.; Prossnitz, E.R.; Arterburn, J.B. Highly efficient synthesis and characterization of the GPR30-selective agonist G-1 and related tetrahydroquinoline analogs. Org. Biomol. Chem. 2010, 8, 2252–2259. [Google Scholar] [CrossRef]
- Dennis, M.K.; Burai, R.; Ramesh, C.; Petrie, W.K.; Alcon, S.N.; Nayak, T.K.; Bologa, C.G.; Leitao, A.; Brailoiu, E.; Deliu, E.; et al. In vivo effects of a GPR30 antagonist. Nat. Chem. Biol. 2009, 5, 421–427. [Google Scholar] [CrossRef] [Green Version]
- Dennis, M.K.; Field, A.S.; Burai, R.; Ramesh, C.; Petrie, W.K.; Bologa, C.G.; Oprea, T.I.; Yamaguchi, Y.; Hayashi, S.-I.; Sklar, L.A.; et al. Identification of a GPER/GPR30 antagonist with improved estrogen receptor counterselectivity. J. Steroid Biochem. Mol. Biol. 2011, 127, 358–366. [Google Scholar] [CrossRef] [Green Version]
- Guan, B.-Z.; Yan, R.-L.; Huang, J.-W.; Rui-Ling, Y.; Zhong, Y.-X.; Chen, Y.; Liu, F.-N.; Hu, B.; Huang, S.-B.; Yin, L.-H. Activation of G protein coupled estrogen receptor (GPER) promotes the migration of renal cell carcinoma via the PI3K/AKT/MMP-9 signals. Cell Adhes. Migr. 2018, 12, 109–117. [Google Scholar] [CrossRef] [Green Version]
- Chaturantabut, S.; Shwartz, A.; Evason, K.J.; Cox, A.G.; Labella, K.; Schepers, A.G.; Yang, S.; Acuña, M.; Houvras, Y.; Mancio-Silva, L.; et al. Estrogen Activation of G-Protein–Coupled Estrogen Receptor 1 Regulates Phosphoinositide 3-Kinase and mTOR Signaling to Promote Liver Growth in Zebrafish and Proliferation of Human Hepatocytes. Gastroenterology 2019, 156, 1788–1804. [Google Scholar] [CrossRef] [Green Version]
- Cortes, E.; Sarper, M.; Robinson, B.; Lachowski, D.; Chronopoulos, A.; Thorpe, S.D.; Lee, D.A.; Hernández, A.E.D.R. GPER is a mechanoregulator of pancreatic stellate cells and the tumor microenvironment. EMBO Rep. 2019, 20, 46556. [Google Scholar] [CrossRef]
- Wang, C.; Lv, X.; Jiang, C.; Davis, J.S. The putative G-protein coupled estrogen receptor agonist G-1 suppresses proliferation of ovarian and breast cancer cells in a GPER-independent manner. Am. J. Transl. Res. 2012, 4, 390–402. [Google Scholar]
- Wang, C.; Lv, X.Y.; He, C.; Hua, G.; Tsai, M.-Y.; Davis, J.S. The G-protein-coupled estrogen receptor agonist G-1 suppresses proliferation of ovarian cancer cells by blocking tubulin polymerization. Cell Death Dis. 2013, 4, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Rosano, C.; Ponassi, M.; Santolla, M.F.; Pisano, A.; Felli, L.; Vivacqua, A.; Maggiolini, M.; Lappano, R. Macromolecular Modelling and Docking Simulations for the Discovery of Selective GPER Ligands. AAPS J. 2015, 18, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Méndez-Luna, D.; Martínez-Archundia, M.; Maroun, R.C.; Ceballos-Reyes, G.; Fragoso-Vázquez, M.; González-Juárez, D.; Correa-Basurto, J. Deciphering the GPER/GPR30-agonist and antagonists interactions using molecular modeling studies, molecular dynamics, and docking simulations. J. Biomol. Struct. Dyn. 2015, 33, 2161–2172. [Google Scholar] [CrossRef]
- Méndez-Luna, D.; Bello, M.; Correa-Basurto, J. Understanding the molecular basis of agonist/antagonist mechanism of GPER1/GPR30 through structural and energetic analyses. J. Steroid Biochem. Mol. Biol. 2016, 158, 104–116. [Google Scholar] [CrossRef]
- Zacarias-Lara, O.J.; Mendez-Luna, D.; Martinez-Ruiz, G.; Garcia-Sanchez, J.R.; Fragoso-Vazquez, M.J.; Bello, M.; Becerra-Martinez, E.; Garcia-Vazquez, J.B.; Correa-Basurto, J. Synthesis and In Vitro Evaluation of Tetrahydroquinoline Derivatives as Antiproliferative Compounds of Breast Cancer via Targeting the GPER. Anti Cancer Agents Med. Chem. 2019, 19, 760–771. [Google Scholar] [CrossRef]
- Martínez-Muñoz, A.; Prestegui-Martel, B.; Mendez-Luna, D.; Fragoso-Vazquez, M.J.; García-Sánchez, J.R.; Bello, M.; Martinez-Archundia, M.; Chávez-Blanco, A.D.; Duenas-Gonzalez, A.; Mendoza-Lujambio, I.; et al. Selection of a GPER1 Ligand via Ligand-based Virtual Screening Coupled to Molecular Dynamics Simulations and Its Anti-proliferative Effects on Breast Cancer Cells. Anti Cancer Agents Med. Chem. 2019, 18, 1629–1638. [Google Scholar] [CrossRef]
- Moreno-Ulloa, A.; Mendez-Luna, D.; Beltrán-Partida, E.; Castillo, C.; Guevara, G.; Ramírez-Sánchez, I.; Correa-Basurto, J.; Ceballos, G.; Villarreal, F. The effects of (-)-epicatechin on endothelial cells involve the G protein-coupled estrogen receptor (GPER). Pharmacol. Res. 2015, 100, 309–320. [Google Scholar] [CrossRef] [Green Version]
- Qian, H.; Xuan, J.; Liu, Y.; Shi, G. Function of G-Protein-Coupled Estrogen Receptor-1 in Reproductive System Tumors. J. Immunol. Res. 2016, 2016, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Jacenik, D.; Cygankiewicz, A.I.; Krajewska, W.M. The G protein-coupled estrogen receptor as a modulator of neoplastic transformation. Mol. Cell. Endocrinol. 2016, 429, 10–18. [Google Scholar] [CrossRef]
- Abe, M.; Yamashita, J.; Ogawa, M. Medroxyprogesterone acetate inhibits human pancreatic carcinoma cell growth by inducing apoptosis in association with Bcl-2 phosphorylation. Cancer 2000, 88, 2000–2009. [Google Scholar] [CrossRef]
- Guo, J.-M.; Xiao, B.-X.; Dai, D.-J.; Liu, Q.; Ma, H.-H. Effects of daidzein on estrogen-receptor-positive and negative pancreatic cancer cellsin vitro. World J. Gastroenterol. 2004, 10, 860–863. [Google Scholar] [CrossRef]
- Lonser, R.R.; Glenn, G.M.; Walther, M.; Chew, E.Y.; Libutti, S.K.; Linehan, W.M.; Oldfield, E.H. von Hippel-Lindau disease. Lancet 2003, 361, 2059–2067. [Google Scholar] [CrossRef]
- Cautain, B.; De Pedro, N.; De Escalona, M.M.; Tormo, J.R.; Genilloud, O.; Vicente, F. HCS strategy targeting dysregulation of the VHL/HIF pathway for drug discovery. Adv. Biosci. Biotechnol. 2013, 4, 398–405. [Google Scholar] [CrossRef] [Green Version]
- Rigiracciolo, D.C.; Scarpelli, A.; Lappano, R.; Pisano, A.; Santolla, M.F.; De Marco, P.; Cirillo, F.; Cappello, A.R.; Dolce, V.; Belfiore, A.; et al. Copper activates HIF-1α/GPER/VEGF signalling in cancer cells. Oncotarget 2015, 6, 34158–34177. [Google Scholar] [CrossRef] [Green Version]
- De Francesco, E.M.; Lappano, R.; Santolla, M.F.; Marsico, S.; Caruso, A.; Maggiolini, M. HIF-1α/GPER signaling mediates the expression of VEGF induced by hypoxia in breast cancer associated fibroblasts (CAFs). Breast Cancer Res. 2013, 15, 1–18. [Google Scholar] [CrossRef] [Green Version]
- De Francesco, E.M.; Pellegrino, M.; Santolla, M.F.; Lappano, R.; Ricchio, E.; Abonante, S.; Maggiolini, M. GPER Mediates Activation of HIF1/VEGF Signaling by Estrogens. Cancer Res. 2014, 74, 4053–4064. [Google Scholar] [CrossRef] [Green Version]
- ChemBioDraw Ultra 12.0. Available online: http://www.cambridgesoft.com/software/overview.aspx (accessed on 12 November 2020).
- Denningtion, R.; Roy, T.; Millam, J. GaussView, Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- DeLano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific: San Carlos, CA, USA, 2002; Available online: http://www.pymol.org (accessed on 12 November 2020).
- Molinspiration Cheminformatics, Bratislava, Slovak Republic. Available online: http://www.molinspiration.com (accessed on 12 November 2020).
- Sander, T.; Freyss, J.; Von Korff, M.; Rufener, C. DataWarrior: An Open-Source Program for Chemistry Aware Data Visualization and Analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Lomize, M.A.; Lomize, A.L.; Pogozheva, I.D.; Mosberg, H.I. OPM: Orientations of Proteins in Membranes database. Bioinformatics 2006, 22, 623–625. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Im, W. Automated Builder and Database of Protein/Membrane Complexes for Molecular Dynamics Simulations. PLoS ONE 2007, 2, e880. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Lim, J.B.; Klauda, J.B.; Im, W. CHARMM-GUI Membrane Builder for Mixed Bilayers and Its Application to Yeast Membranes. Biophys. J. 2009, 97, 50–58. [Google Scholar] [CrossRef] [Green Version]
- Woolf, T.B.; Roux, B. Structure, energetics, and dynamics of lipid-protein interactions: A molecular dynamics study of the gramicidin A channel in a DMPC bilayer. Proteins 1996, 24, 92–114. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Skjevik, Å.A.; Madej, B.D.; Walker, R.C.; Eigen, K.T. LIPID11: A Modular Framework for Lipid Simulations Using Amber. J. Phys. Chem. B 2012, 116, 11124–11136. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Darden, T.A.; York, D.; Pedersen, L. Particle mesh Ewald: An N-log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Van Gunsteren, W.; Berendsen, H. Algorithms for macromolecular dynamics and constraint dynamics. Mol. Phys. 1977, 34, 1311–1327. [Google Scholar] [CrossRef]
- Lindahl, E.; Hess, B.; Van Der Spoel, D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Model. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Amadei, A.; Linssen, A.B.M.; Berendsen, H.J.C. Essential dynamics of proteins. Proteins Struct. Funct. Bioinform. 1993, 17, 412–425. [Google Scholar] [CrossRef]
- Berendsen, H.; Van Der Spoel, D.; Van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Gohlke, H.; Case, D.A. Converging free energy estimates: MM-PB(GB)SA studies on the protein-protein complex Ras-Raf. J. Comput. Chem. 2004, 25, 238–250. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Accounts Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the molecular mechanics/Poisson Boltzmann surface area and molecular mechanics/generalized Born surface area methods. II. The accuracy of ranking poses generated from docking. J. Comput. Chem. 2011, 32, 866–877. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Sun, H.; Li, Y.; Wang, J.; Hou, T. Assessing the Performance of MM/PBSA and MM/GBSA Methods. 3. The Impact of Force Fields and Ligand Charge Models. J. Phys. Chem. B 2013, 117, 8408–8421. [Google Scholar] [CrossRef]
- Sun, H.; Li, Y.; Tian, S.; Xu, L.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. Phys. Chem. Chem. Phys. 2014, 16, 16719–16729. [Google Scholar] [CrossRef]
- Hou, T.; Li, N.; Li, Y.; Wang, W. Characterization of Domain–Peptide Interaction Interface: Prediction of SH3 Domain-Mediated Protein–Protein Interaction Network in Yeast by Generic Structure-Based Models. J. Proteome Res. 2012, 11, 2982–2995. [Google Scholar] [CrossRef]
- Sun, H.; Li, Y.; Shen, M.; Tian, S.; Xu, L.; Pan, P.; Guan, Y.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 5. Improved docking performance using high solute dielectric constant MM/GBSA and MM/PBSA rescoring. Phys. Chem. Chem. Phys. 2014, 16, 22035–22045. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, Y.; Wang, Y. Phosphine-free Palladium acetate catalyzed Suzuki reaction in water. J. Org. Chem. 2005, 70, 6122–6125. [Google Scholar] [CrossRef]
- Miyaura, N.; Yamada, K.; Suzuki, A. A new stereospecific cross-coupling by the palladium-catalyzed reaction of 1-alkenylboranes with 1-alkenyl or 1-alkynyl halides. Tetrahedron Lett. 1979, 20, 3437–3440. [Google Scholar] [CrossRef] [Green Version]
- Verhulst, C.; Coiffard, C.; Coiffard, L.J.; Rivalland, P.; De Roeck-Holtzhauer, Y. In vitro correlation between two colorimetric assays and the pyruvic acid consumption by fibroblasts cultured to determine the sodium laurylsulfate cytotoxicity. J. Pharmacol. Toxicol. Methods 1998, 39, 143–146. [Google Scholar] [CrossRef]
- Liu, Y.; Nair, M.G. An Efficient and Economical MTT Assay for Determining the Antioxidant Activity of Plant Natural Product Extracts and Pure Compounds. J. Nat. Prod. 2010, 73, 1193–1195. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
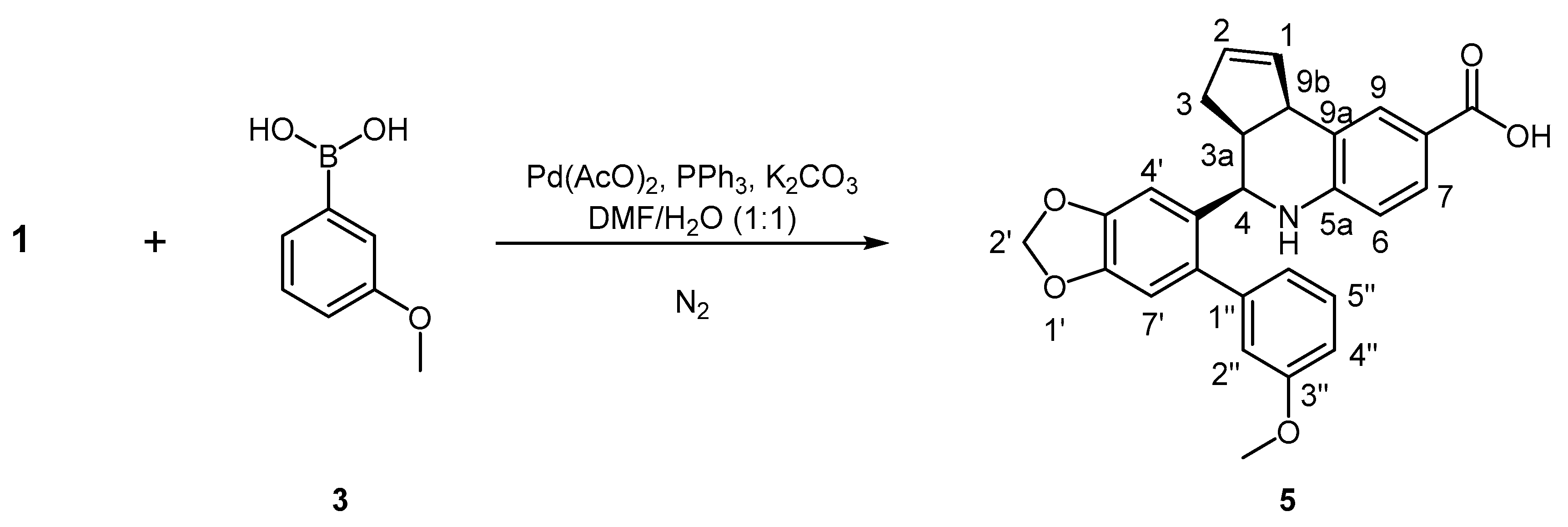{kind=link}
{kind=link}
| Ligand | Binding Energy | Nonbonded Interactions |
|---|---|---|
| 4 | −8.46 | N44, H52, Q54 *, L59, L119, R122 *, F206 |
| 5 | −8.36 | Q53, Q54 *, G58, L59, H120*, F278 *, I279, H302 *, P303, G306, H307, N310 |
| 7 | −8.21 | E51, Q53 *, Q54, G58, L59, H120 *, F278 *, I279, H282 *, H302 *, P303, N310 |
| Ligand | MW | Log P | HBD | HBA | Mut | Ter | Irri | Rep |
|---|---|---|---|---|---|---|---|---|
| 4 | 456.45 | 5.62 | 2 | 8 | None | None | None | None |
| 5 | 441.48 | 5.71 | 2 | 6 | None | None | None | None |
| 7 | 514.37 | 5.97 | 1 | 7 | None | None | None | None |
| System | ΔEvdw | ΔEele | ΔGele,sol | ΔGnpol,sol | ΔEpolar | ΔEnonpolar | ΔGmm/gbsa | TDS | ΔGbind |
|---|---|---|---|---|---|---|---|---|---|
| GPER-4 | −51.66 (2.9) | 15.72 (4.2) | 3.50 (0.10) | −6.44 (0.22) | 19.22 | −58.10 | −38.88 (4.0) | −26.43 (1.4) | −12.43 |
| GPER-5 | −47.3 (2.78) | 19.15 (3.0) | 4.70 (0.45) | −6.10 (0.20) | 23.85 | −53.40 | −29.55 (3.2) | −22.34 (2.6) | −7.21 |
| GPER-7 | −41.73 (2.5) | −3.90 (0.3) | 22.94 (0.3) | −5.52 (0.10) | 19.04 | −47.25 | −28.21 (2.5) | −24.14 (1.3) | −4.07 |
| IC50 | ||||
|---|---|---|---|---|
| Ligand | RCC4-VA | RCC4-VHL | MIA Paca-2 | Hep G2 |
| 7 | 24.46 | >50 | 14.78 | >50 |
| 4 | >50 | >50 | 18.18 | >50 |
| 5 | 39.29 | >50 | 21.15 | 31.58 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Méndez-Luna, D.; Morelos-Garnica, L.A.; García-Vázquez, J.B.; Bello, M.; Padilla-Martínez, I.I.; Fragoso-Vázquez, M.J.; Dueñas González, A.; De Pedro, N.; Gómez-Vidal, J.A.; Mendoza-Figueroa, H.L.; et al. Modifications on the Tetrahydroquinoline Scaffold Targeting a Phenylalanine Cluster on GPER as Antiproliferative Compounds against Renal, Liver and Pancreatic Cancer Cells. Pharmaceuticals 2021, 14, 49. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14010049
Méndez-Luna D, Morelos-Garnica LA, García-Vázquez JB, Bello M, Padilla-Martínez II, Fragoso-Vázquez MJ, Dueñas González A, De Pedro N, Gómez-Vidal JA, Mendoza-Figueroa HL, et al. Modifications on the Tetrahydroquinoline Scaffold Targeting a Phenylalanine Cluster on GPER as Antiproliferative Compounds against Renal, Liver and Pancreatic Cancer Cells. Pharmaceuticals. 2021; 14(1):49. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14010049
Chicago/Turabian StyleMéndez-Luna, David, Loreley Araceli Morelos-Garnica, Juan Benjamín García-Vázquez, Martiniano Bello, Itzia Irene Padilla-Martínez, Manuel Jonathan Fragoso-Vázquez, Alfonso Dueñas González, Nuria De Pedro, José Antonio Gómez-Vidal, Humberto Lubriel Mendoza-Figueroa, and et al. 2021. "Modifications on the Tetrahydroquinoline Scaffold Targeting a Phenylalanine Cluster on GPER as Antiproliferative Compounds against Renal, Liver and Pancreatic Cancer Cells" Pharmaceuticals 14, no. 1: 49. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14010049