Hot-Melt Extruded Amorphous Solid Dispersion for Solubility, Stability, and Bioavailability Enhancement of Telmisartan

 ,
, 
Abstract
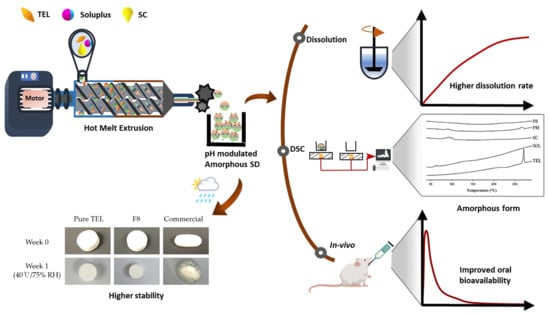
:
1. Introduction
2. Results and Discussion
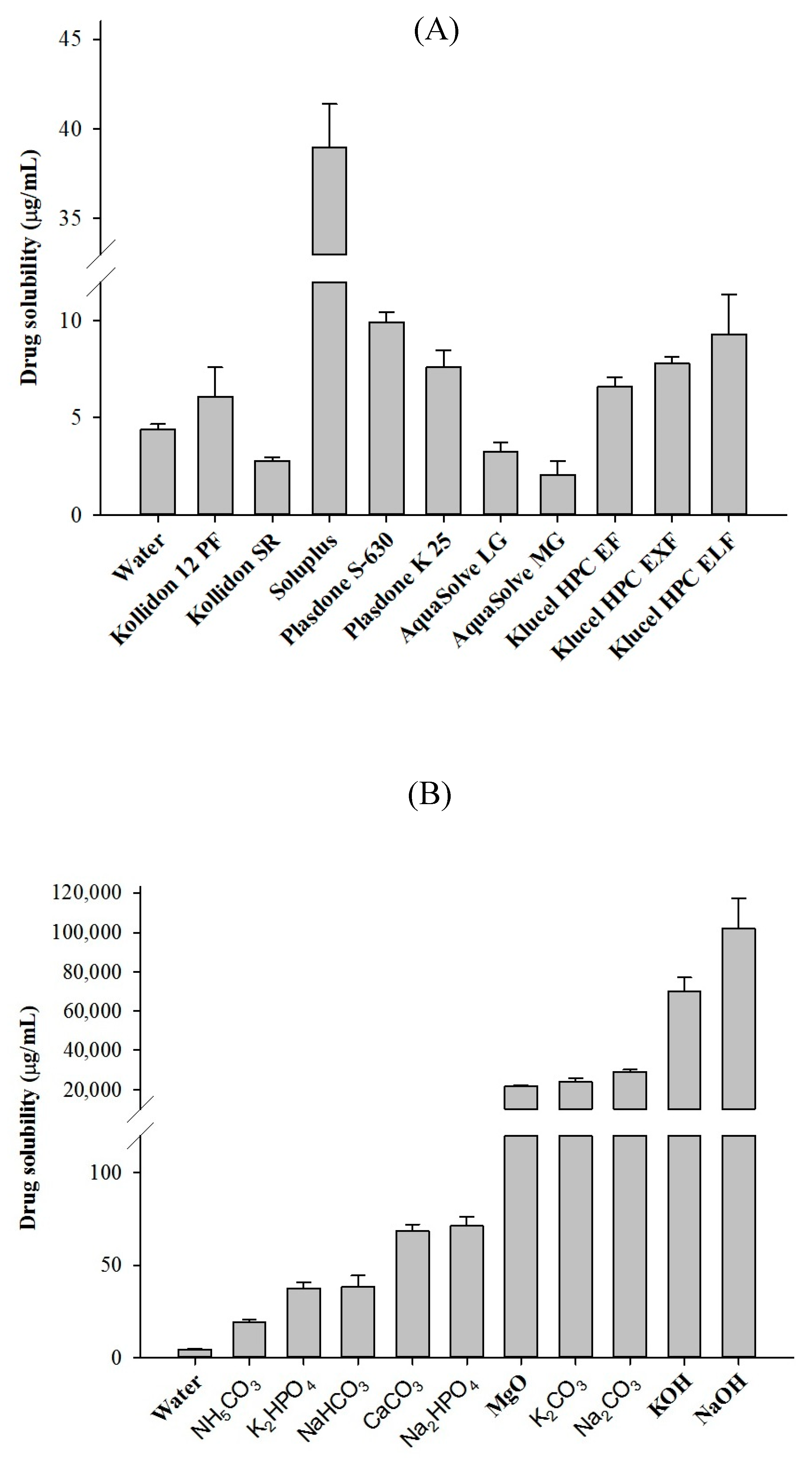
2.1. Solubility of TEL in Different pH
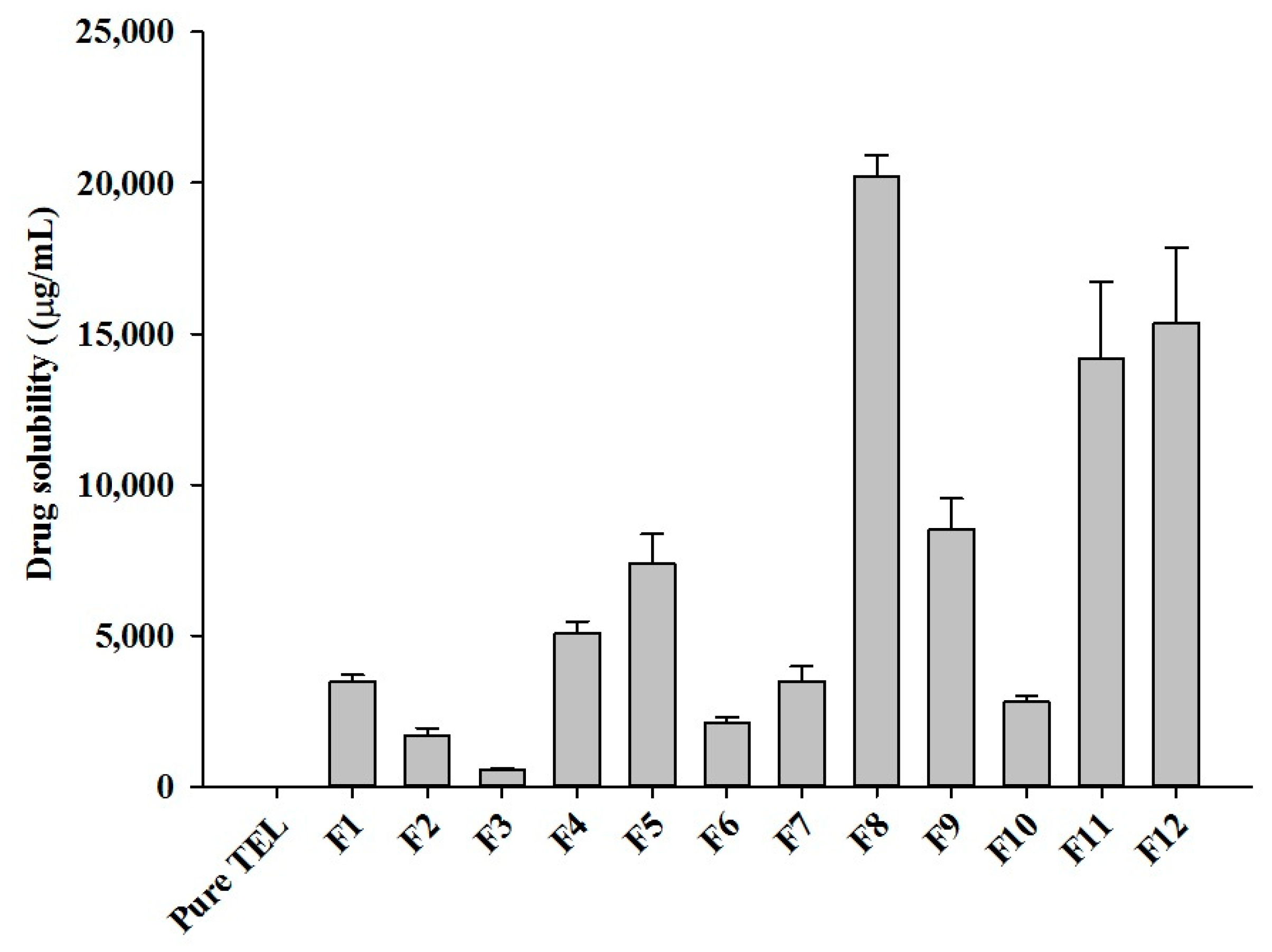
2.2. Formulation and Optimization of TEL-Loaded ASD
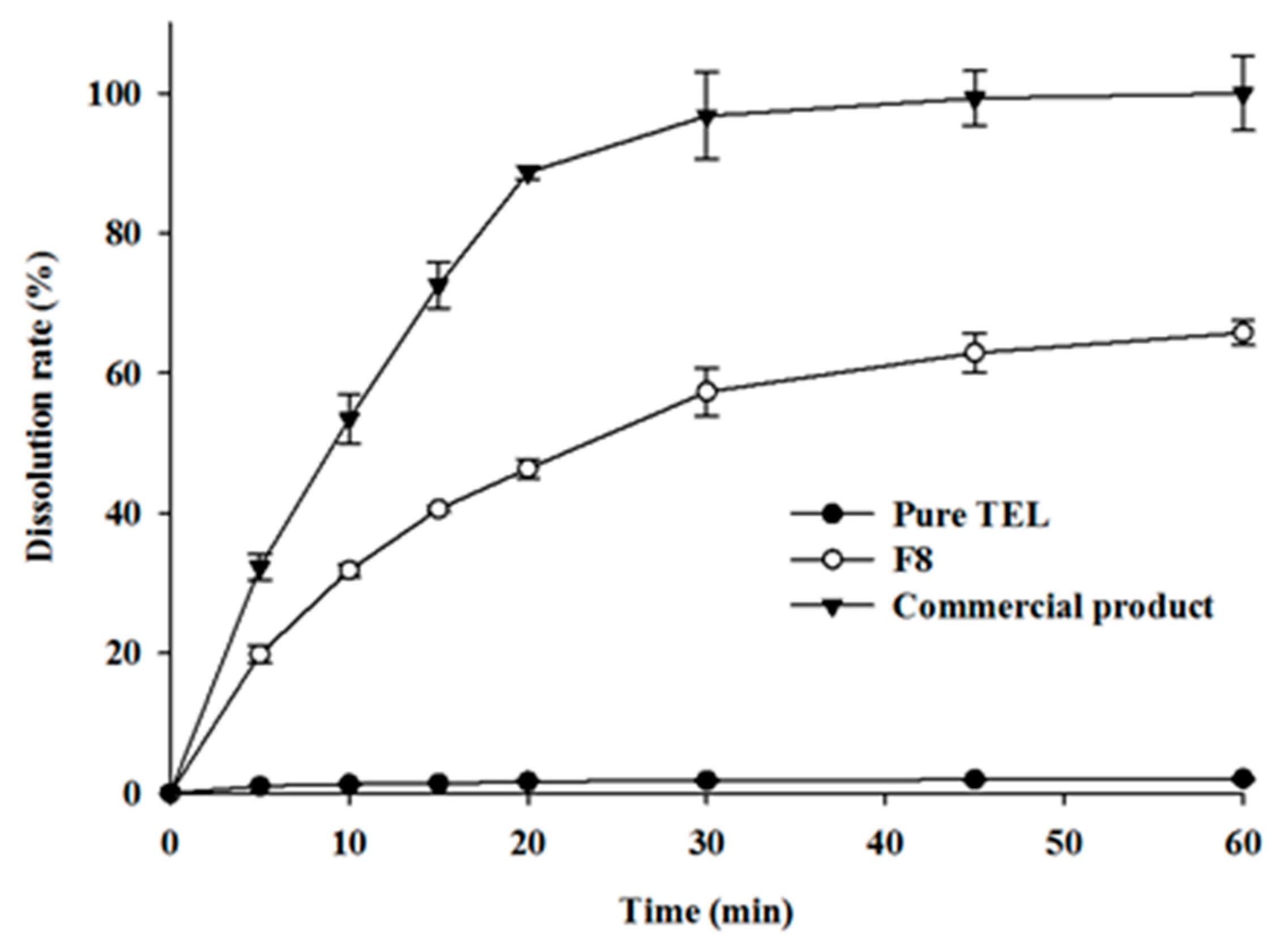
2.3. Evaluation of TEL-Loaded ASD
2.4. Solid-State Characterization
2.4.1. Scanning Electron Microscopy (SEM)
2.4.2. Differential Scanning Calorimetry (DSC)
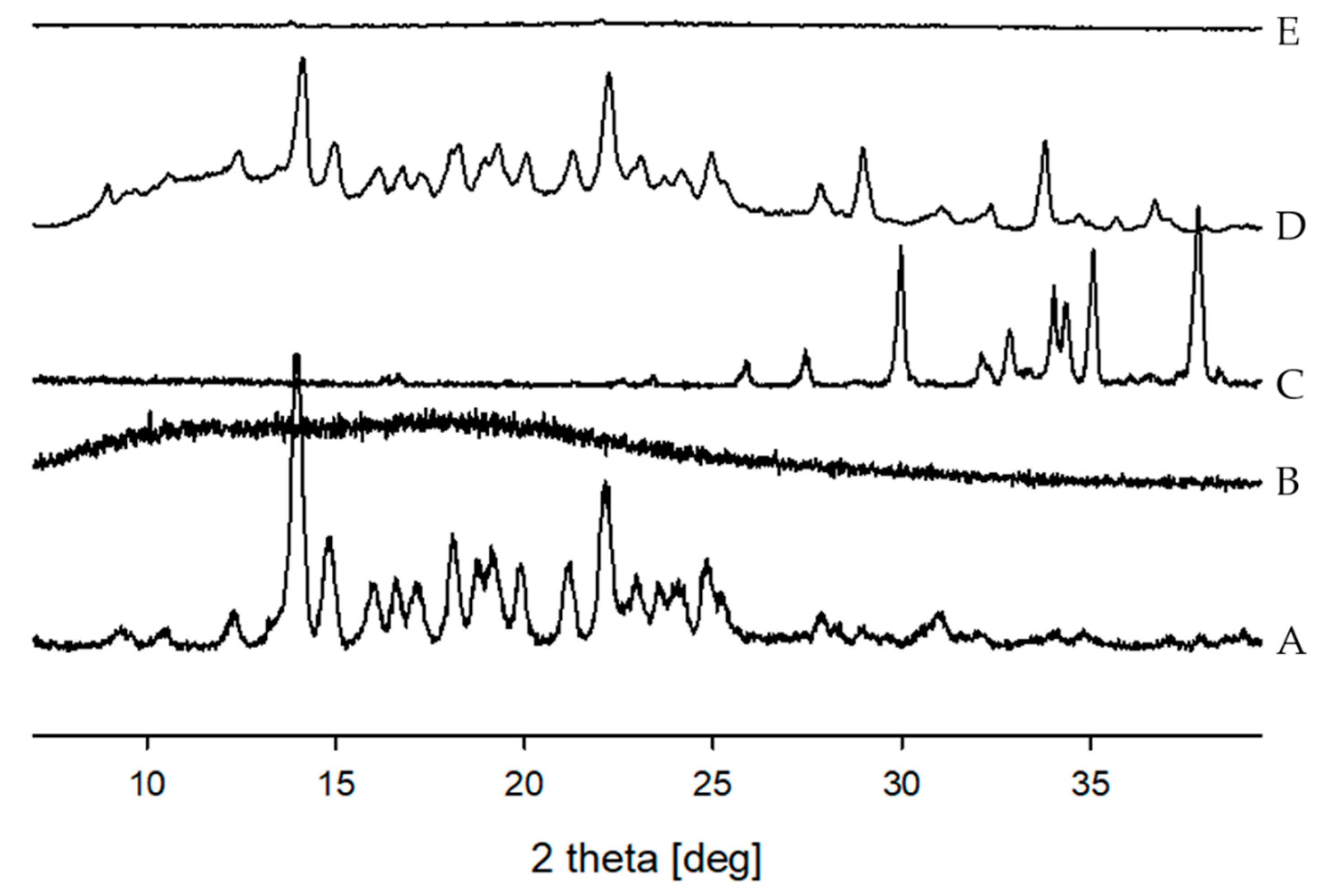
2.4.3. Powder X-ray Diffraction (PXRD)
2.5. Stability Studies
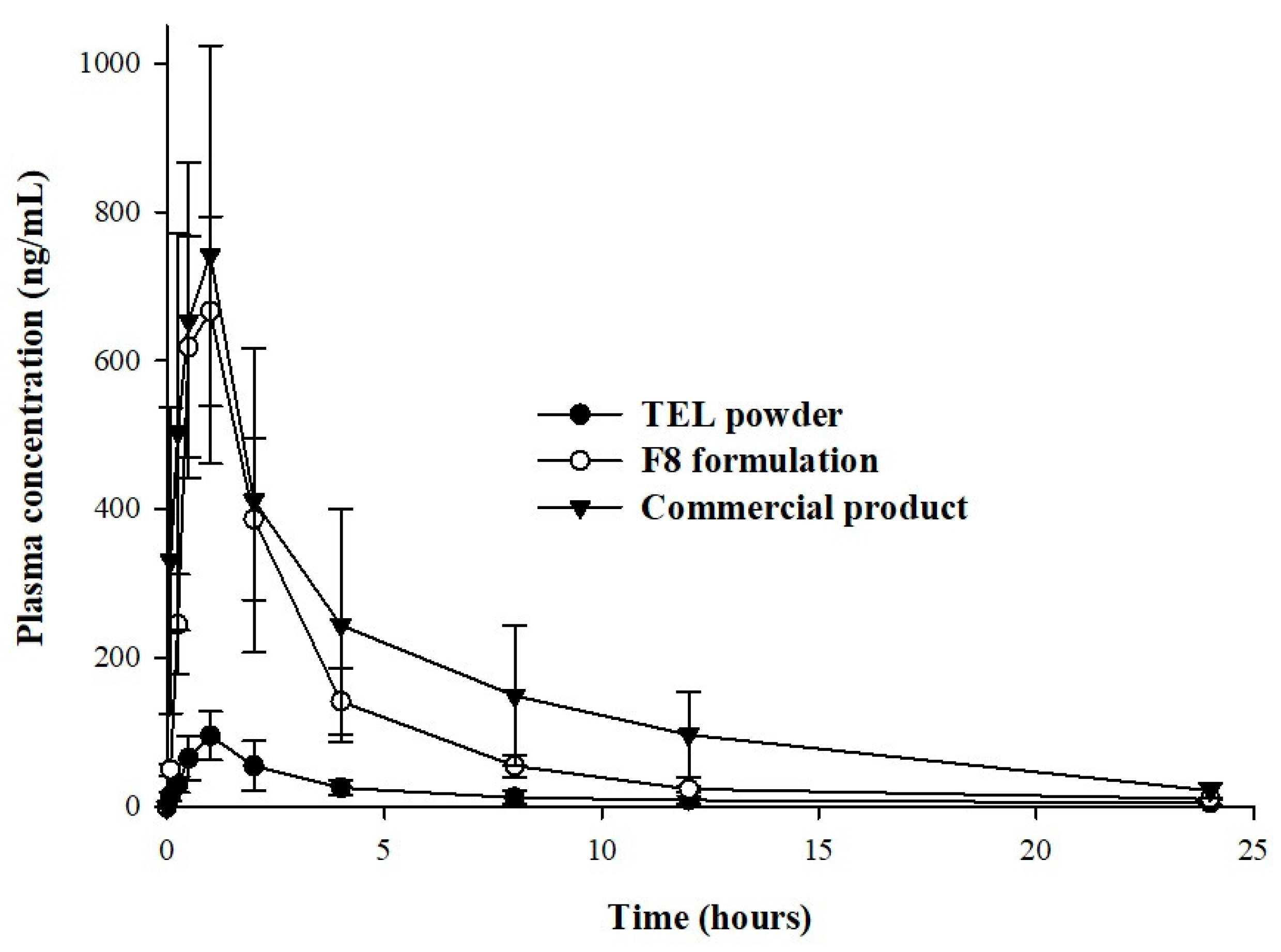
2.6. In Vivo Pharmacokinetic Studies
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Solubility Study of TEL
3.2.2. Solubility Screening of Polymers and Alkalizers
3.2.3. Preparation and Optimization of pH-Modulated Solid Dispersions and Tablets
3.2.4. In Vitro Dissolution Study
3.3. Solid-State Characterizations
3.3.1. Scanning Electron Microscopy (SEM)
3.3.2. Differential Scanning Calorimetry (DSC)
3.3.3. Powder X-ray Diffraction (PXRD)
3.4. Stability Studies
3.5. Pharmacokinetic Studies
3.5.1. Animals
3.5.2. In Vivo Plasma Sample Preparation
3.5.3. Statistical Data Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kearney, P.M.; Whelton, M.; Reynolds, K.; Whelton, P.K.; He, J. Worldwide prevalence of hypertension. J. Hypertens. 2004, 22, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Galzerano, D. New standards in hypertension and cardiovascular risk management: Focus on telmisartan. Vasc. Health Risk Manag. 2010, 6, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velasquez, M.T. Angiotensin II receptor blockers. A new class of antihypertensive drugs. Arch. Fam. Med. 1996, 5, 351. [Google Scholar] [CrossRef]
- Gosse, P. A review of telmisartan in the treatment of hypertension: Blood pressure control in the early morning hours. Vasc. Health Risk Manag. 2006, 2, 195–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmermans, P.B.M.W.M. Angiotensin II receptor antagonists: An emerging new class of cardiovascular therapeutics. Hypertens. Res. 1999, 22, 147–153. [Google Scholar] [CrossRef] [Green Version]
- Tran, P.H.L.; Tran, H.T.T.; Lee, B.-J. Modulation of microenvironmental pH and crystallinity of ionizable telmisartan using alkalizers in solid dispersions for controlled release. J. Control. Release 2008, 129, 59–65. [Google Scholar] [CrossRef]
- Wienen, W.; Entzeroth, M.; Meel, J.C.A.; Stangier, J.; Busch, U.; Ebner, T.; Schmid, J.; Lehmann, H.; Matzek, K.; Kempthorne-Rawson, J.; et al. A review on Telmisartan: A novel, long-acting angiotensin II-receptor antagonist. Cardiovasc. Drug Rev. 2006, 18, 127–154. [Google Scholar] [CrossRef]
- Patel, B.B.; Patel, J.K.; Chakraborty, S.; Shukla, D. Revealing facts behind spray dried solid dispersion technology used for solubility enhancement. Saudi Pharm. J. 2015, 23, 352–365. [Google Scholar] [CrossRef] [Green Version]
- Stella, V.J.; Nti-Addae, K.W. Prodrug strategies to overcome poor water solubility. Adv. Drug Deliv. Rev. 2007, 59, 677–694. [Google Scholar] [CrossRef]
- Kim, D.S.; Cho, J.H.; Park, J.H.; Kim, J.S.; Song, E.S.; Kwon, J.; Giri, B.R.; Jin, S.G.; Kim, K.S.; Choi, H.-G.; et al. Self-microemulsifying drug delivery system (SMEDDS) for improved oral delivery and photostability of methotrexate. Int. J. Nanomed. 2019, 14, 4949–4960. [Google Scholar] [CrossRef] [Green Version]
- Serajuddin, A.T.M. Salt formation to improve drug solubility. Adv. Drug Deliv. Rev. 2007, 59, 603–616. [Google Scholar] [CrossRef]
- Khadka, P.; Ro, J.; Kim, H.; Kim, I.; Kim, J.T.; Kim, H.; Cho, J.M.; Yun, G.; Lee, J. Pharmaceutical particle technologies: An approach to improve drug solubility, dissolution and bioavailability. Asian J. Pharm. Sci. 2014, 9, 304–316. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.A.; Harwansh, R.; Mirza, M.A.; Hussain, S.; Hussain, A. Oral lipid based drug delivery system (LBDDS): Formulation, characterization and application: A review. Curr. Drug Deliv. 2011, 8, 330–345. [Google Scholar] [CrossRef]
- Brewster, M.E.; Loftsson, T. Cyclodextrins as pharmaceutical solubilizers. Adv. Drug Deliv. Rev. 2007, 59, 645–666. [Google Scholar] [CrossRef]
- Kwon, J.; Giri, B.R.; Song, E.S.; Bae, J.; Lee, J.; Kim, D.W. Spray-dried amorphous solid dispersions of atorvastatin calcium for improved supersaturation and oral bioavailability. Pharmaceutics 2019, 11, 461. [Google Scholar] [CrossRef] [Green Version]
- Luu, T.D.; Lee, B.-J.; Tran, P.H.L.; Tran, T.T.D. Modified sprouted rice for modulation of curcumin crystallinity and dissolution enhancement by solid dispersion. J. Pharm. Investig. 2019, 49, 127–134. [Google Scholar] [CrossRef]
- Byeon, J.C.; Ahn, J.B.; Jang, W.S.; Lee, S.-E.; Choi, J.-S.; Park, J.-S. Recent formulation approaches to oral delivery of herbal medicines. J. Pharm. Investig. 2019, 49, 17–26. [Google Scholar] [CrossRef]
- McFall, H.; Sarabu, S.; Shankar, V.; Bandari, S.; Murthy, S.N.; Kolter, K.; Langley, N.; Kim, D.W.; Repka, M.A. Formulation of aripiprazole-loaded pH-modulated solid dispersions via hot-melt extrusion technology: In vitro and in vivo studies. Int. J. Pharm. 2019, 554, 302–311. [Google Scholar] [CrossRef]
- Agrawal, A.M.; Dudhedia, M.S.; Zimny, E. Hot melt extrusion: Development of an amorphous solid dispersion for an insoluble drug from mini-scale to clinical scale. AAPS PharmSciTech 2016, 17, 133–147. [Google Scholar] [CrossRef]
- Repka, M.A.; Bandari, S.; Kallakunta, V.R.; Vo, A.Q.; McFall, H.; Pimparade, M.B.; Bhagurkar, A.M. Melt extrusion with poorly soluble drugs—An integrated review. Int. J. Pharm. 2018, 535, 68–85. [Google Scholar] [CrossRef]
- Bajaj, A.; Rao, M.R.P.; Pardeshi, A.; Sali, D. Nanocrystallization by evaporative antisolvent technique for solubility and bioavailability enhancement of telmisartan. AAPS PharmSciTech 2012, 13, 1331–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Shao, Y.; Han, H.-K. Improved pH-dependent drug release and oral exposure of telmisartan, a poorly soluble drug through the formation of drug-aminoclay complex. Int. J. Pharm. 2014, 471, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Sangwai, M.; Vavia, P. Amorphous ternary cyclodextrin nanocomposites of telmisartan for oral drug delivery: Improved solubility and reduced pharmacokinetic variability. Int. J. Pharm. 2013, 453, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Zhu, X.; Luo, X.; Su, W. Dissolution properties and physical characterization of telmisartan–chitosan solid dispersions prepared by mechanochemical activation. AAPS PharmSciTech 2013, 14, 541–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Cho, W.; Cha, K.H.; Ahn, J.; Han, K.; Hwang, S.J. Solubilization of the poorly water soluble drug, telmisartan, using supercritical anti-solvent (SAS) process. Int. J. Pharm. 2013, 441, 50–55. [Google Scholar] [CrossRef]
- Dukeck, R.; Sieger, P.; Karmwar, P. Investigation and correlation of physical stability, dissolution behaviour and interaction parameter of amorphous solid dispersions of telmisartan: A drug development perspective. Eur. J. Pharm. Sci. 2013, 49, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Lepek, P.; Sawicki, W.; Wlodarski, K.; Wojnarowska, Z.; Paluch, M.; Guzik, L. Effect of amorphization method on telmisartan solubility and the tableting process. Eur. J. Pharm. Biopharm. 2013, 83, 114–121. [Google Scholar] [CrossRef]
- Shi, X.; Xu, T.; Huang, W.; Fan, B.; Sheng, X. Stability and bioavailability enhancement of telmisartan ternary solid dispersions: The synergistic effect of polymers and drug-polymer(s) interactions. AAPS PharmSciTech 2019, 20, 143. [Google Scholar] [CrossRef]
- Marasini, N.; Tran, T.H.; Poudel, B.K.; Cho, H.J.; Choi, Y.K.; Chi, S.-C.; Choi, H.-G.; Yong, C.S.; Kim, J.O. Fabrication and evaluation of pH-modulated solid dispersion for telmisartan by spray-drying technique. Int. J. Pharm. 2013, 441, 424–432. [Google Scholar] [CrossRef]
- Chae, J.S.; Chae, B.R.; Shin, D.J.; Goo, Y.T.; Lee, E.S.; Yoon, H.Y.; Kim, C.H.; Choi, Y.W. Tablet Formulation of a Polymeric Solid Dispersion Containing Amorphous Alkalinized Telmisartan. AAPS PharmSciTech 2018, 19, 2990–2999. [Google Scholar] [CrossRef]
- Cheow, W.S.; Kiew, T.Y.; Yang, Y.; Hadinoto, K. Amorphization Strategy Affects the Stability and Supersaturation Profile of Amorphous Drug Nanoparticles. Mol. Pharm. 2014, 11, 1611–1620. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, C.; Kawabata, Y.; Wada, K.; Yamada, S.; Onoue, S. Microenvironmental pH-modification to improve dissolution behavior and oral absorption for drugs with pH-dependent solubility. Expert Opin. Drug Deliv. 2014, 11, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Djuris, J.; Nikolakakis, I.; Ibric, S.; Djuric, Z.; Kachrimanis, K. Preparation of carbamazepine–Soluplus® solid dispersions by hot-melt extrusion, and prediction of drug–polymer miscibility by thermodynamic model fitting. Eur. J. Pharm. Biopharm. 2013, 84, 228–237. [Google Scholar] [CrossRef]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Polymeric amorphous solid dispersions: A review of amorphization, crystallization, stabilization, solid-state characterization, and aqueous solubilization of Biopharmaceutical Classification System Class II Drugs. J. Pharm. Sci. 2016, 105, 2527–2544. [Google Scholar] [CrossRef] [Green Version]
- Fule, R.; Amin, P. Development and evaluation of lafutidine solid dispersion via hot melt extrusion: Investigating drug-polymer miscibility with advanced characterisation. Asian J. Pharm. Sci. 2014, 9, 92–106. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Y.; Jing, G.; Tian, B.; Huang, H.; Zhang, Y.; Gou, J.; Tang, X.; He, H.; Wang, Y. Supersaturation induced by Itraconazole/Soluplus® micelles provided high GI absorption in vivo. Asian J. Pharm. Sci. 2016, 11, 255–264. [Google Scholar] [CrossRef] [Green Version]
- Enose, A.A.; Dasan, P. Formulation, Characterization and Pharmacokinetic Evaluation of Telmisartan Solid Dispersions. J. Mol. Pharm. Org. Process Res. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Alshahrani, S.M.; Lu, W.; Park, J.-B.; Morott, J.T.; Alsulays, B.B.; Majumdar, S.; Langley, N.; Kolter, K.; Gryczke, A.; Repka, M.A. Stability-enhanced hot-melt extruded amorphous solid dispersions via combinations of soluplus® and HPMCAS-HF. AAPS PharmSciTech 2015, 16, 824–834. [Google Scholar] [CrossRef] [Green Version]
- Farag Badawy, S.I.; Hussain, M.A. Microenvironmental pH modulation in solid dosage forms. J. Pharm. Sci. 2007, 96, 948–959. [Google Scholar] [CrossRef]
- Tran, P.H.-L.; Tran, T.T.-D.; Lee, K.-H.; Kim, D.-J.; Lee, B.-J. Dissolution-modulating mechanism of pH modifiers in solid dispersion containing weakly acidic or basic drugs with poor water solubility. Expert Opin. Drug Deliv. 2010, 7, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Hardung, H.; Djuric, D.; Ali, S. Combining HME & solubilization: Soluplus®—The solid solution. Drug Deliv. Technol. 2010, 10, 20–27. [Google Scholar]
- Patel, H.; Patel, H.; Gohel, M.; Tiwari, S. Dissolution rate improvement of telmisartan through modified MCC pellets using 32 full factorial design. Saudi Pharm. J. 2016, 24, 579–587. [Google Scholar] [CrossRef] [Green Version]
- Dinnebier, R.E.; Sieger, P.; Nar, H.; Shankland, K.; David, W.I.F. Structural characterization of three crystalline modifications of telmisartan by single crystal and high-resolution X-ray powder diffraction. J. Pharm. Sci. 2000, 89, 1465–1479. [Google Scholar] [CrossRef] [Green Version]
- Theil, F.; Anantharaman, S.; Kyeremateng, S.O.; van Lishaut, H.; Dreis-Kühne, S.H.; Rosenberg, J.; Mägerlein, M.; Woehrle, G.H. Frozen in time: Kinetically stabilized amorphous solid dispersions of nifedipine stable after a quarter century of storage. Mol. Pharm. 2017, 14, 183–192. [Google Scholar] [CrossRef]
- Sareen, S.; Joseph, L.; Mathew, G. Improvement in solubility of poor water-soluble drugs by solid dispersion. Int. J. Pharm. Investig. 2012, 2, 12. [Google Scholar] [CrossRef] [Green Version]
- Laitinen, R.; Löbmann, K.; Strachan, C.J.; Grohganz, H.; Rades, T. Emerging trends in the stabilization of amorphous drugs. Int. J. Pharm. 2013, 453, 65–79. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhi, Z.; Jiang, T.; Zhang, J.; Wang, Z.; Wang, S. Spherical mesoporous silica nanoparticles for loading and release of the poorly water-soluble drug telmisartan. J. Control. Release 2010, 145, 257–263. [Google Scholar] [CrossRef]
- Shamma, R.N.; Basha, M. Soluplus®: A novel polymeric solubilizer for optimization of Carvedilol solid dispersions: Formulation design and effect of method of preparation. Powder Technol. 2013, 237, 406–414. [Google Scholar] [CrossRef]
- Paaver, U.; Tamm, I.; Laidmäe, I.; Lust, A.; Kirsimäe, K.; Veski, P.; Kogermann, K.; Heinämäki, J. Soluplus graft copolymer: Potential novel carrier polymer in electrospinning of nanofibrous drug delivery systems for wound therapy. Biomed Res. Int. 2014, 2014, 1–7. [Google Scholar] [CrossRef]
- Rowe, R.C.; Sheskey, P.J.; Quinn, M.E. Handbook-of-Pharmaceutical-Excipients, 6th ed.; Rowe, R.C., Sheskey, P.J., Quinn, M.E., Eds.; Pharmaceutical Press: London, UK, 2009; ISBN 978 1 58212 135 2. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pH Level | Drug Solubility (µg/mL) |
|---|---|
| Water | 4.42 ± 0.25 |
| pH 1.2 | 97.18 ± 18.24 |
| pH 4.0 | 2.46 ± 0.39 |
| pH 6.8 | 1.26 ± 0.37 |
| pH 10.0 | 2557.77 ± 171.02 |
| Parameters | TEL | TEL-Loaded pHM-ASD | Commercial Formulation |
|---|---|---|---|
| AUC0–∞ (h·ng/mL) | 423.69 ± 114.82 | 2275.21 ± 776.84 * | 3425.42 ± 1553.04 * |
| Cmax (ng/mL) | 105.46 ± 21.07 | 697.51 ± 92.65 * | 757.27 ± 244.72 * |
| Tmax (h) | 0.83 ± 0.26 | 0.75 ± 0.27 | 1.00 ± 0.55 |
| T1/2 (h) | 6.62 ± 2.20 | 4.53 ± 0.974 | 5.97 ± 1.96 |
| Kel (h−1) | 0.11 ± 0.04 | 0.15 ± 0.04 | 0.12 ± 0.07 |
| Structure | Properties | Uses | Ref. |
|---|---|---|---|
 Telmisartan (Mol. Wt. = 514.6 Da) | Weak base, low aqueous solubility (0.09 µg/mL), and hygroscopic in nature | Blood pressure lowering agent (Antihypertensive) | [7] |
 Soluplus® (Mol. Wt. = 90–140 KDa) | Amphiphilic characteristics, low glass transition temperature (Tg), and low hygroscopicity | Matrix former, solubility enhancer, and stabilizer for SDs | [48,49] |
 Sodium carbonate (Mol. Wt. = 105.988 Da) | Freely water-soluble white powder with strong alkaline property | Excipients for pharmaceutical formulations | [50] |
| Formulations | TEL | SOL | SC | Formulations | TEL | SOL | SC |
|---|---|---|---|---|---|---|---|
| F1 | 10 | 90 | 0 | F7 | 40 | 60 | 0 |
| F2 | 10 | 85 | 5 | F8 | 40 | 55 | 5 |
| F3 | 10 | 80 | 10 | F9 | 40 | 50 | 10 |
| F4 | 20 | 80 | 0 | F10 | 60 | 40 | 0 |
| F5 | 20 | 75 | 5 | F11 | 60 | 35 | 5 |
| F6 | 20 | 70 | 10 | F12 | 60 | 30 | 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giri, B.R.; Kwon, J.; Vo, A.Q.; Bhagurkar, A.M.; Bandari, S.; Kim, D.W. Hot-Melt Extruded Amorphous Solid Dispersion for Solubility, Stability, and Bioavailability Enhancement of Telmisartan. Pharmaceuticals 2021, 14, 73. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14010073
Giri BR, Kwon J, Vo AQ, Bhagurkar AM, Bandari S, Kim DW. Hot-Melt Extruded Amorphous Solid Dispersion for Solubility, Stability, and Bioavailability Enhancement of Telmisartan. Pharmaceuticals. 2021; 14(1):73. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14010073
Chicago/Turabian StyleGiri, Bhupendra Raj, Jaewook Kwon, Anh Q. Vo, Ajinkya M. Bhagurkar, Suresh Bandari, and Dong Wuk Kim. 2021. "Hot-Melt Extruded Amorphous Solid Dispersion for Solubility, Stability, and Bioavailability Enhancement of Telmisartan" Pharmaceuticals 14, no. 1: 73. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14010073