
Discovery of a Novel Tetrapeptide against Influenza A Virus: Rational Design, Synthesis, Bioactivity Evaluation and Computational Studies


 ,
,  ,
,  and
and
Abstract
:1. Introduction
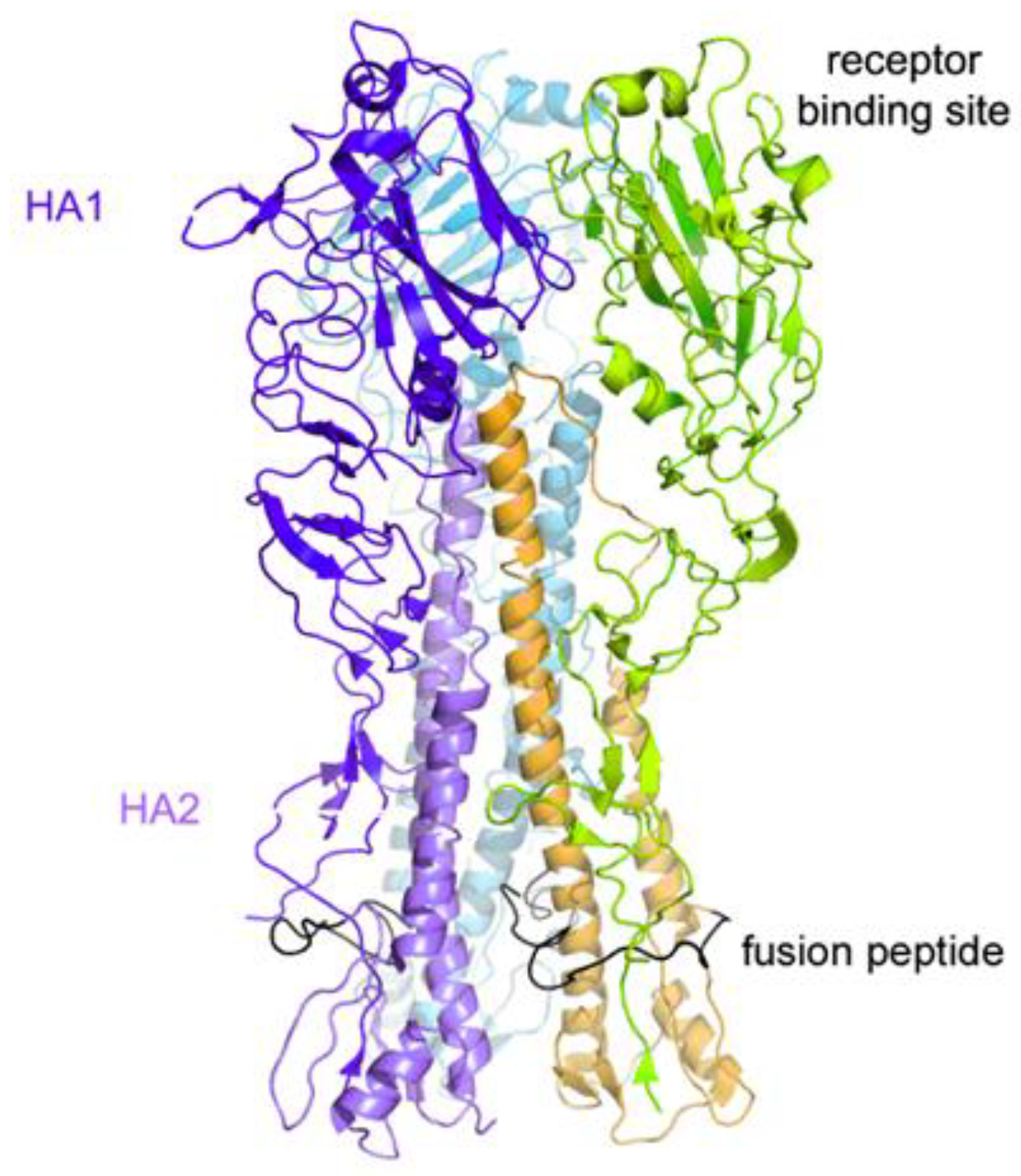
2. Results and Discussion
2.1. Design and Synthesis
2.2. Direct Binding Assays
2.2.1. Microscale Thermophoresis (MST)
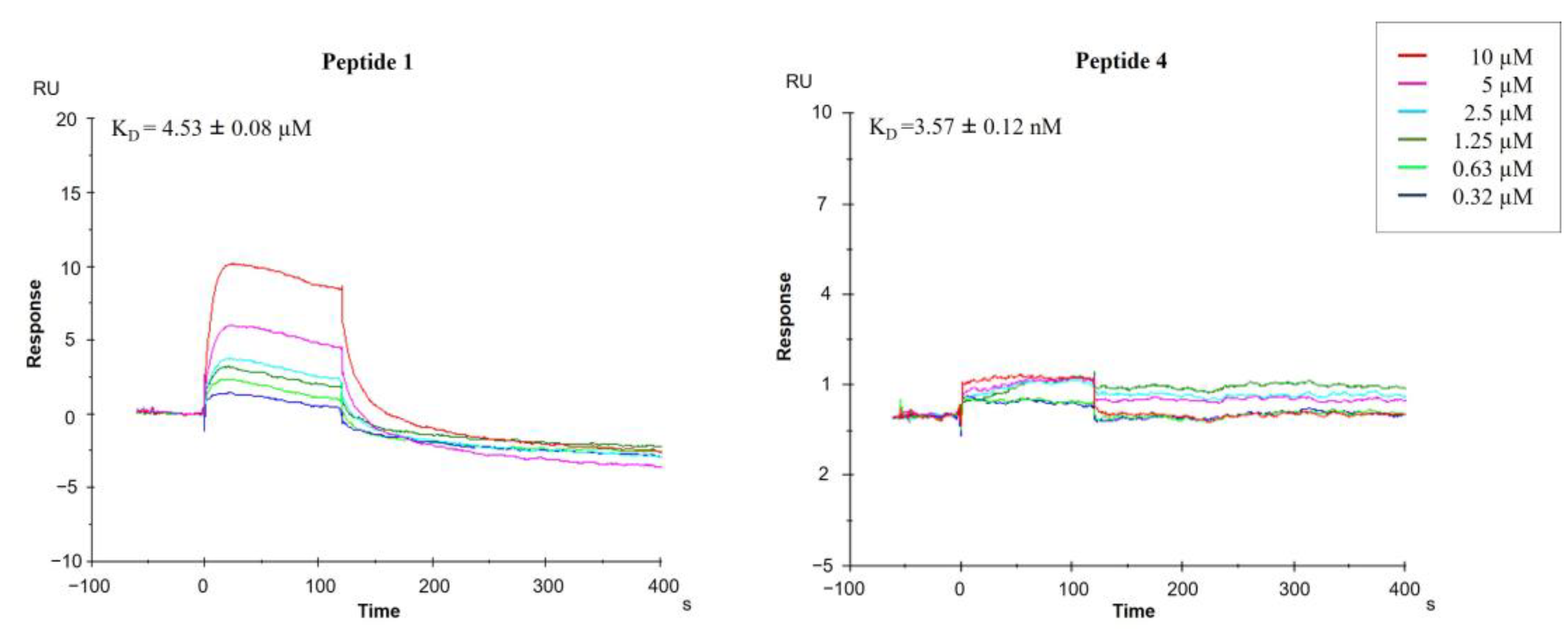
2.2.2. Surface Plasmon Resonance (SPR)
2.3. Antiviral Activity
2.3.1. Hemagglutination Inhibition Assay (HI)
2.3.2. Neutralization Assay (NT)
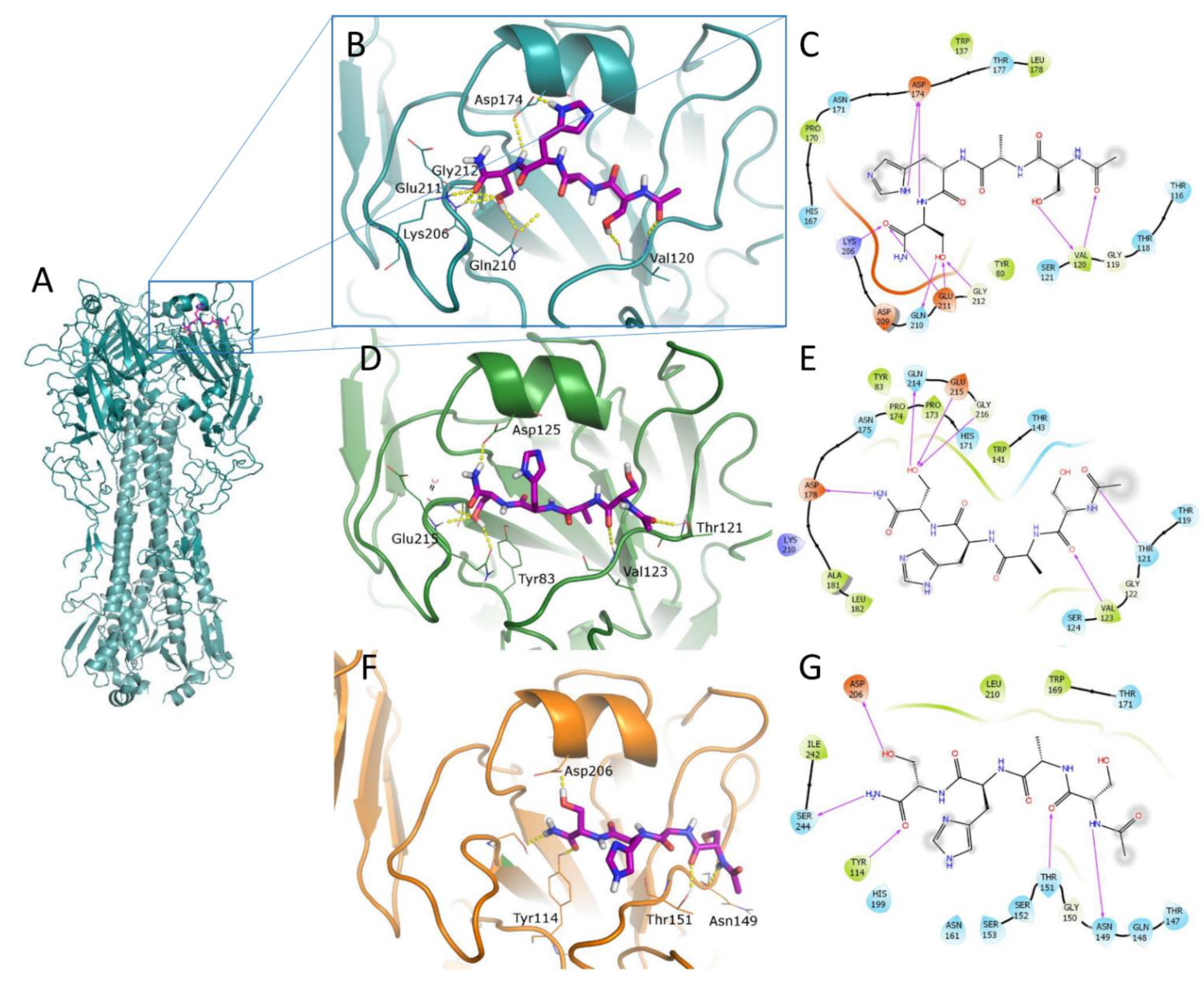
2.4. Computational Studies
3. Materials and Methods
3.1. Synthesis
3.1.1. Peptide Synthesis
3.1.2. Purification and Characterization
3.2. Direct Binding Assay
3.2.1. Microscale Thermophoresis (MST)
3.2.2. Surface Plasmon Resonance (SPR)
3.3. Biological Assay
3.3.1. Cells and Viral Strains
3.3.2. Cytotoxicity Assay
3.3.3. Hemagglutination Inhibition Assay (HI)
3.3.4. Neutralization Assay (NT)
3.4. Computational Studies
3.4.1. Homology Modeling
3.4.2. Protein Preparation
3.4.3. Binding Site Identification and Analysis
3.4.4. Receptor Grid Generation
3.4.5. Ligand Preparation
3.4.6. Docking Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hale, B.G.; Albrecht, R.A.; García-Sastre, A. Innate immune evasion strategies of influenza viruses. Future Microbiol. 2010, 5, 23–41. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.; Gao, G.F. Emerging H5N8 avian influenza viruses. Science 2021, 372, 784–786. [Google Scholar] [CrossRef]
- Hayden, F.G. Respiratory viral threats. Curr. Opin. Infect. Dis. 2006, 19, 169–178. [Google Scholar] [CrossRef]
- de St Maurice, A.; Halasa, N. Preparing for the 2019–2020 influenza season. Pediatr. Transplant. 2020, 24, e13645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves Galvão, M.G.; Rocha Crispino Santos, M.A.; Alves da Cunha, A.J. Amantadine and rimantadine for influenza A in children and the elderly. Cochrane Database Syst. Rev. 2014, 11, CD002745. [Google Scholar] [CrossRef]
- Kamali, A.; Holodniy, M. Influenza treatment and prophylaxis with neuraminidase inhibitors: A review. Infect. Drug. Resist. 2013, 6, 187–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shie, J.J.; Fang, J.M. Development of effective anti-influenza drugs: Congeners and conjugates—A review. J Biomed Sci. 2019, 26, 84. [Google Scholar] [CrossRef] [Green Version]
- Aschenbrenner, D.S. Xofluza Now Indicated to Prevent Influenza. Am. J. Nurs. 2021, 121, 26–27. [Google Scholar] [CrossRef] [PubMed]
- Belshe, R.B.; Burk, B.; Newman, F.; Cerruti, R.L.; Sim, I.S. Resistance of influenza A virus to amantadine and rimantadine: Results of one decade of surveillance. J. Infect. Dis. 1989, 159, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Bertram, S.; Glowacka, I.; Steffen, I.; Kühl, A.; Pöhlmann, S. Novel insights into proteolytic cleavage of influenza virus hemagglutinin. Rev. Med. Virol. 2010, 20, 298–310. [Google Scholar] [CrossRef]
- Bullard, B.L.; Weaver, E.A. Strategies Targeting Hemagglutinin as a Universal Influenza Vaccine. Vaccines 2021, 9, 257. [Google Scholar] [CrossRef]
- Ammendolia, M.G.; Agamennone, M.; Pietrantoni, A.; Lannutti, F.; Siciliano, R.A.; De Giulio, B.; Amici, C.; Superti, F. Bovine lactoferrin-derived peptides as novel broad-spectrum inhibitors of influenza virus. Pathog. Glob. Health 2012, 106, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Superti, F.; Agamennone, M.; Pietrantoni, A.; Ammendolia, M.G. Bovine Lactoferrin Prevents Influenza A Virus Infection by Interfering with the Fusogenic Function of Viral Hemagglutinin. Viruses 2019, 11, 51. [Google Scholar] [CrossRef] [Green Version]
- Scala, M.C.; Sala, M.; Pietrantoni, A.; Spensiero, A.; Di Micco, S.; Agamennone, M.; Bertamino, A.; Novellino, E.; Bifulco, G.; Gomez-Monterrey, I.M.; et al. Lactoferrin-derived Peptides Active towards Influenza: Identification of Three Potent Tetrapeptide Inhibitors. Sci. Rep. 2017, 7, 10593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Monterrey, I.; Sala, M.; Rusciano, M.R.; Monaco, S.; Maione, A.S.; Iaccarino, G.; Tortorella, P.; D’Ursi, A.M.; Scrima, M.; Carotenuto, A.; et al. Characterization of a selective CaMKII peptide inhibitor. Eur. J. Med. Chem. 2013, 62, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Renaud, J.P.; Chung, C.W.; Danielson, U.H.; Egner, U.; Hennig, M.; Hubbard, R.E.; Nar, H. Biophysics in drug discovery: Impact, challenges and opportunities. Nat. Rev. Drug. Discov. 2016, 15, 679–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duhr, S.; Braun, D. Why molecules move along a temperature gradient. Proc. Natl. Acad. Sci. USA 2006, 103, 19678–19682. [Google Scholar] [CrossRef] [Green Version]
- Navratilova, I.; Hopkins, A.L. Fragment screening by surface plasmon resonance. ACS Med. Chem. Lett. 2010, 1, 44–48. [Google Scholar] [CrossRef] [Green Version]
- Weis, W.; Brown, J.H.; Cusack, S.; Paulson, J.C.; Skehel, J.J.; Wiley, D.C. Structure of the influenza virus haemagglutinin complexed with its receptor, sialic acid. Nature 1988, 333, 426–431. [Google Scholar] [CrossRef]
- Kadam, R.U.; Wilson, I.A. A small-molecule fragment that emulates binding of receptor and broadly neutralizing antibodies to influenza A hemagglutinin. Proc. Natl. Acad. Sci. USA 2018, 115, 4240–4245. [Google Scholar] [CrossRef] [Green Version]
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger LLC. Schrödinger Release, 2020–2024: Maestro, Glide, LigPrep, MacroModel, SiteMap; Schrödinger, LLC: New York, NY, USA, 2020. [Google Scholar]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; McBride, R.; Nycholat, C.M.; Paulson, J.C.; Wilson, I.A. Structural characterization of the hemagglutinin receptor specificity from the 2009 H1N1 influenza pandemic. J. Virol. 2012, 86, 982–990. [Google Scholar] [CrossRef] [Green Version]
- Atherton, E.; Sheppard, R.C. Solid-Phase Peptide Synthesis: A Practical Approach; Oxford University Press: Oxford, UK, 1989. [Google Scholar]
- Pescina, S.; Sala, M.; Padula, C.; Scala, M.C.; Spensiero, A.; Belletti, S.; Gatti, R.; Novellino, E.; Campiglia, P.; Santi, P.; et al. Design and Synthesis of New Cell Penetrating Peptides: Diffusion and Distribution Inside the Cornea. Mol. Pharm. 2016, 13, 3876–3883. [Google Scholar] [CrossRef] [Green Version]
- Monolith NT Protein Labeling Kit RED-NHS. User Manual. Available online: https://manualzz.com/doc/7334293/monolith-nt%E2%84%A2-protein-labeling-kit-red-nhs (accessed on 29 October 2020).
- Milite, C.; Feoli, A.; Horton, J.R.; Rescigno, D.; Cipriano, A.; Pisapia, V.; Viviano, M.; Pepe, G.; Amendola, G.; Novellino, E.; et al. Discovery of a Novel Chemotype of Histone Lysine Methyltransferase EHMT1/2 (GLP/G9a) Inhibitors: Rational Design, Synthesis, Biological Evaluation, and Co-crystal Structure. J. Med. Chem. 2019, 62, 2666–2689. [Google Scholar] [CrossRef]
- Gaush, C.R.; Smith, T.F. Replication and plaque assay of influenza virus in an established line of canine kidney cells. Appl. Microbiol. 1968, 16, 588–594. [Google Scholar] [CrossRef]
- Rimmelzwaan, G.F.; Baars, M.; Claas, E.C.; Osterhaus, A.D. Comparison of RNA hybridization, hemagglutination assay, titration of infectious virus and immunofluorescence as methods for monitoring influenza virus replication in vitro. J. Virol. Methods 1998, 74, 57–66. [Google Scholar] [CrossRef]
- Pietrantoni, A.; Dofrelli, E.; Tinari, A.; Ammendolia, M.G.; Puzelli, S.; Fabiani, C.; Donatelli, I.; Superti, F. Bovine lactoferrin inhibits influenza A virus induced programmed cell death in vitro. Biometals 2010, 23, 465–475. [Google Scholar] [CrossRef]
- Marchetti, M.; Trybala, E.; Superti, F.; Johansson, M.; Bergström, T. Inhibition of herpes simplex virus infection by lactoferrin is dependent on interference with the virus binding to glycosaminoglycans. Virology 2004, 318, 405–413. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2012, 9, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Gallo Cassarino, T.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef] [PubMed]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Bertoni, M.; Kiefer, F.; Biasini, M.; Bordoli, L.; Schwede, T. Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci. Rep. 2017, 7, 10480. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
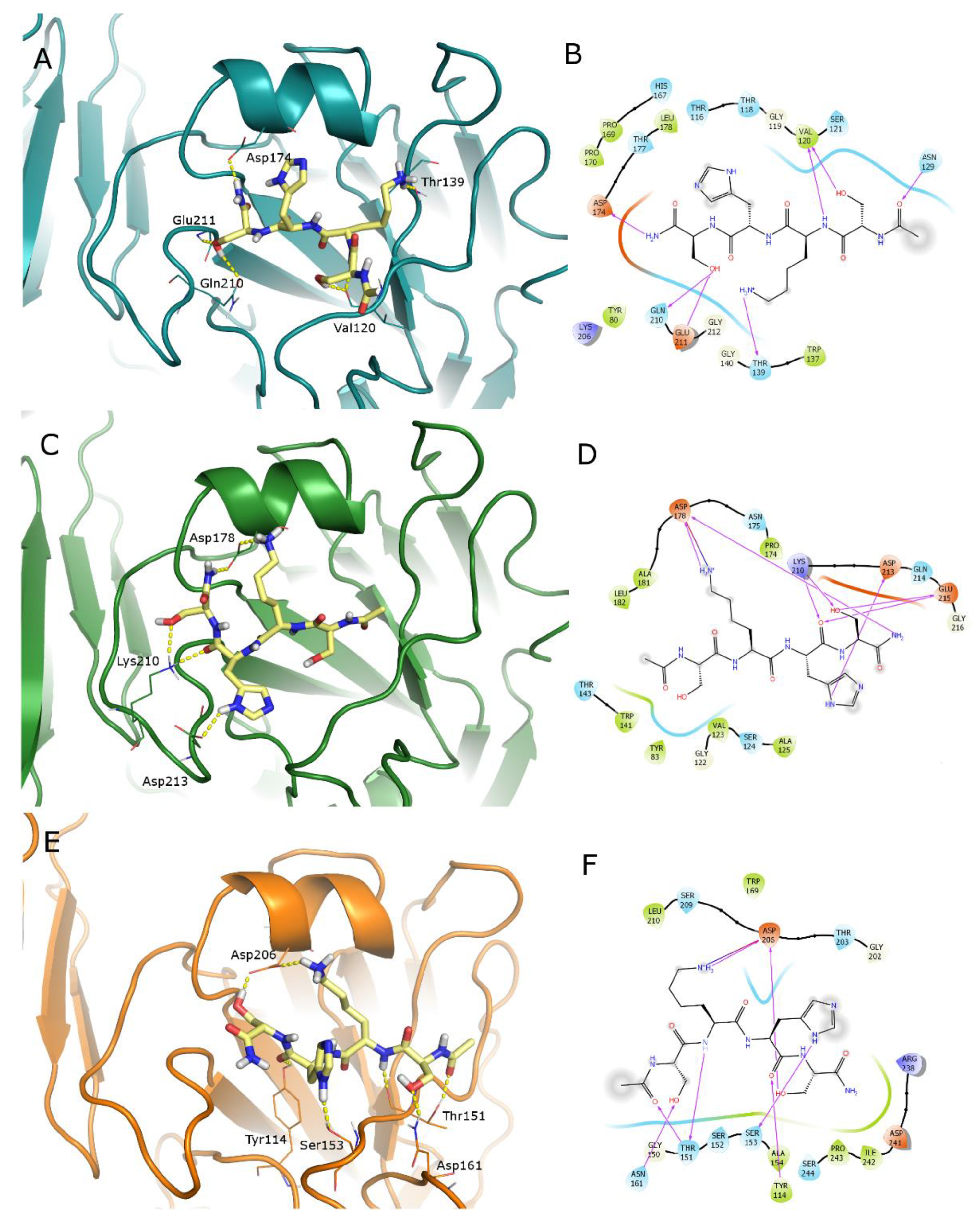{kind=link}
| Pep. | Seq. | MST KD (μM) | SPR KD (μM) | HI Titer a (nM) | ||
|---|---|---|---|---|---|---|
| A/Roma-ISS/02/08 H1N1 | A/Parma/24/09 H1N1 | A/Parma/05/06 H3N2 | ||||
| 1 a | SKHS | 7.26 ± 0.06 | 4.53 ± 0.08 | 0.1 | 1.5 | 12 |
| 3 | AKHS | 3.12 ± 0.11 | 2.7 ± 0.04 | 0.6 | N.D. b | 12 |
| 4 | SAHS | 0.0082 ± 0.0001 | 0.0035 ± 0.00012 | 1.8 × 10−6 | 0.5 | 2.4 × 10−6 |
| 5 | SKAS | 7.01 ± 0.09 | 1.03 ± 0.01 | 1.8 × 10−6 | 2.9 × 10−3 | N.D. b |
| 6 | SKHA | 11.4 ± 0.17 | 6.75 ± 0.81 | 5 × 10−7 | N.D. b | N.D. b |
| 2a | SLDC | 10.4 ± 0.23 | 7.12 ± 0.26 | 1.4 × 10−6 | 6 | 1.5 |
| 7 | ALDC | 21.2 ± 0.41 | 0.0277 ± 0.0017 | N.D. b | 9 × 10−7 | 3.6 × 10−7 |
| 8 | SADC | 6.38 ± 0.21 | 2.19 ± 0.51 | N.D. b | 2.2 × 10−6 | 5 × 10−7 |
| 9 | SLAC | 0.0058 ± 0.0003 | 2.57 ± 0.34 | N.D. b | 5 × 10−7 | 9 × 10−7 |
| 10 | SLDA | 2.69 ± 0.09 | 0.343 ± 0.019 | N.D. b | 6.1 | 2.1 × 10−3 |
| Pep. | Seq. | A/Roma-ISS/02/08 H1N1 | A/Parma/24/09 H1N1 | A/Parma/05/06 H3N2 | |||
|---|---|---|---|---|---|---|---|
| EC50 a (μM) | SI | EC50 a (μM) | SI | EC50 a (μM) | SI | ||
| 1 b | SKHS | 3 ± 0.61 × 10−6 | >8.33 × 106 | 4.8 ± 0.12 × 10−8 | >5.2 × 108 | 5 ± 0.02 × 10−6 | >5 × 106 |
| 2b | SLDC | 5 ± 0.01 × 10−7 | >5 × 107 | 4.6 ± 0.05 × 10−6 | >5.4 × 106 | 4.3 ± 0.03 × 10−6 | >5.8 × 107 |
| 4 | SAHS | 5.77 ± 0.01 × 10−7 | >4.33 × 107 | 4.3 ± 0.3 × 10−10 | >5.81 × 1010 | 9.36 ± 0.1 × 10−7 | >2.67 × 107 |
| HA Subtype | Pep. | Strain Energy | Docking Score |
|---|---|---|---|
| Roma/H1N1 | 1 | 6.682 | −5.174 |
| 4 | 2.525 | −7.554 | |
| Parma/H1N1 | 1 | 9.014 | −5.266 |
| 4 | 4.050 | −6.259 | |
| Parma/H3N2 | 1 | 6.958 | −5.230 |
| 4 | 2.081 | −6.137 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scala, M.C.; Agamennone, M.; Pietrantoni, A.; Di Sarno, V.; Bertamino, A.; Superti, F.; Campiglia, P.; Sala, M. Discovery of a Novel Tetrapeptide against Influenza A Virus: Rational Design, Synthesis, Bioactivity Evaluation and Computational Studies. Pharmaceuticals 2021, 14, 959. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14100959
Scala MC, Agamennone M, Pietrantoni A, Di Sarno V, Bertamino A, Superti F, Campiglia P, Sala M. Discovery of a Novel Tetrapeptide against Influenza A Virus: Rational Design, Synthesis, Bioactivity Evaluation and Computational Studies. Pharmaceuticals. 2021; 14(10):959. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14100959
Chicago/Turabian StyleScala, Maria Carmina, Mariangela Agamennone, Agostina Pietrantoni, Veronica Di Sarno, Alessia Bertamino, Fabiana Superti, Pietro Campiglia, and Marina Sala. 2021. "Discovery of a Novel Tetrapeptide against Influenza A Virus: Rational Design, Synthesis, Bioactivity Evaluation and Computational Studies" Pharmaceuticals 14, no. 10: 959. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14100959