
Mechanisms, Pathophysiology and Currently Proposed Treatments of Chronic Obstructive Pulmonary Disease

 and
and 
Abstract
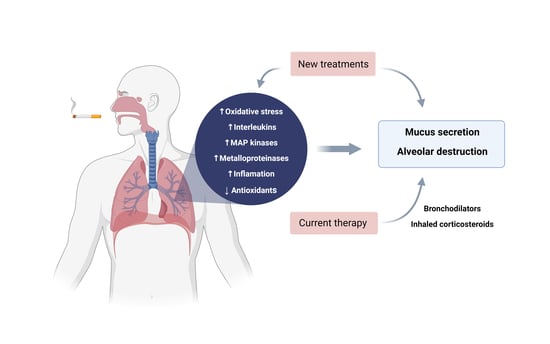
:
1. Introduction
2. Epidemiology
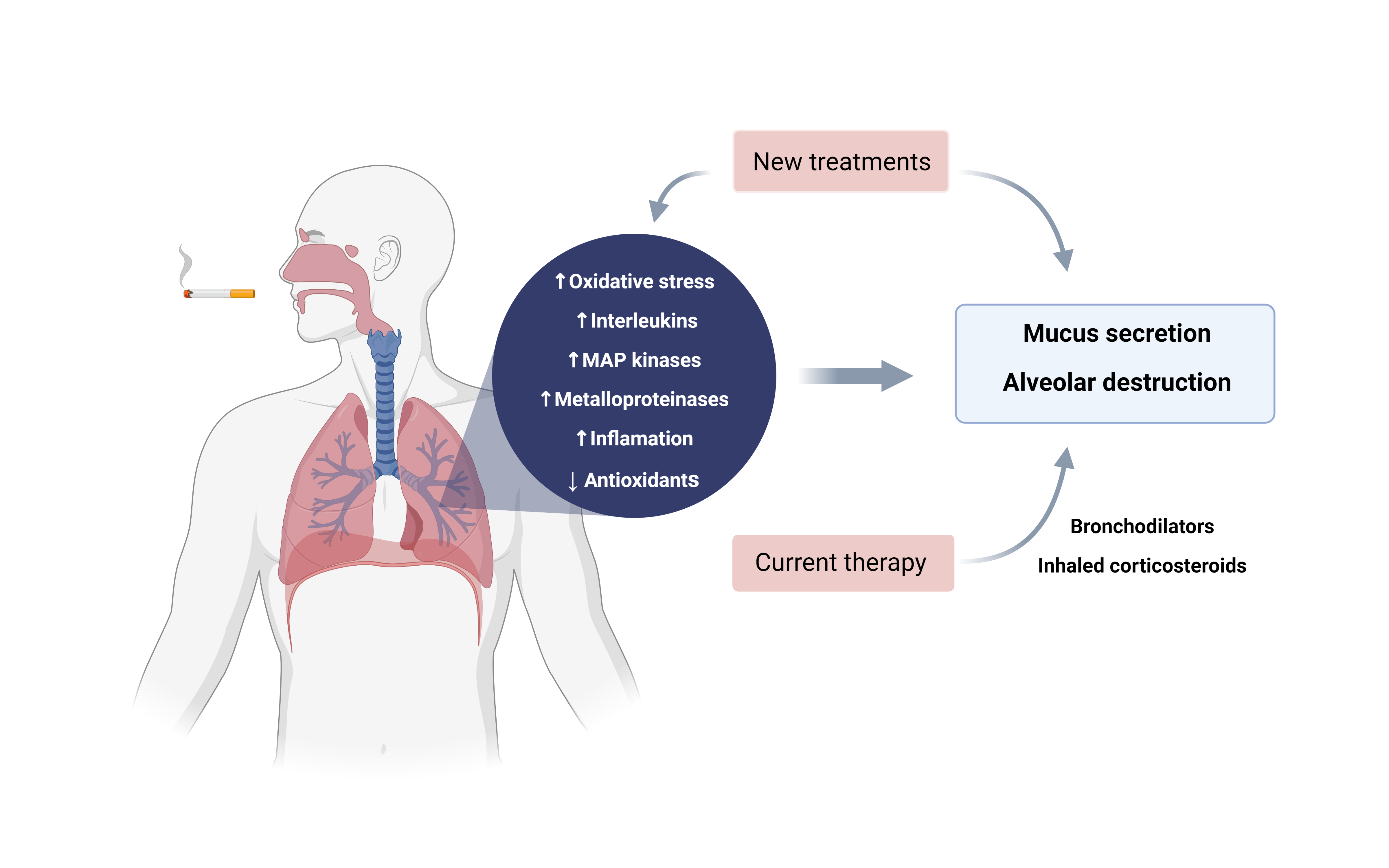
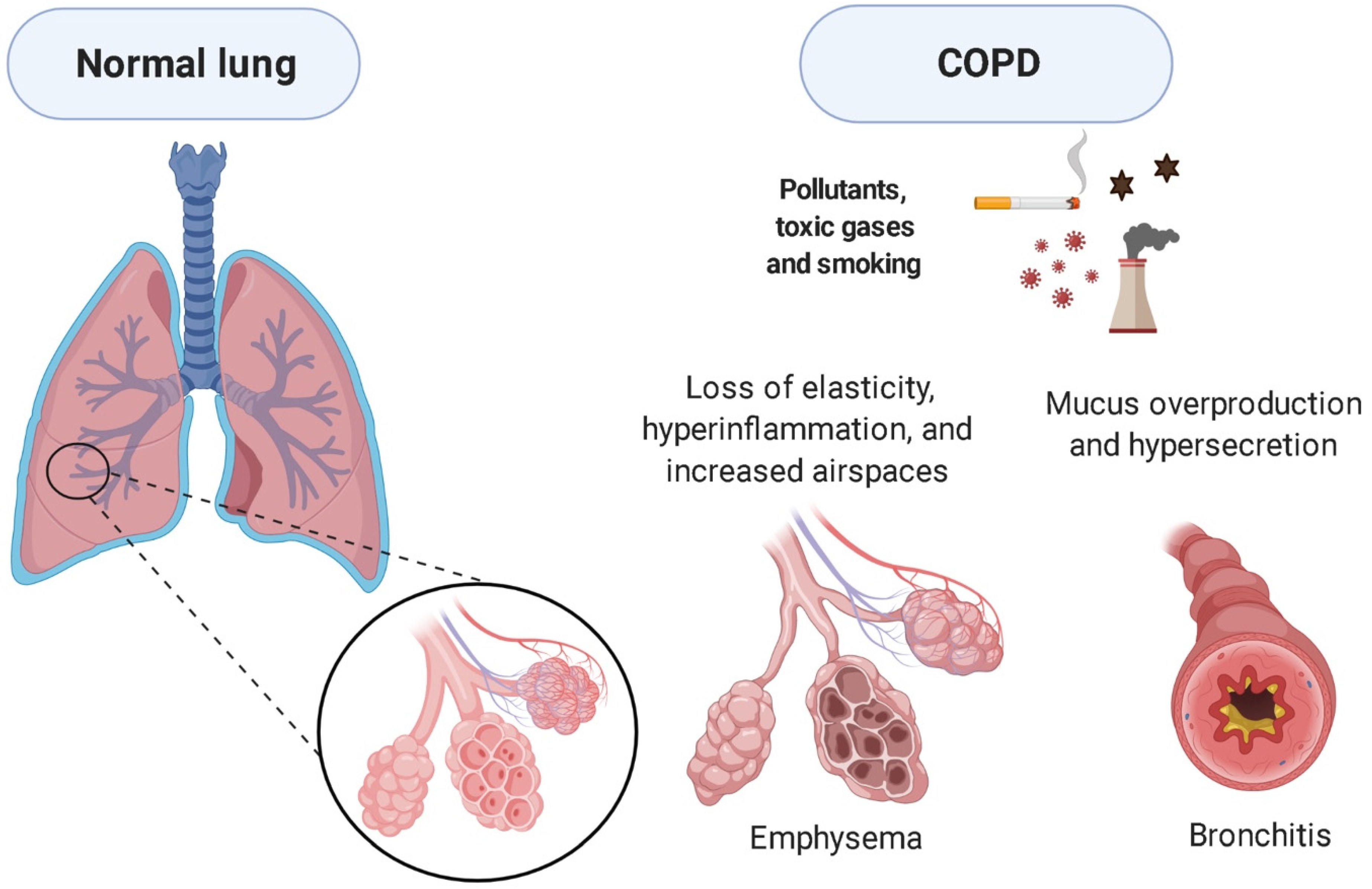
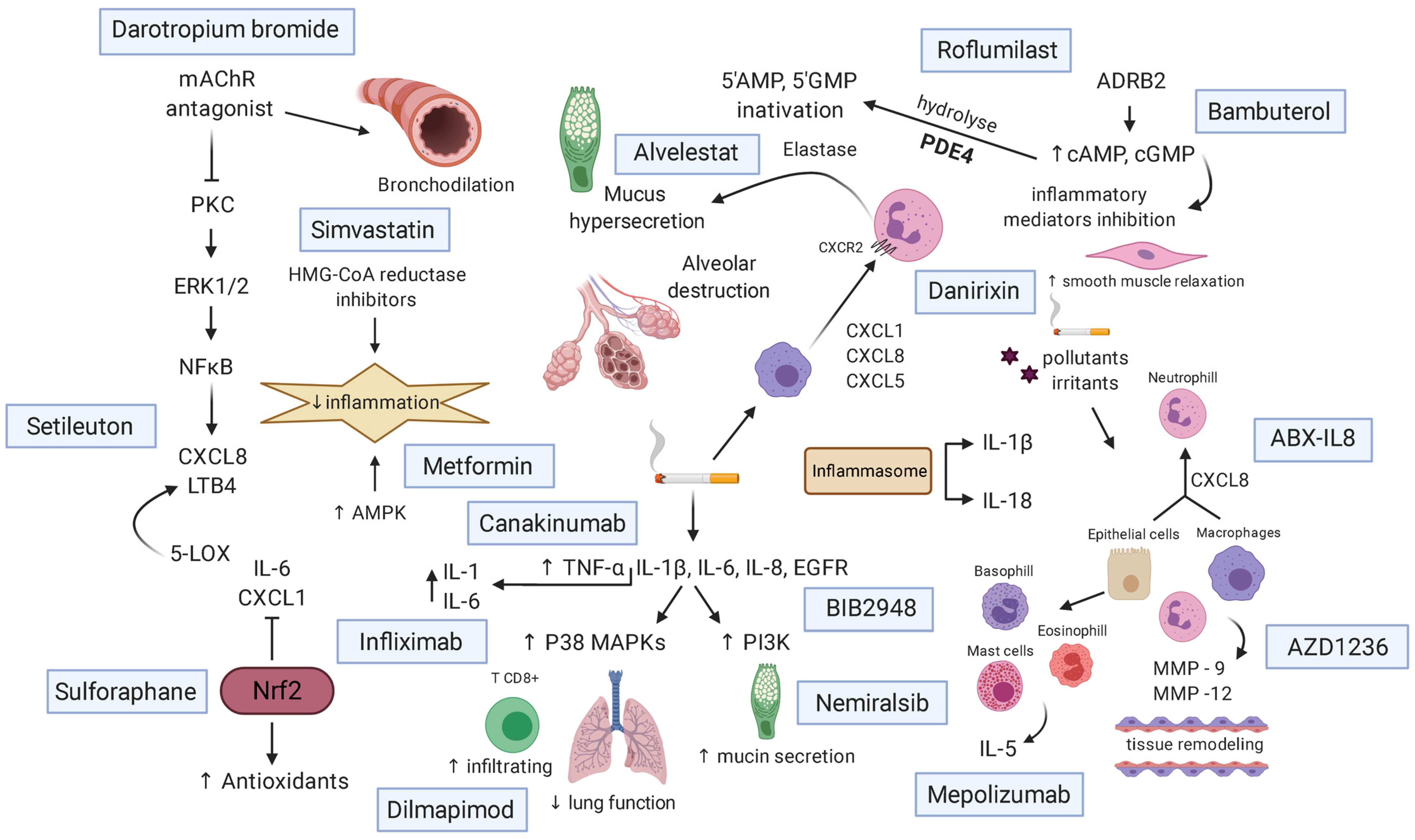
3. Pathophysiology
3.1. Diaphragm Dysfunction and COPD
3.2. Pulmonary Arterial Hypertension and COPD
3.3. Reactive Oxygen Species and COPD
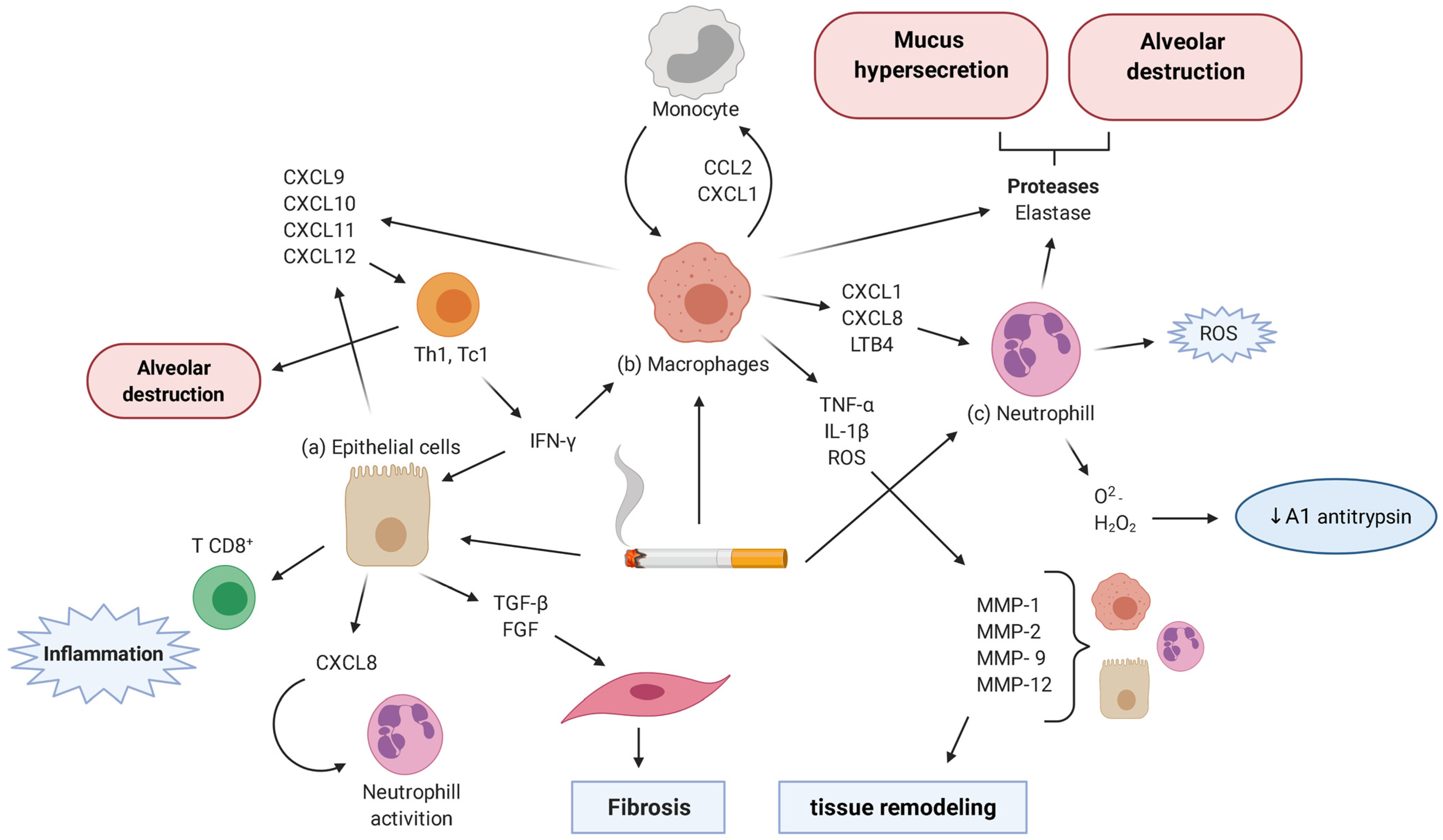
4. Inflammatory Cells and Mediators
4.1. Alveolar Epithelial Cells
4.2. Goblet Cells
4.3. Alveolar Macrophages
4.4. Lymphocytes
4.5. Neutrophils
5. Genetic and Epigenetic Regulation
6. Treatments and New Therapeutic Approaches
Role of Medications of Each Drug in Patients with COPD
7. COPD and COVID-19
8. Prevention
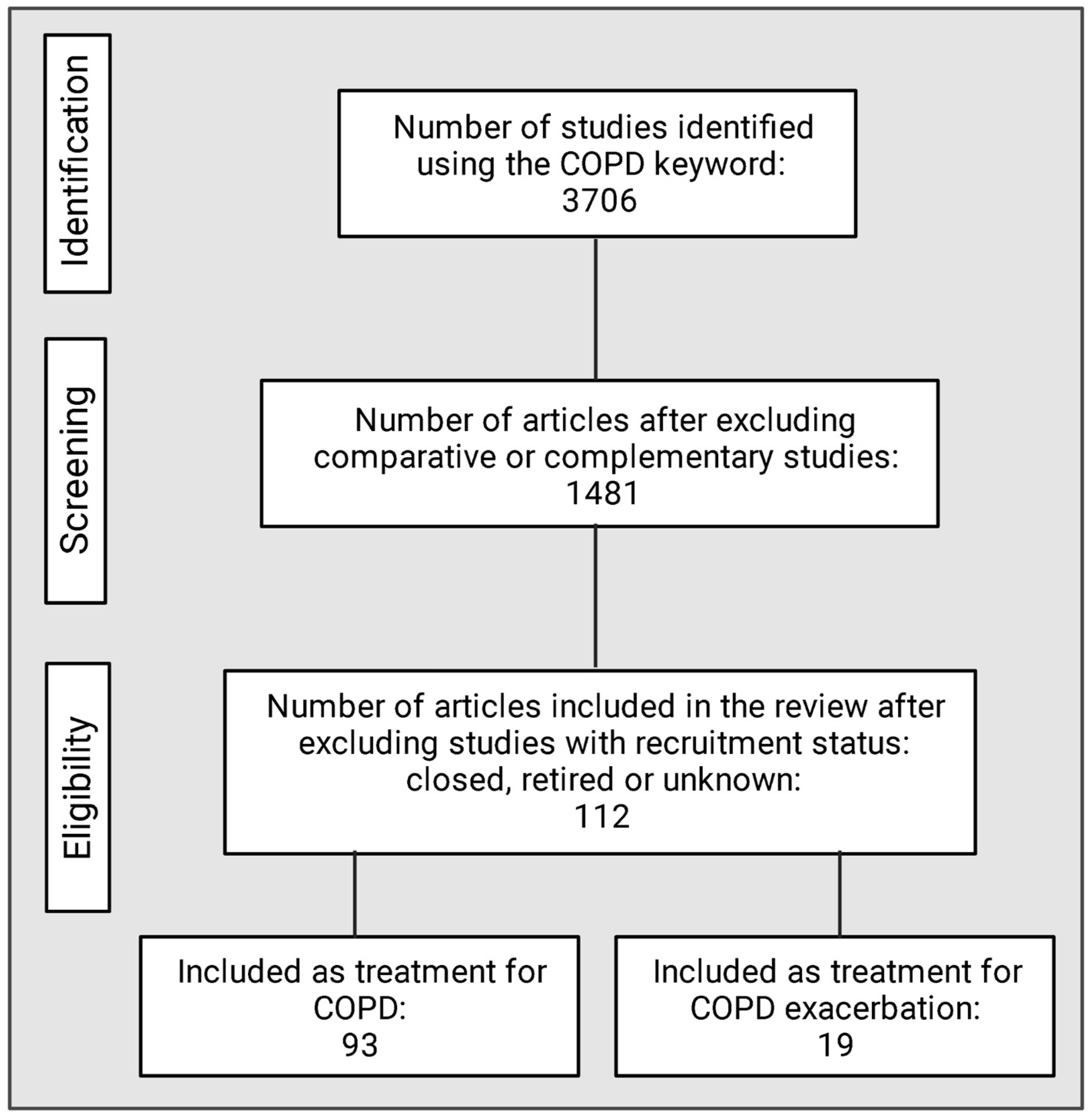
9. Methodology
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mathers, C.D.; Loncar, D. Projections of Global Mortality and Burden of Disease from 2002 to 2030. PLoS Med. 2006, 3, e442. [Google Scholar] [CrossRef] [Green Version]
- GOLD. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease 2021 Repost. Global Initiative for Chronic Obstructive Lung Disease—GOLD. 2021. Available online: https://goldcopd.org/2021-gold-reports/ (accessed on 30 August 2021).
- Senior, R.M.; Anthonisen, N.R. Chronic Obstructive Pulmonary Disease (COPD). Am. J. Respir. Crit. Care Med. 1998, 157, S139–S147. [Google Scholar] [CrossRef]
- Labaki, W.W.; Rosenberg, S.R. Chronic Obstructive Pulmonary Disease. Ann. Intern. Med. 2020, 173, ITC17–ITC32. [Google Scholar] [CrossRef]
- Halpin, D.M.G.; Celli, B.R.; Criner, G.J.; Frith, P.; Varela, M.V.L.; Salvi, S.; Vogelmeier, C.F.; Chen, R.; Mortimer, K.; De Oca, M.M.; et al. The GOLD Summit on chronic obstructive pulmonary disease in low- and middle-income countries. Int. J. Tuberc. Lung Dis. 2019, 23, 1131–1141. [Google Scholar] [CrossRef]
- Halbert, R.J.; Natoli, J.L.; Gano, A.; Badamgarav, E.; Buist, A.S.; Mannino, D. Global burden of COPD: Systematic review and meta-analysis. Eur. Respir. J. 2006, 28, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Burney, P.; Silverman, E.K.; Celli, B.R.; Vestbo, J.; Wedzicha, J.A.; Wouters, E.F.M. Chronic obstructive pulmonary disease. Nat. Rev. Dis. Prim. 2015, 1, 15076. [Google Scholar] [CrossRef]
- Matsunaga, K.; Harada, M.; Suizu, J.; Oishi, K.; Asami-Noyama, M.; Hirano, T. Comorbid Conditions in Chronic Obstructive Pulmonary Disease: Potential Therapeutic Targets for Unmet Needs. J. Clin. Med. 2020, 9, 3078. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2000, 343, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Celli, B.R. Predictors of mortality in COPD. Respir. Med. 2010, 104, 773–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gharib, S.A.; Manicone, A.M.; Parks, W.C. Matrix metalloproteinases in emphysema. Matrix Biol. 2018, 73, 34–51. [Google Scholar] [CrossRef]
- Saetta, M.; Turato, G.; Maestrelli, P.; Mapp, C.E.; Fabbri, L.M. Cellular and Structural Bases of Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2001, 163, 1304–1309. [Google Scholar] [CrossRef]
- De Oliveira, M.V.; Silva, P.L.; Rocco, P.R.M. Animal Models of Chronic Obstructive Pulmonary Disease Exacerbations: A Review of the Current Status. J. Biomed. Sci. 2016, 5. [Google Scholar] [CrossRef]
- Kim, V.; Oros, M.; Durra, H.; Kelsen, S.; Aksoy, M.; Cornwell, W.D.; Rogers, T.J.; Criner, G.J. Chronic Bronchitis and Current Smoking Are Associated with More Goblet Cells in Moderate to Severe COPD and Smokers without Airflow Obstruction. PLoS ONE 2015, 10, e0116108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. Chronic Obstructive Pulmonary Disease (COPD). Available online: https://www.who.int/news-room/fact-sheets/detail/chronic-obstructive-pulmonary-disease-(copd) (accessed on 25 December 2020).
- Adeloye, D.; Chua, S.; Lee, C.; Basquill, C.; Papana, A.; Theodoratou, E.; Nair, H.; Gasevic, D.; Sridhar, D.; Campbell, H.; et al. Global and regional estimates of COPD prevalence: Systematic review and meta–analysis. J. Glob. Health 2015, 5, 020415. [Google Scholar] [CrossRef]
- Ruvuna, L.; Sood, A. Epidemiology of Chronic Obstructive Pulmonary Disease. Clin. Chest Med. 2020, 41, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Woldeamanuel, G.G.; Mingude, A.B.; Geta, T.G. Prevalence of chronic obstructive pulmonary disease (COPD) and its associated factors among adults in Abeshge District, Ethiopia: A cross sectional study. BMC Pulm. Med. 2019, 19, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stolbrink, M.; Mortimer, K. Collision of communicable and non-communicable disease epidemics—The case of HIV and COPD. Lancet Glob. Health 2018, 6, e126–e127. [Google Scholar] [CrossRef] [Green Version]
- Rabe, K.F.; Watz, H. Chronic obstructive pulmonary disease. Lancet 2017, 389, 1931–1940. [Google Scholar] [CrossRef]
- Lamprecht, B.; McBurnie, M.A.; Vollmer, W.M.; Gudmundsson, G.; Welte, T.; Nizankowska-Mogilnicka, E.; Studnicka, M.; Bateman, E.; Anto, J.M.; Burney, P.; et al. COPD in Never Smokers. Chest 2011, 139, 752–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Center for Health Statistics. Chronic Obstructive Pulmonary Disease (COPD) Includes: Chronic Bronchitis and Emphysema. Available online: https://www.cdc.gov/nchs/fastats/copd.htm (accessed on 17 March 2021).
- Lopez, A.D.; Shibuya, K.; Rao, C.; Mathers, C.D.; Hansell, A.; Held, L.S.; Schmid, V.; Buist, S. Chronic obstructive pulmonary disease: Current burden and future projections. Eur. Respir. J. 2006, 27, 397–412. [Google Scholar] [CrossRef] [Green Version]
- Wheaton, A.G.; Liu, Y.; Croft, J.B.; VanFrank, B.; Croxton, T.L.; Punturieri, A.; Postow, L.; Greenlund, K.J. Chronic Obstructive Pulmonary Disease and Smoking Status—United States, 2017. Morb. Mortal. Wkly. Rep. 2019, 68, 533–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nugmanova, D.; Feshchenko, Y.; Iashyna, L.; Gyrina, O.; Malynovska, K.; Mammadbayov, E.; Akhundova, I.; Nurkina, N.; Tariq, L.; Makarova, J.; et al. The prevalence, burden and risk factors associated with chronic obstructive pulmonary disease in Commonwealth of Independent States (Ukraine, Kazakhstan and Azerbaijan): Results of the CORE study. BMC Pulm. Med. 2018, 18, 26. [Google Scholar] [CrossRef]
- Lange, P.; Ahmed, E.; Lahmar, Z.M.; Martinez, F.J.; Bourdin, A. Natural history and mechanisms of COPD. Respirology 2021, 26, 298–321. [Google Scholar] [CrossRef]
- Duan, R.-R.; Hao, K.; Yang, T. Air pollution and chronic obstructive pulmonary disease. Chronic Dis. Transl. Med. 2020, 6, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Chapman, K.R.; Mannino, D.M.; Soriano, J.B.; Vermeire, P.A.; Buist, A.S.; Thun, M.J.; Connell, C.; Jemal, A.; Lee, T.A.; Miravitlles, M.; et al. Epidemiology and costs of chronic obstructive pulmonary disease. Eur. Respir. J. 2006, 27, 188–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, T.; Cusack, R.P.; Chaudhary, N.; Satia, I.; Kurmi, O.P. Under- and over-diagnosis of COPD: A global perspective. Breathe 2019, 15, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Ejaz, H.; Alsrhani, A.; Zafar, A.; Javed, H.; Junaid, K.; Abdalla, A.E.; Abosalif, K.O.; Ahmed, Z.; Younas, S. COVID-19 and comorbidities: Deleterious impact on infected patients. J. Infect. Public Health 2020, 13, 1833–1839. [Google Scholar] [CrossRef]
- Olloquequi, J. COVID-19 Susceptibility in chronic obstructive pulmonary disease. Eur. J. Clin. Investig. 2020, 50, e13382. [Google Scholar] [CrossRef]
- Halpin, D.M.G.; Criner, G.J.; Papi, A.; Singh, D.; Anzueto, A.; Martinez, F.J.; Agusti, A.A.; Vogelmeier, C.F. Global Initiative for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease. The 2020 GOLD Science Committee Report on COVID-19 and Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2021, 203, 24–36. [Google Scholar] [CrossRef]
- Rocco, P.R.M.; Negri, E.M.; Kurtz, P.M.; Vasconcellos, F.P.; Silva, G.H.; Capelozzi, V.L.; Romero, P.V.; Zin, W.A. Lung Tissue Mechanics and Extracellular Matrix Remodeling in Acute Lung Injury. Am. J. Respir. Crit. Care Med. 2001, 164, 1067–1071. [Google Scholar] [CrossRef]
- Mecham, R.P. Elastin in lung development and disease pathogenesis. Matrix Biol. 2018, 73, 6–20. [Google Scholar] [CrossRef] [PubMed]
- Burgstaller, G.; Oehrle, B.; Gerckens, M.; White, E.S.; Schiller, H.B.; Eickelberg, O. The instructive extracellular matrix of the lung: Basic composition and alterations in chronic lung disease. Eur. Respir. J. 2017, 50, 1601805. [Google Scholar] [CrossRef] [Green Version]
- McDonough, J.; Yuan, R.; Suzuki, M.; Seyednejad, N.; Elliott, W.M.; Sanchez, P.G.; Wright, A.C.; Gefter, W.B.; Litzky, L.; Coxson, H.O.; et al. Small-Airway Obstruction and Emphysema in Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2011, 365, 1567–1575. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xu, J.; Meng, Y.; Adcock, I.M.; Yao, X. Role of inflammatory cells in airway remodeling in COPD. Int. J. Chronic Obstr. Pulm. Dis. 2018, 13, 3341–3348. [Google Scholar] [CrossRef] [Green Version]
- Lareau, S.C.; Fahy, B.; Meek, P.; Wang, A. Chronic Obstructive Pulmonary Disease (COPD). Am. J. Respir. Crit. Care Med. 2019, 199, P1–P2. [Google Scholar] [CrossRef]
- Siddiqui, N.A.; Mansour, M.K.; Nookala, V. Bullous Emphysema. In StatPearls; StatPearls Publishing Copyright © 2021; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2021. [Google Scholar]
- Pahal, P.; Avula, A.; Sharma, S. Emphysema. In StatPearls; StatPearls Publishing Copyright © 2021; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2021. [Google Scholar]
- Houghton, A.M. Matrix metalloproteinases in destructive lung disease. Matrix Biol. 2015, 44–46, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, A.Y.; Kheradmand, F. The Role of Matrix Metalloproteinases in Development, Repair, and Destruction of the Lungs. Prog. Mol. Biol. Transl. Sci. 2017, 148, 1–29. [Google Scholar] [CrossRef]
- Marchioni, A.; Tonelli, R.; Fantini, R.; Tabbì, L.; Castaniere, I.; Livrieri, F.; Bedogni, S.; Ruggieri, V.; Pisani, L.; Nava, S.; et al. Respiratory Mechanics and Diaphragmatic Dysfunction in COPD Patients Who Failed Non-Invasive Mechanical Ventilation. Int. J. Chronic Obstr. Pulm. Dis. 2019, 14, 2575–2585. [Google Scholar] [CrossRef] [Green Version]
- Sharma, B.B.; Singh, V. Diaphragmatic dysfunction in chronic obstructive pulmonary disease. Lung India 2019, 36, 285–287. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.Y.; Suh, H.J.; Hong, S.-B.; Koh, Y.; Lim, C.-M. Diaphragm dysfunction assessed by ultrasonography: Influence on weaning from mechanical ventilation*. Crit. Care Med. 2011, 39, 2627–2630. [Google Scholar] [CrossRef]
- Samanta, S.; Singh, R.K.; Baronia, A.K.; Poddar, B.; Azim, A.; Gurjar, M. Diaphragm thickening fraction to predict weaning-a prospective exploratory study. J. Intensive Care 2017, 5, 62. [Google Scholar] [CrossRef] [Green Version]
- Gan, W.Q.; Man, S.F.P.; Senthilselvan, A.; Sin, D.D. Association between chronic obstructive pulmonary disease and systemic inflammation: A systematic review and a meta-analysis. Thorax 2004, 59, 574–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meduri, G.U.; Annane, D.; Chrousos, G.P.; Marik, P.E.; Sinclair, S.E. Activation and Regulation of Systemic Inflammation in ARDS. Chest 2009, 136, 1631–1643. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, E.; de la Puente, B.; Minguella, J.; Corominas, J.M.; Serrano, S.; Hussain, S.N.A.; Gea, J. Oxidative Stress and Respiratory Muscle Dysfunction in Severe Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2005, 171, 1116–1124. [Google Scholar] [CrossRef]
- Ottenheijm, C.A.C.; Heunks, L.M.A.; Dekhuijzen, P.N.R. Diaphragm Muscle Fiber Dysfunction in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2007, 175, 1233–1240. [Google Scholar] [CrossRef]
- De Troyer, A.; Wilson, T.A. Effect of acute inflation on the mechanics of the inspiratory muscles. J. Appl. Physiol. 2009, 107, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; McManus, M.J.; Gray, J.D.; Nasca, C.; Moffat, C.; Kopinski, P.K.; Seifert, E.L.; McEwen, B.S.; Wallace, D.C. Mitochondrial functions modulate neuroendocrine, metabolic, inflammatory, and transcriptional responses to acute psychological stress. Proc. Natl. Acad. Sci. USA 2015, 112, E6614–E6623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khosravi, S.; Harner, M.E. The MICOS complex, a structural element of mitochondria with versatile functions. Biol. Chem. 2020, 401, 765–778. [Google Scholar] [CrossRef] [PubMed]
- Hoppins, S.; Collins, S.; Cassidy-Stone, A.; Hummel, E.; DeVay, R.M.; Lackner, L.L.; Westermann, B.; Schuldiner, M.; Weissman, J.S.; Nunnari, J. A mitochondrial-focused genetic interaction map reveals a scaffold-like complex required for inner membrane organization in mitochondria. J. Cell Biol. 2011, 195, 323–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Vermulst, M.; Wang, Y.E.; Chomyn, A.; Prolla, T.A.; McCaffery, J.M.; Chan, D.C. Mitochondrial Fusion Is Required for mtDNA Stability in Skeletal Muscle and Tolerance of mtDNA Mutations. Cell 2010, 141, 280–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benard, G.; Bellance, N.; Jose, C.; Melser, S.; Nouette-Gaulain, K.; Rossignol, R. Multi-site control and regulation of mitochondrial energy production. Biochim. et Biophys. Acta (BBA) Bioenerg. 2010, 1797, 698–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic Phosphorylation of Dynamin-related GTPase Drp1 Participates in Mitochondrial Fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Lee, H.-Y.; Hanna, R.A.; Gustafsson, Å.B. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1924–H1931. [Google Scholar] [CrossRef]
- Gao, F.; Reynolds, M.B.; Passalacqua, K.D.; Sexton, J.Z.; Abuaita, B.H.; O’Riordan, M.X.D. The Mitochondrial Fission Regulator DRP1 Controls Post-Transcriptional Regulation of TNF-α. Front. Cell. Infect. Microbiol. 2021, 10. [Google Scholar] [CrossRef]
- Horn, A.; Raavicharla, S.; Shah, S.; Cox, D.; Jaiswal, J.K. Mitochondrial fragmentation enables localized signaling required for cell repair. J. Cell Biol. 2020, 219, e201909154. [Google Scholar] [CrossRef] [Green Version]
- Dai, W.; Jiang, L. Dysregulated Mitochondrial Dynamics and Metabolism in Obesity, Diabetes, and Cancer. Front. Endocrinol. 2019, 10, 570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, I.; Tura-Ceide, O.; Peinado, V.I.; Barberà, J.A. Updated Perspectives on Pulmonary Hypertension in COPD. Int. J. Chronic Obstr. Pulm. Dis. 2020, 15, 1315–1324. [Google Scholar] [CrossRef]
- Opitz, I.; Ulrich, S. Pulmonary hypertension in chronic obstructive pulmonary disease and emphysema patients: Prevalence, therapeutic options and pulmonary circulatory effects of lung volume reduction surgery. J. Thorac. Dis. 2018, 10, S2763–S2774. [Google Scholar] [CrossRef]
- Mathioudakis, A.G.; Janssens, W.; Sivapalan, P.; Singanayagam, A.; Dransfield, M.T.; Jensen, J.-U.S.; Vestbo, J. Acute exacerbations of chronic obstructive pulmonary disease: In search of diagnostic biomarkers and treatable traits. Thorax 2020, 75, 520–527. [Google Scholar] [CrossRef] [Green Version]
- Bouquet, J.; Tabor, D.E.; Silver, J.S.; Nair, V.; Tovchigrechko, A.; Griffin, M.P.; Esser, M.T.; Sellman, B.R.; Jin, H. Microbial burden and viral exacerbations in a longitudinal multicenter COPD cohort. Respir. Res. 2020, 21, 77. [Google Scholar] [CrossRef] [Green Version]
- Tuder, R.M. Pulmonary vascular remodeling in pulmonary hypertension. Cell Tissue Res. 2017, 367, 643–649. [Google Scholar] [CrossRef] [Green Version]
- Bós, D.D.S.G.; Van Der Bruggen, C.E.E.; Kurakula, K.; Sun, X.-Q.; Casali, K.R.; Casali, A.G.; Rol, N.; Szulcek, R.; Dos Remedios, C.; Guignabert, C.; et al. Contribution of Impaired Parasympathetic Activity to Right Ventricular Dysfunction and Pulmonary Vascular Remodeling in Pulmonary Arterial Hypertension. Circulation 2018, 137, 910–924. [Google Scholar] [CrossRef]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmüller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53, 1801887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparks, M.A.; Makhanova, N.A.; Griffiths, R.C.; Snouwaert, J.N.; Koller, B.H.; Coffman, T.M. Thromboxane Receptors in Smooth Muscle Promote Hypertension, Vascular Remodeling, and Sudden Death. Hypertension 2013, 61, 166–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, B.; Voynow, J.; Ghio, A. COPD: Balancing oxidants and antioxidants. Int. J. Chronic Obstr. Pulm. Dis. 2015, 10, 261–276. [Google Scholar] [CrossRef] [Green Version]
- MacNee, W. ABC of chronic obstructive pulmonary disease. Pathology, pathogenesis, and pathophysiology. BMJ 2006, 332, 1202–1204. [Google Scholar] [CrossRef] [Green Version]
- Austin, V.; Crack, P.; Bozinovski, S.; Miller, A.A.; Vlahos, R. COPD and stroke: Are systemic inflammation and oxidative stress the missing links? Clin. Sci. 2016, 130, 1039–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, I. Pharmacological antioxidant strategies as therapeutic interventions for COPD. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2012, 1822, 714–728. [Google Scholar] [CrossRef] [Green Version]
- Schuliga, M. NF-kappaB Signaling in Chronic Inflammatory Airway Disease. Biomolecules 2015, 5, 1266–1283. [Google Scholar] [CrossRef]
- Rahman, I.; MacNee, W. Antioxidant pharmacological therapies for COPD. Curr. Opin. Pharmacol. 2012, 12, 256–265. [Google Scholar] [CrossRef] [Green Version]
- Marginean, C.; Popescu, M.S.; Vladaia, M.; Tudorascu, D.; Pirvu, D.C.; Petrescu, F. Involvement of Oxidative Stress in COPD. Curr. Health Sci. J. 2018, 44, 48–54. [Google Scholar] [CrossRef]
- Liu, Q.; Gao, Y.; Ci, X. Role of Nrf2 and Its Activators in Respiratory Diseases. Oxidative Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- Vlahos, R.; Bozinovski, S. Recent advances in pre-clinical mouse models of COPD. Clin. Sci. 2013, 126, 253–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bautista, M.V.; Chen, Y.; Ivanova, V.S.; Rahimi, M.K.; Watson, A.M.; Rose, M.C. IL-8 Regulates Mucin Gene Expression at the Posttranscriptional Level in Lung Epithelial Cells. J. Immunol. 2009, 183, 2159–2166. [Google Scholar] [CrossRef] [Green Version]
- Inui, T.; Watanabe, M.; Nakamoto, K.; Sada, M.; Hirata, A.; Nakamura, M.; Honda, K.; Ogawa, Y.; Takata, S.; Yokoyama, T.; et al. Bronchial epithelial cells produce CXCL1 in response to LPS and TNFα: A potential role in the pathogenesis of COPD. Exp. Lung Res. 2018, 44, 323–331. [Google Scholar] [CrossRef]
- Shukla, S.D.; Mahmood, M.Q.; Weston, S.; Latham, R.; Muller, H.K.; Sohal, S.S.; Walters, E.H. The main rhinovirus respiratory tract adhesion site (ICAM-1) is upregulated in smokers and patients with chronic airflow limitation (CAL). Respir. Res. 2017, 18, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucich, U.; Christner, P.; Lippmann, M.; Fein, A.; Goldberg, A.; Kimbel, P.; Weinbaum, G.; Rosenbloom, J. Immunologic measurement of elastin-derived peptides in human serum. Am. Rev. Respir. Dis. 1983, 127, S28–S30. [Google Scholar] [CrossRef] [PubMed]
- Lam, D.C.; Chan, S.C.; Mak, J.C.; Freeman, C.; Ip, M.S.; Shum, D.K. S-maltoheptaose targets syndecan-bound effectors to reduce smoking-related neutrophilic inflammation. Sci. Rep. 2015, 5, 12945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeffery, P.K. Remodeling and Inflammation of Bronchi in Asthma and Chronic Obstructive Pulmonary Disease; American Thoracic Society: New York, NY, USA, 2004; Volume 1, pp. 176–183. [Google Scholar]
- Linden, S.K.; Sutton, P.; Karlsson, N.G.; Korolik, V.; McGuckin, M.A. Mucins in the mucosal barrier to infection. Mucosal Immunol. 2008, 1, 183–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, D.F. Airway mucus hypersecretion in asthma: An undervalued pathology? Curr. Opin. Pharmacol. 2004, 4, 241–250. [Google Scholar] [CrossRef]
- Wedzicha, J. Airway Mucins in Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2017, 377, 986–987. [Google Scholar] [CrossRef]
- Brusselle, G.G.; Joos, G.F.; Bracke, K.R. New insights into the immunology of chronic obstructive pulmonary disease. Lancet 2011, 378, 1015–1026. [Google Scholar] [CrossRef]
- Gao, W.; Li, L.; Wang, Y.; Zhang, S.; Adcock, I.; Barnes, P.J.; Huang, M.; Yao, X. Bronchial epithelial cells: The key effector cells in the pathogenesis of chronic obstructive pulmonary disease? Respirology 2015, 20, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2016, 138, 16–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanazawa, H.; Tochino, Y.; Asai, K.; Hirata, K. Simultaneous Assessment of Hepatocyte Growth Factor and Vascular Endothelial Growth Factor in Epithelial Lining Fluid From Patients With COPD. Chest 2014, 146, 1159–1165. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Horie, M.; Nagase, T. TGF-β Signaling in Lung Health and Disease. Int. J. Mol. Sci. 2018, 19, 2460. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Khalil, R.A. Matrix Metalloproteinases, Vascular Remodeling, and Vascular Disease. Adv. Pharmacol. 2018, 81, 241–330. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.-Z.; Fang, X.-C.; Wang, D.; Tang, F.-D.; Wang, X.-D. Involvement of type II pneumocytes in the pathogenesis of chronic obstructive pulmonary disease. Respir. Med. 2010, 104, 1391–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shokolenko, I.; Venediktova, N.; Bochkareva, A.; Wilson, G.L.; Alexeyev, M.F. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res. 2009, 37, 2539–2548. [Google Scholar] [CrossRef] [Green Version]
- Wiegman, C.H.; Michaeloudes, C.; Haji, G.; Narang, P.; Clarke, C.J.; Russell, K.E.; Bao, W.; Pavlidis, S.; Barnes, P.J.; Kanerva, J.; et al. Oxidative stress–induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2015, 136, 769–780. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Luan, G.; Xu, Y.; Shen, S.; Qian, S.; Zhu, Z.; Zhang, X.; Yin, S.; Ye, J. Cigarette smoke extract increases mitochondrial membrane permeability through activation of adenine nucleotide translocator (ANT) in lung epithelial cells. Biochem. Biophys. Res. Commun. 2020, 525, 733–739. [Google Scholar] [CrossRef]
- Anseth, J.W.; Goffin, A.J.; Fuller, G.G.; Ghio, A.J.; Kao, P.N.; Upadhyay, D. Lung Surfactant Gelation Induced by Epithelial Cells Exposed to Air Pollution or Oxidative Stress. Am. J. Respir. Cell Mol. Biol. 2005, 33, 161–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lommatzsch, M.; Cicko, S.; Müller, T.; Lucattelli, M.; Bratke, K.; Stoll, P.; Grimm, M.; Dürk, T.; Zissel, G.; Ferrari, D.; et al. Extracellular Adenosine Triphosphate and Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2010, 181, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Shaykhiev, R.; Otaki, F.; Bonsu, P.; Dang, D.T.; Teater, M.; Strulovici-Barel, Y.; Salit, J.; Harvey, B.-G.; Crystal, R.G. Cigarette smoking reprograms apical junctional complex molecular architecture in the human airway epithelium in vivo. Cell. Mol. Life Sci. 2011, 68, 877–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comstock, A.T.; Ganesan, S.; Chattoraj, A.; Faris, A.N.; Margolis, B.L.; Hershenson, M.B.; Sajjan, U.S. Rhinovirus-Induced Barrier Dysfunction in Polarized Airway Epithelial Cells Is Mediated by NADPH Oxidase 1. J. Virol. 2011, 85, 6795–6808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldkorn, T.; Filosto, S. Lung Injury and Cancer. Am. J. Respir. Cell Mol. Biol. 2010, 43, 259–268. [Google Scholar] [CrossRef]
- Yang, T.; Luo, F.; Shen, Y.; An, J.; Li, X.; Liu, X.; Ying, B.; Liao, Z.; Dong, J.; Guo, L.; et al. Quercetin attenuates airway inflammation and mucus production induced by cigarette smoke in rats. Int. Immunopharmacol. 2012, 13, 73–81. [Google Scholar] [CrossRef]
- Anagnostis, A.; Neofytou, E.; Soulitzis, N.; Kampas, D.; Drositis, I.; Dermitzaki, D.; Tzanakis, N.; Schiza, S.; Siafakas, N.M.; Tzortzaki, E.G. Molecular profiling of EGFR family in chronic obstructive pulmonary disease: Correlation with airway obstruction. Eur. J. Clin. Investig. 2013, 43, 1299–1306. [Google Scholar] [CrossRef] [PubMed]
- Ridley, C.; Thornton, D.J. Mucins: The frontline defence of the lung. Biochem. Soc. Trans. 2018, 46, 1099–1106. [Google Scholar] [CrossRef] [Green Version]
- Fujisawa, T.; Chang, M.M.-J.; Velichko, S.; Thai, P.; Hung, L.-Y.; Huang, F.; Phuong, N.; Chen, Y.; Wu, R. NF-κB Mediates IL-1β– and IL-17A–InducedMUC5BExpression in Airway Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2011, 45, 246–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kesimer, M.; Ford, A.A.; Ceppe, A.; Radicioni, G.; Cao, R.; Davis, C.W.; Doerschuk, C.M.; Alexis, N.E.; Anderson, W.H.; Henderson, A.G.; et al. Airway Mucin Concentration as a Marker of Chronic Bronchitis. N. Engl. J. Med. 2017, 377, 911–922. [Google Scholar] [CrossRef]
- Lin, V.Y.; Kaza, N.; Birket, S.E.; Kim, H.; Edwards, L.; Lafontaine, J.; Liu, L.; Mazur, M.; Byzek, S.A.; Hanes, J.; et al. Excess mucus viscosity and airway dehydration impact COPD airway clearance. Eur. Respir. J. 2019, 55, 1900419. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.; Thorley, A.; Culpitt, S.V.; Dodd, S.; Donnelly, L.; Demattos, C.; Fitzgerald, M.; Barnes, P.J. Alveolar macrophage-mediated elastolysis: Roles of matrix metalloproteinases, cysteine, and serine proteases. Am. J. Physiol. Cell. Mol. Physiol. 2002, 283, L867–L873. [Google Scholar] [CrossRef] [Green Version]
- Bernardo, I.; Bozinovski, S.; Vlahos, R. Targeting oxidant-dependent mechanisms for the treatment of COPD and its comorbidities. Pharmacol. Ther. 2015, 155, 60–79. [Google Scholar] [CrossRef] [Green Version]
- Trocme, C.; Deffert, C.; Cachat, J.; Donati, Y.; Tissot, C.; Papacatzis, S.; Braunersreuther, V.; Pache, J.-C.; Krause, K.-H.; Holmdahl, R.; et al. Macrophage-specific NOX2 contributes to the development of lung emphysema through modulation of SIRT1/MMP-9 pathways. J. Pathol. 2014, 235, 65–78. [Google Scholar] [CrossRef] [Green Version]
- Atri, C.; Guerfali, F.Z.; Laouini, D. Role of Human Macrophage Polarization in Inflammation during Infectious Diseases. Int. J. Mol. Sci. 2018, 19, 1801. [Google Scholar] [CrossRef] [Green Version]
- Parisi, L.; Gini, E.; Baci, D.; Tremolati, M.; Fanuli, M.; Bassani, B.; Farronato, G.; Bruno, A.; Mortara, L. Macrophage Polarization in Chronic Inflammatory Diseases: Killers or Builders? J. Immunol. Res. 2018, 2018. [Google Scholar] [CrossRef]
- Vlahos, R. Role of alveolar macrophages in chronic obstructive pulmonary disease. Front. Immunol. 2014, 5, 435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chana, K.K.; Fenwick, P.S.; Nicholson, A.G.; Barnes, P.J.; Donnelly, L.E. Identification of a distinct glucocorticosteroid-insensitive pulmonary macrophage phenotype in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2014, 133, 207–216.e11. [Google Scholar] [CrossRef]
- Traves, S.L.; Culpitt, S.V.; Russell, R.; Barnes, P.J.; Donnelly, L. Increased levels of the chemokines GROalpha and MCP-1 in sputum samples from patients with COPD. Thorax 2002, 57, 590–595. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, K.; Van Eeden, S.F. Lung Macrophage Phenotypes and Functional Responses: Role in the Pathogenesis of COPD. Int. J. Mol. Sci. 2018, 19, 582. [Google Scholar] [CrossRef] [Green Version]
- Belchamber, K.B.; Singh, R.; Batista, C.M.; Whyte, M.K.; Dockrell, D.H.; Kilty, I.; Robinson, M.J.; Wedzicha, J.; Barnes, P.J.; Donnelly, L.E. Defective bacterial phagocytosis is associated with dysfunctional mitochondria in COPD macrophages. Eur. Respir. J. 2019, 54, 1802244. [Google Scholar] [CrossRef] [PubMed]
- Naito, K.; Yamasaki, K.; Yatera, K.; Akata, K.; Noguchi, S.; Kawanami, T.; Fukuda, K.; Kido, T.; Ishimoto, H.; Mukae, H. Bacteriological incidence in pneumonia patients with pulmonary emphysema: A bacterial floral analysis using the 16S ribosomal RNA gene in bronchoalveolar lavage fluid. Int. J. Chronic Obstr. Pulm. Dis. 2017, 12, 2111–2120. [Google Scholar] [CrossRef] [Green Version]
- Kirkham, P.A.; Barnes, P.J. Oxidative Stress in COPD. Chest 2013, 144, 266–273. [Google Scholar] [CrossRef]
- Shibata, S.; Miyake, K.; Tateishi, T.; Yoshikawa, S.; Yamanishi, Y.; Miyazaki, Y.; Inase, N.; Karasuyama, H. Basophils trigger emphysema development in a murine model of COPD through IL-4–mediated generation of MMP-12–producing macrophages. Proc. Natl. Acad. Sci. USA 2018, 115, 13057–13062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akata, K.; Van Eeden, S.F. Lung Macrophage Functional Properties in Chronic Obstructive Pulmonary Disease. Int. J. Mol. Sci. 2020, 21, 853. [Google Scholar] [CrossRef] [Green Version]
- Guiedem, E.; Ikomey, G.M.; Nkenfou, C.; Walter, P.-Y.E.; Mesembe, M.; Chegou, N.N.; Jacobs, G.B.; Assoumou, M.C.O. Chronic obstructive pulmonary disease (COPD): Neutrophils, macrophages and lymphocytes in patients with anterior tuberculosis compared to tobacco related COPD. BMC Res. Notes 2018, 11, 192. [Google Scholar] [CrossRef] [Green Version]
- Di Stefano, A.; Caramori, G.; Gnemmi, I.; Contoli, M.; Vicari, C.; Capelli, A.; Magno, F.; D’Anna, S.E.; Zanini, A.; Brun, P.; et al. T helper type 17-related cytokine expression is increased in the bronchial mucosa of stable chronic obstructive pulmonary disease patients. Clin. Exp. Immunol. 2009, 157, 316–324. [Google Scholar] [CrossRef]
- Grundy, S.; Plumb, J.; Lea, S.; Kaur, M.; Ray, D.; Singh, D. Down Regulation of T Cell Receptor Expression in COPD Pulmonary CD8 Cells. PLoS ONE 2013, 8, e71629. [Google Scholar] [CrossRef] [Green Version]
- Hodge, G.; Nairn, J.; Holmes, M.; Reynolds, P.N.; Hodge, S. Increased intracellular T helper 1 proinflammatory cytokine production in peripheral blood, bronchoalveolar lavage and intraepithelial T cells of COPD subjects. Clin. Exp. Immunol. 2007, 150, 22–29. [Google Scholar] [CrossRef]
- Hodge, G.; Mukaro, V.; Reynolds, P.N.; Hodge, S. Role of increased CD8/CD28null T cells and alternative co-stimulatory molecules in chronic obstructive pulmonary disease. Clin. Exp. Immunol. 2011, 166, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Demedts, I.K.; Demoor, T.; Bracke, K.R.; Joos, G.F.; Brusselle, G.G. Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema. Respir. Res. 2006, 7, 53. [Google Scholar] [CrossRef] [Green Version]
- Dumitriu, I.E. The life (and death) of CD4+CD28null T cells in inflammatory diseases. Immunology 2015, 146, 185–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambers, C.; Hacker, S.; Posch, M.; Hoetzenecker, K.; Pollreisz, A.; Lichtenauer, M.; Klepetko, W.; Ankersmit, H.J. T cell senescence and contraction of T cell repertoire diversity in patients with chronic obstructive pulmonary disease. Clin. Exp. Immunol. 2009, 155, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Hodge, G.; Hodge, S. Steroid Resistant CD8+CD28null NKT-Like Pro-inflammatory Cytotoxic Cells in Chronic Obstructive Pulmonary Disease. Front. Immunol. 2016, 7, 617. [Google Scholar] [CrossRef] [Green Version]
- Hodge, G.; Jersmann, H.; Tran, H.B.; Roscioli, E.; Holmes, M.; Reynolds, P.N.; Hodge, S. Lymphocyte senescence in COPD is associated with decreased histone deacetylase 2 expression by pro-inflammatory lymphocytes. Respir. Res. 2015, 16, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baniyash, M. TCR ζ-chain downregulation: Curtailing an excessive inflammatory immune response. Nat. Rev. Immunol. 2004, 4, 675–687. [Google Scholar] [CrossRef]
- Okeke, E.B.; Uzonna, J.E. The Pivotal Role of Regulatory T Cells in the Regulation of Innate Immune Cells. Front. Immunol. 2019, 10, 680. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Li, D.; Tsun, A.; Li, B. FOXP3+ regulatory T cells and their functional regulation. Cell. Mol. Immunol. 2015, 12, 558–565. [Google Scholar] [CrossRef]
- Kalathil, S.G.; Lugade, A.A.; Pradhan, V.; Miller, A.; Parameswaran, G.I.; Sethi, S.; Thanavala, Y. T-Regulatory Cells and Programmed Death 1+T Cells Contribute to Effector T-Cell Dysfunction in Patients with Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2014, 190, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Olloquequi, J.; Ferrer, J.; Montes, J.F.; Rodríguez, E.; Montero, M.A.; García-Valero, J. Differential lymphocyte infiltration in small airways and lung parenchyma in COPD patients. Respir. Med. 2010, 104, 1310–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seys, L.J.M.; Verhamme, F.M.; Schinwald, A.; Hammad, H.; Cunoosamy, D.M.; Bantsimba-Malanda, C.; Sabirsh, A.; McCall, E.; Flavell, L.; Herbst, R.; et al. Role of B Cell–Activating Factor in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2015, 192, 706–718. [Google Scholar] [CrossRef] [PubMed]
- Kirkham, P.A.; Caramori, G.; Casolari, P.; Papi, A.; Edwards, M.; Shamji, B.; Triantaphyllopoulos, K.; Hussain, F.; Pinart, M.; Khan, Y.; et al. Oxidative Stress–induced Antibodies to Carbonyl-modified Protein Correlate with Severity of Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2011, 184, 796–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.-H.; Goswami, S.; Grudo, A.; Song, L.-Z.; Bandi, V.; Goodnight-White, S.; Green, L.; Hacken-Bitar, J.; Huh, J.; Bakaeen, F.; et al. Antielastin autoimmunity in tobacco smoking–induced emphysema. Nat. Med. 2007, 13, 567–569. [Google Scholar] [CrossRef]
- Wen, L.; Krauss-Etschmann, S.; Petersen, F.; Yu, X. Autoantibodies in Chronic Obstructive Pulmonary Disease. Front. Immunol. 2018, 9, 66. [Google Scholar] [CrossRef]
- Klareskog, L.; Catrina, A.I. Lungs and citrullination. Nat. Rev. Rheumatol. 2015, 11, 261–262. [Google Scholar] [CrossRef] [PubMed]
- Olloquequi, J.; Montes, J.F.; Prats, A.; Rodriguez, E.; Montero, M.A.; Garcia-Valero, J.; Ferrer, J. Significant increase of CD57+ cells in pulmonary lymphoid follicles of COPD patients. Eur. Respir. J. 2010, 37, 289–298. [Google Scholar] [CrossRef]
- Sullivan, J.-L.; Bagevalu, B.; Glass, C.; Sholl, L.; Kraft, M.; Martinez, F.D.; Bastarrika, G.; de Torres, J.P.; Estepar, R.S.J.; Guerra, S.; et al. B Cell–Adaptive Immune Profile in Emphysema-Predominant Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2019, 200, 1434–1439. [Google Scholar] [CrossRef]
- Pascoe, S.J.; Papi, A.; Midwinter, D.; Lettis, S.; Barnes, N. Circulating neutrophils levels are a predictor of pneumonia risk in chronic obstructive pulmonary disease. Respir. Res. 2019, 20, 195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caramori, G.; Casolari, P.; Barczyk, A.; Durham, A.L.; Di Stefano, A.; Adcock, I. COPD immunopathology. Semin. Immunopathol. 2016, 38, 497–515. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Um, S.-J.; Kim, Y.S.; Kim, D.K.; Jang, A.S.; Choi, H.S.; Kim, Y.H.; Kim, T.E.; Yoo, K.H.; Jung, K.-S. Association of the Neutrophil-to-Lymphocyte Ratio with Lung Function and Exacerbations in Patients with Chronic Obstructive Pulmonary Disease. PLoS ONE 2016, 11, e0156511. [Google Scholar] [CrossRef]
- Paliogiannis, P.; Fois, A.G.; Sotgia, S.; Mangoni, A.A.; Zinellu, E.; Pirina, P.; Negri, S.; Carru, C.; Zinellu, A. Neutrophil to lymphocyte ratio and clinical outcomes in COPD: Recent evidence and future perspectives. Eur. Respir. Rev. 2018, 27, 170113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zheng, H.; Zhang, H.; Ma, W.; Wang, F.; Liu, C.; He, S. Increased interleukin (IL)-8 and decreased IL-17 production in chronic obstructive pulmonary disease (COPD) provoked by cigarette smoke. Cytokine 2011, 56, 717–725. [Google Scholar] [CrossRef]
- Strzelak, A.; Ratajczak, A.; Adamiec, A.; Feleszko, W. Tobacco Smoke Induces and Alters Immune Responses in the Lung Triggering Inflammation, Allergy, Asthma and Other Lung Diseases: A Mechanistic Review. Int. J. Environ. Res. Public Health 2018, 15, 1033. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T.; Kobayashi, S.; Fujino, N.; Suzuki, T.; Ota, C.; He, M.; Yamada, M.; Suzuki, S.; Yanai, M.; Kurosawa, S.; et al. Increased circulating endothelial microparticles in COPD patients: A potential biomarker for COPD exacerbation susceptibility. Thorax 2012, 67, 1067–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, A.; Ge, D.; Zhang, J.; Teng, Y.; Yuan, C.; Huang, M.; Adcock, I.M.; Barnes, P.J.; Yao, X. Sputum myeloperoxidase in chronic obstructive pulmonary disease. Eur. J. Med Res. 2014, 19, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlahos, R.; Wark, P.; Anderson, G.P.; Bozinovski, S. Glucocorticosteroids Differentially Regulate MMP-9 and Neutrophil Elastase in COPD. PLoS ONE 2012, 7, e33277. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Wang, F.-P.; Wang, G.; Mao, H. Role of Neutrophil Extracellular Traps in Asthma and Chronic Obstructive Pulmonary Disease. Chin. Med. J. 2017, 130, 730–736. [Google Scholar] [CrossRef]
- Porto, B.N.; Stein, R. Neutrophil Extracellular Traps in Pulmonary Diseases: Too Much of a Good Thing? Front. Immunol. 2016, 7, 311. [Google Scholar] [CrossRef] [Green Version]
- Noguera, A.; Batle, S.; Miralles, C.; Iglesias, J.; Busquets, X.; MacNee, W.; Agusti, A. G Enhanced neutrophil response in chronic obstructive pulmonary disease. Thorax 2001, 56, 432–437. [Google Scholar] [CrossRef]
- Milara, J.; Juan, G.; Peiró, T.; Serrano, A.; Cortijo, J. Neutrophil Activation in Severe, Early-Onset COPD Patients versus Healthy Non-Smoker Subjects in vitro: Effects of Antioxidant Therapy. Respiration 2012, 83, 147–158. [Google Scholar] [CrossRef]
- Kuiper, J.W.P.; Sun, C.; Magalhães, M.A.O.; Glogauer, M. Rac regulates PtdInsP3 signaling and the chemotactic compass through a redox-mediated feedback loop. Blood 2011, 118, 6164–6171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front. Cell. Infect. Microbiol. 2017, 7, 373. [Google Scholar] [CrossRef]
- McGuinness, A.J.A.; Sapey, E. Oxidative Stress in COPD: Sources, Markers, and Potential Mechanisms. J. Clin. Med. 2017, 6, 21. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Wang, F.; Cui, L.; Zhou, J.; Xu, Z. Diagnostic value of serum neutrophil gelatinase-associated lipocalin, interleukin-6 and anti-citrullinated alpha-enolase peptide 1 for lower respiratory tract infections. Clin. Biochem. 2019, 75, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jia, M.; Yan, X.; Cao, L.; Barnes, P.J.; Adcock, I.M.; Huang, M.; Yao, X. Increased neutrophil gelatinase-associated lipocalin (NGAL) promotes airway remodelling in chronic obstructive pulmonary disease. Clin. Sci. 2017, 131, 1147–1159. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Shukla, M.; Cowland, J.B.; Malemud, C.J.; Haqqi, T. Neutrophil gelatinase–associated lipocalin is expressed in osteoarthritis and forms a complex with matrix metalloproteinase 9. Arthritis Rheum. 2007, 56, 3326–3335. [Google Scholar] [CrossRef] [PubMed]
- Tsantikos, E.; Lau, M.; Castelino, C.M.; Maxwell, M.J.; Passey, S.L.; Hansen, M.J.; McGregor, N.E.; Sims, N.; Steinfort, D.; Irving, L.B.; et al. Granulocyte-CSF links destructive inflammation and comorbidities in obstructive lung disease. J. Clin. Investig. 2018, 128, 2406–2418. [Google Scholar] [CrossRef] [PubMed]
- Becher, B.; Tugues, S.; Greter, M. GM-CSF: From Growth Factor to Central Mediator of Tissue Inflammation. Immunity 2016, 45, 963–973. [Google Scholar] [CrossRef] [Green Version]
- Jasper, A.E.; McIver, W.; Sapey, E.; Walton, G.M. Understanding the role of neutrophils in chronic inflammatory airway disease. F1000Research 2019, 8, 557. [Google Scholar] [CrossRef]
- Sundar, I.K.; Yao, H.; Rahman, I. Oxidative Stress and Chromatin Remodeling in Chronic Obstructive Pulmonary Disease and Smoking-Related Diseases. Antioxid. Redox Signal. 2013, 18, 1956–1971. [Google Scholar] [CrossRef]
- Aoshiba, K.; Zhou, F.; Tsuji, T.; Nagai, A. DNA damage as a molecular link in the pathogenesis of COPD in smokers. Eur. Respir. J. 2012, 39, 1368–1376. [Google Scholar] [CrossRef] [Green Version]
- Sidhaye, V.K.; Nishida, K.; Martinez, F.J. Precision medicine in COPD: Where are we and where do we need to go? Eur. Respir. Rev. 2018, 27, 180022. [Google Scholar] [CrossRef] [Green Version]
- Rajendrasozhan, S.; Yang, S.-R.; Edirisinghe, I.; Yao, H.; Adenuga, D.; Rahman, I. Deacetylases and NF-κB in Redox Regulation of Cigarette Smoke-Induced Lung Inflammation: Epigenetics in Pathogenesis of COPD. Antioxid. Redox Signal. 2008, 10, 799–812. [Google Scholar] [CrossRef] [Green Version]
- Caramori, G.; Romagnoli, M.; Casolari, P.; Bellettato, C.; Casoni, G.L.; Boschetto, P.; Chung, K.F.; Barnes, P.J.; Adcock, I.; Ciaccia, A.; et al. Nuclear localisation of p65 in sputum macrophages but not in sputum neutrophils during COPD exacerbations. Thorax 2003, 58, 348–351. [Google Scholar] [CrossRef] [Green Version]
- Cosio, B.G.; Tsaprouni, L.; Ito, K.; Jazrawi, E.; Adcock, I.; Barnes, P.J. Theophylline Restores Histone Deacetylase Activity and Steroid Responses in COPD Macrophages. J. Exp. Med. 2004, 200, 689–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, P.J. Role of HDAC2 in the Pathophysiology of COPD. Annu. Rev. Physiol. 2009, 71, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Mercado, N.; Thimmulappa, R.; Thomas, C.M.; Fenwick, P.S.; Chana, K.K.; Donnelly, L.; Biswal, S.; Ito, K.; Barnes, P.J. Decreased histone deacetylase 2 impairs Nrf2 activation by oxidative stress. Biochem. Biophys. Res. Commun. 2011, 406, 292–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saco, T.V.; Breitzig, M.T.; Lockey, R.F.; Kolliputi, N. Epigenetics of Mucus Hypersecretion in Chronic Respiratory Diseases. Am. J. Respir. Cell Mol. Biol. 2018, 58, 299–309. [Google Scholar] [CrossRef]
- Izzotti, A.; Calin, G.; Arrigo, P.; Steele, V.E.; Croce, C.M.; De Flora, S. Downregulation of microRNA expression in the lungs of rats exposed to cigarette smoke. FASEB J. 2008, 23, 806–812. [Google Scholar] [CrossRef] [PubMed]
- Finicelli, M.; Squillaro, T.; Galderisi, U.; Peluso, G. Micro-RNAs: Crossroads between the Exposure to Environmental Particulate Pollution and the Obstructive Pulmonary Disease. Int. J. Mol. Sci. 2020, 21, 7221. [Google Scholar] [CrossRef] [PubMed]
- Omote, N.; Sauler, M. Non-coding RNAs as Regulators of Cellular Senescence in Idiopathic Pulmonary Fibrosis and Chronic Obstructive Pulmonary Disease. Front. Med. 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Cañas, J.A.; Rodrigo-Muñoz, J.M.; Sastre, B.; Gil-Martinez, M.; Redondo, N.; Del Pozo, V. MicroRNAs as Potential Regulators of Immune Response Networks in Asthma and Chronic Obstructive Pulmonary Disease. Front. Immunol. 2021, 11, 608666. [Google Scholar] [CrossRef] [PubMed]
- Gon, Y.; Shimizu, T.; Mizumura, K.; Maruoka, S.; Hikichi, M. Molecular techniques for respiratory diseases: MicroRNA and extracellular vesicles. Respirology 2019, 25, 149–160. [Google Scholar] [CrossRef] [Green Version]
- Matera, M.G.; Cazzola, M.; Page, C. Prospects for COPD treatment. Curr. Opin. Pharmacol. 2021, 56, 74–84. [Google Scholar] [CrossRef]
- Celli, B.; MacNee, W.; Agusti, A.; Anzueto, A.; Berg, B.; Buist, A.; Calverley, P.; Chavannes, N.; Dillard, T.; Fahy, B.; et al. Standards for the diagnosis and treatment of patients with COPD: A summary of the ATS/ERS position paper. Eur. Respir. J. 2004, 23, 932–946. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, D.E. Hyperinflation, Dyspnea, and Exercise Intolerance in Chronic Obstructive Pulmonary Disease; American Thoracic Society: New York, NY, USA, 2006; Volume 3, pp. 180–184. [Google Scholar]
- Similowski, T.; Yan, S.; Gauthier, A.P.; Macklem, P.T.; Bellemare, F. Contractile Properties of the Human Diaphragm during Chronic Hyperinflation. New Engl. J. Med. 1991, 325, 917–923. [Google Scholar] [CrossRef]
- Garvey, C.; Tiep, B.; Carter, R.; Barnett, M.; Hart, M.; Casaburi, R. Severe Exercise-Induced Hypoxemia. Respir. Care 2012, 57, 1154–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodduluri, S.; Reinhardt, J.M.; Hoffman, E.A.; Newell, J.D.; Bhatt, S.P.; John, D.N., Jr. Recent Advances in Computed Tomography Imaging in Chronic Obstructive Pulmonary Disease. Ann. Am. Thorac. Soc. 2018, 15, 281–289. [Google Scholar] [CrossRef]
- Decramer, M.; Janssens, W.; Miravitlles, M. Chronic obstructive pulmonary disease. Lancet 2012, 379, 1341–1351. [Google Scholar] [CrossRef]
- Beiko, T.; Strange, C. Treatment of Alpha-1 Antitrypsin Deficiency. Semin. Respir. Crit. Care Med. 2015, 36, 470–477. [Google Scholar] [CrossRef] [PubMed]
- López-Campos, J.L.; Hernandez, L.C.; Eraso, C.C. Implications of a Change of Paradigm in Alpha1 Antitrypsin Deficiency Augmentation Therapy: From Biochemical to Clinical Efficacy. J. Clin. Med. 2020, 9, 2526. [Google Scholar] [CrossRef]
- Malhotra, D.; Thimmulappa, R.K.; Mercado, N.; Ito, K.; Kombairaju, P.; Kumar, S.; Ma, J.; Feller-Kopman, D.; Wise, R.; Barnes, P.; et al. Denitrosylation of HDAC2 by targeting Nrf2 restores glucocorticosteroid sensitivity in macrophages from COPD patients. J. Clin. Investig. 2011, 121, 4289–4302. [Google Scholar] [CrossRef] [Green Version]
- Kirkham, P. Oxidative stress and macrophage function: A failure to resolve the inflammatory response. Biochem. Soc. Trans. 2007, 35, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Xuan, L.; Cao, S.; Yu, G.; Hou, Q.; Wang, H. Decreased Histone Deacetylase 2 (HDAC2) in Peripheral Blood Monocytes (PBMCs) of COPD Patients. PLoS ONE 2016, 11, e0147380. [Google Scholar] [CrossRef]
- Ito, K.; Ito, M.; Elliott, W.M.; Cosio, B.G.; Caramori, G.; Kon, O.M.; Barczyk, A.; Hayashi, S.; Adcock, I.; Hogg, J.C.; et al. Decreased Histone Deacetylase Activity in Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2005, 352, 1967–1976. [Google Scholar] [CrossRef] [Green Version]
- Liao, W.; Lim, A.Y.H.; Tan, W.S.D.; Abisheganaden, J.; Wong, W.S.F. Restoration of HDAC2 and Nrf2 by andrographolide overcomes corticosteroid resistance in chronic obstructive pulmonary disease. Br. J. Pharmacol. 2020, 177, 3662–3673. [Google Scholar] [CrossRef]
- Izquierdo, J.L.; Cosio, B.G. The dose of inhaled corticosteroids in patients with COPD: When less is better. Int. J. Chronic Obstr. Pulm. Dis. 2018, 13, 3539–3547. [Google Scholar] [CrossRef]
- Yawn, B.; Wollan, P.; Rank, M. Exacerbations in the pre- and post-COPD diagnosis periods. Pragmatic Obs. Res. 2013, 4, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Restrepo, M.I.; Sibila, O.; Anzueto, A. Pneumonia in Patients with Chronic Obstructive Pulmonary Disease. Tuberc. Respir. Dis. 2018, 81, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Hwang, J.W.; Kirkham, P.A.; Rahman, I. Pharmacological and Dietary Antioxidant Therapies for Chronic Obstructive Pulmonary Disease. Curr. Med. Chem. 2013, 20, 1496–1530. [Google Scholar] [CrossRef]
- Barnes, P.J. Oxidative stress-based therapeutics in COPD. Redox Biol. 2020, 33, 101544. [Google Scholar] [CrossRef]
- Menzel, M.; Ramu, S.; Calvén, J.; Olejnicka, B.; Sverrild, A.; Porsbjerg, C.; Tufvesson, E.; Bjermer, L.; Akbarshahi, H.; Uller, L. Oxidative Stress Attenuates TLR3 Responsiveness and Impairs Anti-viral Mechanisms in Bronchial Epithelial Cells From COPD and Asthma Patients. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- de Groot, L.; Van Der Veen, T.A.; Martinez, F.O.; Hamann, J.; Lutter, R.; Melgert, B.N. Oxidative stress and macrophages: Driving forces behind exacerbations of asthma and chronic obstructive pulmonary disease? Am. J. Physiol. Cell. Mol. Physiol. 2019, 316, L369–L384. [Google Scholar] [CrossRef] [Green Version]
- Dekhuijzen, P.N.R.; Van Beurden, W.J.C. The role for N-acetylcysteine in the management of COPD. Int. J. Chronic Obstr. Pulm. Dis. 2006, 1, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Van Antwerpen, P.; Boudjeltia, K.Z.; Babar, S.; Legssyer, I.; Moreau, P.; Moguilevsky, N.; Vanhaeverbeek, M.; Ducobu, J.; Nève, J. Thiol-containing molecules interact with the myeloperoxidase/H2O2/chloride system to inhibit LDL oxidation. Biochem. Biophys. Res. Commun. 2005, 337, 82–88. [Google Scholar] [CrossRef]
- Moretti, M. Pharmacology and clinical efficacy of erdosteine in chronic obstructive pulmonary disease. Expert Rev. Respir. Med. 2007, 1, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Rhee, C.K.; Kang, C.M.; You, M.B.; Yoon, H.K.; Kim, Y.K.; Kim, K.H.; Moon, H.S.; Park, S.H.; Song, J.S. Effect of fudosteine on mucin production. Eur. Respir. J. 2008, 32, 1195–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, B.B.; Sundaram, C.; Malani, N.; Ichikawa, H. Curcumin: The indian solid gold. Adv. Exp. Med. Biol. 2007, 595, 1–75. [Google Scholar] [CrossRef]
- Suzuki, M.; Betsuyaku, T.; Ito, Y.; Nagai, K.; Odajima, N.; Moriyama, C.; Nasuhara, Y.; Nishimura, M. Curcumin attenuates elastase- and cigarette smoke-induced pulmonary emphysema in mice. Am. J. Physiol. Cell. Mol. Physiol. 2009, 296, L614–L623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beijers, R.J.; Gosker, H.R.; Schols, A.M. Resveratrol for patients with chronic obstructive pulmonary disease. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Campos, K.K.D.; Ramos, C.D.O.; Martins, T.L.; Costa, G.D.P.; Talvani, A.; Garcia, C.C.M.; Oliveira, L.A.M.; Cangussú, S.D.; Costa, D.C.; Bezerra, F.S. Lycopene mitigates pulmonary emphysema induced by cigarette smoke in a murine model. J. Nutr. Biochem. 2018, 65, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Gumus, S.; Yucel, O.; Gamsizkan, M.; Eken, A.; Deniz, O.; Tozkoparan, E.; Genc, O.; Bilgic, H. The role of oxidative stress and effect of alpha-lipoic acid in reexpansion pulmonary edema—An experimental study. Arch. Med Sci. 2010, 6, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Oostwoud, L.C.; Gunasinghe, P.; Seow, H.; Ye, J.M.; Selemidis, S.; Bozinovski, S.; Vlahos, R. Apocynin and ebselen reduce influenza A virus-induced lung inflammation in cigarette smoke-exposed mice. Sci. Rep. 2016, 6, 20983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.C.; Huang, Y.C.; Hsu, S.Y.; Wang, Y.C.; Yeh, S.L. Vitamin E and Vitamin C Supplementation in Patients with Chronic Obstructive Pulmonary Disease. Int. J. Vitam. Nutr. Res. 2007, 77, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Hesslinger, C.; Strub, A.; Boer, R.; Ulrich, W.-R.; Lehner, M.D.; Braun, C. Inhibition of inducible nitric oxide synthase in respiratory diseases. Biochem. Soc. Trans. 2009, 37, 886–891. [Google Scholar] [CrossRef]
- Wang, S.; Lantz, R.C.; Vermeulen, M.W.; Chen, G.J.; Breceda, V.; Robledo, R.F.; Hays, A.M.; Young, S.; Witten, M.L. Functional alterations of alveolar macrophages subjected to smoke exposure and antioxidant lazaroids. Toxicol. Ind. Heal. 1999, 15, 464–469. [Google Scholar] [CrossRef]
- Pizzini, A.; Lunger, L.; Sonnweber, T.; Weiss, G.; Tancevski, I. The Role of Omega-3 Fatty Acids in the Setting of Coronary Artery Disease and COPD: A Review. Nutrients 2018, 10, 1864. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.; Yu, G.; Wu, Y.; Wang, H. Role of β2-adrenergic receptors in chronic obstructive pulmonary disease. Life Sci. 2021, 265, 118864. [Google Scholar] [CrossRef]
- Zhai, T.; Li, S.; Hu, W.; Li, D.; Leng, S. Potential Micronutrients and Phytochemicals against the Pathogenesis of Chronic Obstructive Pulmonary Disease and Lung Cancer. Nutrients 2018, 10, 813. [Google Scholar] [CrossRef] [Green Version]
- Whyand, T.; Hurst, J.R.; Beckles, M.; Caplin, M.E. Pollution and respiratory disease: Can diet or supplements help? A review. Respir. Res. 2018, 19, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Eeden, S.F.; Sin, D.D. Oxidative Stress in Chronic Obstructive Pulmonary Disease: A Lung and Systemic Process. Can. Respir. J. 2013, 20, 27–29. [Google Scholar] [CrossRef] [Green Version]
- Malerba, M.; Foci, V.; Patrucco, F.; Pochetti, P.; Nardin, M.; Pelaia, C.; Radaeli, A. Single Inhaler LABA/LAMA for COPD. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Kerwin, E.; Stiler, T.M.; Korenblat, P.; White, A.; Eckert, J.H.; Henley, M.; Patalano, F.; D’Andrea, P. Efficacy and Safety of Twice-Daily Glycopyrrolate Versus Placebo in Patients With COPD: The GEM2 Study. Chronic Obstr. Pulm. Dis. J. COPD Found. 2016, 3, 549–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takami, S.; Mizuno, T.; Oyanagi, T.; Tadaki, H.; Suzuki, T.; Muramatsu, K.; Takizawa, T.; Arakawa, H. Glucocorticoids Inhibit MUC5AC Production Induced by Transforming Growth Factor-α in Human Respiratory Cells. Allergol. Int. 2012, 61, 451–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agusti, A.; Fabbri, L.M.; Singh, D.; Vestbo, J.; Celli, B.; Franssen, F.M.; Rabe, K.F.; Papi, A. Inhaled corticosteroids in COPD: Friend or foe? Eur. Respir. J. 2018, 52, 1801219. [Google Scholar] [CrossRef]
- Rider, C.; Altonsy, M.O.; Mostafa, M.; Shah, S.V.; Sasse, S.; Manson, M.; Yan, D.; Kärrman-Mårdh, C.; Miller-Larsson, A.; Gerber, A.N.; et al. Long-Acting β2-Adrenoceptor Agonists Enhance Glucocorticoid Receptor (GR)–Mediated Transcription by Gene-Specific Mechanisms Rather Than Generic Effects via GR. Mol. Pharmacol. 2018, 94, 1031–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suissa, S.; Dell’Aniello, S.; Ernst, P. Comparative Effects of LAMA-LABA-ICS vs LAMA-LABA for COPD. Chest 2020, 157, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Papaioannou, A.I.; Loukides, S.; Bakakos, P.; Kosmas, E.N.; Rovina, N.; Steiropoulos, P.; Fouka, E.; Hillas, G.; Patentalakis, G.; Kouvela, M.; et al. Dual Bronchodilator in the Era of Triple Therapy. Int. J. Chronic Obstr. Pulm. Dis. 2020, 15, 2695–2705. [Google Scholar] [CrossRef]
- Cazzola, M.; Lopez-Campos, J.-L.; Puente-Maestu, L. The MABA approach: A new option to improve bronchodilator therapy. Eur. Respir. J. 2013, 42, 885–887. [Google Scholar] [CrossRef]
- Pinho-Ribeiro, V.; Melo, A.C.; Kennedy-Feitosa, E.; Graca-Reis, A.; Barroso, M.V.; Cattani-Cavalieri, I.; Carvalho, G.M.C.; Zin, W.A.; Porto, L.C.; Gitirana, L.B.; et al. Atorvastatin and Simvastatin Promoted Mouse Lung Repair After Cigarette Smoke-Induced Emphysema. Inflammation 2017, 40, 965–979. [Google Scholar] [CrossRef] [PubMed]
- Melo, A.C.; Cattani-Cavalieri, I.; Barroso, M.V.; Quesnot, N.; Gitirana, L.B.; Lanzetti, M.; Valença, S.S. Atorvastatin dose-dependently promotes mouse lung repair after emphysema induced by elastase. Biomed. Pharmacother. 2018, 102, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Bao, J.; Shi, Y.; Zhang, B.; Yuan, L.; Li, J.; Zhang, L.; Sun, M.; Zhang, L.; Sun, W. Effect of simvastatin on MMPs and TIMPs in cigarette smoke-induced rat COPD model. Int. J. Chronic Obstr. Pulm. Dis. 2017, 12, 717–724. [Google Scholar] [CrossRef] [Green Version]
- Juergens, L.J.; Worth, H.; Juergens, U.R. New Perspectives for Mucolytic, Anti-inflammatory and Adjunctive Therapy with 1,8-Cineole in COPD and Asthma: Review on the New Therapeutic Approach. Adv. Ther. 2020, 37, 1737–1753. [Google Scholar] [CrossRef] [Green Version]
- Worth, H.; Schacher, C.; Dethlefsen, U. Concomitant therapy with Cineole (Eucalyptole) reduces exacerbations in COPD: A placebo-controlled double-blind trial. Respir. Res. 2009, 10, 69. [Google Scholar] [CrossRef] [Green Version]
- Fischer, J.; Dethlefsen, U. Efficacy of cineole in patients suffering from acute bronchitis: A placebo-controlled double-blind trial. Cough 2013, 9, 25. [Google Scholar] [CrossRef] [Green Version]
- Yu, N.; Sun, Y.; Su, X.; He, M.; Dai, B.; Kang, J. Eucalyptol protects lungs against bacterial invasion through attenuating ciliated cell damage and suppressing MUC5AC expression. J. Cell. Physiol. 2019, 234, 5842–5850. [Google Scholar] [CrossRef]
- Kennedy-Feitosa, E.; Cattani-Cavalieri, I.; Barroso, M.V.; Romana-Souza, B.; Brito-Gitirana, L.; Valenca, S.S. Eucalyptol promotes lung repair in mice following cigarette smoke-induced emphysema. Phytomedicine 2019, 55, 70–79. [Google Scholar] [CrossRef]
- Boo, H.-J.; Park, S.J.; Noh, M.; Min, H.-Y.; Jeong, L.S.; Lee, H.-Y. LJ-2698, an Adenosine A3 Receptor Antagonist, Alleviates Elastase-Induced Pulmonary Emphysema in Mice. Biomol. Ther. 2020, 28, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Boo, H.-J.; Park, S.J.; Noh, M.; Min, H.-Y.; Jeong, L.S.; Lee, H.-Y. LJ-529, a partial peroxisome proliferator-activated receptor gamma (PPARγ) agonist and adenosine A3 receptor agonist, ameliorates elastase-induced pulmonary emphysema in mice. Arch. Pharmacal Res. 2020, 43, 540–552. [Google Scholar] [CrossRef]
- Antunes, M.A.; Silva, J.R.L.E.; Rocco, P.R. Mesenchymal stromal cell therapy in COPD: From bench to bedside. Int. J. Chronic Obstr. Pulm. Dis. 2017, 12, 3017–3027. [Google Scholar] [CrossRef] [Green Version]
- Mohammadipoor, A.; Antebi, B.; Batchinsky, A.I.; Cancio, L.C. Therapeutic potential of products derived from mesenchymal stem/stromal cells in pulmonary disease. Respir. Res. 2018, 19, 218. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhou, J.; Wang, J.; Li, S.; Fukunaga, A.; Yodoi, J.; Tian, H. Progress in the mechanism and targeted drug therapy for COPD. Signal Transduct. Target. Ther. 2020, 5, 248. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, C.; Wu, J.; Fukunaga, A.; Cheng, Z.; Wang, J.; Yamauchi, A.; Yodoi, J.; Tian, H. Anti-Allergic and Anti-Inflammatory Effects and Molecular Mechanisms of Thioredoxin on Respiratory System Diseases. Antioxid. Redox Signal. 2020, 32, 785–801. [Google Scholar] [CrossRef]
- Kinoshita, T.; Hoshino, T.; Imaoka, H.; Ichiki, H.; Okamoto, M.; Kawayama, T.; Yodoi, J.; Kato, S.; Aizawa, H. Thioredoxin prevents the development and progression of elastase-induced emphysema. Biochem. Biophys. Res. Commun. 2007, 354, 712–719. [Google Scholar] [CrossRef]
- Tanabe, N.; Hoshino, Y.; Marumo, S.; Kiyokawa, H.; Sato, S.; Kinose, D.; Uno, K.; Muro, S.; Hirai, T.; Yodoi, J.; et al. Thioredoxin-1 Protects against Neutrophilic Inflammation and Emphysema Progression in a Mouse Model of Chronic Obstructive Pulmonary Disease Exacerbation. PLoS ONE 2013, 8, e79016. [Google Scholar] [CrossRef] [Green Version]
- Blair, H.A. Tiotropium/Olodaterol: A Review in COPD. Drugs 2019, 79, 997–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babu, K.S.; Morjaria, J.B. Umeclidinium in chronic obstructive pulmonary disease: Latest evidence and place in therapy. Ther. Adv. Chronic Dis. 2017, 8, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Rogliani, P.; Calzetta, L.; Ora, J.; Matera, M.G. Canakinumab for the treatment of chronic obstructive pulmonary disease. Pulm. Pharmacol. Ther. 2015, 31, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Rennard, S.I.; Fogarty, C.; Kelsen, S.; Long, W.; Ramsdell, J.; Allison, J.; Mahler, D.; Saadeh, C.; Siler, T.; Snell, P.; et al. The Safety and Efficacy of Infliximab in Moderate to Severe Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2007, 175, 926–934. [Google Scholar] [CrossRef] [PubMed]
- Mkorombindo, T.; Dransfield, M.T. Mepolizumab in the treatment of eosinophilic chronic obstructive pulmonary disease. Int. J. Chronic Obstr. Pulm. Dis. 2019, 14, 1779–1787. [Google Scholar] [CrossRef]
- Janjua, S.; Fortescue, R.; Poole, P. Phosphodiesterase-4 inhibitors for chronic obstructive pulmonary disease. Cochrane Database Syst. Rev. 2020, 5, CD002309. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.J.; Rabe, K.F.; Lipworth, B.J.; Arora, S.; Jenkins, M.; Martin, U.J.; Reisner, C. Glycopyrrolate/Formoterol Fumarate Metered Dose Inhaler Improves Lung Function versus Monotherapies in GOLD Category A Patients with COPD: Pooled Data from the Phase III PINNACLE Studies. Int. J. Chronic Obstr. Pulm. Dis. 2020, 15, 99–106. [Google Scholar] [CrossRef]
- Worldometers. COVID-19 Coronavirus Pandemic. 2021. Available online: https://www.worldometers.info/coronavirus/ (accessed on 26 April 2021).
- Leung, J.M.; Yang, C.X.; Tam, A.; Shaipanich, T.; Hackett, T.-L.; Singhera, G.K.; Dorscheid, D.R.; Sin, D.D. ACE-2 expression in the small airway epithelia of smokers and COPD patients: Implications for COVID-19. Eur. Respir. J. 2020, 55, 2000688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alqahtani, J.S.; Oyelade, T.; Aldhahir, A.M.; Alghamdi, S.M.; Almehmadi, M.; Alqahtani, A.S.; Quaderi, S.; Mandal, S.; Hurst, J.R. Prevalence, Severity and Mortality associated with COPD and Smoking in patients with COVID-19: A Rapid Systematic Review and Meta-Analysis. PLoS ONE 2020, 15, e0233147. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Meng, M.; Kumar, R.; Wu, Y.; Huang, J.; Lian, N.; Deng, Y.; Lin, S. The impact of COPD and smoking history on the severity of COVID-19: A systemic review and meta-analysis. J. Med Virol. 2020, 92, 1915–1921. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Higham, A.; Mathioudakis, A.; Vestbo, J.; Singh, D. COVID-19 and COPD: A narrative review of the basic science and clinical outcomes. Eur. Respir. Rev. 2020, 29, 200199. [Google Scholar] [CrossRef]
- Toru, U.; Ayada, C.; Genç, O.; Sahin, S.; Arik, Ö.; Bulut, I.; Şahın, S. Serum levels of RAAS components in COPD. Monit. Airw. Dis. 2015, 46. [Google Scholar] [CrossRef]
- Higham, A.; Quinn, A.M.; Cançado, J.E.D.; Singh, D. The pathology of small airways disease in COPD: Historical aspects and future directions. Respir. Res. 2019, 20, 49. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Sin, D.D. COVID-19 in COPD: A growing concern. EClinicalMedicine 2020, 26, 100546. [Google Scholar] [CrossRef]
- Dagens, A.; Sigfrid, L.; Cai, E.; Lipworth, S.; Cheung, V.; Harris, E.; Bannister, P.; Rigby, I.; Horby, P. Scope, quality, and inclusivity of clinical guidelines produced early in the covid-19 pandemic: Rapid review. BMJ 2020, 369, m1936. [Google Scholar] [CrossRef] [PubMed]
- Demeyer, H.; Burtin, C.; Hornikx, M.; Camillo, C.A.; Van Remoortel, H.; Langer, D.; Janssens, W.; Troosters, T. The Minimal Important Difference in Physical Activity in Patients with COPD. PLoS ONE 2016, 11, e0154587. [Google Scholar] [CrossRef] [PubMed]
- Ambrosino, N.; Bertella, E. Lifestyle interventions in prevention and comprehensive management of COPD. Breathe 2018, 14, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Silva, A.R.; Moraes, B.P.T.; Gonçalves-De-Albuquerque, C.F. Mediterranean Diet: Lipids, Inflammation, and Malaria Infection. Int. J. Mol. Sci. 2020, 21, 4489. [Google Scholar] [CrossRef]
- Kopsaftis, Z.; Wood-Baker, R.; Poole, P. Influenza vaccine for chronic obstructive pulmonary disease (COPD). Cochrane Database Syst. Rev. 2018, 2018, CD002733. [Google Scholar] [CrossRef] [PubMed]
- Froes, F.; Roche, N.; Blasi, F. Pneumococcal vaccination and chronic respiratory diseases. Int. J. Chronic Obstr. Pulm. Dis. 2017, 12, 3457–3468. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Other Names | Use | Target/Action | Identification | Phase | Status | Approved COPD Treatment | Approved Treatment for Another Disease |
|---|---|---|---|---|---|---|---|
| Canakinumab ACZ885 | Antibody | IL-1β inhibitor | NCT00581945 | 1/2 | Completed | Adult-onset Still’s disease, Gouty arthritis, and others | |
| ABX-IL8 | Antibody | IL-8 inhibitor | NCT00035828 | 2 | Completed | ||
| Infliximab Remicade, TA-650 | Antibody | TNF-α inhibitor | NCT00056264 | 3 | Completed | Ankylosing spondylitis, Crohn’s disease, and others | |
| Mepolizumab | Antibody | IL-5 inhibitor | NCT04075331 | 2/3 | Recruiting | Asthma | |
| SB-240563, Bosatria, Nucala | NCT01463644 | 3 | Completed | ||||
| Ensifentrine | Uninformed | PDE3/PDE4 inhibitor | NCT03443414 | 2 | Completed | ||
| RPL554 | NCT04091360 | 2 | Completed | ||||
| Roflumilast Daliresp | Oral | PDE4 inhibitor | NCT01509677 | 3 | Completed | Yes | |
| AZD2115 | Inhaled | MABA | NCT01498081 | 2 | Completed | ||
| NCT02109406 | 2 | Completed | |||||
| MEDI8968 AMG-108 | IL-1 antagonist | NCT01448850 | 2 | Completed | |||
| Benralizumab | Antibody | IL-5 inhibitor | NCT01227278 | 2 | Completed | Asthma | |
| NCT04053634 | 3 | Recruiting | |||||
| AZD1236 | MMP-9/12 inhibitor | NCT00758706 | 2 | Completed | |||
| Glycopyrrolate SUN-101, Glycopyrrolate bromide, glycopyrronium bromide, NVA-237, AD-237 Seebri Breezhaler, CHF-5259 | Inhaled | M3 receptor antagonists | NCT00545311 | 1 | Completed | Yes | |
| NCT00242333 | 2 | Completed | |||||
| NCT00856193 | 2 | Completed | |||||
| NCT02680197 | 2 | Completed | |||||
| NCT02189577 | 2 | Completed | |||||
| NCT02347761 | 3 | Completed | |||||
| NCT01566604 | 3 | Completed | |||||
| NCT01154127 | 3 | Completed | |||||
| NCT01715298 | 3 | Completed | |||||
| NCT01005901 | 3 | Completed | |||||
| NCT02371629 | 4 | Completed | |||||
| Glycopyrrolate-formoterol Bevespi Aerosphere | Inhaled | MABA | NCT01854645 | 3 | Completed | Yes | |
| NCT01854658 | 3 | Completed | |||||
| NCT01970878 | 3 | Completed | |||||
| Glycopyrrolate-indacaterol Utibron Neohaler | Inhaled | MABA | NCT01727141 | 3 | Completed | Yes | |
| NCT01712516 | 3 | Completed | |||||
| NCT01682863 | 3 | Completed | |||||
| Simvastatin | Oral | HMG-CoA reductase inhibitors Nitric oxide synthase type II Inhibitors IL-17 | NCT01944176 | 3 | Completed | Diabetic cardiomiopathy, HCL 1, hyperlipidemia, and others | |
| NCT02070133 | 3 | Completed | |||||
| Rhodiola Crenulata | Oral | Anti-inflammation and anti-oxidation | NCT02242461 | 2 | Completed | ||
| Revefenacin GSK1160724, TD-4208 | Inhaled | mAChR antagonist | NCT00555022 | 1 | Completed | Yes | |
| NCT02040792 | 2 | Completed | |||||
| NCT02109172 | 2 | Completed | |||||
| NCT02512510 | 3 | Completed | |||||
| Sulforaphane SFX-01, Broccoli-sprout-extract | Oral | Nrf2 stimulator | NCT01335971 | 2 | Completed | ||
| QVA149 Indacaterol maleate/ glycopyrronium bromide, NVA-237/QAB-149 | Inhaled | MABA | NCT01996319 | 3 | Completed | Yes | |
| NCT01120717 | 3 | Completed | |||||
| Indacanterol | Inhaled | LABA | NCT00636961 | 2 | Completed | Yes | |
| NCT00792805 | 3 | Completed | |||||
| NCT01543828 | 4 | Completed | |||||
| CHF6523 | Uninformed | PI3K inhibitor | NCT04032535 | 1 | Recruiting | ||
| AZD8683 | Inhaled | M3 receptor antagonist | NCT01205269 | 2 | Completed | ||
| Tiotropium bromide Ba 679 BR, Spiriva, PUR-0200 | Inhaled | M1 and M3 receptor antagonist | NCT01921712 | 1 | Completed | Yes | Asthma |
| NCT02671825 | 1 | Completed | |||||
| NCT02172352 | 2 | Completed | |||||
| NCT02175342 | 2 | Completed | |||||
| NCT00292448 | 2 | Completed | |||||
| NCT02172391 | 3 | Completed | |||||
| NCT00144339 | 3 | Completed | |||||
| NCT00274573 | 3 | Completed | |||||
| NCT00274547 | 3 | Completed | |||||
| NCT02172378 | 3 | Completed | |||||
| NCT00274053 | 3 | Completed | |||||
| NCT00168831 | 3 | Completed | |||||
| NCT04061161 | 4 | Recruiting | |||||
| NCT00523991 | 4 | Completed | |||||
| NCT01072396 | 4 | Completed | |||||
| NCT00274079 | 4 | Completed | |||||
| Bambuterol Bambec, KWD 2183 | Oral | ADRB2 agonist | NCT01796730 | 4 | Completed | Yes | Asthma and bronchitis |
| AZD3199 | Inhaled | ADRB2 agonist | NCT00929708 | 2 | Completed | ||
| PT003 Formoterol/ glycopyrrolate, PT005/PT001, GFF MDI, Bevespi | Inhaled | MABA | NCT04087590 | 2 | Recruiting | Yes | |
| NCT02347085 | 3 | Completed | |||||
| NCT02643082 | 3 | Completed | |||||
| Astegolimab MSTT1041A, AMG 282, Anti-ST2, RO 7187807 | Antibody | IL-33 inhibitor | NCT03615040 | 2 | Completed | ||
| AZD1981 | Oral | CRTH2 antagonist | NCT00690482 | 2 | Completed | ||
| Abetiterol LAS100977, AZD-0548 | Inhaled | LABA | NCT01425814 | 2 | Completed | ||
| NCT01425801 | 2 | Completed | |||||
| Formoterol fumarate CHF 1531 | Inhaled | LABA | NCT00215436 | 3 | Completed | Yes | Asthma |
| CHF 6001 Tanimilast | Inhaled | PDE4 inhibitor | NCT01703052 | 1 | Completed | ||
| NCT02386761 | 1 | Completed | |||||
| NCT01730404 | 2 | Completed | |||||
| NCT03004417 | 2 | Completed | |||||
| DNK333 | Uninformed | NK1/NK2 antagonist | NCT01287325 | ½ | Completed | ||
| Aclidinium Bromide LAS 34273, KRP-AB1102, Bretaris Genuair, Eklira Genuair, Tudorza | Inhaled | M3 receptor inhibitor | NCT03276052 | 1 | Not yet recruiting | Yes | |
| NCT01471171 | 3 | Completed | |||||
| NCT00970268 | 3 | Completed | |||||
| NCT00891462 | 3 | Completed | |||||
| NCT00358436 | 3 | Completed | |||||
| NCT00500318 | 3 | Completed | |||||
| NCT01966107 | 4 | Completed | |||||
| Quercetin | Oral | Inflammation and oxidative stress | NCT01708278 | 1 | Completed | ||
| NCT03989271 | 1/2 | Recruiting | |||||
| Arformoterol tartrate Brovana | Inhaled | LABA | NCT00691405 | 2 | Completed | Yes | |
| NCT00250679 | 3 | Completed | |||||
| NCT00909779 | 3 | Completed | |||||
| BIO-11006 | Inhaled | MARCKS inhibitor | NCT00648245 | 2 | Completed | ||
| Bimosiamose | Inhaled | Pan-selectin antagonist | NCT01108913 | 2 | Completed | ||
| QBW251 | Oral | CFTR stimulant | NCT02449018 | 2 | Completed | ||
| NCT04268823 | 2 | Recruiting | |||||
| Bufei Jianpi granule | Oral | Elaying pulmonar function decline | NCT03976700 | 3 | Not yet recruiting | ||
| Alvelestat MPH966, AZD9668 | Oral | Neutrophil elastase inhibitor | NCT01035411 | 1 | Completed | ||
| NCT03679598 | 2 | Recruiting | |||||
| NCT00703391 | 2 | Completed | |||||
| NCT01054170 | 2 | Completed | |||||
| Tetomilast OPC-6535 | Oral | PDE4 inhibitor | NCT00917150 | 2 | Completed | ||
| Ipratropium Bromide | Inhaled | LAMA | NCT02236182 | 2 | Completed | Yes | |
| NCT00202176 | 4 | Completed | |||||
| Setileuton MK-0633 | 5-LOX inhibitor | NCT00418613 | 2 | Completed | |||
| Cyclosporine | Oral | Calcineurin inhibitor | NCT00974142 | 1/2 | Completed | ||
| PT010 Budesonide/ formoterol/glycopyrrolate, BGF-MDI, Budesonide/PT 003 | Inhaled | ICS/LAMA/ LABA | NCT03906045 | 1 | Completed | Yes | |
| Lovastatin | Oral | HMG-CoA reductase inhibitor | NCT00700921 | 2 | Completed | HCL 1 and hyperlipidemia | |
| Reldesemtiv CK-107, CK-2127107 | Oral | Troponin stimulant | NCT02662582 | 2 | Completed | ||
|
Symbicort Budesonide-formoterol | Inhaled | ICS/LABA | NCT00206154 | 3 | Completed | Yes | Asthma, Crohn’s disease, and ulcerative colitis |
| NCT00206167 | 3 | Completed | |||||
| Rosuvastatin | Oral | HMG-CoA reductase inhibitor | NCT00929734 | 2 | Completed | Atherosclerosis, cardiovascular disorders, HCL1, and others | |
| Dilmapimod GSK 681323, SB681323 | Uninformed | p38 MAPK inhibitor | NCT00144859 | 2 | Completed | ||
| Losartan | Oral | AT1 receptor antagonist | NCT00720226 | 4 | Completed | Diabetic nephropathies, heart failure, and hypertension | |
| NCT02696564 | 4 | Active, not recruiting | |||||
| Levalbuterol Xopenex HFA | Inhaled | SAMA | NCT00665600 | 3 | Completed | Yes | Asthma |
| Albuterol Salbutamol | Spray aerosol, injectable or inhaled | SABA | NCT00440245 | 4 | Completed | Yes | Asthma |
| Albuterol-ipratrópio Combivent Respimat | Inhaled | MABA | NCT00400153 | 3 | Completed | Yes | |
| AZD2423 | Oral | CCR2 antagonist | NCT01153321 | 2 | Completed | ||
| PH-797804 | Oral | p38 MAPK inhibitor | NCT00559910 | 2 | Completed | ||
| NCT01321463 | 2 | Completed | |||||
| CHF6366 | Inhaled | MABA | NCT03378648 | 1/2 | Completed | ||
| Indacaterol Arcapta | Inhaled | LABA | NCT00624286 | 3 | Completed | Yes | |
| MEDI2338 CERC 007 | Intravenous | IL-18 inhibitor | NCT01322594 | 1 | Completed | ||
| AZD5069 | Oral | CXCR2 antagonist | NCT01233232 | 2 | Completed | ||
| UMC119-06 | Intravenous | Cell replacements | NCT04206007 | 1 | Recruiting | ||
| ION-827359 | Inhaled | Epithelial sodium channel antagonist | NCT04441788 | 2 | Recruiting | ||
| Erdosteine | Oral | Glycoprotein inhibitor | NCT00338507 | 2 | Completed | Yes | Bronchitis |
| RV1162 PUR 1800 | Inhaled | Narrow-spectrum kinase inhibitor | NCT01970618 | 1 | Completed | ||
| JNJ 49095397 RV568 | Inhaled | PTS inhibitors | NCT01867762 | 2 | Completed | ||
| Selenium | Oral | GPx-1 levels | NCT00186706 | 4 | Completed | ||
| Epeleuton DS102, 15-HEPE, AF-102 | Oral | 5-LOX inhibitor | NCT03414541 | 2 | Completed | ||
| AZD8871 | Inhaled | MABA | NCT02814656 | 1 | Completed | ||
| NCT02971293 | 2 | Completed | |||||
| CHF 5993 Beclometasone/formoterol/glycopyrrolate, BDP/FF/GB | Inhaled | ICS/LABA/ LAMA | NCT02743013 | 1 | Completed | Yes | Asthma |
| Vilanterol GW642444 | Inhaled | LABA | NCT00372112 | 2 | Completed | ||
| NCT00606684 | 2 | Completed | |||||
| GSK256066 | Inhaled | Type 4 cyclic nucleotide PDE inhibitors | NCT00549679 | 2 | Completed | ||
| PF00610355 | Inhaled | ADRB2 agonist | NCT00808288 | 2 | Completed | ||
| Umeclidinium GSK573719, Incruse Ellipta | Inhaled | LAMA | NCT01110018 | 1 | Completed | Yes | |
| NCT00950807 | 2 | Completed | |||||
| NCT00732472 | 2 | Completed | |||||
| NCT01030965 | 2 | Completed | |||||
| NCT01387230 | 3 | Completed | |||||
| NCT02184611 | 3 | Completed | |||||
| Umeclidinium-vilanterol Anoro Elipta | Inhaled | MABA | NCT01899742 | 3 | Completed | Yes | |
| Darotropium bromide GSK233705 | Inhaled | mAChR antagonist | NCT00676052 | 2 | Completed | ||
| NCT00376714 | 2 | Completed | |||||
| NCT00453479 | 2 | Completed | |||||
| Tiotropium-Olodaterol Stiolto Respimat | Inhaled | MABA | NCT01431274 | 3 | Completed | Yes | |
| NCT01431287 | 3 | Completed | |||||
| AZD8871 | Inhaled | MABA | NCT03159442 | 1 | Completed | ||
| Oglemilast GRC 3886, GRC-3836 | Oral | PD4 inhibitor | NCT00671073 | 2 | Completed | ||
| Danirixin GSK-1325756 | Oral | CXCR2 antagonist | NCT01209052 | 1 | Completed | ||
| NCT03034967 | 2 | Completed | |||||
| NCT02130193 | 2 | Completed | |||||
| Fluticasone Propionate/ salmeterol Advair HFA | Inhaled | ICS/LABA Arachidonic acid inhibitors Lipocortin synthesis agonists | NCT00633217 | 4 | Completed | Yes | Asthma |
| Batefenterol GSK961081 | Inhaled | MABA | NCT00887406 | 1 | Completed | ||
| NCT02663089 | 1 | Completed | |||||
| NCT00478738 | 2 | Completed | |||||
| NCT02570165 | 2 | Completed | |||||
| Remestemcel-L RYONCIL | Intravenous | Stem cell therapies | NCT00683722 | 2 | Completed | Graft-versus-host disease | |
| Budesonida Pulmicort | Inhaled | ICS | NCT00232674 | 4 | Completed | Yes | Asthma |
| N-acetylcysteine | Oral | Antioxidant | NCT02579772 | 4 | Completed | Yes | Bronchiectasis, cystic fibrosis, dry eyes, and poisoning |
| MK-0873 | Oral | PDE4 inhibitor | NCT00132730 | 2 | Terminated | ||
| BI 1026706 | Oral | Bradykinin B1 receptor antagonist | NCT02642614 | 1 | Completed | ||
| BIBW 2948 | Inhaled | EGFR inhibitor | NCT00423137 | 2 | Completed | ||
| Cilomilast SB 207499, AL-38583 | Oral | PDE4 inhibitor | NCT00103922 | 3 | Completed | ||
| Olodaterol BI 1744 CL, Striverdi Respimat | Inhaled | LABA | NCT00824382 | 2 | Completed | Yes | |
| NCT00452400 | 2 | Completed | |||||
| NCT01809262 | 2 | Completed | |||||
| NCT00793624 | 3 | Completed | |||||
| PH-797804 | Oral | p38 inhibitor | NCT01543919 | 2 | Completed | ||
| CCI15106 | Inhaled | Undefined mechanism | NCT03235726 | 1 | Completed | ||
| Losmapimod GW856553X, FTX-1821 | Oral | p38α/β MAPK inhibitor | NCT01218126 | 2 | Completed | ||
| BI 113608 | Oral | Undefined mechanism | NCT01958008 | 1 | Completed | ||
| TRN-157 | Inhaled | M3 receptor antagonists | NCT02133339 | 1 | Completed | ||
| PF03635659 | Inhaled | Undefined mechanism | NCT00864786 | 1 | Completed | ||
| CNTO 6785 | Intravenous | IL17A protein inhibitor | NCT01966549 | 2 | Completed | ||
| Sildenafil Viagra | Oral | PDE5 inhibitor | NCT00104637 | 2 | Completed | Erectile dysfunction and pulmonary arterial hypertension | |
| AZD7594 AZ13189620 | Inhaled | Glucocorticoid receptor modulators | NCT02645253 | 1 | Completed | ||
| Tofimilast CP 325366 | Inhaled | PDE4 inhibitor | NCT00219622 | 2 | Completed | ||
| Fluticasone-furoate/vilanterol | Inhaled | LABA/ICS | NCT01691885 | 3 | Completed | Yes | Asthma |
| Retinoic Acid | RAR agonists | NCT00000621 | 2 | Completed | Acne, acute promyelocytic leukaemia, photodamage, and warts | ||
| Lebrikizumab | Subcutaneous | IL-13 inhibitor | NCT02546700 | 2 | Completed | ||
| Fluticasone- umeclidinium Trelegy Ellipta | Inhaled | MABA | NCT02345161 | 3 | Completed | Yes | Asthma |
| NCT02729051 | 3 | Completed |
| Drug Other Names | Use | Target/Action | Identification | Phase | Status | Approved COPD Treatment | Approved Treatment for Another Disease |
|---|---|---|---|---|---|---|---|
| Ciprofloxacin | Oral | DNA gyrase Inhibitor DNA topoisomerase inhibitor | NCT02300220 | 3 | Completed | Acute sinusitis, gonorrhoea, Intestinal infections, respiratory tract infections, and others | |
| Tezepelumab MEDI-9929 | Subcutaneous | Hymic stromal lymphopoietin inhibitor | NCT04039113 | 2 | Recruiting | ||
| Ismigen Antibacterial vaccine sublingual, Provax, Pulmigen, Respibron, Bactovax, Bromunyl. | Sublingual | Immunostimulants | NCT02417649 | 4 | Completed | Yes | Respiratory tract infections |
| Doxycycline | Oral | Protein 30S ribosomal subunit inhibitors | NCT02305940 | 3 | Completed | ||
| Levofloxacin MP-376, Quinsair, Aeroquin | Inhaled | DNA gyrase inhibitor DNA topoisomerase type IV and type II inhibitor | NCT00739648 | 2 | Completed | Bacterial infections, pneumonia, Sinusitis, tuberculosis, and others | |
| Roflumilast | Oral | PDE4 inhibitor | NCT00076089 NCT00430729 | 3 3 | Completed | Yes | |
| Benralizumab | Subcutaneous | Anti-IL5Rα antibody | NCT04098718 | 2 | Not yet recruiting | Asthma | |
| NCT04053634 | 3 | Recruiting | |||||
| NCT02155660 | 3 | Completed | |||||
| Aclidinium Bromide LAS 34273, KRP-AB1102, Bretaris Genuair, Eklira Genuair, Tudorza | Inhaled | M3 receptor inhibitor | NCT01966107 | 4 | Completed | Yes | |
| Metformin | Oral | AMPK stimulants Gluconeogenesis inhibitors | NCT01247870 | 4 | Completed | Type 2 diabetes mellitus | |
| Enoximone | Intravenous | PDE3 inhibitor | NCT04420455 | 4 | Not yet recruiting | Heart failure | |
| QBW251 | Oral | CFTR stimulant | NCT04268823 | 2 | Recruiting | ||
| Losmapimod GW856553X, FTX-1821 | Oral | p38α/β MAPK inhibitor | NCT02299375 | 2 | Completed | ||
| Nemiralisib GSK2269557 | Inhaled | PI3Kδ inhibitor | NCT02294734 | 2 | Completed | ||
| Acumapimod BCT197 | Oral | P38 inhibitor | NCT02700919 | 2 | Completed | ||
| Mepolizumab SB-240563, Bosatria, Nucala | Subcutaneous | IL-5 inhibitor | NCT04133909 | 3 | Recruiting | Asthma | |
| Azithromycin | Oral | Protein 50S ribosomal subunit inhibitors | NCT04319705 | Recruiting | Acute exacerbations of chronic bronchitis, acute sinusitis, pneumonia, pharyngitis, and respiratory tract infections | ||
| Arbidol | Oral | DNA and RNA synthesis inhibiting | NCT03851991 | Recruiting | |||
| Anti-ST2 MSTT1041A, AMG 282 | Subcutaneous | IL33 inhibitors | NCT03615040 | Not yet recruiting |
| Study | Treatment | Model Species | Experimental Intervention | Results |
|---|---|---|---|---|
| Horio et al., 2017 | Galectina (Gal) −9 administered subcutaneously once daily from 1 day before PPE instillation to day 5 | Female C57BL/6 mice (8–10 weeks old) | Lungs were intratracheally instilled with two units of PPE diluted in 50 μL of saline via 24-gauge catheter on day 0 | Infiltration of neutrophils was inhibited and MMP levels decreased |
| Melo et al, 2018 | Atorvastatin, 1, 5, and 20 mg, treated from day 33 until day 64 via inhalation for 10 min once a day | Male C57BL/6 mice (8 weeks old/18–22 g) | Administered intranasally 4 × 0.6 U of porcine pancreatic elastase (PPE) every other day (days 1, 3, 5 and 7). | Induced lung tissue repair in mice with emphysema |
| Pinho-Ribeiro et al., 2017 | Atorvastatin and simvastatin administered via inhalation for 15 min (1 mg/mL, once/day) | Male C57BL/6 mice (8–10 weeks old) | Mice exposed to 12 cigarettes a day for 60 days, then treated for another 60 days | Improved lung repair after cigarette smoke-induced emphysema, accompanied by a reduction in oxidative stress markers. |
| Sun et al., 2017 | Simvastatin administered intra-gastrically at a dose of 5 mg/kg/day followed by CS | Male Sprague Dawley (SD) rats (6 weeks old/110–20 g) | Animals were passively exposed (whole body) to smoke from 20 cigarettes in a box for 1 hour, twice a day, 5 days a week, for 16 weeks | Partial blockage of airway inflammation, and MMP production |
| Susuki et al., 2009 | Curcumin (100 mg/kg) administrated daily by oral gavage throughout a 21-day period | Male C57BL/6J mice (9 weeks old) | Administered intratracheal porcine pancreatic elastase (PPE), or exposed to CS (60 min/day for 10 consecutive days, or 5 days/week for 12 weeks) | Inhibited PPE-induced increase in neutrophils, inhibited increase in neutrophils and macrophages in BAL, and attenuated increase in air space induced by CS |
| Kennedy-Feitosa et al., 2018 | Inhalation of 1 mg/mL or 10 mg/mL eucalyptol for 15 min per day | Male C57BL/6 mice (8 weeks old/18–25 g) | Mice exposed to 12 cigarettes a day for 60 days, then treated for another 60 days without exposure to smoke | Lung repair, reduced inflammatory cytokines and NE levels, and increased elastin and TIMP-1 levels. |
| Boo et al., 2020 | LJ-2698 (50 μg/kg) administrated by oral gavage six times per week for 5 weeks. | FVB mice (8 weeks old) | One week after drug treatment, 0.25 units of PPE was intratracheally instilled into the lungs of the mice | Induction of anti-inflammatory cytokine production and recruitment of M2 macrophages |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodrigues, S.d.O.; Cunha, C.M.C.d.; Soares, G.M.V.; Silva, P.L.; Silva, A.R.; Gonçalves-de-Albuquerque, C.F. Mechanisms, Pathophysiology and Currently Proposed Treatments of Chronic Obstructive Pulmonary Disease. Pharmaceuticals 2021, 14, 979. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14100979
Rodrigues SdO, Cunha CMCd, Soares GMV, Silva PL, Silva AR, Gonçalves-de-Albuquerque CF. Mechanisms, Pathophysiology and Currently Proposed Treatments of Chronic Obstructive Pulmonary Disease. Pharmaceuticals. 2021; 14(10):979. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14100979
Chicago/Turabian StyleRodrigues, Sarah de Oliveira, Carolina Medina Coeli da Cunha, Giovanna Martins Valladão Soares, Pedro Leme Silva, Adriana Ribeiro Silva, and Cassiano Felippe Gonçalves-de-Albuquerque. 2021. "Mechanisms, Pathophysiology and Currently Proposed Treatments of Chronic Obstructive Pulmonary Disease" Pharmaceuticals 14, no. 10: 979. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14100979