
Endothelial Thioredoxin-Interacting Protein Depletion Reduces Hemorrhagic Transformation in Hyperglycemic Mice after Embolic Stroke and Thrombolytic Therapy

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
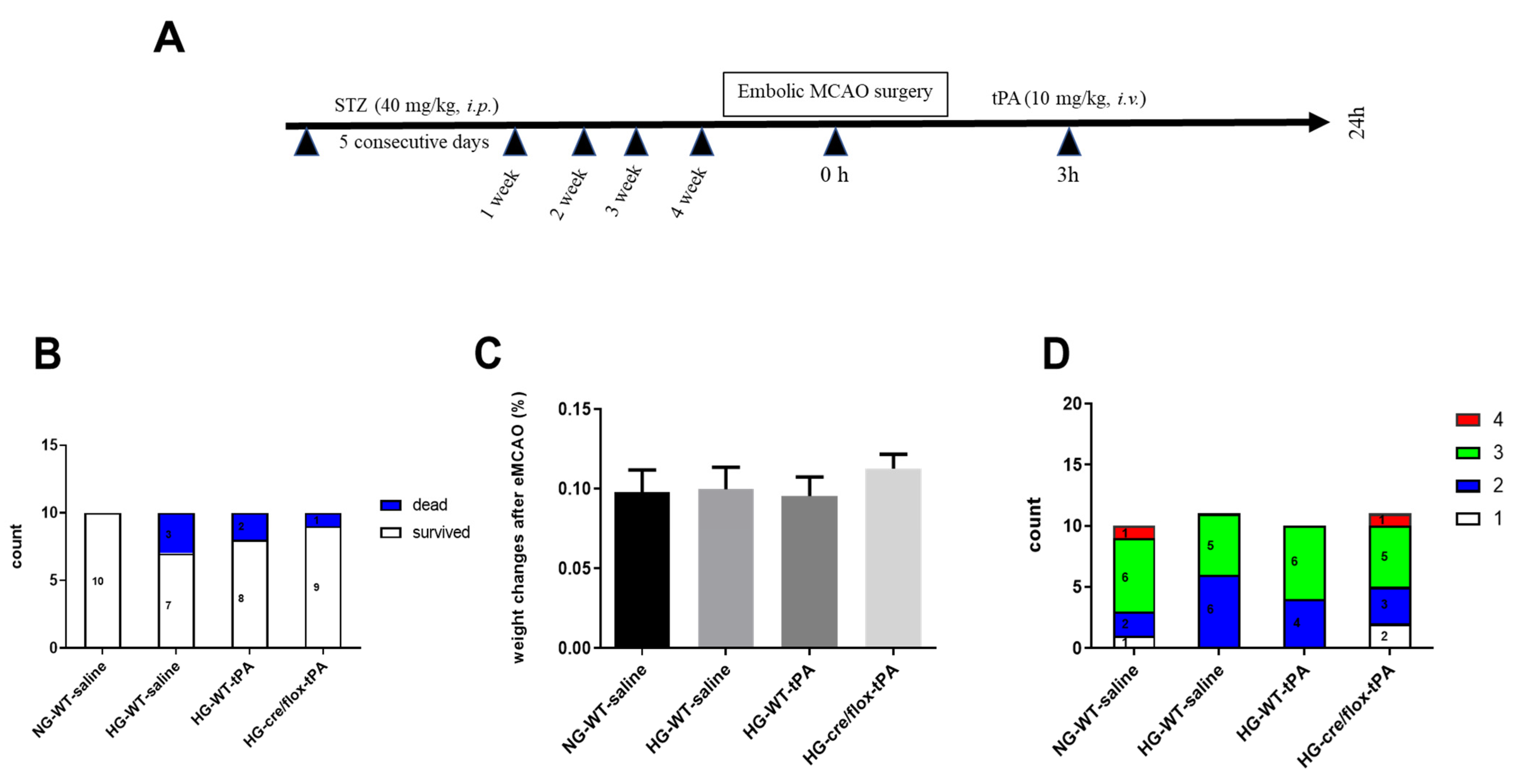
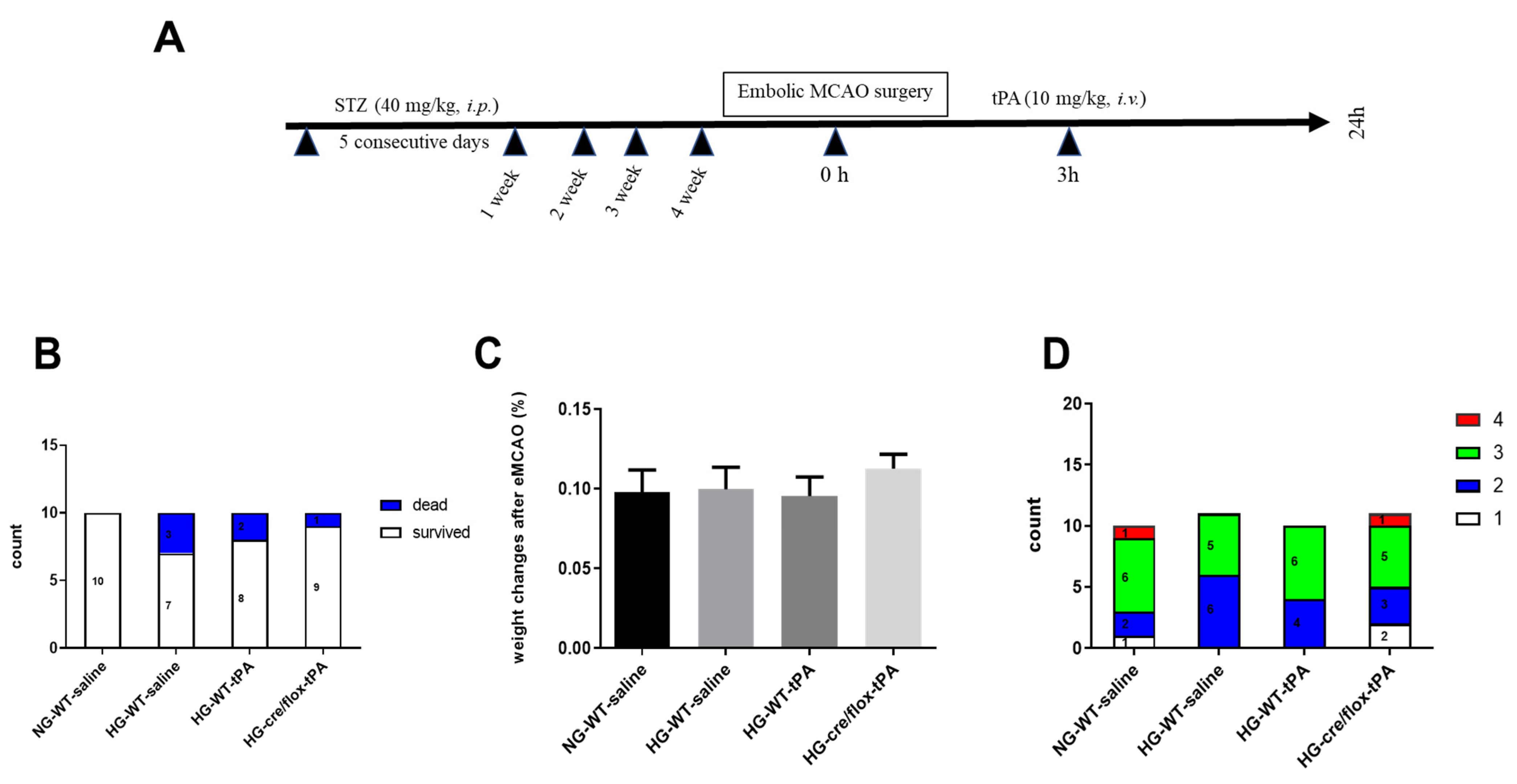
2.1. Post-Stroke Assessment of Weight Loss, Mortality Rate, and Neurological Deficits
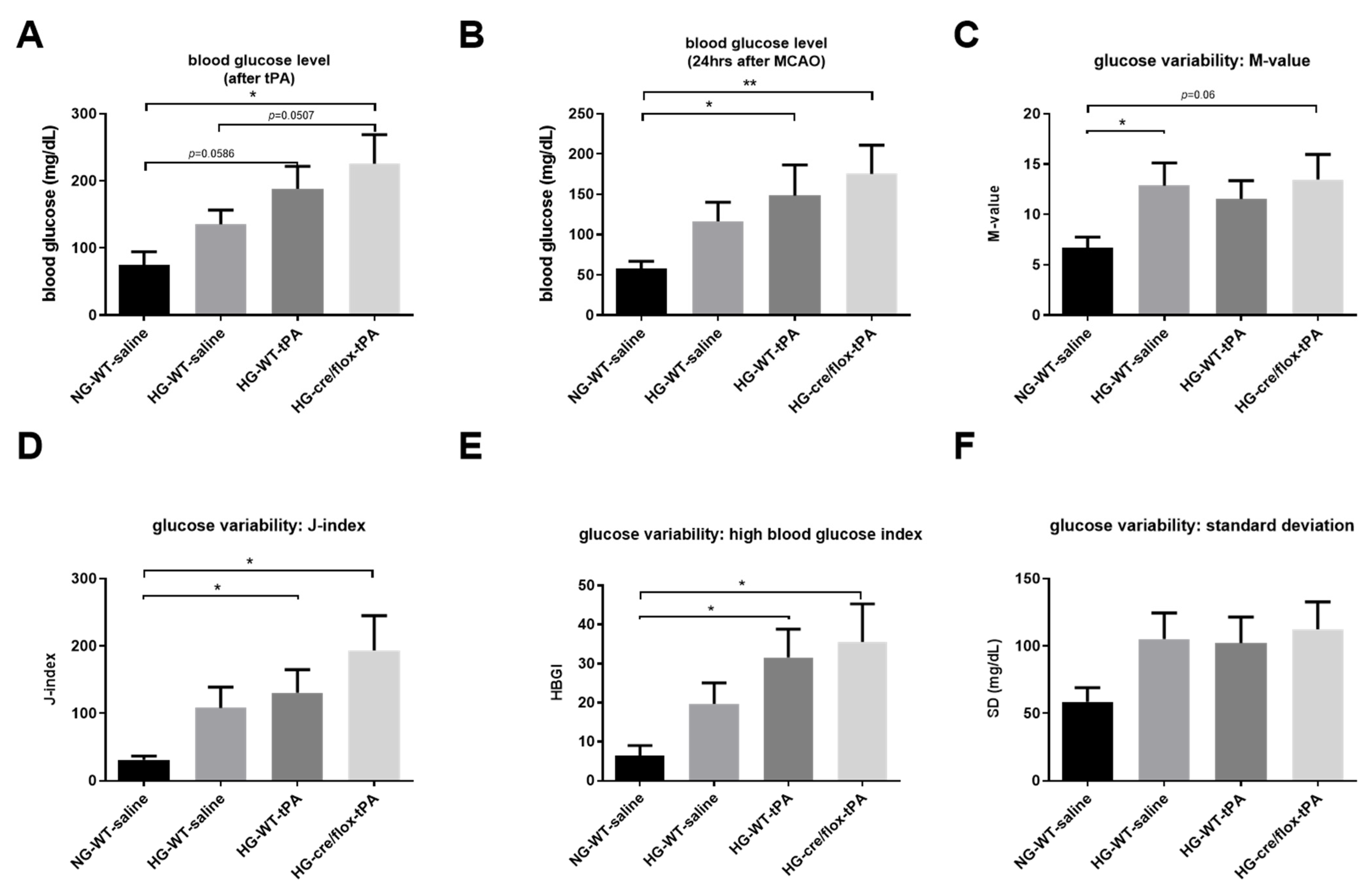
2.2. STZ-Based Hyperglycemic Profiling and Analysis of Glucose Variability upon Stroke with tPA-Reperfusion
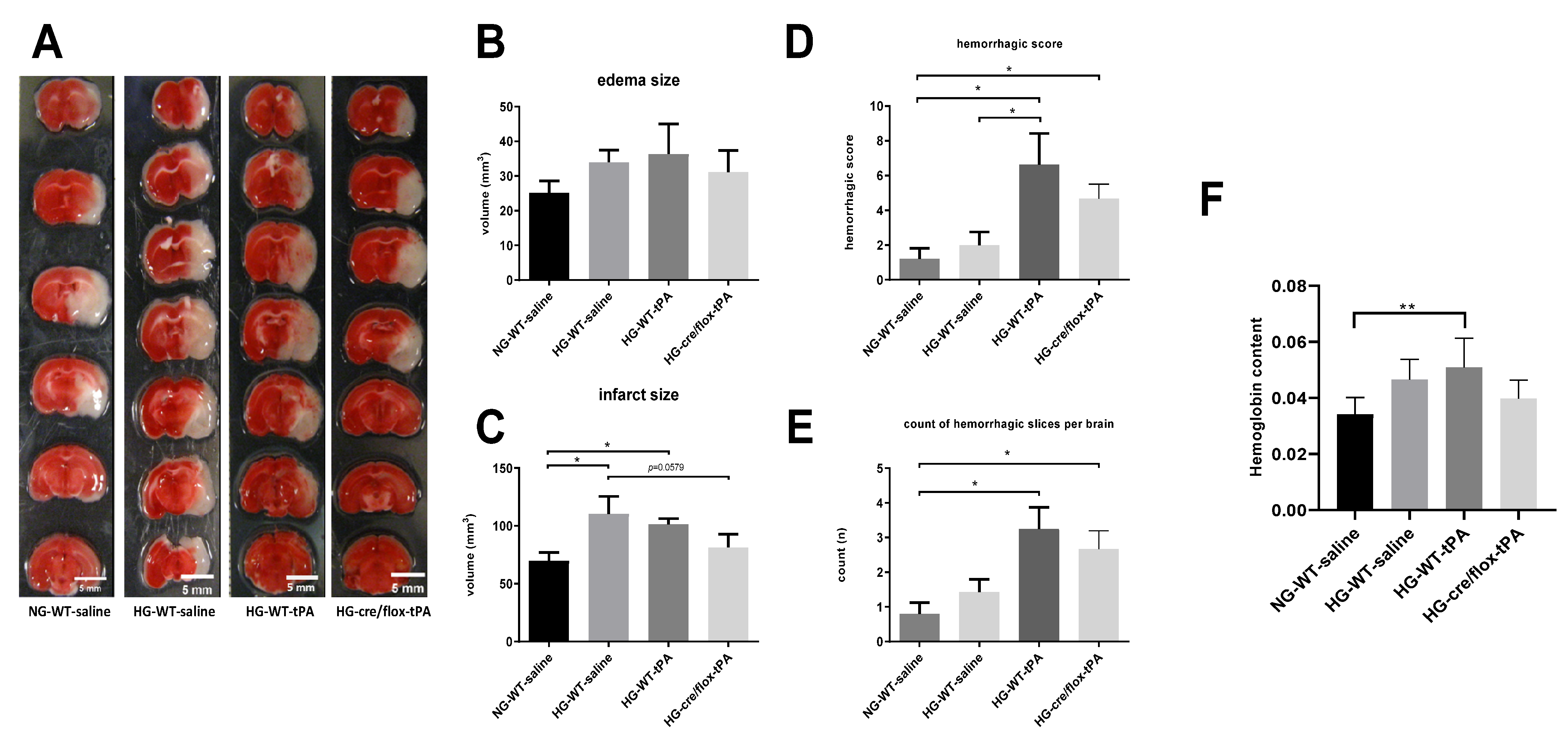
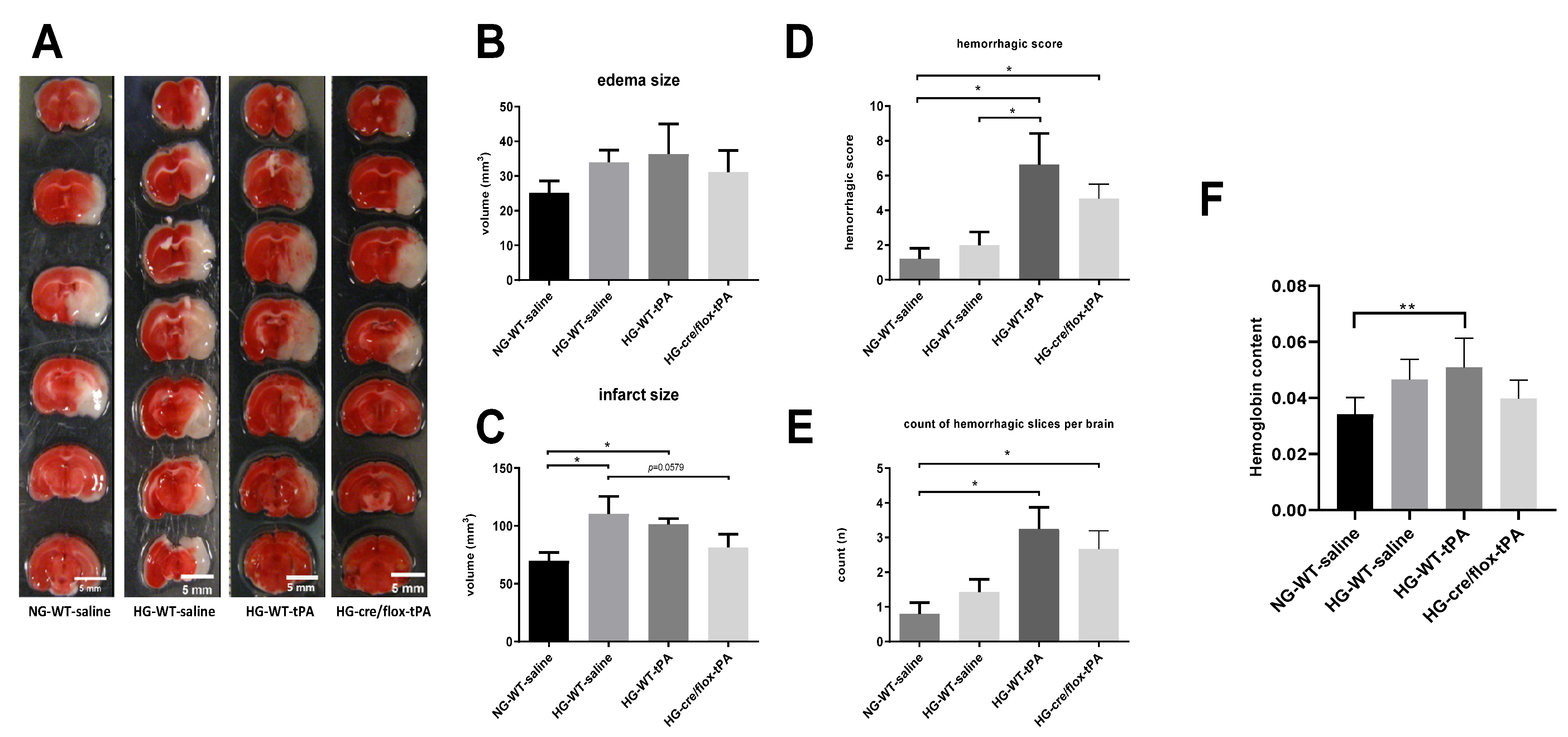
2.3. Effects of Sub-Acute HG on Infract Volume, Edema, and HT after eMCAO with tPA-Reperfusion
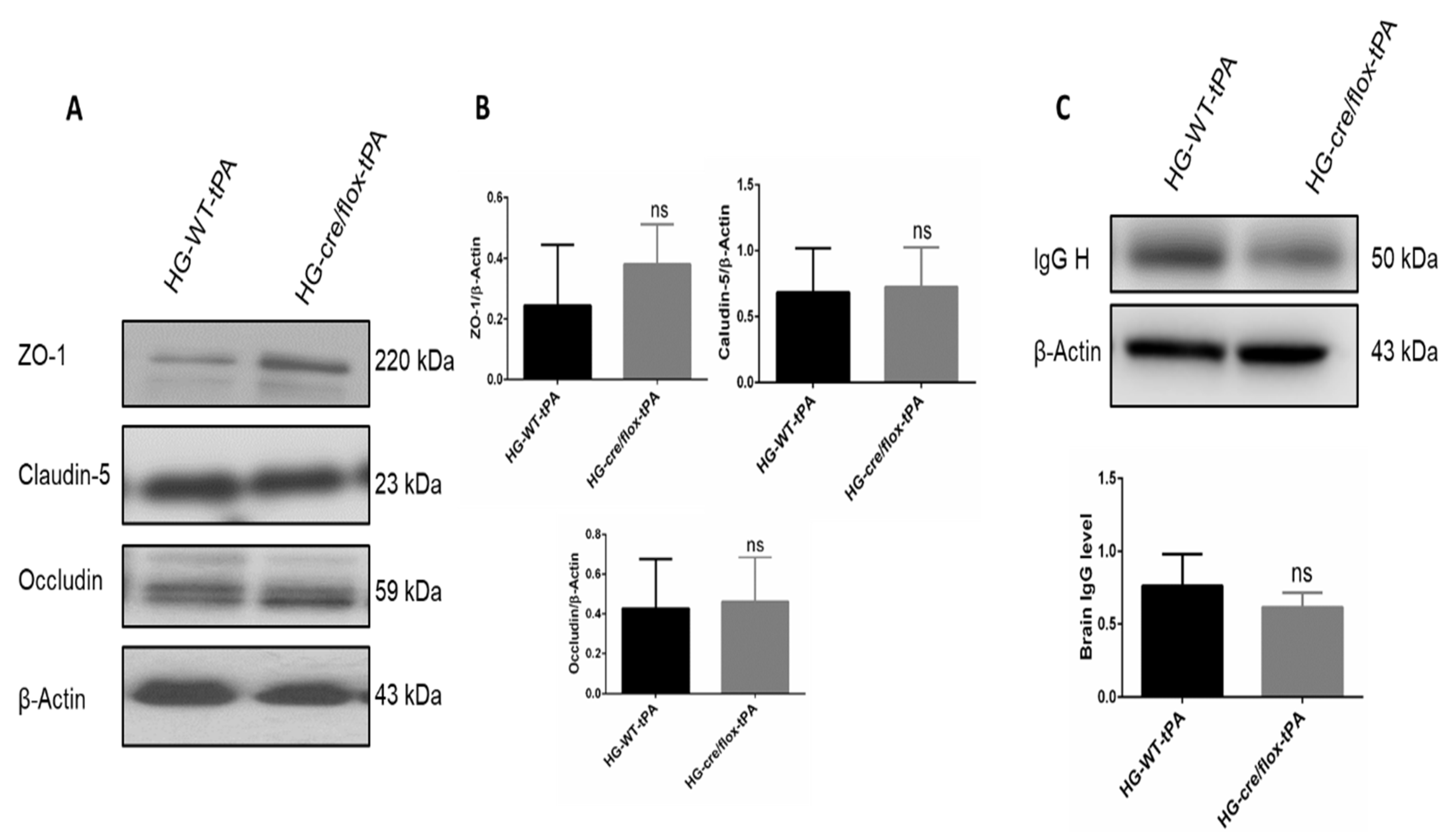
2.4. Endothelial TXNIP Deletion Upregulates Junction Proteins Expression in HG Mice after eMCAO and tPA-Reperfusion
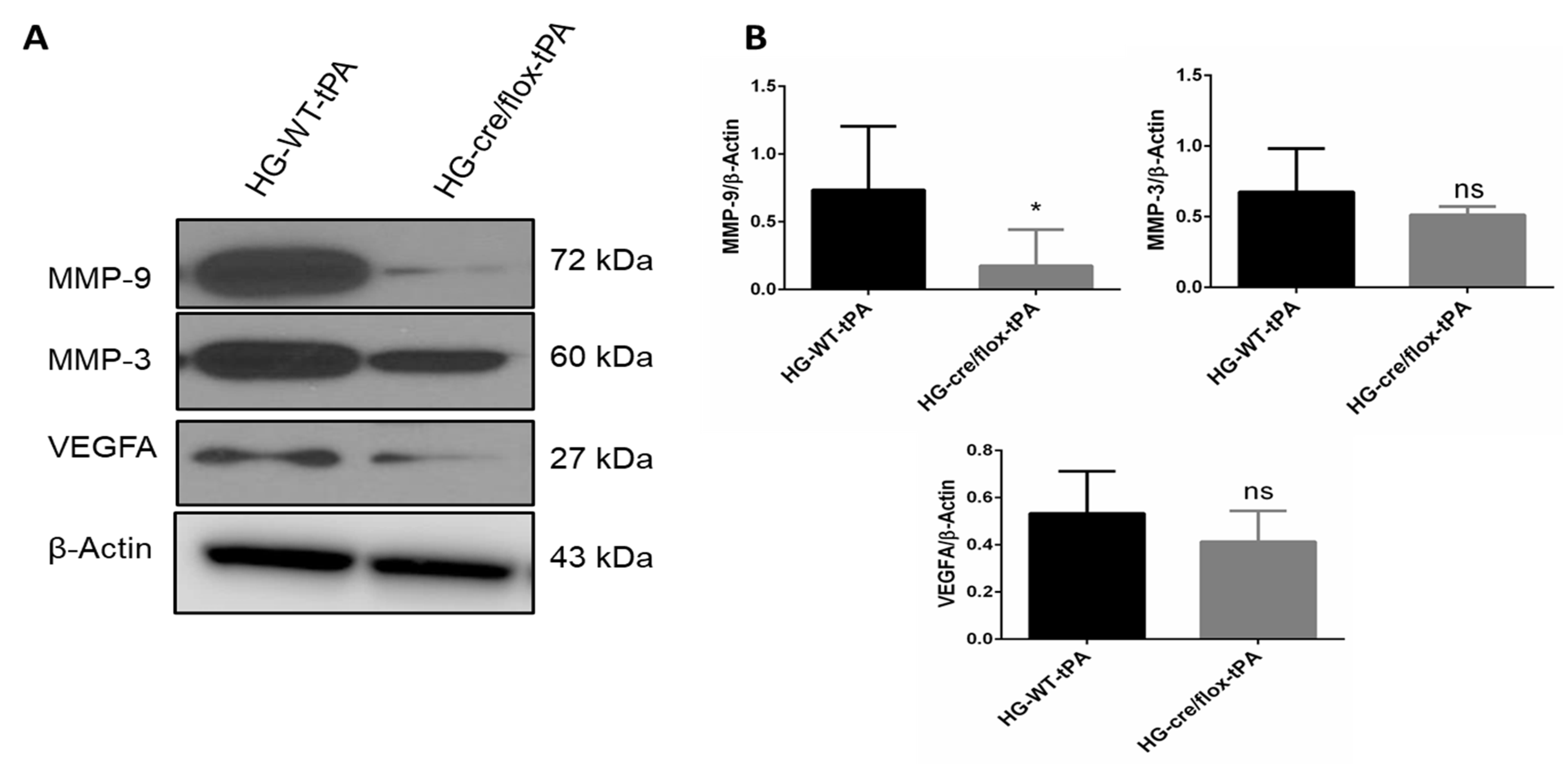
2.5. Endothelial TXNIP Deletion Downregulates the Expression of MMP-3/9 and VEGFA in HG Mice after eMCAO and tPA-Reperfusion
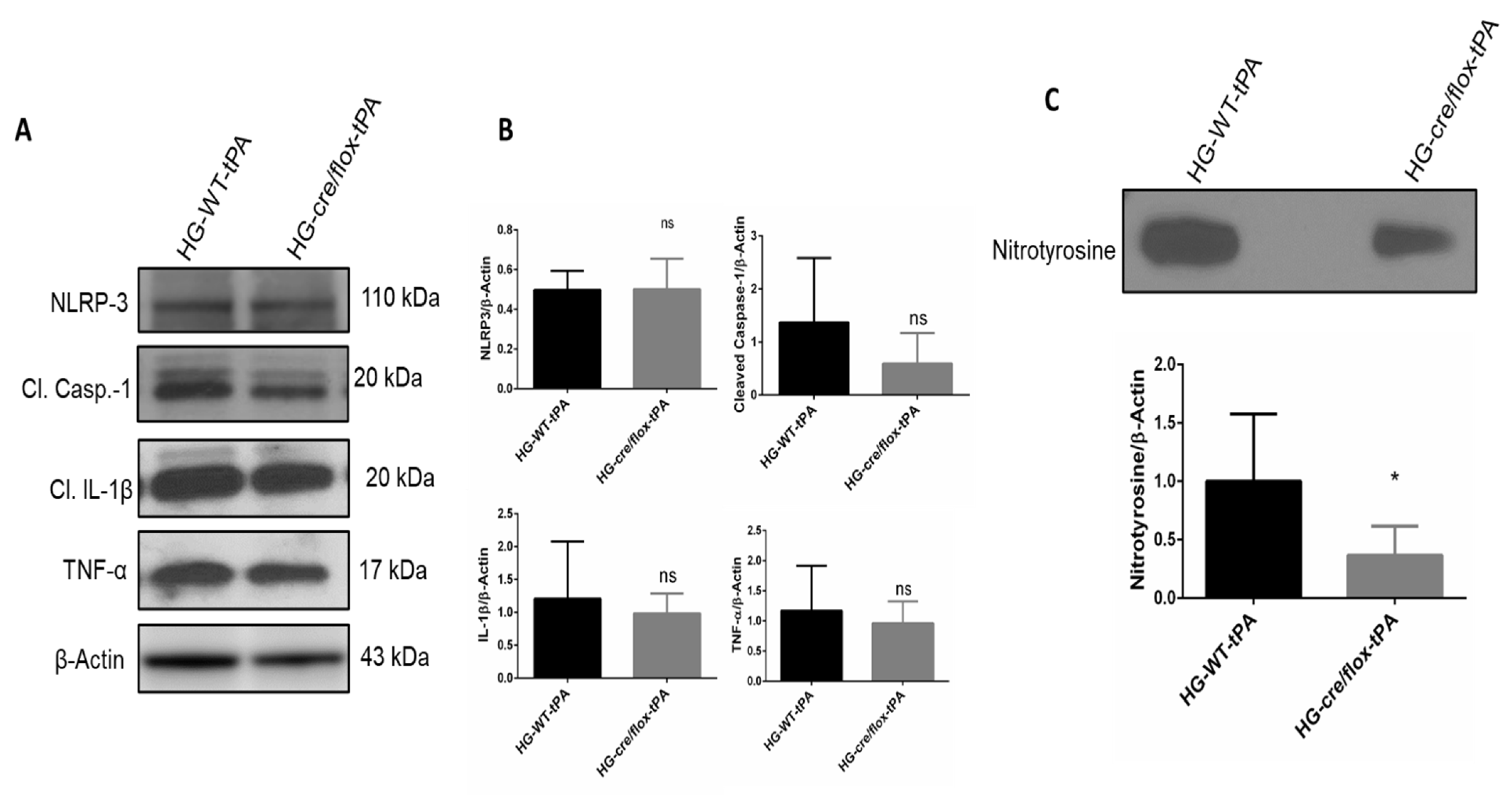
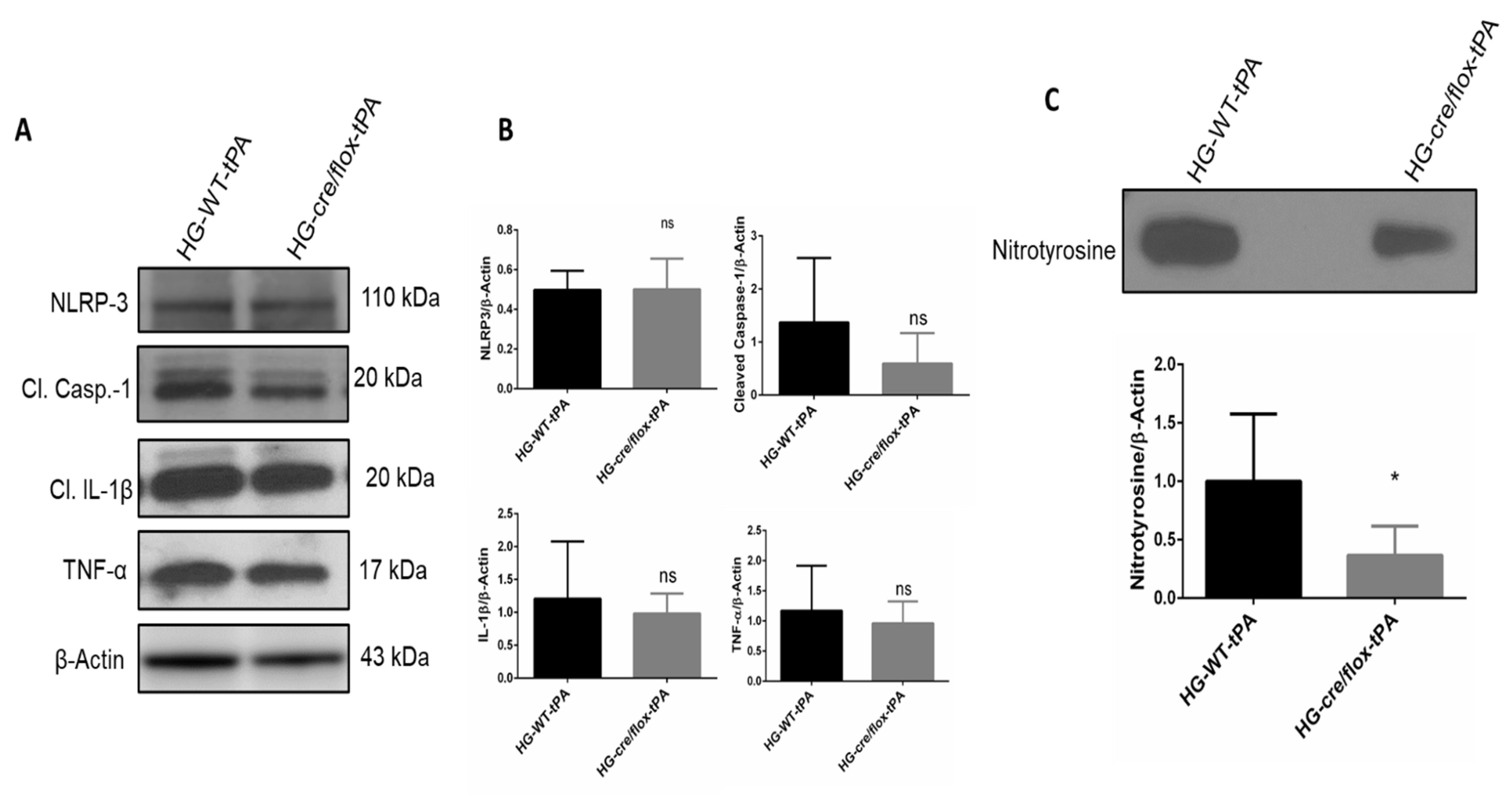
2.6. Endothelial TXNIP Deletion Reduces Inflammation in HG after eMCAO and tPA-Reperfusion
2.7. Endothelial TXNIP Deletion Attenuates Nitrotyrosine Formation in Brains of HG Mice, after eMCAO and tPA-Reperfusion
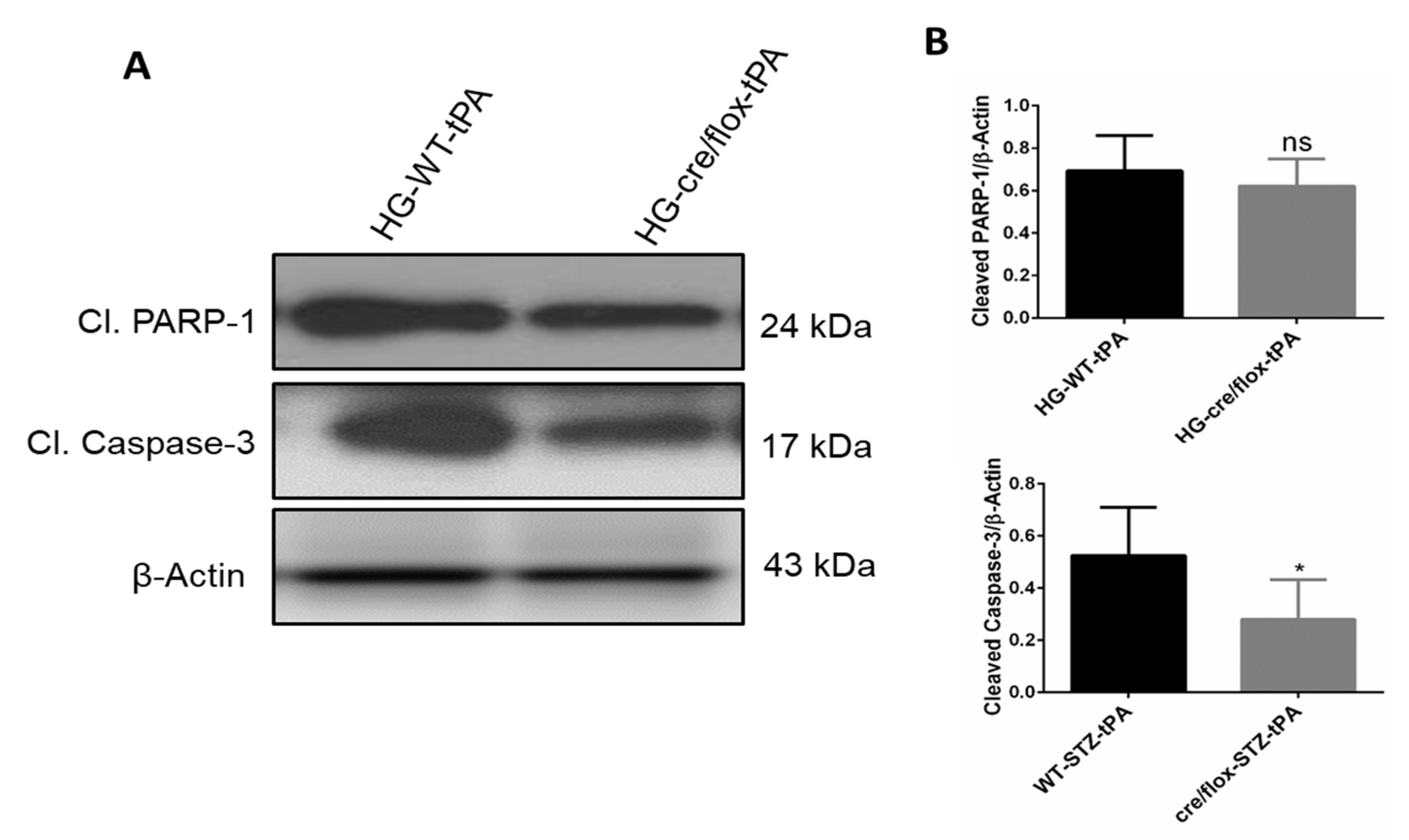
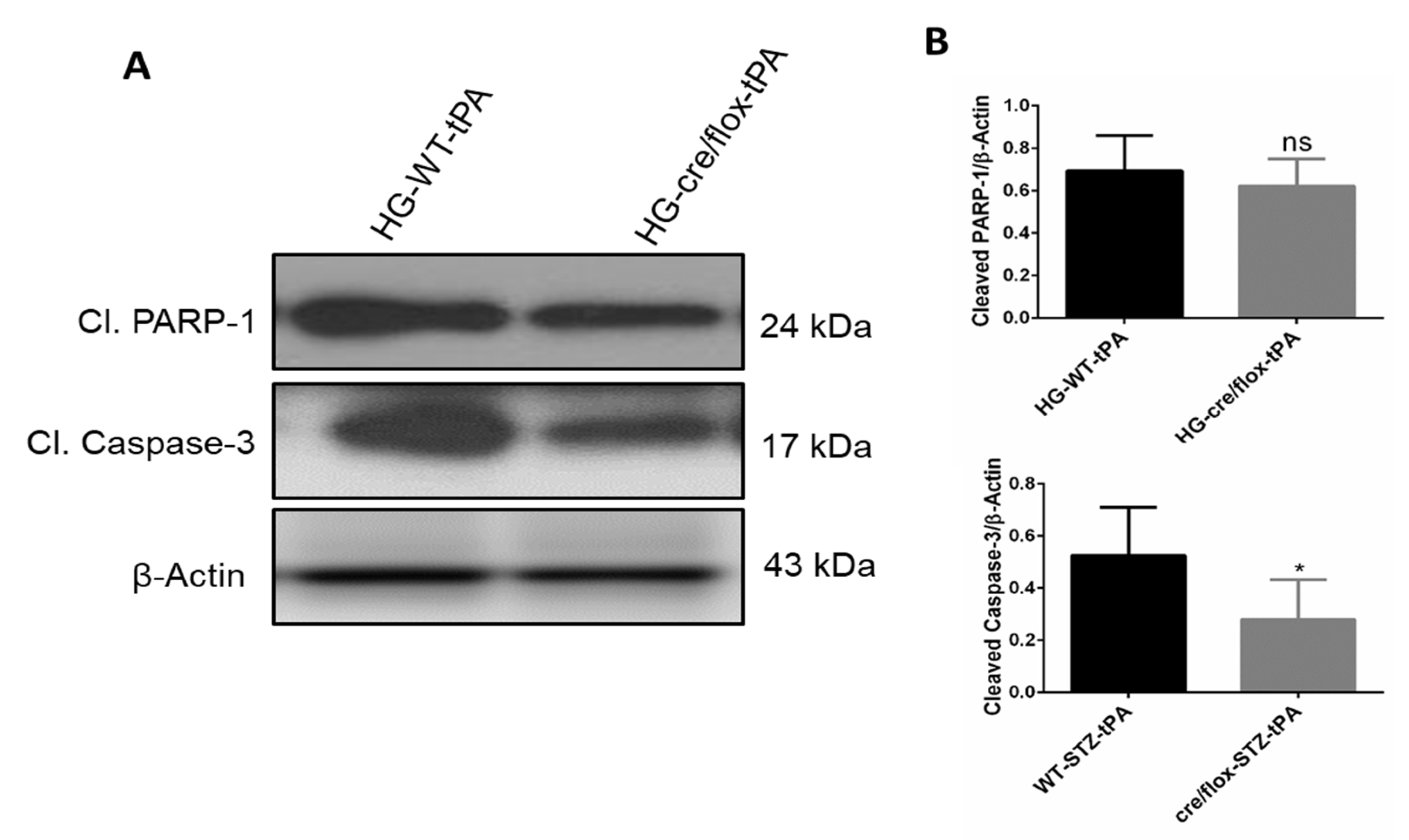
2.8. Endothelial TXNIP Deletion Downregulates the Protein Expression Levels of Cleaved-caspase-3, and Cleaved PARP-1 in Brains of HG Mice, after eMCAO and tPA-Reperfusion
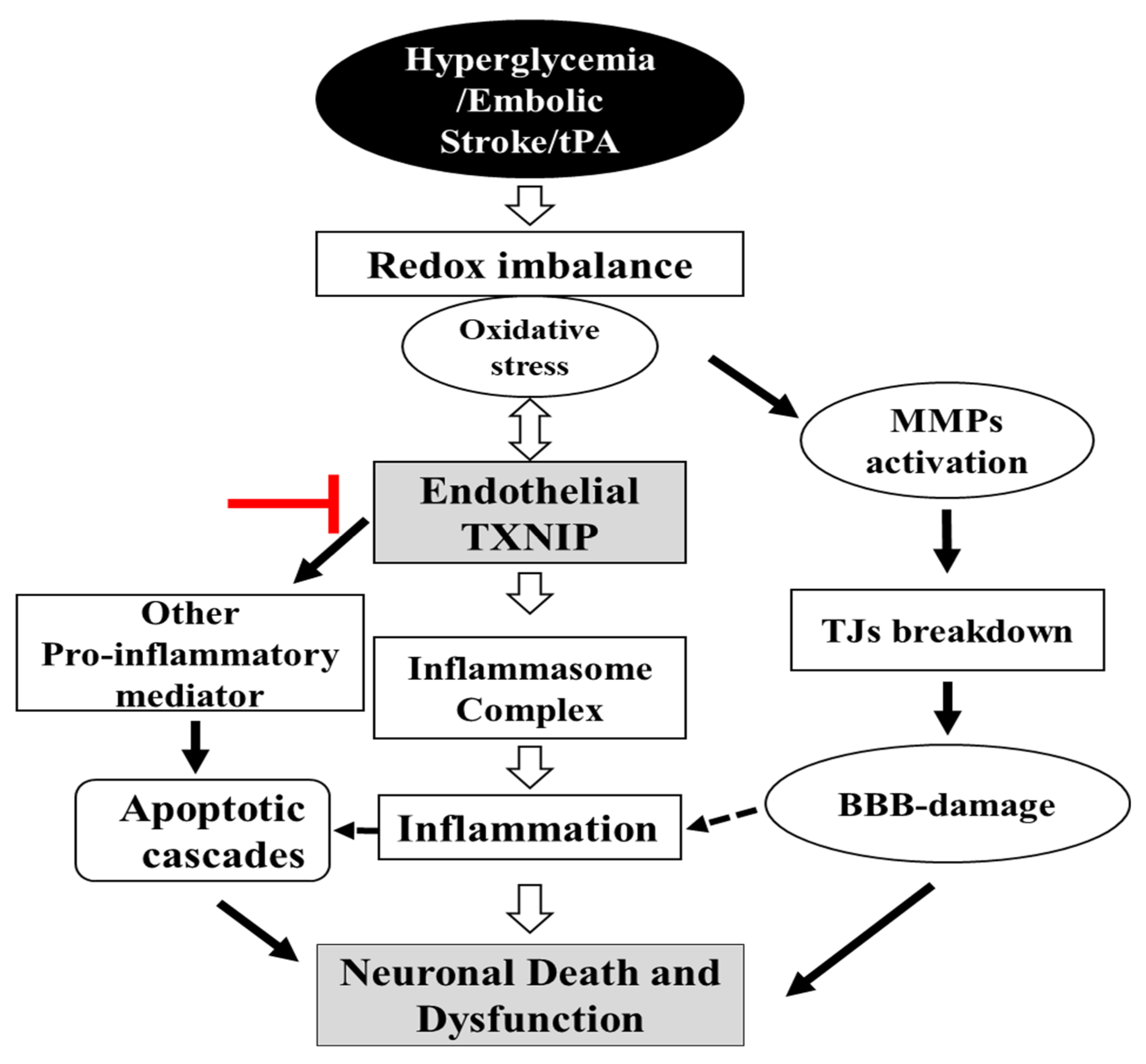
3. Discussion
4. Materials and Methods
4.1. The Animals and Study Design
4.2. Induction of Focal Embolic Stroke Model
4.3. Neurobehavioral Assessment
4.4. Assessment of Infarct Size and Edema
4.5. Evaluation of Hemoglobin Content
4.6. Slot Blot for Nitrotyrosine
4.7. Western Blot Analysis
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gaillard, T.; Miller, E. Guidelines for Stroke Survivors With Diabetes Mellitus. Stroke 2018, 49, e215–e217. [Google Scholar] [CrossRef]
- Zhang, G.; Kim, S.; Gu, X.; Yu, S.P.; Wei, L. DPP-4 Inhibitor Linagliptin is Neuroprotective in Hyperglycemic Mice with Stroke via the AKT/mTOR Pathway and Anti-apoptotic Effects. Neurosci. Bull. 2020, 36, 407–418. [Google Scholar] [CrossRef]
- Capes, S.E.; Hunt, D.; Malmberg, K.; Pathak, P.; Gerstein, H.C. Stress Hyperglycemia and Prognosis of Stroke in Nondiabetic and Diabetic Patients. Stroke 2001, 32, 2426–2432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nedergaard, M. Transient focal ischemia in hyperglycemic rats is associated with increased cerebral infarction. Brain Res. 1987, 408, 79–85. [Google Scholar] [CrossRef]
- Ismael, S.; Nasoohi, S.; Yoo, A.; Ahmed, H.A.; Ishrat, T. Tissue Plasminogen Activator Promotes TXNIP-NLRP3 Inflammasome Activation after Hyperglycemic Stroke in Mice. Mol. Neurobiol. 2020, 57, 2495–2508. [Google Scholar] [CrossRef] [PubMed]
- IDRIS, I.; THOMSON, G.A.; SHARMA, J.C. Diabetes mellitus and stroke. Int. J. Clin. Pract. 2005, 60, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Luitse, M.J.; Biessels, G.J.; Rutten, G.E.; Kappelle, L.J. Diabetes, hyperglycaemia, and acute ischaemic stroke. Lancet Neurol. 2012, 11, 261–271. [Google Scholar] [CrossRef]
- Hafez, S.; Coucha, M.; Bruno, A.; Fagan, S.C.; Ergul, A. Hyperglycemia, Acute Ischemic Stroke, and Thrombolytic Therapy. Transl. Stroke Res. 2014, 5, 442–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, J.; Zhao, Z.; Klein, G.M.; Lo, E.H.; Buchan, A.M. The Neurotoxicity of Tissue Plasminogen Activator? J. Cereb. Blood Flow Metab. 2004, 24, 945–963. [Google Scholar] [CrossRef]
- Gong, P.; Li, M.; Zou, C.; Tian, Q.; Xu, Z. Tissue Plasminogen Activator Causes Brain Microvascular Endothelial Cell Injury After Oxygen Glucose Deprivation by Inhibiting Sonic Hedgehog Signaling. Neurochem. Res. 2019, 44, 441–449. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Guo, Q.; Hossain, M.; Fazio, V.; Zeynalov, E.; Janigro, D.; Mayberg, M.R.; Namura, S. Bone marrow-derived cells are the major source of MMP-9 contributing to blood–brain barrier dysfunction and infarct formation after ischemic stroke in mice. Brain Res. 2009, 1294, 183–192. [Google Scholar] [CrossRef] [Green Version]
- del Zoppo, G.J.; Hallenbeck, J.M. Advances in the Vascular Pathophysiology of Ischemic Stroke. Thromb. Res. 2000, 98, 73–81. [Google Scholar] [CrossRef]
- Chow, B.W.; Gu, C. The Molecular Constituents of the Blood–Brain Barrier. Trends Neurosci. 2015, 38, 598–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Y.-B.; Gao, Q.; Tan, X.-X.; Huang, X.-W.; Ma, Y.-Z.; Fang, C.; Wang, S.-N.; Qiu, L.-H.; Cheng, Y.-X.; Guo, F.-Y.; et al. Lithium alleviates blood-brain barrier breakdown after cerebral ischemia and reperfusion by upregulating endothelial Wnt/β-catenin signaling in mice. Neuropharmacology 2021, 186, 108474. [Google Scholar] [CrossRef] [PubMed]
- Ishrat, T.; Pillai, B.; Ergul, A.; Hafez, S.; Fagan, S.C. Candesartan Reduces the Hemorrhage Associated with Delayed Tissue Plasminogen Activator Treatment in Rat Embolic Stroke. Neurochem. Res. 2013, 38, 2668–2677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ismael, S.; Nasoohi, S.; Yoo, A.; Mirzahosseini, G.; Ahmed, H.A.; Ishrat, T. Verapamil as an Adjunct Therapy to Reduce tPA Toxicity in Hyperglycemic Stroke: Implication of TXNIP/NLRP3 Inflammasome. Mol. Neurobiol. 2021, 58, 3792–3804. [Google Scholar] [CrossRef] [PubMed]
- Shalev, A. Minireview: Thioredoxin-Interacting Protein: Regulation and Function in the Pancreatic β-Cell. Mol. Endocrinol. 2014, 28, 1211–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsubaki, H.; Tooyama, I.; Walker, D.G. Thioredoxin-Interacting Protein (TXNIP) with Focus on Brain and Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 9357. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Masutani, H.; Nakamura, H.; Nishinaka, Y.; Yodoi, J. Redox Regulation by Thioredoxin and Thioredoxin-Binding Proteins. IUBMB Life 2001, 52, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Ishrat, T.; Mohamed, I.N.; Pillai, B.; Soliman, S.; Fouda, A.Y.; Ergul, A.; El-Remessy, A.B.; Fagan, S.C. Thioredoxin-Interacting Protein: A Novel Target for Neuroprotection in Experimental Thromboembolic Stroke in Mice. Mol. Neurobiol. 2015, 51, 766–778. [Google Scholar] [CrossRef] [Green Version]
- Nasoohi, S.; Ismael, S.; Ishrat, T. Thioredoxin-Interacting Protein (TXNIP) in Cerebrovascular and Neurodegenerative Diseases: Regulation and Implication. Mol. Neurobiol. 2018, 55, 7900–7920. [Google Scholar] [CrossRef]
- Kim, G.S.; Jung, J.E.; Narasimhan, P.; Sakata, H.; Chan, P.H. Induction of thioredoxin-interacting protein is mediated by oxidative stress, calcium, and glucose after brain injury in mice. Neurobiol. Dis. 2012, 46, 440–449. [Google Scholar] [CrossRef] [Green Version]
- Kelly-Cobbs, A.I.; Prakash, R.; Li, W.; Pillai, B.; Hafez, S.; Coucha, M.; Johnson, M.H.; Ogbi, S.N.; Fagan, S.C.; Ergul, A. Targets of vascular protection in acute ischemic stroke differ in type 2 diabetes. Am. J. Physiol. Circ. Physiol. 2013, 304, H806–H815. [Google Scholar] [CrossRef]
- Couret, D.; Bourane, S.; Catan, A.; Nativel, B.; Planesse, C.; Dorsemans, A.-C.; Ait-Arsa, I.; Cournot, M.; Rondeau, P.; Patche, J.; et al. A hemorrhagic transformation model of mechanical stroke therapy with acute hyperglycemia in mice. J. Comp. Neurol. 2018, 526, 1006–1016. [Google Scholar] [CrossRef]
- Deng, J.; Zhao, F.; Zhang, Y.; Zhou, Y.; Xu, X.; Zhang, X.; Zhao, Y. Neutrophil extracellular traps increased by hyperglycemia exacerbate ischemic brain damage. Neurosci. Lett. 2020, 738, 135383. [Google Scholar] [CrossRef] [PubMed]
- Ribo, M.; Molina, C.A.; Delgado, P.; Rubiera, M.; Delgado-Mederos, R.; Rovira, A.; Munuera, J.; Alvarez-Sabin, J. Hyperglycemia during Ischemia Rapidly Accelerates Brain Damage in Stroke Patients Treated with tPA. J. Cereb. Blood Flow Metab. 2007, 27, 1616–1622. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.F.; Tsirka, S.E.; Strickland, S.; Stieg, P.E.; Soriano, S.G.; Lipton, S.A. Tissue plasminogen activator (tPA) increase neuronal damage after focal cerebral ischemia in wild-type and tPA-deficient mice. Nat. Med. 1998, 4, 228–231. [Google Scholar] [CrossRef]
- Lee, C.Z.; Xue, Z.; Zhu, Y.; Yang, G.-Y.; Young, W.L. Matrix Metalloproteinase-9 Inhibition Attenuates Vascular Endothelial Growth Factor-Induced Intracerebral Hemorrhage. Stroke 2007, 38, 2563–2568. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.-T.; Zhang, P.; Gao, Y.; Li, C.-L.; Wang, H.-J.; Chen, L.-C.; Feng, Y.; Li, R.-Y.; Li, Y.-L.; Jiang, C.-L. Early VEGF inhibition attenuates blood-brain barrier disruption in ischemic rat brains by regulating the expression of MMPs. Mol. Med. Rep. 2017, 15, 57–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelsaid, M.A.; Matragoon, S.; El-Remessy, A.B. Thioredoxin-Interacting Protein Expression Is Required for VEGF-Mediated Angiogenic Signal in Endothelial Cells. Antioxid. Redox Signal. 2013, 19, 2199–2212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abulafia, D.P.; de Rivero Vaccari, J.P.; Lozano, J.D.; Lotocki, G.; Keane, R.W.; Dietrich, W.D. Inhibition of the Inflammasome Complex Reduces the Inflammatory Response after Thromboembolic Stroke in Mice. J. Cereb. Blood Flow Metab. 2009, 29, 534–544. [Google Scholar] [CrossRef] [Green Version]
- Kuroki, T.; Tanaka, R.; Shimada, Y.; Yamashiro, K.; Ueno, Y.; Shimura, H.; Urabe, T.; Hattori, N. Exendin-4 Inhibits Matrix Metalloproteinase-9 Activation and Reduces Infarct Growth After Focal Cerebral Ischemia in Hyperglycemic Mice. Stroke 2016, 47, 1328–1335. [Google Scholar] [CrossRef] [Green Version]
- Shao, B.; Bayraktutan, U. Hyperglycaemia promotes cerebral barrier dysfunction through activation of protein kinase C-β. Diabetes Obes. Metab. 2013, 15, 993–999. [Google Scholar] [CrossRef]
- Huang, J.; Liu, B.; Yang, C.; Chen, H.; Eunice, D.; Yuan, Z. Acute hyperglycemia worsens ischemic stroke-induced brain damage via high mobility group box-1 in rats. Brain Res. 2013, 1535, 148–155. [Google Scholar] [CrossRef]
- Srivastava, K.; Shao, B.; Bayraktutan, U. PKC- β Exacerbates in vitro Brain Barrier Damage in Hyperglycemic Settings via Regulation of RhoA/Rho-kinase/MLC2 Pathway. J. Cereb. Blood Flow Metab. 2013, 33, 1928–1936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romanic, A.M.; White, R.F.; Arleth, A.J.; Ohlstein, E.H.; Barone, F.C. Matrix Metalloproteinase Expression Increases after Cerebral Focal Ischemia in Rats. Stroke 1998, 29, 1020–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosell, A.; Ortega-Aznar, A.; Alvarez-Sabiín, J.; Fernaíndez-Cadenas, I.; Riboí, M.; Molina, C.A.; Lo, E.H.; Montaner, J. Increased Brain Expression of Matrix Metalloproteinase-9 After Ischemic and Hemorrhagic Human Stroke. Stroke 2006, 37, 1399–1406. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.G.; Zhang, L.; Jiang, Q.; Zhang, R.; Davies, K.; Powers, C.; van Bruggen, N.; Chopp, M. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J. Clin. Investig. 2000, 106, 829–838. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Yan, J.; Shi, H. Role of Hypoxia Inducible Factor 1 in Hyperglycemia-Exacerbated Blood-Brain Barrier Disruption in Ischemic Stroke. Neurobiol. Dis. 2016, 95, 82–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekerdag, E.; Solaroglu, I.; Gursoy-Ozdemir, Y. Cell Death Mechanisms in Stroke and Novel Molecular and Cellular Treatment Options. Curr. Neuropharmacol. 2018, 16, 1396–1415. [Google Scholar] [CrossRef]
- Portt, L.; Norman, G.; Clapp, C.; Greenwood, M.; Greenwood, M.T. Anti-apoptosis and cell survival: A review. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 238–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duris, K.; Splichal, Z.; Jurajda, M. The Role of Inflammatory Response in Stroke Associated Programmed Cell Death. Curr. Neuropharmacol. 2018, 16, 1365–1374. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, S.; Yokoo, H.; Tomita, K.; Kageyama-Yahara, N.; Uchimido, R.; Matsuda, N.; Yamamoto, S.; Hattori, Y. High glucose-induced apoptosis in human coronary artery endothelial cells involves up-regulation of death receptors. Cardiovasc. Diabetol. 2011, 10, 73. [Google Scholar] [CrossRef] [Green Version]
- Gong, L.; Liu, F.; Wang, J.; Wang, X.; Hou, X.; Sun, Y.; Qin, W.; Wei, S.; Zhang, Y.; Chen, L.; et al. Hyperglycemia induces apoptosis of pancreatic islet endothelial cells via reactive nitrogen species-mediated Jun N-terminal kinase activation. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1211–1219. [Google Scholar] [CrossRef] [Green Version]
- Ren, M.; Lin, Z.-J.; Qian, H.; Choudhury, G.R.; Liu, R.; Liu, H.; Yang, S.-H. Embolic middle cerebral artery occlusion model using thrombin and fibrinogen composed clots in rat. J. Neurosci. Methods 2012, 211, 296–304. [Google Scholar] [CrossRef] [Green Version]
- Bederson, J.B.; Pitts, L.H.; Tsuji, M.; Nishimura, M.C.; Davis, R.L.; Bartkowski, H. Rat middle cerebral artery occlusion: Evaluation of the model and development of a neurologic examination. Stroke 1986, 17, 472–476. [Google Scholar] [CrossRef] [Green Version]
- Crumrine, R.C.; Marder, V.J.; Taylor, G.M.; LaManna, J.C.; Tsipis, C.P.; Scuderi, P.; Petteway, S.R.; Arora, V. Intra-arterial administration of recombinant tissue-type plasminogen activator (rt-PA) causes more intracranial bleeding than does intravenous rt-PA in a transient rat middle cerebral artery occlusion model. Exp. Transl. Stroke Med. 2011, 3, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swanson, R.A.; Morton, M.T.; Tsao-Wu, G.; Savalos, R.A.; Davidson, C.; Sharp, F.R. A Semiautomated Method for Measuring Brain Infarct Volume. J. Cereb. Blood Flow Metab. 1990, 10, 290–293. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salman, M.; Ismael, S.; Li, L.; Ahmed, H.A.; Puchowicz, M.A.; Ishrat, T. Endothelial Thioredoxin-Interacting Protein Depletion Reduces Hemorrhagic Transformation in Hyperglycemic Mice after Embolic Stroke and Thrombolytic Therapy. Pharmaceuticals 2021, 14, 983. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14100983
Salman M, Ismael S, Li L, Ahmed HA, Puchowicz MA, Ishrat T. Endothelial Thioredoxin-Interacting Protein Depletion Reduces Hemorrhagic Transformation in Hyperglycemic Mice after Embolic Stroke and Thrombolytic Therapy. Pharmaceuticals. 2021; 14(10):983. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14100983
Chicago/Turabian StyleSalman, Mohd., Saifudeen Ismael, Lexiao Li, Heba A. Ahmed, Michelle A. Puchowicz, and Tauheed Ishrat. 2021. "Endothelial Thioredoxin-Interacting Protein Depletion Reduces Hemorrhagic Transformation in Hyperglycemic Mice after Embolic Stroke and Thrombolytic Therapy" Pharmaceuticals 14, no. 10: 983. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14100983