
A Rational Design of α-Helix-Shaped Peptides Employing the Hydrogen-Bond Surrogate Approach: A Modulation Strategy for Ras-RasGRF1 Interaction in Neuropsychiatric Disorders


Abstract
:
1. Introduction
1.1. Mutational Studies on Sos and Design of a Peptide-Based Ras Inhibitor
- GRKKRRQRRR—PPCVPYLGMYLTDLVFIEEGTPNYTEDGLVN
- TAT sequence RasGRF1 interacting region
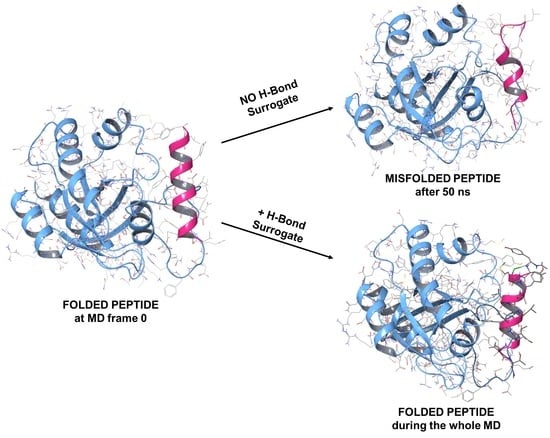
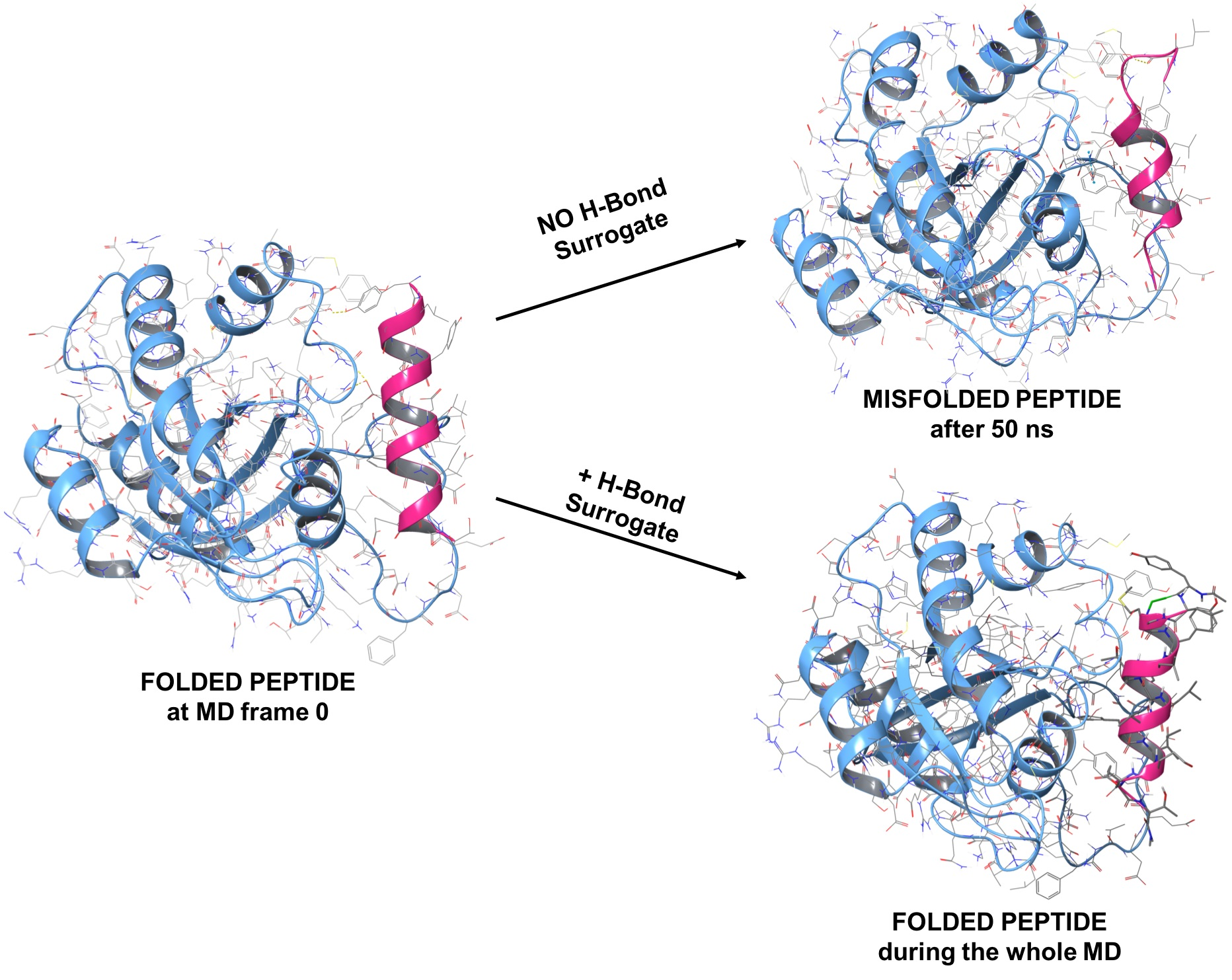
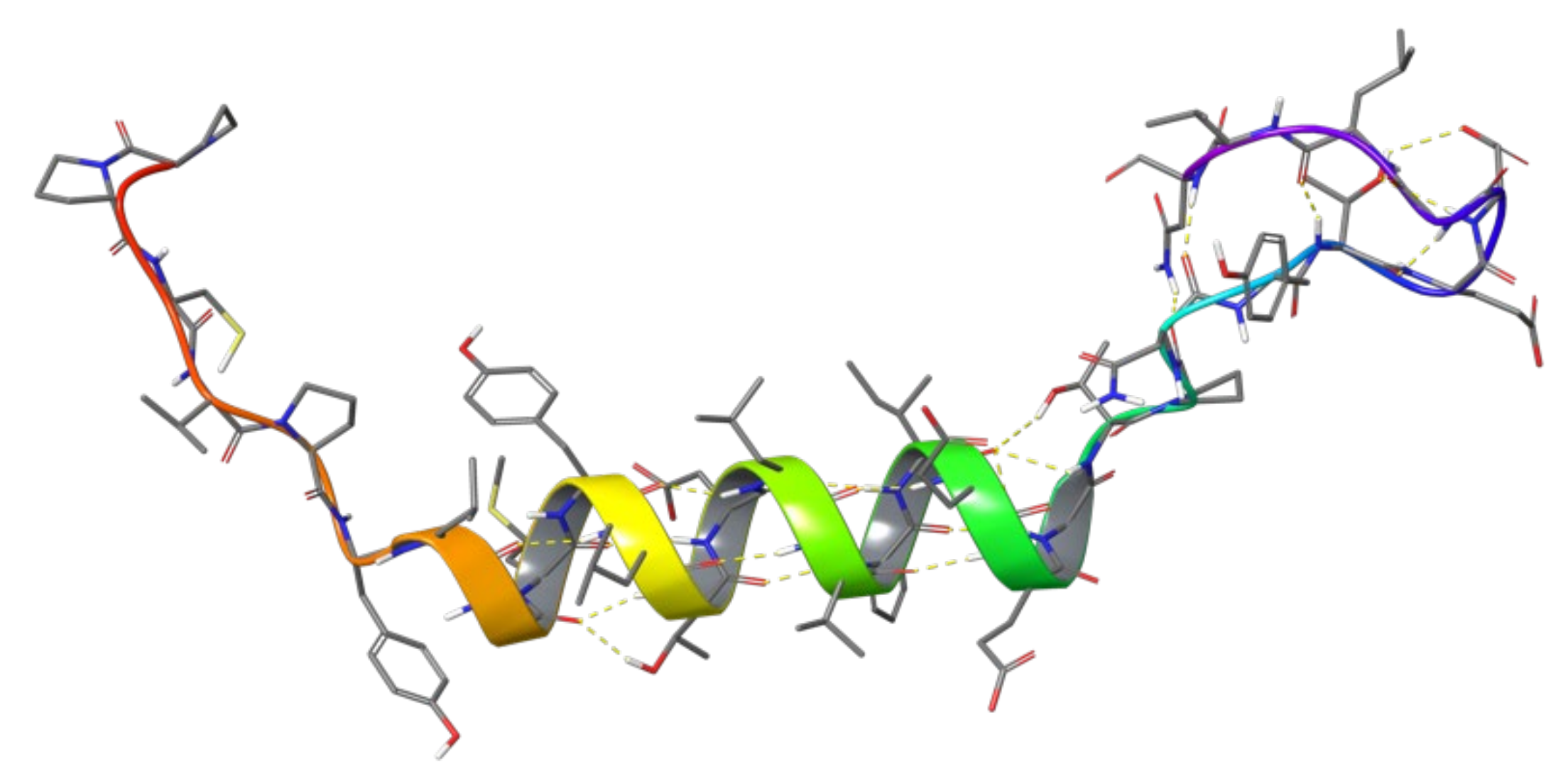
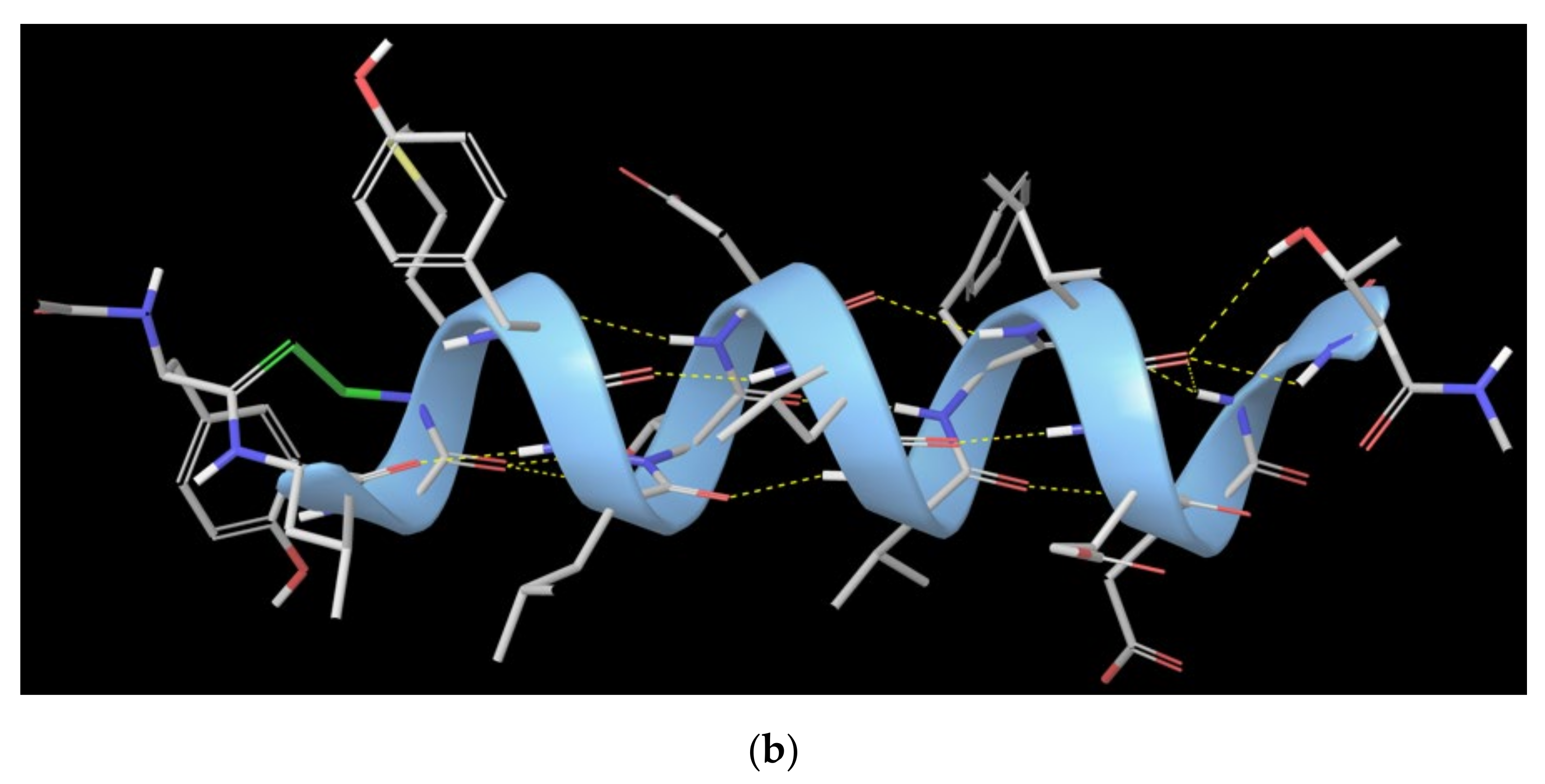
1.2. RB3 Peptide Modifications by Using Hydrogen-Bond Surrogates
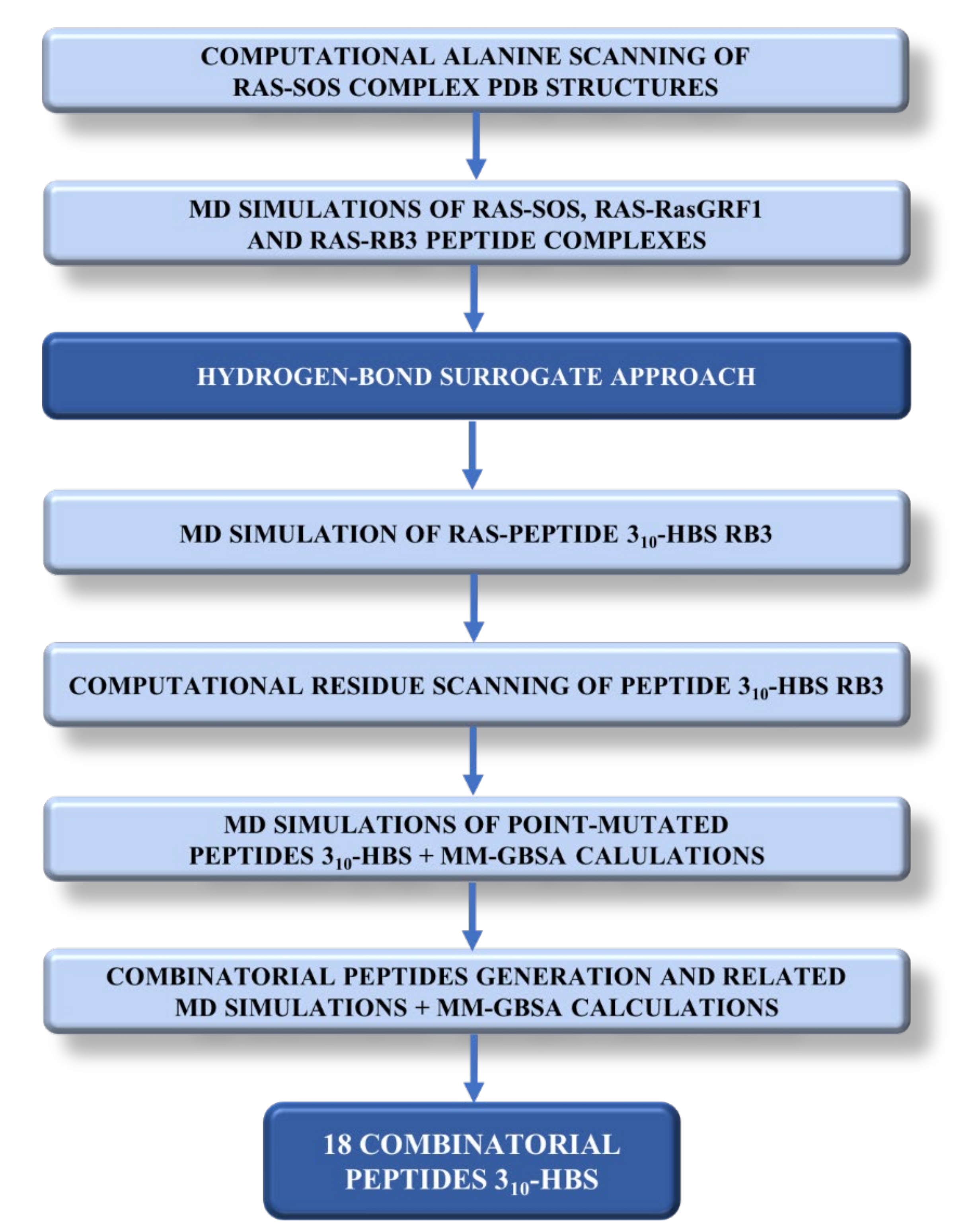
2. Results and Discussion
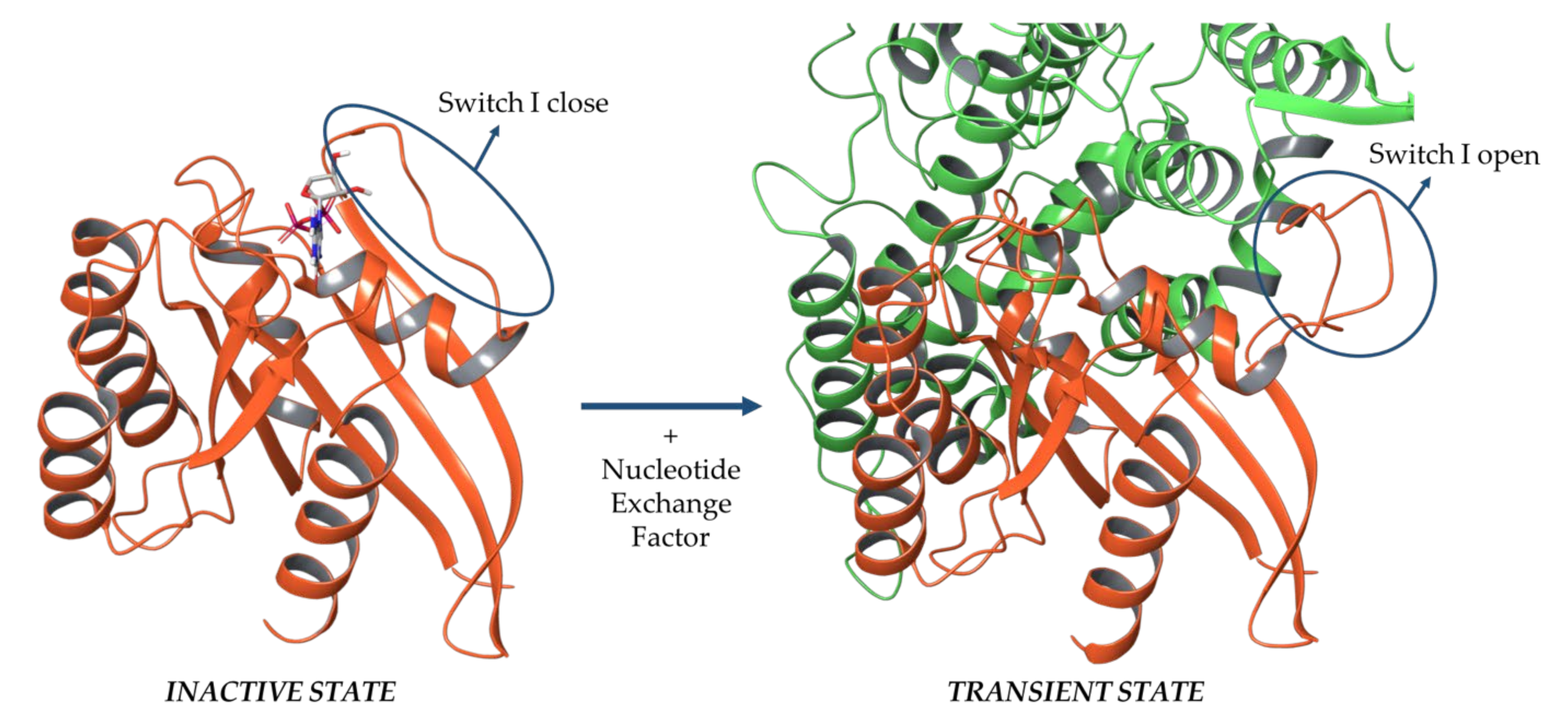
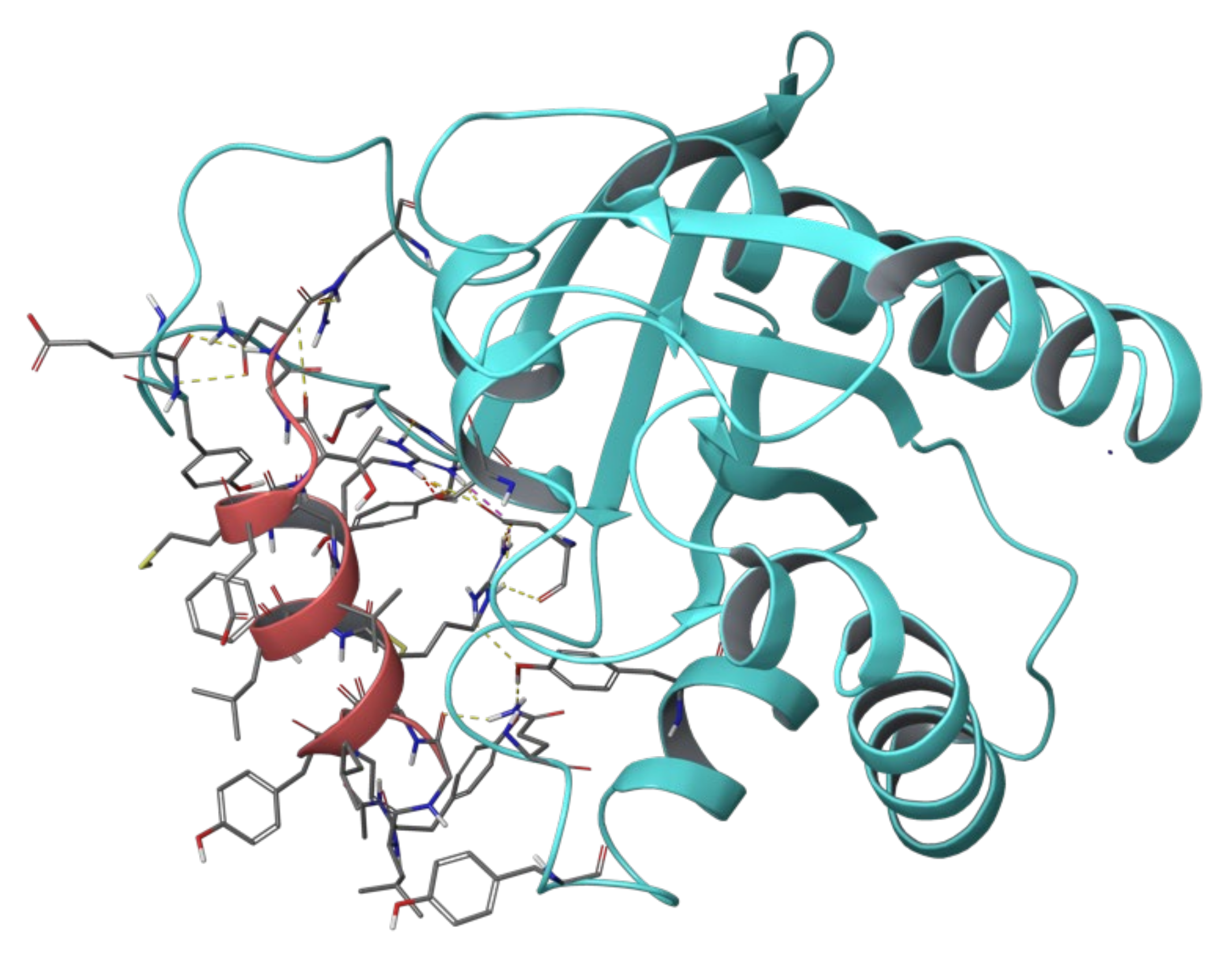
2.1. Sos and RasGRF1 Binding Interfaces Analysis
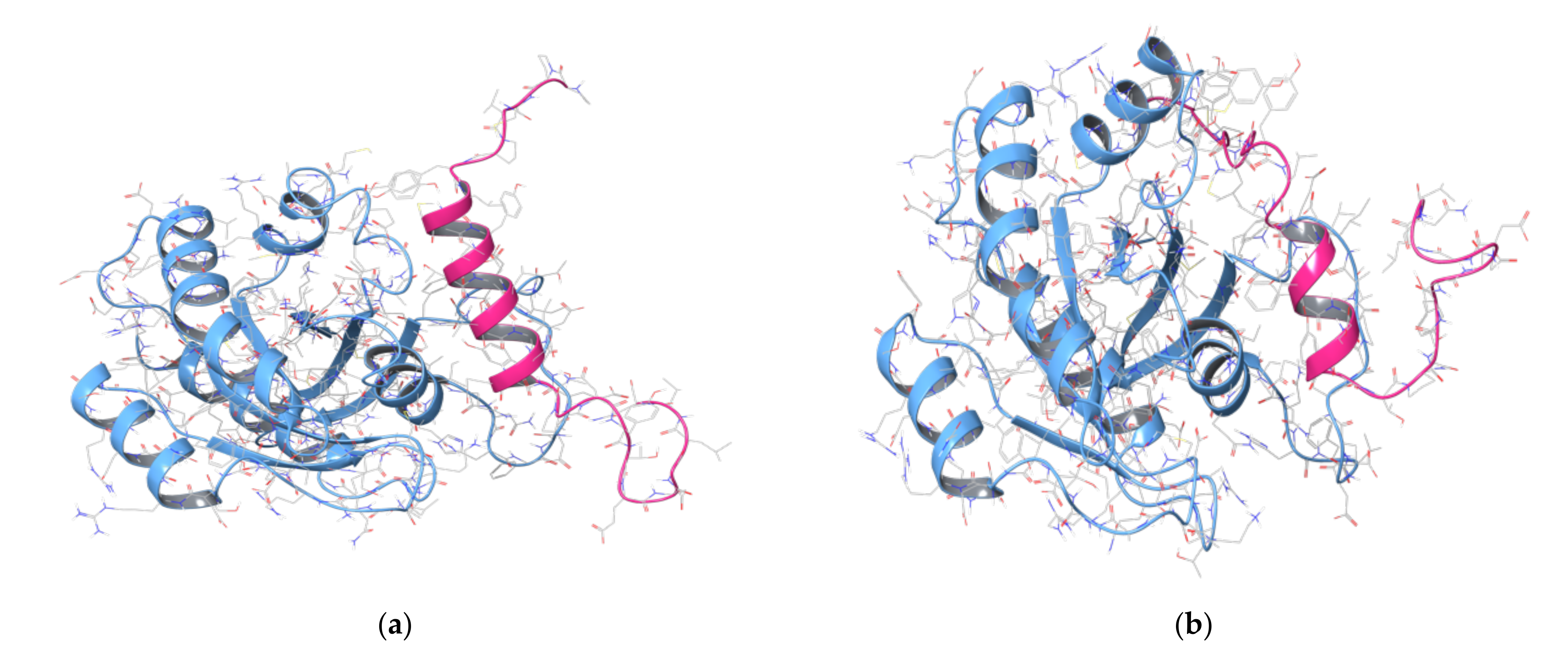
2.2. MD Simulations of Ras in Complex with Sos and RasGRF1 Binding Fragments
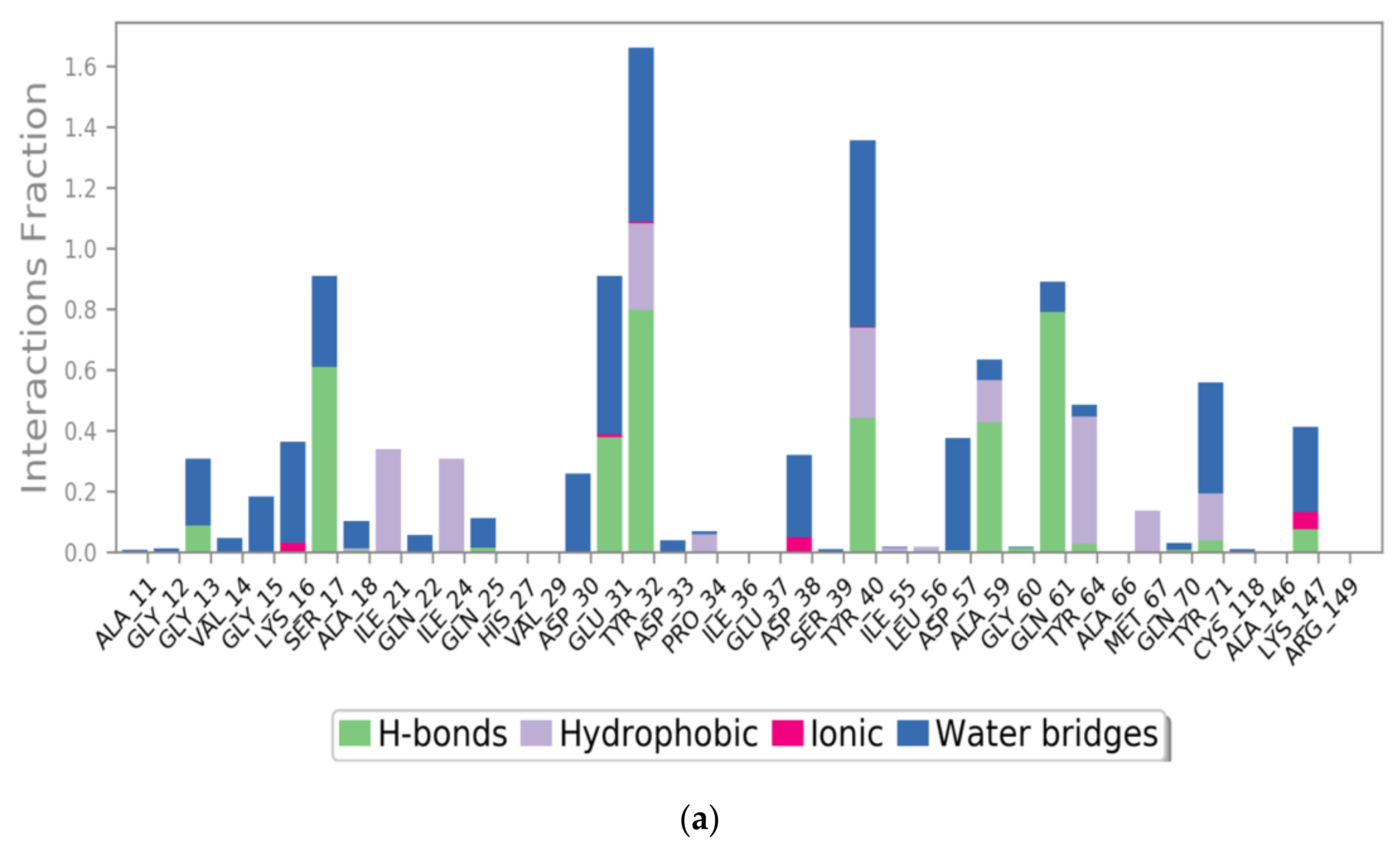
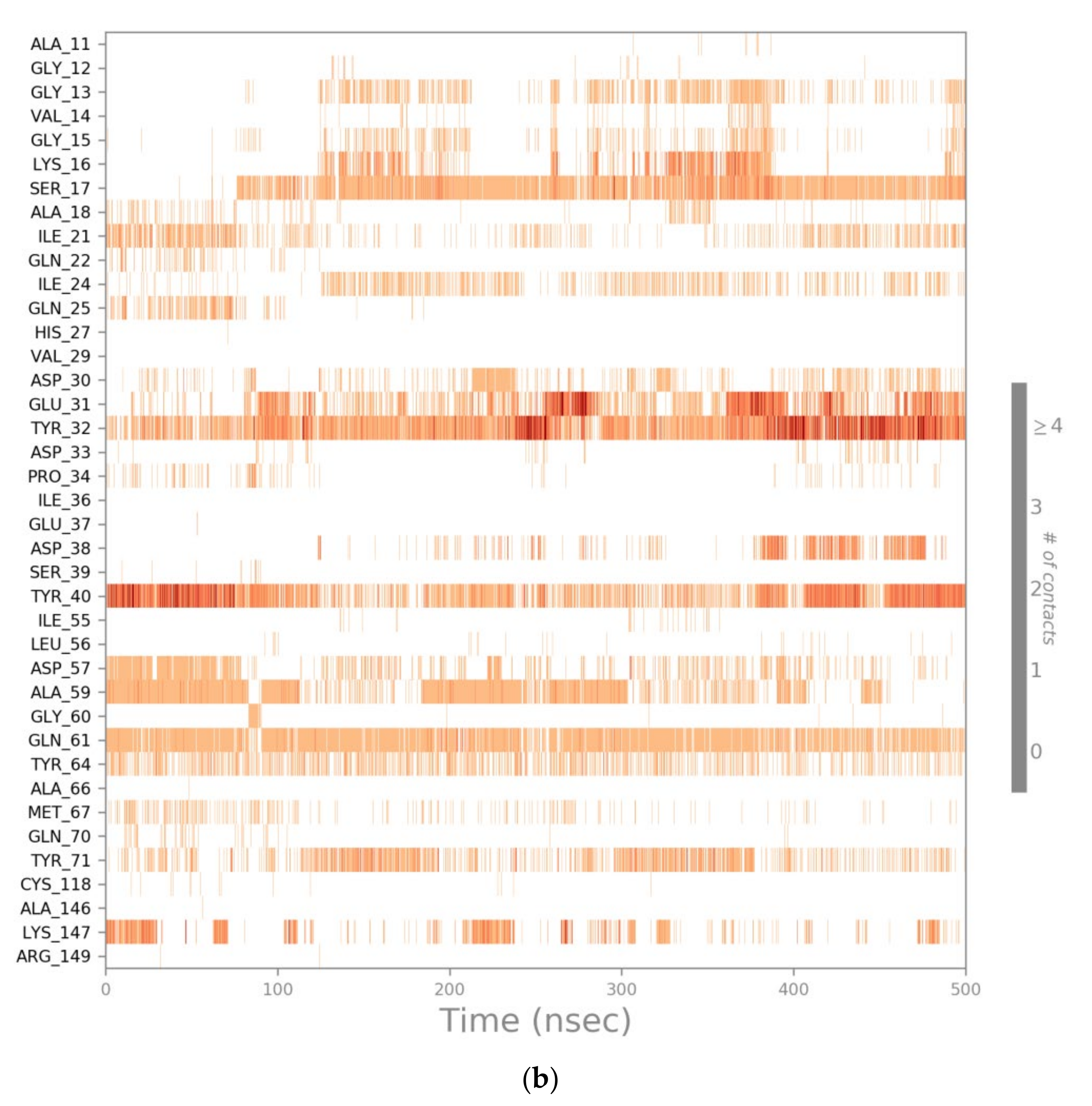
2.3. MD Simulations of the Ras-RB3 Peptide Complex
2.4. Computational Residue Scanning of the 310-HBS RB3 Peptide and MD Simulations of Point-Mutated Peptides
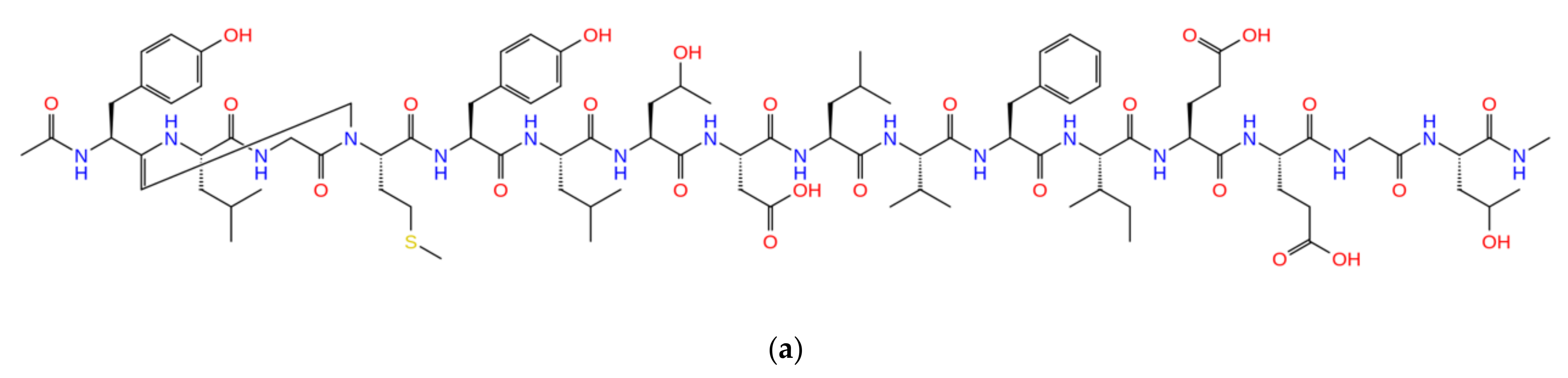
2.5. Combinatorial Peptides Using 310-HBS RB3: Creation and MD Simulations
- ΔGbinding value higher than the reference one (−79.70 kcal/mol);
- Loss of helical conformation.
3. Methods
3.1. Protein Preparation
3.2. MD Simulations of Ras Protein in Complex with Sos, RasGRF1, RB3 Peptide, and the Designed 310-HBS Peptides
- Minimization with the solute restrained;
- Minimization without restraints;
- 12 ps in the NVT ensemble with a Berendsen thermostat, temperature of 10 K, a fast temperature relaxation constant, velocity resampling every 1 ps, and nonhydrogen solute atoms restrained;
- 12 ps in the NPT ensemble in a Berendsen thermostat and barostat, temperature equal to 10 K and a pressure of 1 atm, a fast temperature relaxation constant, a slow pressure relaxation constant, velocity resampling every 1ps, and nonhydrogen solute atoms restrained;
- 24 ps in the NPT ensemble with a Berendsen thermostat and barostat, temperature of 300 K and a pressure of 1 atm, a fast temperature relaxation constant, a slow pressure relaxation constant, velocity resampling every 1 ps, and nonhydrogen solute atoms restrained;
- Final step of 24 ps of relaxation in NPT ensemble using a Berendsen thermostat and barostat, a temperature of 300 K and a pressure of 1 atm, a fast temperature relaxation constant, and a normal pressure relaxation constant.
3.3. MD Frame Clustering
3.4. Computational Residue Scanning of Peptide 310-HBS RB3 in Complex with Ras
3.5. MM-GBSA Calculations of All the Complexes Used to Perform MD
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Everitt, B.J. Neural and psychological mechanisms underlying compulsive drug seeking habits and drug memories—Indications for novel treatments of addiction. Eur. J. Neurosci. 2014, 40, 2163–2182. [Google Scholar] [CrossRef] [Green Version]
- Berhow, M.T.; Hiroi, N.; Nestler, E.J. Regulation of ERK (Extracellular Signal Regulated Kinase), Part of the Neurotrophin Signal Transduction Cascade, in the Rat Mesolimbic Dopamine System by Chronic Exposure to Morphine or Cocaine. J. Neurosci. 1996, 16, 4707–4715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nestler, E.J. Molecular mechanisms of drug addiction. Neuropharmacology 2004, 47, 24–32. [Google Scholar] [CrossRef]
- Miller, C.A.; Marshall, J.F. Molecular Substrates for Retrieval and Reconsolidation of Cocaine-Associated Contextual Memory. Neuron 2005, 47, 873–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torregrossa, M.M.; Corlett, P.R.; Taylor, J.R. Aberrant learning and memory in addiction. Neurobiol. Learn. Mem. 2011, 96, 609–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itzhak, Y.; Perez-Lanza, D.; Liddie, S. The strength of aversive and appetitive associations and maladaptive behaviors. IUBMB Life 2014, 66, 559–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belin, D.; Belin-Rauscent, A.; Murray, J.E.; Everitt, B.J. Addiction: Failure of control over maladaptive incentive habits. Curr. Opin. Neurobiol. 2013, 23, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Sanchis-Segura, C.; Spanagel, R. Behavioural assessment of drug reinforcement and addictive features in rodents: An overview. Addict. Biol. 2006, 11, 2–38. [Google Scholar] [CrossRef] [PubMed]
- Fasano, S.; Brambilla, R. Ras-ERK Signaling in Behavior: Old Questions and New Perspectives. Front. Behav. Neurosci. 2011, 5, 79. [Google Scholar] [CrossRef] [Green Version]
- Pascoli, V.; Besnard, A.; Hervé, D.; Pagès, C.; Heck, N.; Girault, J.-A.; Caboche, J.; Vanhoutte, P. Cyclic Adenosine Monophosphate–Independent Tyrosine Phosphorylation of NR2B Mediates Cocaine-Induced Extracellular Signal-Regulated Kinase Activation. Biol. Psychiatry 2011, 69, 218–227. [Google Scholar] [CrossRef]
- Pascoli, V.; Turiault, M.; Lüscher, C. Reversal of cocaine-evoked synaptic potentiation resets drug-induced adaptive behaviour. Nature 2012, 481, 71–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascoli, V.; Cahill, E.; Bellivier, F.; Caboche, J.; Vanhoutte, P. Extracellular Signal-Regulated Protein Kinases 1 and 2 Activation by Addictive Drugs: A Signal Toward Pathological Adaptation. Biol. Psychiatry 2014, 76, 917–926. [Google Scholar] [CrossRef]
- Cahill, E.; Salery, M.; Vanhoutte, P.; Caboche, J. Convergence of dopamine and glutamate signaling onto striatal ERK activation in response to drugs of abuse. Front. Pharmacol. 2014, 4, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Pardo, M.P.; Roger-Sanchez, C.; Rodríguez-Arias, M.; Miñarro, J.; Aguilar, M.A. Pharmacological modulation of protein kinases as a new approach to treat addiction to cocaine and opiates. Eur. J. Pharmacol. 2016, 781, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Valjent, E.; Corvol, J.-C.; Pagès, C.; Besson, M.-J.; Maldonado, R.; Caboche, J. Involvement of the Extracellular Signal-Regulated Kinase Cascade for Cocaine-Rewarding Properties. J. Neurosci. 2000, 20, 8701–8709. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Hope, B.T.; Dempsey, J.; Liu, S.Y.; Bossert, J.M.; Shaham, Y. Central amygdala ERK signaling pathway is critical to incubation of cocaine craving. Nat. Neurosci. 2005, 8, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Valjent, E.; Corbille, A.-G.; Bertran-Gonzalez, J.; Herve, D.; Girault, J.-A. Inhibition of ERK pathway or protein synthesis during reexposure to drugs of abuse erases previously learned place preference. Proc. Natl. Acad. Sci. USA 2006, 103, 2932–2937. [Google Scholar] [CrossRef] [Green Version]
- Valjent, E.; Corvol, J.-C.; Trzaskos, J.M.; Girault, J.-A.; Hervé, D. Role of the ERK pathway in psychostimulant-induced locomotor sensitization. BMC Neurosci. 2006, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Koya, E.; Zhai, H.; Hope, B.T.; Shaham, Y. Role of ERK in cocaine addiction. Trends Neurosci. 2006, 29, 695–703. [Google Scholar] [CrossRef]
- Ferguson, S.M.; Fasano, S.; Yang, P.; Brambilla, R.; Robinson, T.E. Knockout of ERK1 Enhances Cocaine-Evoked Immediate Early Gene Expression and Behavioral Plasticity. Neuropsychopharmacology 2006, 31, 2660–2668. [Google Scholar] [CrossRef]
- Girault, J.; Valjent, E.; Caboche, J.; Herve, D. ERK2: A logical AND gate critical for drug-induced plasticity? Curr. Opin. Pharmacol. 2007, 7, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Fasano, S.; D’Antoni, A.; Orban, P.C.; Valjent, E.; Putignano, E.; Vara, H.; Pizzorusso, T.; Giustetto, M.; Yoon, B.; Soloway, P.; et al. Ras-Guanine Nucleotide-Releasing Factor 1 (Ras-GRF1) Controls Activation of Extracellular Signal-Regulated Kinase (ERK) Signaling in the Striatum and Long-Term Behavioral Responses to Cocaine. Biol. Psychiatry 2009, 66, 758–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papale, A.; D’Isa, R.; Menna, E.; Cerovic, M.; Solari, N.; Hardingham, N.; Cambiaghi, M.; Cursi, M.; Barbacid, M.; Leocani, L.; et al. Severe Intellectual Disability and Enhanced Gamma-Aminobutyric Acidergic Synaptogenesis in a Novel Model of Rare RASopathies. Biol. Psychiatry 2017, 81, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Pucilowska, J.; Vithayathil, J.; Pagani, M.; Kelly, C.; Karlo, J.C.; Robol, C.; Morella, I.; Gozzi, A.; Brambilla, R.; Landreth, G.E. Pharmacological Inhibition of ERK Signaling Rescues Pathophysiology and Behavioral Phenotype Associated with 16p11.2 Chromosomal Deletion in Mice. J. Neurosci. 2018, 38, 6640–6652. [Google Scholar] [CrossRef] [PubMed]
- Kyosseva, S.V. Mitogen-Activated Protein Kinase Signaling. Int. Rev. Neurobiol. 2004, 59, 201–220. [Google Scholar] [PubMed]
- Kim, E.K.; Choi, E.-J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta-Mol. Basis Dis. 2010, 1802, 396–405. [Google Scholar] [CrossRef] [Green Version]
- Seger, R.; Krebs, E.G. The MAPK signaling cascade. FASEB J. 1995, 9, 726–735. [Google Scholar] [CrossRef]
- Vetter, I.R.; Wittinghofer, A. The Guanine Nucleotide-Binding Switch in Three Dimensions. Science 2001, 294, 1299–1304. [Google Scholar] [CrossRef] [Green Version]
- Mattingly, R.R.; Macara, I.G. Phosphorylation-dependent activation of the Ras-GRF/CDC25Mm exchange factor by muscarinic receptors and G-protein βγ subunits. Nature 1996, 382, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Baouz, S.; Jacquet, E.; Bernardi, A.; Parmeggiani, A. The N-terminal Moiety of CDC25 Mm, a GDP/GTP Exchange Factor of Ras Proteins, Controls the Activity of the Catalytic Domain. J. Biol. Chem. 1997, 272, 6671–6676. [Google Scholar] [CrossRef] [Green Version]
- Farnsworth, C.L.; Freshney, N.W.; Rosen, L.B.; Ghosh, A.; Greenberg, M.E.; Feig, L.A. Calcium activation of Ras mediated by neuronal exchange factor Ras-GRF. Nature 1995, 376, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Schweighoffer, F.; Faure, M.; Fath, I.; Chevallier-Multon, M.; Apiou, F.; Dutrillaux, B.; Sturani, E.; Jacquet, M.; Tocque, B. Identification of a human guanine nucleotide-releasing factor (H-GRF55) specific for Ras proteins. Oncogene 1993, 8, 1477–1485. [Google Scholar] [PubMed]
- Lenzen, C.; Cool, R.H.; Wittinghofer, A. Analysis of intrinsic and CDC25-stimulated guanine nucleotide exchange of p21ras-nucleotide complexes by fluorescence measurements. Methods Enzymol. 1995, 255, 95–109. [Google Scholar] [PubMed]
- Tian, X.; Gotoh, T.; Tsuji, K.; Lo, E.H.; Huang, S.; Feig, L.A. Developmentally regulated role for Ras-GRFs in coupling NMDA glutamate receptors to Ras, Erk and CREB. EMBO J. 2004, 23, 1567–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-Sagi, D. The Sos (Son of sevenless) protein. Trends Endocrinol. Metab. 1994, 5, 165–169. [Google Scholar] [CrossRef]
- Tinhofer, I.; Maly, K.; Dietl, P.; Hochholdinger, F.; Mayr, S.; Obermeier, A.; Grunicke, H.H. Differential Ca2+ Signaling Induced by Activation of the Epidermal Growth Factor and Nerve Growth Factor Receptors. J. Biol. Chem. 1996, 271, 30505–30509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, S.E.; Giddings, B.W.; Brooks, M.W.; Buday, L.; Sizeland, A.M.; Weinberg, R.A. Association of Sos Ras exchange protein with Grb2 is implicated in tyrosine kinase signal transduction and transformation. Nature 1993, 363, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Gale, N.W.; Kaplan, S.; Lowenstein, E.J.; Schlessinger, J.; Bar-Sagi, D. Grb2 mediates the EGF-dependent activation of guanine nucleotide exchange on Ras. Nature 1993, 363, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Batzer, A.; Daly, R.; Yajnik, V.; Skolnik, E.; Chardin, P.; Bar-Sagi, D.; Margolis, B.; Schlessinger, J. Guanine-nucleotide-releasing factor hSos1 binds to Grb2 and links receptor tyrosine kinases to Ras signalling. Nature 1993, 363, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Buday, L.; Downward, J. Epidermal growth factor regulates p21ras through the formation of a complex of receptor, Grb2 adapter protein, and Sos nucleotide exchange factor. Cell 1993, 73, 611–620. [Google Scholar] [CrossRef]
- Lenzen, C.; Cool, R.H.; Prinz, H.; Kuhlmann, J.; Wittinghofer, A. Kinetic Analysis by Fluorescence of the Interaction between Ras and the Catalytic Domain of the Guanine Nucleotide Exchange Factor Cdc25 Mm. Biochemistry 1998, 37, 7420–7430. [Google Scholar] [CrossRef] [PubMed]
- Zarich, N.; Oliva, J.L.; Jorge, R.; Santos, E.; Rojas, J.M. The isoform-specific stretch of hSos1 defines a new Grb2-binding domain. Oncogene 2000, 19, 5872–5883. [Google Scholar] [CrossRef] [Green Version]
- Boriack-Sjodin, P.A.; Margarit, S.M.; Bar-Sagi, D.; Kuriyan, J. The structural basis of the activation of Ras by Sos. Nature 1998, 394, 337–343. [Google Scholar] [CrossRef]
- Chardin, P.; Mattei, M.-G. Chromosomal localization of two genes encoding human ras exchange factors: SOS1 maps to the 2p22 -> p16 region and SOS2 to the 14q21 -> q22 region of the human genome. Cytogenet. Genome Res. 1994, 66, 68–69. [Google Scholar] [CrossRef]
- Boguski, M.S.; McCormick, F. Proteins regulating Ras and its relatives. Nature 1993, 366, 643–654. [Google Scholar] [CrossRef]
- Liu, B.X.; Wei, W.; Broek, D. The catalytic domain of the mouse sos1 gene product activates Ras proteins in vivo and in vitro. Oncogene 1993, 8, 3081–3084. [Google Scholar] [PubMed]
- Margarit, S.M.; Sondermann, H.; Hall, B.E.; Nagar, B.; Hoelz, A.; Pirruccello, M.; Bar-Sagi, D.; Kuriyan, J. Structural Evidence for Feedback Activation by Ras·GTP of the Ras-Specific Nucleotide Exchange Factor SOS. Cell 2003, 112, 685–695. [Google Scholar] [CrossRef] [Green Version]
- Sondermann, H.; Soisson, S.M.; Boykevisch, S.; Yang, S.-S.; Bar-Sagi, D.; Kuriyan, J. Structural Analysis of Autoinhibition in the Ras Activator Son of Sevenless. Cell 2004, 119, 393–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willumsen, B.M.; Papageorge, A.G.; Kung, H.F.; Bekesi, E.; Robins, T.; Johnsen, M.; Vass, W.C.; Lowy, D.R. Mutational analysis of a ras catalytic domain. Mol. Cell. Biol. 1986, 6, 2646–2654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fasano, O.; Crechet, J.B.; De Vendittis, E.; Zahn, R.; Feger, G.; Vitelli, A.; Parmeggiani, A. Yeast mutants temperature-sensitive for growth after random mutagenesis of the chromosomal RAS2 gene and deletion of the RAS1 gene. EMBO J. 1988, 7, 3375–3383. [Google Scholar] [CrossRef] [PubMed]
- Mistou, M.Y.; Jacquet, E.; Poullet, P.; Rensland, H.; Gideon, P.; Schlichting, I.; Wittinghofer, A.; Parmeggiani, A. Mutations of Ha-ras p21 that define important regions for the molecular mechanism of the SDC25 C-domain, a guanine nucleotide dissociation stimulator. EMBO J. 1992, 11, 2391–2397. [Google Scholar] [CrossRef] [PubMed]
- Howe, L.R.; Marshall, C.J. Identification of amino acids in p21ras involved in exchange factor interaction. Oncogene 1993, 8, 2583–2590. [Google Scholar] [PubMed]
- Segal, M.; Willumsen, B.M.; Levitzki, A. Residues crucial for Ras interaction with GDP-GTP exchangers. Proc. Natl. Acad. Sci. USA 1993, 90, 5564–5568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosteller, R.D.; Han, J.; Broek, D. Identification of residues of the H-ras protein critical for functional interaction with guanine nucleotide exchange factors. Mol. Cell. Biol. 1994, 14, 1104–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, W.; Mosteller, R.D.; Broek, D. Amino acid residues in the CDC25 guanine nucleotide exchange factor critical for interaction with Ras. Mol. Cell. Biol. 1994, 14, 8117–8122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quilliam, L.A.; Kato, K.; Rabun, K.M.; Hisaka, M.M.; Huff, S.Y.; Campbell-Burk, S.; Der, C.J. Identification of residues critical for Ras(17N) growth-inhibitory phenotype and for Ras interaction with guanine nucleotide exchange factors. Mol. Cell. Biol. 1994, 14, 1113–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quilliam, L.A.; Hisaka, M.M.; Zhong, S.; Lowry, A.; Mosteller, R.D.; Han, J.; Drugan, J.K.; Broek, D.; Campbell, S.L.; Der, C.J. Involvement of the Switch 2 Domain of Ras in Its Interaction with Guanine Nucleotide Exchange Factors. J. Biol. Chem. 1996, 271, 11076–11082. [Google Scholar] [CrossRef] [Green Version]
- Day, G.-J.; Mosteller, R.D.; Broek, D. Distinct Subclasses of Small GTPases Interact with Guanine Nucleotide Exchange Factors in a Similar Manner. Mol. Cell. Biol. 1998, 18, 7444–7454. [Google Scholar] [CrossRef] [Green Version]
- Freedman, T.S.; Sondermann, H.; Friedland, G.D.; Kortemme, T.; Bar-Sagi, D.; Marqusee, S.; Kuriyan, J. A Ras-induced conformational switch in the Ras activator Son of sevenless. Proc. Natl. Acad. Sci. USA 2006, 103, 16692–16697. [Google Scholar] [CrossRef] [Green Version]
- Protein Data Bank. Available online: https://pdb101.rcsb.org (accessed on 21 May 2020).
- Hall, B.E.; Yang, S.S.; Boriack-Sjodin, P.A.; Kuriyan, J.; Bar-Sagi, D. Structure-based Mutagenesis Reveals Distinct Functions for Ras Switch 1 and Switch 2 in Sos-catalyzed Guanine Nucleotide Exchange. J. Biol. Chem. 2001, 276, 27629–27637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Bryan, J.P. Pharmacological targeting of RAS: Recent success with direct inhibitors. Pharmacol. Res. 2019, 139, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Papale, A.; Morella, I.M.; Indrigo, M.T.; Bernardi, R.E.; Marrone, L.; Marchisella, F.; Brancale, A.; Spanagel, R.; Brambilla, R.; Fasano, S. Impairment of cocaine-mediated behaviours in mice by clinically relevant Ras-ERK inhibitors. eLife 2016, 5, e17111. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, R.; Papale, A.; Fasano, S. Transcription factor modulating terpene biosynthesis. U.S. Patent WO 2012/016963 Al 2012, 13 December 2012. [Google Scholar]
- Ramsey, J.D.; Flynn, N.H. Cell-penetrating peptides transport therapeutics into cells. Pharmacol. Ther. 2015, 154, 78–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raucher, D.; Ryu, J.S. Cell-penetrating peptides: Strategies for anticancer treatment. Trends Mol. Med. 2015, 21, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, R.; Papale, A.; Fasano, S. Peptides for the Treatment of Brain Diseases. U.S. Patent WO/2012/016963, 9 February 2012. [Google Scholar]
- Gump, J.M.; Dowdy, S.F. TAT transduction: The molecular mechanism and therapeutic prospects. Trends Mol. Med. 2007, 13, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Santini, E.; Valjent, E.; Usiello, A.; Carta, M.; Borgkvist, A.; Girault, J.-A.; Herve, D.; Greengard, P.; Fisone, G. Critical Involvement of cAMP/DARPP-32 and Extracellular Signal-Regulated Protein Kinase Signaling in L-DOPA-Induced Dyskinesia. J. Neurosci. 2007, 27, 6995–7005. [Google Scholar] [CrossRef]
- Santini, E.; Alcacer, C.; Cacciatore, S.; Heiman, M.; Hervé, D.; Greengard, P.; Girault, J.-A.; Valjent, E.; Fisone, G. l-DOPA activates ERK signaling and phosphorylates histone H3 in the striatonigral medium spiny neurons of hemiparkinsonian mice. J. Neurochem. 2009, 108, 621–633. [Google Scholar] [CrossRef]
- Darmopil, S.; Martín, A.B.; De Diego, I.R.; Ares, S.; Moratalla, R. Genetic Inactivation of Dopamine D1 but Not D2 Receptors Inhibits L-DOPA–Induced Dyskinesia and Histone Activation. Biol. Psychiatry 2009, 66, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Banerjee, S.; Kumar, A.; Prabhakaran, E.N. H-Bond Surrogate-Stabilized Shortest Single-Turn α-Helices: Sp 2 Constraints and Residue Preferences for the Highest α-Helicities. ACS Omega 2020, 5, 13902–13912. [Google Scholar] [CrossRef]
- Patgiri, A.; Jochim, A.L.; Arora, P.S. A Hydrogen Bond Surrogate Approach for Stabilization of Short Peptide Sequences in α-Helical Conformation. Acc. Chem. Res. 2008, 41, 1289–1300. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.M.; Tsou, L.K.; Hamilton, A.D. Synthetic non-peptide mimetics of α-helices. Chem. Soc. Rev. 2007, 36, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.P.; Gellman, S.H.; DeGrado, W.F. β-Peptides: From Structure to Function. Chem. Rev. 2001, 101, 3219–3232. [Google Scholar] [CrossRef] [PubMed]
- Bautista, A.D.; Craig, C.J.; Harker, E.A.; Schepartz, A. Sophistication of foldamer form and function in vitro and in vivo. Curr. Opin. Chem. Biol. 2007, 11, 685–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garner, J.; Harding, M.M. Design and synthesis of α-helical peptides and mimetics. Org. Biomol. Chem. 2007, 5, 3577. [Google Scholar] [CrossRef] [PubMed]
- Goodman, C.M.; Choi, S.; Shandler, S.; DeGrado, W.F. Foldamers as versatile frameworks for the design and evolution of function. Nat. Chem. Biol. 2007, 3, 252–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, J.K.; Gellman, S.H. Targeting protein–protein interactions: Lessons from p53/MDM2. Biopolymers 2007, 88, 657–686. [Google Scholar] [CrossRef]
- Siedlecka, M.; Goch, G.; Ejchart, A.; Sticht, H.; Bierzynski, A. α-Helix nucleation by a calcium-binding peptide loop. Proc. Natl. Acad. Sci. USA 1999, 96, 903–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Zhao, K.; Gong, Y.; Vologodskii, A.; Kallenbach, N.R. α-Helix Nucleation Constant in Copolypeptides of Alanine and Ornithine or Lysine. J. Am. Chem. Soc. 1998, 120, 10646–10652. [Google Scholar] [CrossRef]
- Wang, D.; Chen, K.; Kulp, J.L.; Arora, P.S. Evaluation of Biologically Relevant Short α-Helices Stabilized by a Main-Chain Hydrogen-Bond Surrogate. J. Am. Chem. Soc. 2006, 128, 9248–9256. [Google Scholar] [CrossRef] [Green Version]
- Kemp, D.S.; Curran, T.P.; Boyd, J.G.; Allen, T.J. Studies of N-terminal templates for.alpha.-helix formation. Synthesis and conformational analysis of peptide conjugates of (2S,5S,8S,11S)-1-acetyl-1,4-diaza-3-keto-5-carboxy-10-thiatricyclo[2.8.1.04,8]tridecane (Ac-Hel1-OH). J. Org. Chem. 1991, 56, 6683–6697. [Google Scholar] [CrossRef]
- Maurer, T.; Garrenton, L.S.; Oh, A.; Pitts, K.; Anderson, D.J.; Skelton, N.J.; Fauber, B.P.; Pan, B.; Malek, S.; Stokoe, D.; et al. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc. Natl. Acad. Sci. USA 2012, 109, 5299–5304. [Google Scholar] [CrossRef] [Green Version]
- Cabezas, E.; Satterthwait, A.C. The Hydrogen Bond Mimic Approach: Solid-Phase Synthesis of a Peptide Stabilized as an α-Helix with a Hydrazone Link. J. Am. Chem. Soc. 1999, 121, 3862–3875. [Google Scholar] [CrossRef]
- Pal, S.; Prabhakaran, E.N. Hydrogen bond surrogate stabilized water soluble 310-helix from a disordered pentapeptide containing coded α-amino acids. Tetrahedron Lett. 2018, 59, 2515–2519. [Google Scholar] [CrossRef]
- Kortemme, T.; Kim, D.E.; Baker, D. Computational Alanine Scanning of Protein-Protein Interfaces. Sci. Signal. 2004, 2004, pl2. [Google Scholar] [CrossRef] [Green Version]
- Kortemme, T.; Baker, D. A simple physical model for binding energy hot spots in protein-protein complexes. Proc. Natl. Acad. Sci. USA 2002, 99, 14116–14121. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Protein BLAST Sequence Alignment. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastp&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome (accessed on 25 August 2020).
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the SC ’06: 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; Association for Computing Machinery: Tampa, FL, USA, 2006. [Google Scholar]
- Beard, H.; Cholleti, A.; Pearlman, D.; Sherman, W.; Loving, K.A. Applying Physics-Based Scoring to Calculate Free Energies of Binding for Single Amino Acid Mutations in Protein-Protein Complexes. PLoS ONE 2013, 8, e82849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided. Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical p K a Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Mekni, N.; De Rosa, M.; Cipollina, C.; Gulotta, M.R.; De Simone, G.; Lombino, J.; Padova, A.; Perricone, U. In Silico Insights towards the Identification of NLRP3 Druggable Hot Spots. Int. J. Mol. Sci. 2019, 20, 4974. [Google Scholar] [CrossRef] [Green Version]
- Perricone, U.; Gulotta, M.R.; Lombino, J.; Parrino, B.; Cascioferro, S.; Diana, P.; Cirrincione, G.; Padova, A. An overview of recent molecular dynamics applications as medicinal chemistry tools for the undruggable site challenge. MedChemComm 2018, 9, 920–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulotta, M.R.; Lombino, J.; Perricone, U.; De Simone, G.; Mekni, N.; De Rosa, M.; Diana, P.; Padova, A. Targeting SARS-CoV-2 RBD Interface: A Supervised Computational Data-Driven Approach to Identify Potential Modulators. ChemMedChem 2020, 15, 1921–1931. [Google Scholar] [CrossRef] [PubMed]
- Gulotta, M.R.; De Simone, G.; John, J.; Perricone, U.; Brancale, A. A Computer-Based Methodology to Design Non-Standard Peptides Potentially Able to Prevent HOX-PBX1-Associated Cancer Diseases. Int. J. Mol. Sci. 2021, 22, 5670. [Google Scholar] [CrossRef] [PubMed]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the Role of the Crystal Environment in Determining Protein Side-chain Conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins Struct. Funct. Bioinform. 2004, 55, 351–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ΔΔG (kcal/mol) | ||||||
|---|---|---|---|---|---|---|
| Sos aa | 1XD2 | 1BKD | 1NVW | 1NVV | 1NVU | 1NVX |
| Phe929 | 1.54 | 1.48 | 1.64 | 3.00 | 1.53 | 1.47 |
| Thr935 | 2.97 | 1.59 | 1.11 | n.a. | 3.13 | 3.11 |
| Lys939 | n.a. | n.a. | n.a. | 4.21 | n.a. | n.a. |
| Glu942 | n.a. | n.a. | 1.10 | n.a. | n.a. | n.a. |
| Asn944 | 2.51 | 2.63 | 2.35 | 2.66 | 2.63 | 2.70 |
| Sos aa | Corresponding RasGRF1 aa |
|---|---|
| Phe929 | Tyr1178 |
| Thr935 | Thr1184 |
| Lys939 | Phe1188 |
| Glu942 | Glu1191 |
| Asn944 | Thr1193 |
| MD on Ras-Sos Complex | MD on Ras-RasGRF1 Complex | |||
|---|---|---|---|---|
| Ras aa | Sos aa 924–957 | Interaction type | RasGRF1 aa 1173–1203 | Interaction type |
| Tyr64 | Phe929 | Hydrophobic | Tyr1178 | Pi–Pi stacking |
| Phe930 | Hydrophobic | Leu1179 | Hydrophobic | |
| Leu1183 | Hydrophobic | |||
| Tyr40 | - | - | Phe1188 | Hydrophobic |
| Gln61 | Ile932 | Hydrophobic | Tyr1178 | 1 H-bond |
| Thr935 | 1 H-bond | Thr1184 | 1 H-bond | |
| Ala59 | Thr935 | 1 H-bond | Thr1184 | 1 H-bond |
| Leu1183 | Hydrophobic | |||
| Val1187 | Hydrophobic | |||
| Phe1188 | Hydrophobic | |||
| Ser17 | Glu942 | 1 H-bond | Glu1191 | 1 H-bond |
| Val1187 | Hydrophobic | |||
| Ala18 | Glu942 | 1 H-bond | Glu1191 | 1 H-bond |
| Tyr32 | Asn944 | 2 H-bonds | Gly1192 | 1 H-bond |
| Phe1188 | Hydrophobic | |||
| Pro34 | Thr940 | Hydrophobic | - | - |
| Asp57 | Lys939 | 1 H-bond + 1 salt bridge | - | - |
| Gly60 | Leu934 | Hydrophobic | Leu1183 | Hydrophobic |
| Lys147 | - | - | Glu1191 | 1 H-bond + 1 salt bridge |
| First MD | Second MD | ||||
|---|---|---|---|---|---|
| Ras aa | RB3 aa | Interaction type | Ras aa | RB3 aa | Interaction type |
| Tyr40 | Asp1185 | 1 H-bond | Tyr40 | Asp1185 | 1 H-bond |
| Tyr40 | Phe1188 | Pi–Pi stacking | Tyr40 | Phe1188 | Pi–Pi stacking |
| Tyr32 | Gly1192 | 1 H-bond | Glu31 | Gly1192 | 1 H-bond |
| Gln61 | Tyr1182 | 1 H-bond | Arg149 | Glu1198 | 1 H-bond + 1 salt bridge |
| Gln25 | Gly1192 | 1 H-bond | |||
| 310-HBS RB3 Peptide aa | Mutation | ΔΔGaffinity (kcal/mol) | ΔΔGstability (kcal/mol) |
|---|---|---|---|
| Thr1184 | Arg | −19.17 | −3.67 |
| Met | −8.17 | −3.18 | |
| Asp1185 | Trp | −12.56 | −7.64 |
| Tyr | −9.19 | −3.49 | |
| Phe | −8.26 | −4.26 | |
| Leu | −7.77 | −12.61 | |
| Phe1188 | Arg | −9.55 | −4.12 |
| Phe1188 | His | −8.25 | −8.75 |
| Ile1189 | Met | −3.59 | −4.08 |
| Glu1190 | His | −3.29 | −4.13 |
| Glu1191 | Ile | −6.97 | −4.68 |
| Leu | −5.16 | −3.79 | |
| Val | −4.71 | −4.08 | |
| Thr | −4.32 | −4.27 | |
| Thr1193 | Arg | −3.85 | −5.84 |
| Gln | −3.56 | −5.86 |
| First Peptide | Second Peptide | Third Peptide | |
|---|---|---|---|
| Point mutation | T1184R | T1184M | D1185W |
| ΔGbinding average | −89.51 kcal/mol | −92.77 kcal/mol | −103.50 kcal/mol |
| ΔGbinding Std. Dev. | 12.50 | 15.53 | 8.90 |
| ΔGbinding range | −128.52 to −55.50 kcal/mol | −134.237 to −39.51 kcal/mol | −126.30 to −73.35 kcal/mol |
| Fourth Peptide | Fifth Peptide | Sixth Peptide | |
| Point mutation | D1185Y | D1185F | D1185L |
| ΔGbinding average | −102.50 kcal/mol | −94.84 kcal/mol | −82.07 kcal/mol |
| ΔGbinding Std. Dev. | 22.23 | 8.34 | 9.26 |
| ΔGbinding range | −145.44 to −40.67 kcal/mol | −120.13 to −52.39 kcal/mol | −109.19 to −32.63 kcal/mol |
| Seventh Peptide | Eighth Peptide | Ninth Peptide | |
| Point mutation | F1188R | F1188H | I1189M |
| ΔGbinding average | −87.49 kcal/mol | −69.58 kcal/mol | −83.12 kcal/mol |
| ΔGbinding Std. Dev. | 11.60 | 15.23 | 10.89 |
| ΔGbinding range | −120.32 to −57.76 kcal/mol | −111.45 to −24.94 kcal/mol | −122.63 to −44.93 kcal/mol |
| Tenth Peptide | Eleventh Peptide | Twelfth Peptide | |
| Point mutation | E1190H | E1191I | E1191L |
| ΔGbinding average | −73.36 kcal/mol | −78.65 kcal/mol | −95.11 kcal/mol |
| ΔGbinding Std. Dev. | 12.64 | 11.67 | 12.89 |
| ΔGbinding range | −110.79 to −39.82 kcal/mol | −115.33 to −43.76 kcal/mol | −140.50 to −49.67 kcal/mol |
| Thirteenth Peptide | Fourteenth Peptide | Fifteenth Peptide | |
| Point mutation | E1191V | E1191T | T1193R |
| ΔGbinding average | −94.42 kcal/mol | −84.50 kcal /mol | −90.18 kcal/mol |
| ΔGbinding Std. Dev. | 10.69 | 12.71 | 10.85 |
| ΔGbinding range | −121.24 to −58.75 kcal/mol | −116.52 to −47.48 kcal/mol | −119.87 to −59.72 kcal/mol |
| Sixteenth Peptide | |||
| Point mutation | T1193N | ||
| ΔGbinding average | −97.15 kcal/mol | ||
| ΔGbinding Std. Dev. | 11.88 | ||
| ΔGbinding range | −127.15 to −53.43 kcal/mol |
| Combinatorial Peptides | |
|---|---|
|
|
| First Peptide | Third Peptide | Eleventh Peptide | |
|---|---|---|---|
| Peptide sequence | YLGMYLRWLVRMELGR | YLGMYLRYLVRMELGR | YLGMYLRFLVRMEVGR |
| ΔGbinding average | −83.46 kcal/mol | −96.80 kcal/mol | −91.39 kcal/mol |
| ΔGbinding Std. Dev. | 9.00 | 9.84 | 11.55 |
| ΔGbinding range | −117.54 to −55.07 kcal/mol | −123.01 to −52.38 kcal/mol | −119.67 to −61.34 kcal/mol |
| Twelfth Peptide | Fifteenth Peptide | Sixteenth Peptide | |
| Peptide sequence | YLGMYLRLLVRMEVGR | YLGMYLMFLVRMEVGR | YLGMYLMLLVRMEVGR |
| ΔGbinding average | −92.49 kcal/mol | −79.79 kcal/mol | −92.54 kcal/mol |
| ΔGbinding Std. Dev. | 10.15 | 14.46 | 7.93 |
| ΔGbinding range | −129.87 to −56.49 kcal/mol | −112.94 to −41.42 kcal/mol | −116.28 to −65.18 kcal/mol |
| Eighteenth Peptide | Nineteenth Peptide | Twentieth Peptide | |
| Peptide sequence | YLGMYLRYLVRMETGR | YLGMYLRFLVRMETGR | YLGMYLRLLVRMETGR |
| ΔGbinding average | −100.34 kcal/mol | −102.63 kcal/mol | −88.71 kcal/mol |
| ΔGbinding Std. Dev. | 14.21 | 11.01 | 13.50 |
| ΔGbinding range | −137.53 to −65.42 kcal/mol | −130.87 to −62.69 kcal/mol | −125.02 to −50.94 kcal/mol |
| Twenty-third Peptide | Twenty-fourth Peptide | Twenty-fifth Peptide | |
| Peptide sequence | YLGMYLMFLVRMETGR | YLGMYLMLLVRMETGR | YLGMYLRWLVRMELGN |
| ΔGbinding average | −85.53 kcal/mol | −82.31 kcal/mol | −97.24 kcal /mol |
| ΔGbinding Std. Dev. | 11.35 | 10.37 | 14.17 |
| ΔGbinding range | −117.73 to −42.84 kcal/mol | −116.03 to −40.57 kcal/mol | −133.33 to −60.15 kcal/mol |
| Twenty-ninth Peptide | Forty-second Peptide | Forty-third Peptide | |
| Peptide sequence | YLGMYLRFLVRMELGN | YLGMYLRYLVRMETGN | YLGMYLRFLVRMETGN |
| ΔGbinding average | −86.56 kcal/mol | −89.59 kcal/mol | −123.50 kcal/mol |
| ΔGbinding Std. Dev. | 11.67 | 10.32 | 20.97 |
| ΔGbinding range | −124.70 to −56.47 kcal/mol | −128.75 to −60.00 kcal/mol | −161.49 to −75.91 kcal/mol |
| Forty-fourth Peptide | Forty-fifth Peptide | Forty-eighth Peptide | |
| Peptide sequence | YLGMYLRLLVRMETGN | YLGMYLMWLVRMETGN | YLGMYLMLLVRMETGN |
| ΔGbinding average | −96.31 kcal/mol | −86.04 kcal/mol | −91.80 kcal/mol |
| ΔGbinding Std. Dev. | 17.98 | 13.60 | 9.51 |
| ΔGbinding range | −137.97 to −57.81 kcal/mol | −124.08 to −53.76 kcal mol | −122.78 to −60.84 kcal/mol |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gulotta, M.R.; Brambilla, R.; Perricone, U.; Brancale, A. A Rational Design of α-Helix-Shaped Peptides Employing the Hydrogen-Bond Surrogate Approach: A Modulation Strategy for Ras-RasGRF1 Interaction in Neuropsychiatric Disorders. Pharmaceuticals 2021, 14, 1099. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14111099
Gulotta MR, Brambilla R, Perricone U, Brancale A. A Rational Design of α-Helix-Shaped Peptides Employing the Hydrogen-Bond Surrogate Approach: A Modulation Strategy for Ras-RasGRF1 Interaction in Neuropsychiatric Disorders. Pharmaceuticals. 2021; 14(11):1099. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14111099
Chicago/Turabian StyleGulotta, Maria Rita, Riccardo Brambilla, Ugo Perricone, and Andrea Brancale. 2021. "A Rational Design of α-Helix-Shaped Peptides Employing the Hydrogen-Bond Surrogate Approach: A Modulation Strategy for Ras-RasGRF1 Interaction in Neuropsychiatric Disorders" Pharmaceuticals 14, no. 11: 1099. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14111099