
Sweetening Pharmaceutical Radiochemistry by 18F-Fluoroglycosylation: Recent Progress and Future Prospects


Department of Nuclear Medicine, Molecular Imaging and Radiochemistry, Friedrich-Alexander University Erlangen-Nürnberg (FAU), D-91054 Erlangen, Germany
*
Author to whom correspondence should be addressed.
Pharmaceuticals 2021, 14(11), 1175; https://0-doi-org.brum.beds.ac.uk/10.3390/ph14111175
Submission received: 18 October 2021
/
Revised: 11 November 2021
/
Accepted: 15 November 2021
/
Published: 17 November 2021
(This article belongs to the Special Issue Targets, Tracers and Translation, Part 2 - New Horizons in Radiopharmaceutical Development)
Abstract
:In the field of 18F-chemistry for the development of radiopharmaceuticals for positron emission tomography (PET), various labeling strategies by the use of prosthetic groups have been implemented, including chemoselective 18F-labeling of biomolecules. Among those, chemoselective 18F-fluoroglycosylation methods focus on the sweetening of pharmaceutical radiochemistry by offering a highly valuable tool for the synthesis of 18F-glycoconjugates with suitable in vivo properties for PET imaging studies. A previous review covered the various 18F-fluoroglycosylation methods that were developed and applied as of 2014 (Maschauer and Prante, BioMed. Res. Int. 2014, 214748). This paper is an updated review, providing the recent progress in 18F-fluoroglycosylation reactions and the preclinical application of 18F-glycoconjugates, including small molecules, peptides, and high-molecular-weight proteins.

1. Introduction
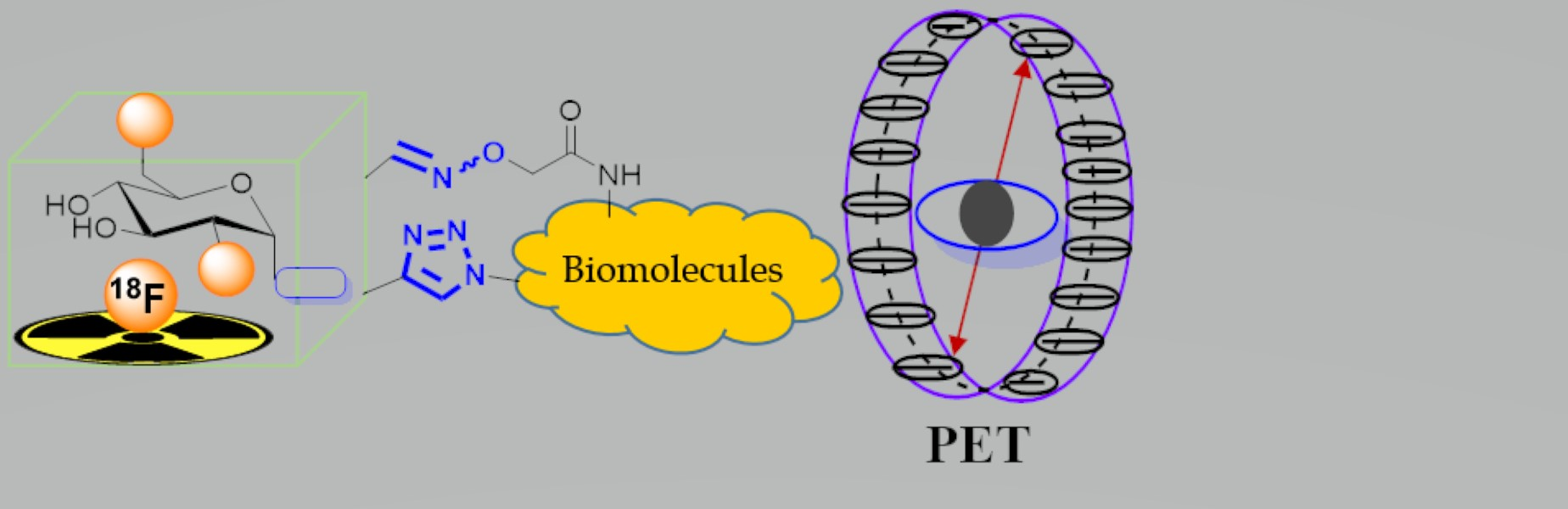
Positron emission tomography (PET) is a highly sensitive medical imaging technique that relies on the use of radioactive tracers for the quantification of biochemical processes in vivo. While various radionuclide positron emitters are suitable for PET, fluorine-18 gained highest interest as the PET radionuclide of choice due to its superior characteristic features of energy (Emax(β+) = 635 keV) and half-life (t1/2 = 109.7 min), which allow for multistep radiochemical syntheses and transportation from production centers to external radiopharmacies [1]. In general, fluorine is one of the highly demanding halogen atoms in medicinal and pharmaceutical chemistry due to its unique physicochemical properties [2]. The exchange of a hydrogen atom by fluorine in a biomolecule can improve the biochemical properties of the molecule significantly, which has, in turn, an effect on the membrane permeability, metabolic stability, (improved) solubility, and receptor-interaction properties [3]. Numerous methods and reaction conditions were developed to facilitate the synthesis of fluorinated molecules by nucleophilic aliphatic and aromatic substitution [4,5], including radiochemical approaches but also non-radioactive chemistry suggesting their application in radiopharmaceutical chemistry [6,7]. Moreover, the introduction of 18F in bioactive molecules by the use of 18F-labeled prosthetic groups is frequently achieved by applying chemoselective strategies for a straightforward design of new PET tracers [8].
Based on Hamacher’s synthesis of β-d-mannopyranose triflate [9] as a precursor for the highly efficient radiosynthesis of 2-[18F]-fluoro-2-deoxy-D-glucose ([18F]FDG, [10]), being the major driving force in the emerging field of PET in nuclear medicine, the idea of using [18F]FDG or derivatives of [18F]FDG for chemoselective 18F-fluoroglycosylation reactions has been followed over the years. The 18F-fluorglycosylation approach aims at a chemoselective and mild labeling method, simultaneously providing the opportunity to influence the biodistribution and tracer uptake characteristics by the introduction of the hydrophilic glycosyl group. It is well known that the glycosylation of biomolecules, such as peptides or proteins, could improve their in vivo stability in blood and accelerate the clearance of distinct glycoconjugates through the kidneys [11,12,13]. Additionally, a series of previous publications have shown that glycosylation prior to radiolabeling was beneficial for improved in vivo properties of several peptide-based PET tracers [12,13,14,15,16]. As one of the most commonly known chemoselective synthetic strategy, the “click chemistry” concept by Sharpless and co-workers [17] was also widely applied in carbohydrate chemistry, facilitating the synthesis of a wide variety of glycoconjugates [18]. Therefore, previous work in our research group was concerned with a click chemistry-based 18F-fluoroglycosylation strategy, starting from a series of mannosyl azide precursors [19] and implementing a convenient approach to the radiosynthesis of 18F-labeled glycopeptides as effective imaging agents for PET [20]. Since then, we and others have frequently applied different 18F-fluoroglycosylation approaches to the radiosynthesis of various 18F-labeled glycoconjugates as PET tracers. A first review article on 18F-fluoroglycosylation reactions was published in 2014 [21]. In the present review, we provide an update on the various 18F-fluoroglycosylation methods and strategies which were developed and adapted to the synthesis of various 18F-glycoconjugate tracers for PET over the past decade.
Table 1 provides an overview of a selection of 18F-labeled glycosyl derivatives that are used as prosthetic groups for the radiosynthesis of 18F-glycoconjugates as potential PET tracers. The following subchapters provide some examples for their application with a focus on recent work published since 2014. In this review, the terms radiochemical yield (RCY) and radioactivity yield (RAY) were used by following the “Consensus nomenclature rules for radiopharmaceutical chemistry” [22]. The RCY is corrected for radioactive decay and usually determined by radio-high performance liquid chromatography (HPLC) analysis of the reaction solution, while the RAY is not decay-corrected and defined as the percentage of radioactivity of the product relative to the starting radioactivity of [18F]fluoride.
2. 2-Deoxy-2-[18F]fluoro-β-glucosyl Azide for Click Chemistry Based 18F-Fluoroglycosylation
The Cu(I)-catalyzed Huisgen 1,3-cycloaddition reaction of an azide and an acyclic alkyne (CuAAC) to yield a 1,2,3-triazole is one of the most prominent reactions belonging to the concept of “click chemistry” [17], defining reactions that are easy to perform, high-yielding, chemoselective, orthogonal and proceed without the formation of by-products. The successful adaption of CuAAC to 18F-chemistry taking advantage of high selectivity, reliability, fast and mild reaction conditions had already been amply documented [29].
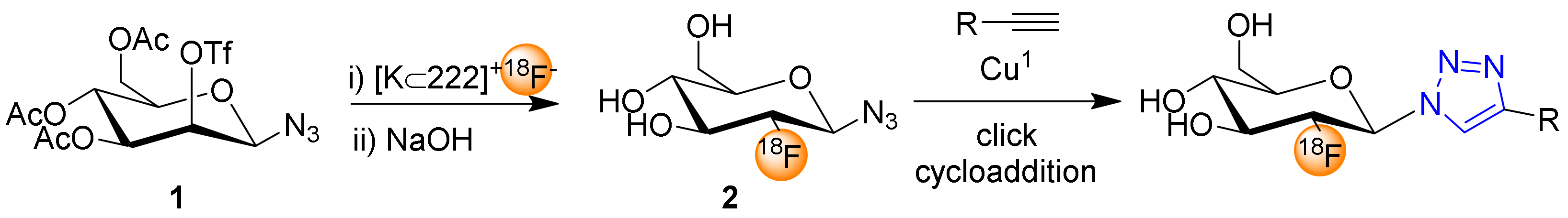
Scheme 1 shows the synthesis of the 18F-fluoroglycosylating agent 3,4,6-tri-O-acetyl-2-deoxy-2-[18F]fluoroglucopyranosyl azide (2), that was achieved by the 18F-labeling of mannosyl precursor 3,4,6-tri-O-acetyl-2-O-trifluoromethanesulfonyl-β-d-mannopyranosyl azide (1, Scheme 1) in a high radiochemical yield (RCY) of 71%, as demonstrated by Maschauer and Prante in 2009 [19]. Interestingly, the RCY of the 18F-substitution depended mainly on the chemical purity of the mannosyl precursor after recrystallization in ethanol, an observation that is similar to the well-known [18F]FDG synthesis. Radiolabeling of β-mannosyl azide 1 was performed under standard conditions (Kryptofix 222, K2CO3) or with K2CO3/KH2PO4 under less basic conditions to reduce the degradation of β-mannosyl azide and thereby simplifying the HPLC purification of 1.
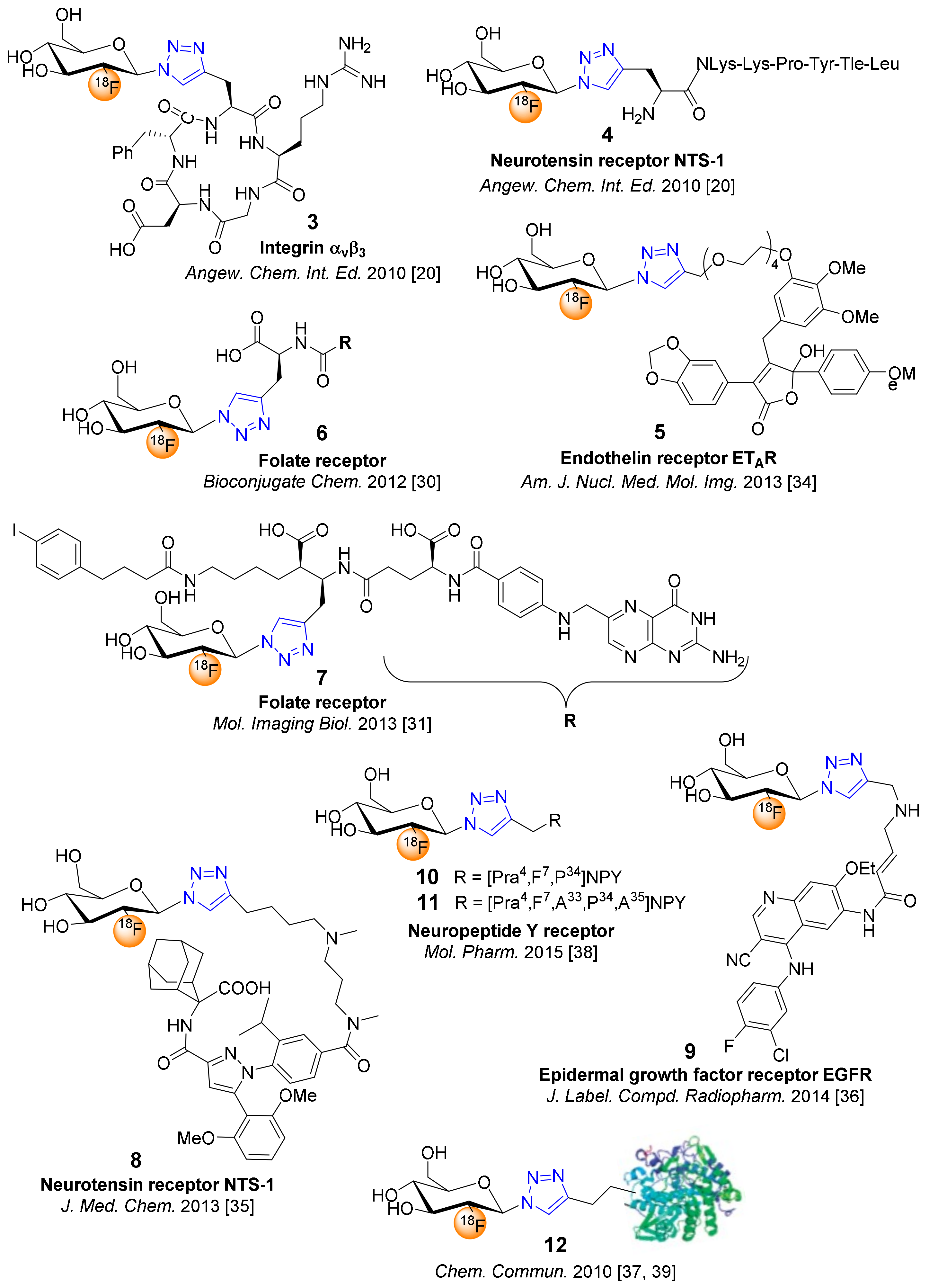
The application of the prosthetic group 2 for CuAAC was initially successfully optimized for alkyne-bearing amino acids [19] and then applied to the radiosynthesis of 18F-glycopeptides, namely an RGD glycopeptide for PET imaging of integrin αvβ3 (3) and a neurotensin peptoid for PET imaging of neurotensin receptor 1 (NTS1)-positive tumors (4) (Figure 1) [20]. The optimized click reaction was performed in PBS/EtOH (10:1) at 60 °C containing 0.2 mM peptide alkyne in the presence of CuSO4 (4 mM) and sodium ascorbate (12 mM). The 18F-glycopeptides were isolated by HPLC in a 17–20% radioactivity yield (RAY) after a total synthesis time of 70–75 min with 55–210 GBq/µmol molar activities and subjected to tumor-bearing nude mice for the successful characterization of in vivo specificity by small animal PET [20].
Moreover, the prosthetic glycosyl azide 2 was also applied to 18F-fluoroglycosylation of various non-peptidic molecules [30,31,32,33,34,35,36]. Interestingly, Fischer et al. reported the radiosynthesis of an 18F-fluoroglycosylated folate, using solid phase extraction of the intermediate 3,4,6-tri-O-acetyl-2-deoxy-2-[18F]fluoroglucopyranosyl azide, thereby omitting the more laborious HPLC purification after the 18F-substitution reaction [30]. The CuAAC with folate alkyne proceeded in aqueous EtOH (38%) in the presence of Cu(OAc)2 (1.2 mM) and sodium ascorbate (2.4 mM) and 18F-glycofolate 6 was achieved after final HPLC purification in RAY of up to 25%, with a molar activity of 90 ± 38 GBq/μmol. Analyses of tissue samples at 30 min post-injection (p.i.) in mice confirmed a high stability of 6 in vivo and small-animal PET studies demonstrated that 6 showed high specific uptake and retention in folate receptor-positive tumors, together with fast blood clearance (tumor-to-blood ratio: 36 ± 15 at 90 min p.i.). The introduction of an albumin binding moiety to the folate precursor, in order to enhance of the blood circulation time of the glycoconjugate tracer, and CuAAC with 2 in the presence of Cu(OAc)2 (1 mM) and sodium ascorbate (3 mM) in water / DMF (60:40) at 50 °C for 15 min gave 7 in a RCY of 15% [31]. The RAY of 7 was only 1–2 % after a total synthesis time of 3 h in molar activities of 20 to 50 GBq/μmol. As expected, 7 revealed a slow blood clearance with tumor uptake values of 11–15 %ID/g at 1–4 h p.i. in PET studies of KB tumor-bearing nude mice and a substantially improved tumor-to-kidney ratio of about 1.
The 18F-fluoroglycosylation by CuAAC applying 2 for small molecules was also used for the radiosynthesis of a subtype-selective glycosylated ligand 5 for the endothelin receptor (ETAR) [34], the non-peptidic neurotensin receptor (NTS1) ligand 8 [35], and the fluoroglycosylated cyanoquinoline 9 as a PET ligand candidate for the epidermal growth factor receptor (EGFR) [36] (Figure 1).
The CuAAC for glycoconjugate 5 (alkyne (0.6 mM), sodium ascorbate (12 mM), CuSO4 (4 mM) in saline/EtOH (3:2)) gave high RAY (20–25%, 70 min) and 5 demonstrated high metabolic stability in vivo, fast blood clearance, low uptake in the kidneys and liver, but a very high uptake in the bile and intestines [34]. Therefore, glycoconjugate 5 is an example of a glycoconjugate that is predominantly excreted via hepatobiliary clearance, such that glycosylation did not significantly change the excretion pathway of analogs of the lead compound PD 156707.
Similarly, the 18F-fluoroglycosylation of a diarylpyrazole, derived from the potent NTS1 antagonist SR142948A, was also successfully performed by CuAAC of 2 with the alkyne-bearing diarylpyrazole precursor (0.3 mM) in saline/THF (3:4) for 10 min at 60 °C [35]. The 18F-glycoconjugate 8 was obtained in a RAY of 20 ± 3% and a molar activity of 35−74 GBq/μmol in a total synthesis time of 70 min. Glycoconjugate 8 displayed excellent NTS1 affinity (Ki = 1 nM) in vitro, high stability in vivo, rapid clearance from blood in vivo, and PET studies in nude mice bearing HT29 tumors demonstrated specific tracer uptake and excellent tumor retention with a tumor-to-blood ratio of 4.4 at 60 min p.i.
The 18F-fluoroglycosylation applying CuAAC with glycosyl azide 2 as a prosthetic group was also adopted to the radiosynthesis of a 4 kDa neuropeptide Y analog (10) and a high-molecular-weight 18F-glycoprotein (12) [37,38]. However, the Davies group employed the click conjugation of 2 with a very low concentration of the alkyne-bearing protein (6 µM) in the presence of Cu(I)Br and TTMA (triethyl 2,2′,2′’-[nitrilotris(methylene-1H-1,2,3-triazole-4,1-diyl)]triacetate) at room temperature, where the RCY of the 18F-glycosylated protein (12) was limited to 4.1% [37]. Thus, 18F-fluoroglycosylation of proteins by CuAAC using 2 is not well suited for proteins that are not readily available. The thionation of [18F]FDG offers [18F]FDG-SH as an alternative prosthetic group for 18F-fluoroglycosylation of proteins [39]; however, 18F-labeled glycoproteins remain particularly rare.
Based on many efforts in the design of neuropeptide Y (NPY) peptide analogs for studying the neuropeptide Y Y1 receptor (Y1R) in breast cancer, Hofmann et al. reported a 18F-fluoroglycosylated peptide for imaging Y1R-positive tumors by small-animal PET [38]. Applying 2 for the click chemistry based strategy of the fluoroglycosylated (FGlc) peptide analogue [Pra4(FGlc),F7,P34]NPY, the alkyne-bearing propargylglycine (Pra) peptide [Pra4,F7,P34]NPY was synthesized and subjected to 18F-fluoroglycosylation, affording a RAY of 20−25% and molar activity of 40−70 GBq/μmol in a total synthesis process of 75 min. The glycosylated peptide [Pra4(FGlc),F7,P34]NPY (10) demonstrated subtype selectivity for Y1R over Y2R and a high potency for the induction of Y1R-mediated inositol accumulation in vitro (EC50 = 3.1 nM). In vitro autoradiography with Y1R-positive MCF-7 tumor tissue slices indicated a high specific binding of the 18F-labeled glycopeptide, when binding was reduced by 95% ([Pra4,F7,P34]NPY) and by 86% (BIBP3226, Y1R antagonist) in competitive binding studies. Small-animal PET studies with [Pra4([18F]FGlc),F7,P34]NPY (10) on MCF-7 breast tumor-bearing nude mice in direct comparison with a scrambled low-affinity peptide (11, Figure 1) revealed specific uptake of 10 in the MCF-7 tumor with increasing tumor-to-blood ratio from 1.2 to 2.4, a tumor retention of 76 % (45−90 min p.i.) and decreased kidney uptake compared to DOTA analogues of this peptide. The 18F-glycopeptide [Pra4([18F]FGlc),F7,P34]NPY (10) can be considered as a lead peptide for the design of improved glycopeptide tracers with shorter amino acid sequences for imaging of Y1R-positve breast tumors by PET [38].
3. 6-Deoxy-6-[18F]fluoro-β-glycosyl Azides for Click Chemistry-Based 18F-Fluoroglycosylation
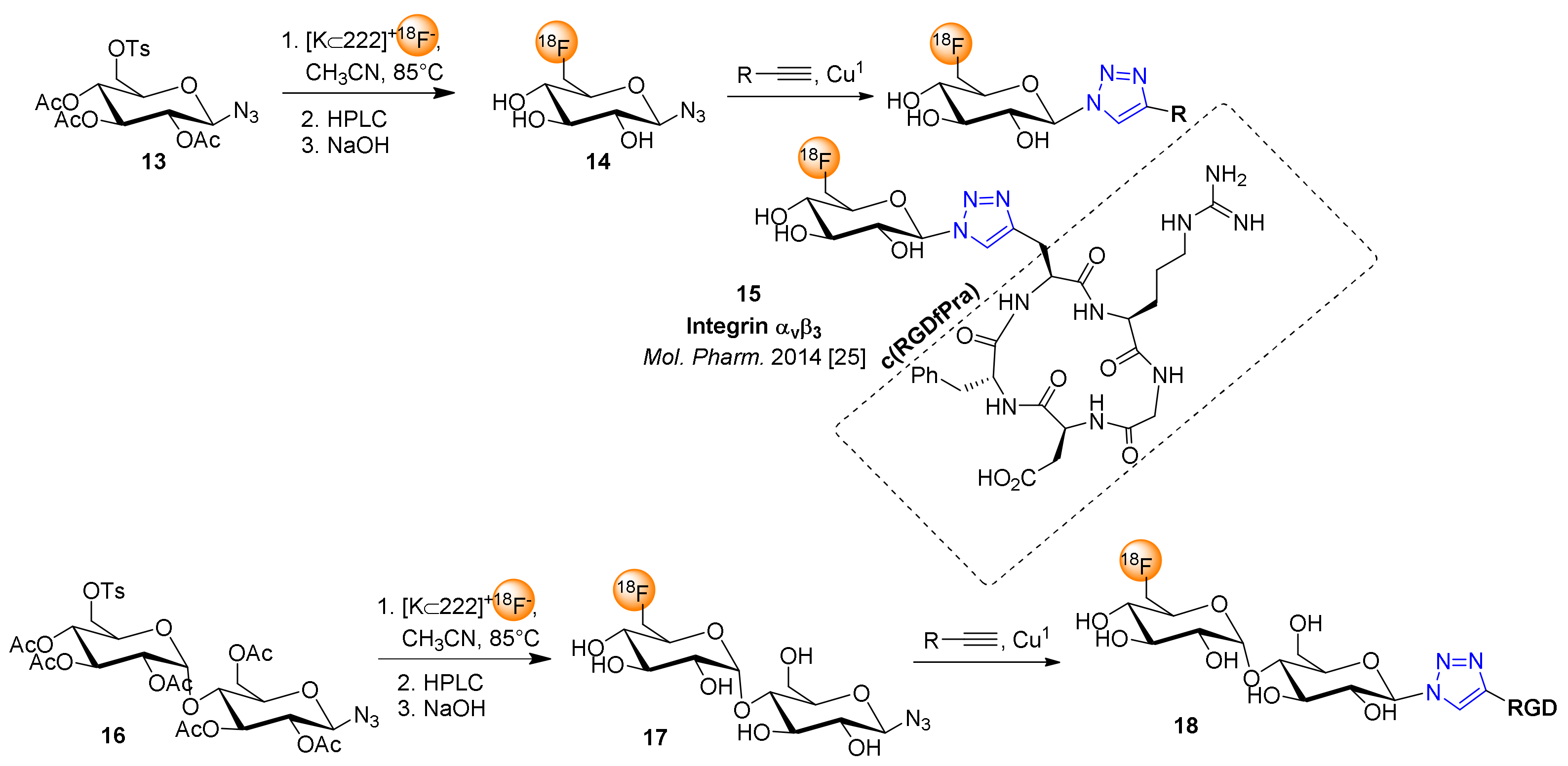
Scheme 2 depicts the application of the prosthetic groups 6-deoxy-6-[18F]fluoroglucopyranosyl azide (14) and 6′-deoxy-6’-[18F]fluoromaltosyl azide (17) for click 18F-fluoroglycosylation of an RGD peptide [26]. Both 18F-glycosides were synthesized from their peracetylated 6-tosylate precursors 13 and 16 in high RCY of 84% and 61%, respectively. The resultant intermediates were purified by HPLC and subsequently hydrolyzed with NaOH (60 mM) to give the glycosyl azides 14 and 17, that were conjugated to the cyclic peptide c(RGDfPra) by CuAAC under similar reaction conditions as described above for 1. Following this strategy, 6-[18F]FGlc-RGD (15) and 6′-[18F]Mlt-RGD (18) were achieved in RAY of 16−24% and molar activities of 50–200 GBq/µmol within 70–75 min. A comparative PET study demonstrated that both 18F-glycopeptides 15 and 18 showed significantly decreased liver and kidney uptake relative to 2-[18F]FGlc-RGD (3) in vivo using U87MG tumor-bearing nude mice [26]. Importantly, the maltosyl peptide 18 revealed substantial tumor uptake and high tumor retention comparable to that of 18F-galacto-RGD [14,40] and high tumor-to-kidney ratios comparable with dimeric RGD peptides [41,42], such that the high tumor uptake and excellent clearance properties in vivo make 18 an alternative glycopeptide tracer for imaging integrin expression by PET.
Inspired by the promising results of 6-[18F]FGlc-RGD (15), especially in terms of clearance through the kidneys, we extended the application of 6-deoxy-6-[18F]fluoroglucopyranosyl azide (14) for 18F-fluoroglycosylation of a series of bioactive compounds shown in Figure 2.
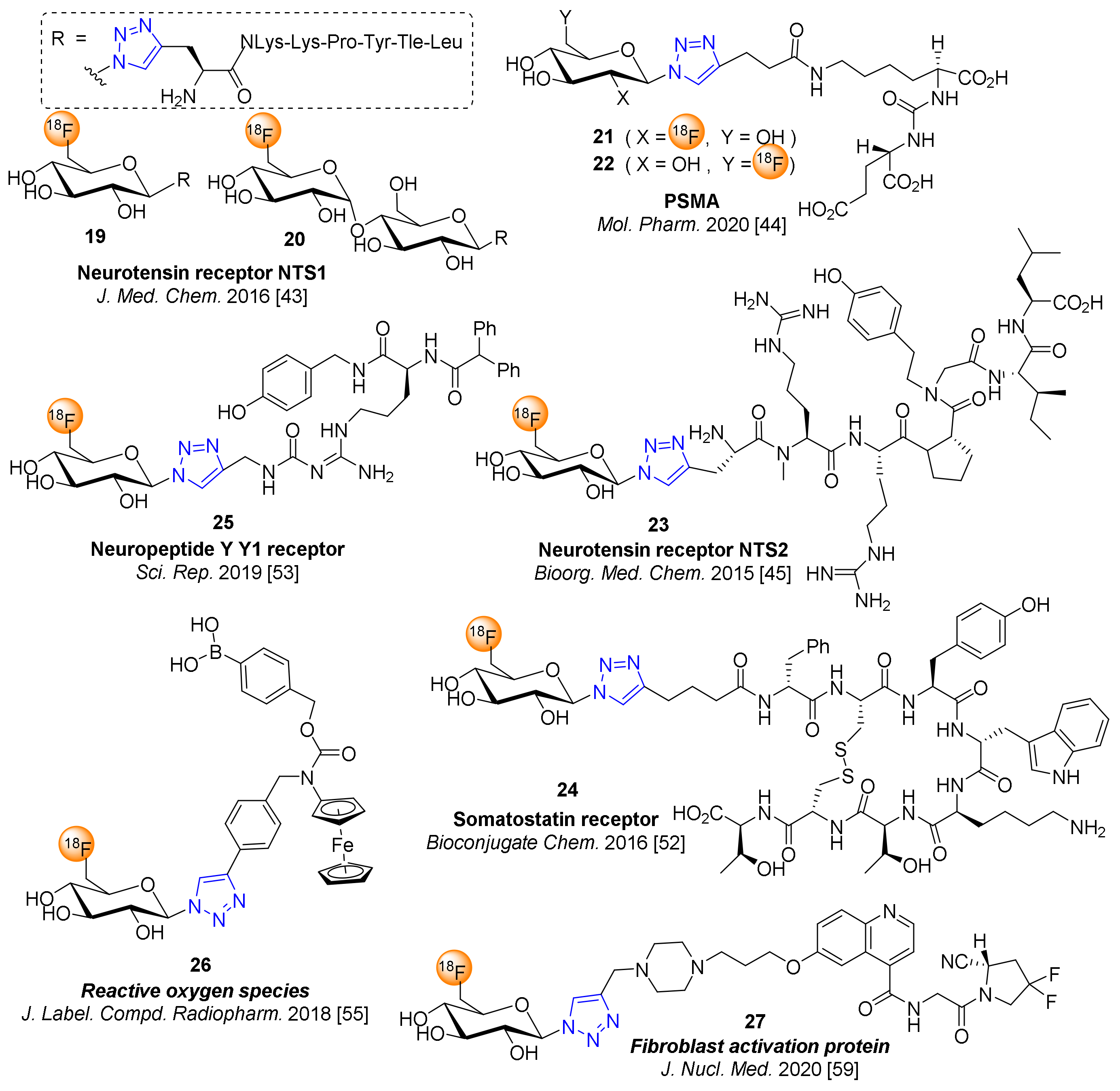
Thus, with the aim of improving the renal clearance of 4 (see Figure 1), the influence of fluoroglycosylation of the NTS1-affine linear peptoid PraNLysLysProTyrTleLeu was investigated [43]. The NTS1 affinity of the target compounds 19 and 20 (Figure 2) were 26 nM and 33 nM, respectively, which compares very well with the best 68Ga-labeled analogues. The 18F-fluoroglycosylation of the N-terminally propargylglycine (Pra)-derivatized peptoids according to Scheme 2 occurred in good RAY of 16–21% after a total synthesis time of 80–85 min. The biodistribution studies of 19 and 20 in HT29 tumor-bearing mice showed significantly better renal clearance compared to 4 and 68Ga-labeled peptoids, but a 40% reduced uptake in the tumor [43].
The click chemistry strategy for 18F-fluoroglcosylation using 2-deoxy-2-[18F]fluoroglucopyranosyl azide (2) or 6-deoxy-6-[18F]fluoroglucopyranosyl azide (14) was applied to prostate-specific membrane antigen (PSMA) inhibitors of the glutamate-urea-lysine type to afforded 2-[18F]FGlc-PSMA (21) and 6-[18F]FGlc-PSMA (22) [44]. The 18F-fluoroglycosylated PSMA inhibitors 21 and 22 were afforded in RAY of 19−22% and with molar activities of 71−136 GBq/μmol. The PSMA inhibitory potencies were moderate for 21 (IC50 = 234 nM) and 22 (IC50 = 59 nM). Small animal PET studies using PSMA-positive PC-3 PIP and PSMA-negative PC-3 tumor-bearing nude mice revealed specific uptake of 21 (13%ID/g) and 22 (6%ID/g) in PC-3 PIP tumors at 60 min p.i. Highly remarkably, 21 had high uptake in the kidneys with very high retention (74 to 72%ID/g at 30 to 60 min p.i.), while 22 showed very low uptake in the kidneys of 7.5%ID/g at 30 min p.i. with rapid clearance (0.9%ID/g at 120 min p.i.). Thus, the 6-fluoroglucosyl analog 22, with an adequate uptake in PSMA-positive tumors, a considerably low kidney uptake and fast clearance from the kidneys, it could be a promising radiotracer for translation into the clinic [44].
The 18F-fluoroglycosylation via the clickable prosthetic group 14 was also applied to the first radiosynthesis of a neurotensin receptor 2 (NTS2)-subtype selective peptide ligand reported by Maschauer et al. [45] NTS2-selective PET ligands had not been previously described, such that the availability of a subtype selective NTS2 radioligand for PET could be a valuable tool for studying the role of this subtype in various tumor types including prostate, pancreas and breast carcinoma [46,47,48,49,50]. Maschauer et al. reported the radiosynthesis of an 18F-glycopeptoid accomplished by a modified CuAAC between the prosthetic group 14 and the alkyne-terminated NT(8–13) analog Pra-N-Me-Arg-Arg-Pro-N-homo-Tyr-Ile-Leu-OH. Very interestingly, the glycopeptide Pra(6FGlc)-N-Me-Arg-Arg-Pro-N-homo-Tyr-Ile-Leu-OH (23) revealed equal NTS2 affinity of Ki = 7 nM relative to the non-glycosylated sequence (N-Me-Arg-Arg-Pro-N-homo-Tyr-Ile-Leu-OH) [45,51]. Remarkably, the use of tris-(3-hydroxypropyltriazolylmethyl)amine (THPTA) in the CuAAC reaction with 14 significantly accelerated the formation of 23 and reduced the necessary amount of alkyne peptide precursor to 20 nmol. In vitro studies on rat brain slices revealed the subtype selectivity of 18F-glycopeptoid 23 for NTS2. As 23 displayed high stability in vitro but fast degradation in vivo, PET imaging experiments using HT29 and PC3 tumor-bearing nude mice revealed only moderate specific uptake of 23 in NTS2-positive tumors [45]. Further studies are needed for the development of more metabolically stable NTS2-selective peptides for PET.
Bioactive peptides are clearly a very important and prominent class of compounds that are highly suitable for the method of 18F-fluoroglcosylation. The synthetic octapeptide analogs derived from the native somatostatin peptides SST-14 and SST-28, namely octreotate (TATE) or octreotide (TOC), are high affinity ligands for the somatostatin receptors (sstr), preferably subtypes 2 and 5, which are overexpressed on neuroendocrine tumors (NET). The 18F-glyco-octreotate analog [18F]FGlc-TATE (24) was achieved by the “click”-18F-fluoroglycosylation using 14 in a RAY 19–22% and molar activities of 32−106 GBq/μmol [52]. The 18F-glycopeptide 24 showed high affinity to somatostatin receptors expressed on AR42J cells with fast and high internalization, and a beneficial logD7.4 of −1.8. In AR42J tumor bearing nude mice, small animal PET studies revealed high uptake of 24 in the tumor and fast clearance of 24 from other organs resulting in an excellent tumor-to-blood ratios of 17 at 60 min p.i. Therefore, 18F-glyco-octreotide 24 could be considered as a reliable alternative 18F-labeled radiopeptide for imaging somatostatin receptor-positive tumors by PET due to excellent in vitro and in vivo properties.
Similarly, 18F-glycoazide 14 was linked to an alkyne derivative of BIBP3226 to afford the fluoroglycosylated derivative 25 as a Y1R radioligand candidate for PET of breast cancer [53]. This study showed that the glycosyl derivative 25 displayed a highly decreased Y1R affinity of 208 nM when compared to the corresponding fluoroethoxyethyl derivative (2.8 nM). Consequently, despite its favorable hydrophilicity, 25 demonstrated low binding to human breast cancer MCF-7-Y1 cells and slices of tumor xenografts in vitro and was not suitable for the in vivo detection of Y1R-positive tumors by PET studies. The comparative study demonstrated that the corresponding 18F-fluoroethoxyethyl and 18F-PEGylated derivatives, despite their higher lipophilicity, were more promising than 25 and showed displaceable and specific binding to Y1R in vitro and in vivo [53].
Tracers for imaging the content of reactive oxygen species (ROS) in tumors could be valuable for PET imaging of tumors and contribute to our knowledge of the biodistribution of anticancer drug candidates that are ROS-dependently trapped in tumor cells [54]. A click chemistry-based 18F-fluoroglycoconjugation of N-alkylaminoferrocene as a potential anticancer agent was optimized by Toms et al., employing 14 and Cu(OAc)2, phosphate buffer/THF, and sodium ascorbate for the CuAAC reaction conditions [55]. Noteworthily, the purification of the 18F-labeled aminoferrocene glycoconjugate was problematic, since the hydrolysis of the boronic acid ester and oxidation of non-carrier-added 26 occurred in the buffered solution. However, the RCY (referred to the CuAAC reaction) of carrier-added 26 was 85% under optimized conditions [55]. Further PET studies in PC3 and AR42J tumor-bearing mice demonstrated that carrier-added 26 showed a 2−3-fold higher tumor uptake at 45–60 min p.i. when compared to background values [56].
Recently, PET imaging of fibrotic diseases, including various types of cancers, by addressing fibrogen activation protein (FAP) by the use of 68Ga-labeled FAP inhibitors (FAPI) has gained enormous interest [57,58]. To provide an 18F-labeled FAPI for translation into the clinic, the 18F-fluoroglycosylation approach by using 14 for click labeling of a FAPI alkyne was reported by Toms et al. [59] The glycoconjugate [18F]FGlc-FAPI (27, Figure 2) was successfully achieved by the two-step 18F-fluoroglycosylation according to Scheme 2, applying optimized reaction conditions for the click labeling step by quenching the deacetylation with phosphate buffer followed by addition of the reactants for 18F-fluoroglycosylation at 60 °C for 15 min (Cu(OAc)2, THPTA, sodium ascorbate and 400 nmol of FAPI alkyne precursor). For the purpose of preclinical evaluation of 27, the radiosynthesis was started with 0.5–1 GBq, providing the formulated tracer with a radioactivity yield of 15%, a radiochemical purity of more than 99%, and a molar activity of 30–200 GBq/mmol. The in vitro and in vivo studies of 27 in tumor-bearing mice demonstrated, in direct comparison with [68Ga]Ga-FAPI-04, a significantly higher blood protein binding of 27 in vitro, comparable tumor uptake with a high tumor retention and a 2-fold higher blood concentration of 27 in vivo over the 60 min period of the PET scan. Interestingly and in accordance to the higher concentration in blood, 27 showed 2-fold higher specific uptake into murine bone structures and joints compared to [68Ga]Ga-FAPI-04. This interesting property could make [18F]FGlc-FAPI a candidate 18F-labeled FAPI tracer for the imaging of bone tissue remodeling in diseases such as rheumatoid arthritis in humans by PET [59]. Currently, a GMP-compliant automated radiosynthesis of 18F-fluoroglycosylated FAPI 27 has been successfully implemented to facilitate first-in-humans PET studies.
4. 18F-Fluoroglyosylation for the Synthesis of Triazolylalkyl-Linked 18F-Glycoconjugates by CuAAC
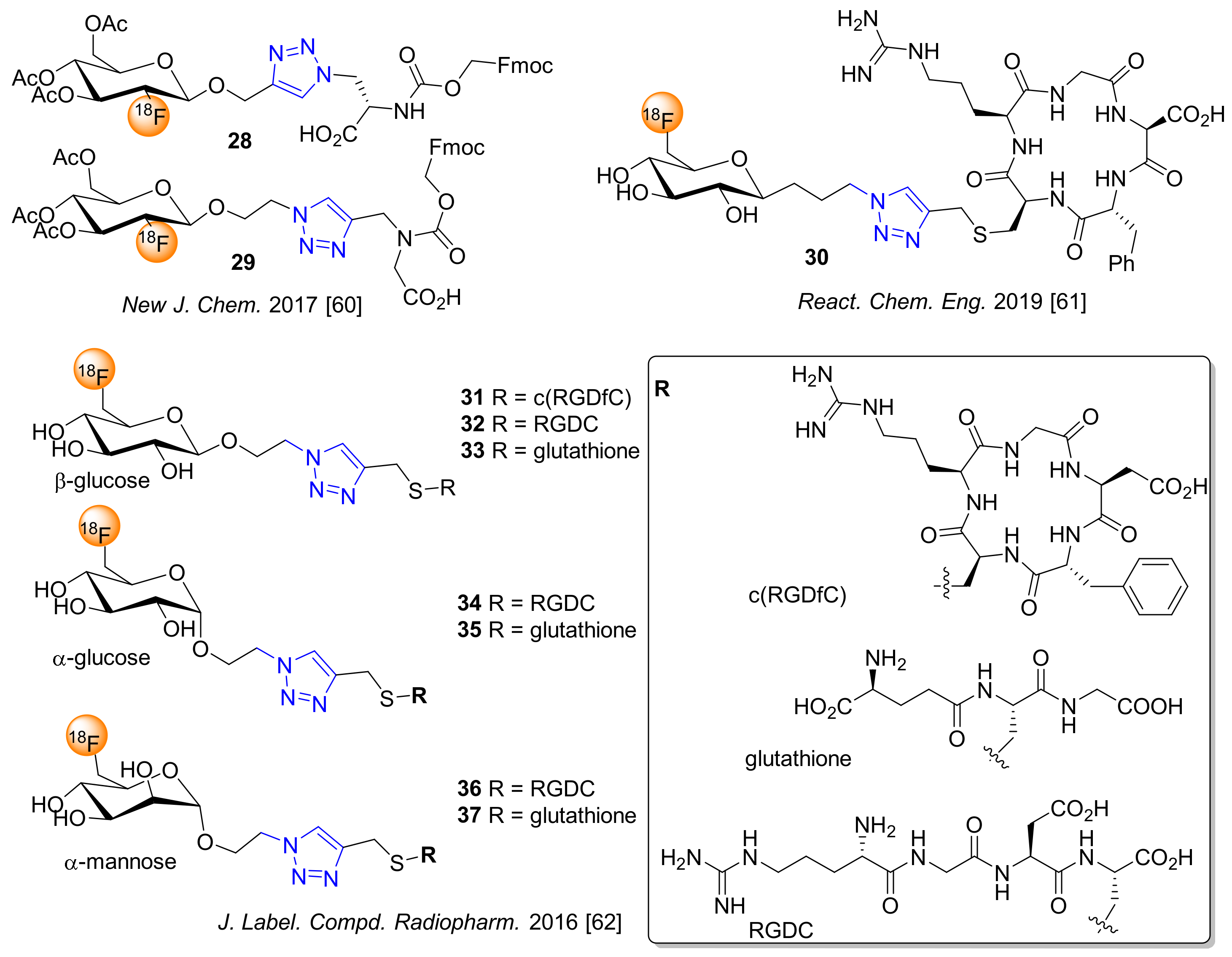
The introduction of an alkyl spacer between 18F-labeled glycosides and various alkyne-bearing bioactive compounds was achieved by CuAAC, resulting in oxyethyl, oxymethyl, or alkyl linked 18F-glycoconjugates (Figure 3). Egland et al. applied the 18F-fluoroglycosylation strategy using O-alkylated β-mannopyranosides functionalized with a terminal azide or alkyne group to conjugate with L-alanine or glycine analogs to give 28 and 29 [60]. The nucleophilic 18F-substitution of the β-mannopyranoside precursors was performed with 77–88 % RCY and the 18F-labeled glycosides as prosthetic groups were subjected to CuAAC reactions with functional Fmoc-3-azido-l-alanine and Fmoc-N-(propargyl)-glycine, which provided the corresponding 18F-fluoroglycosylated amino acid conjugates 28 and 29 in high radiochemical yields. The newly synthesized 18F-fluoroglycosylated amino acids were used as metabolic radiotracers in PET imaging studies [60].
Collet et al. described the fully-automated radiosynthesis of 6-[18F]fluoro-C-glyco-c(RDGfC) (30) in sequential three steps in a one-pot synthesis, affording the 18F-glyco-RGD peptide in high radiochemical purity and a decay-corrected RAY of 3.6% within less than 2.5 h using a fully automated synthesis module. The glycoconjugate 30 showed high stability and hydrophilicity, representing an alternative RGD radiopeptide for imaging integrin expression by PET [61]. Collet et al. further developed a series of 6-[18F]fluoro-carbohydrate-based prosthetic groups and their conjugation to glutathione or RGD peptides via click chemistry. In this study, the authors applied 18F-fluoroglycosylation by CuAAC reaction with various glycosides, such as β-glucosyl, α-glucosyl and α-mannosyl derivatives bearing the anomeric O-ethyl spacer with terminal azide moiety and a thiol-propargyl moiety attached to RGD peptides or glutathione, affording the 18F-glycoconjugates 31–37 (Figure 3) in RCY of up to 76% [62]. A high uptake of 6-[18F]fluoro-O-glyco-c(RDGfC) (31) was shown by PET imaging in rats, revealing the potential of this tracer to monitor integrin expression as part of inflammatory processes and/or angiogenesis.
5. Examples of Non-Radioactive Fluoroglycosylation by Click Chemistry and Effects on Inhibitory Potency or Receptor Affinity
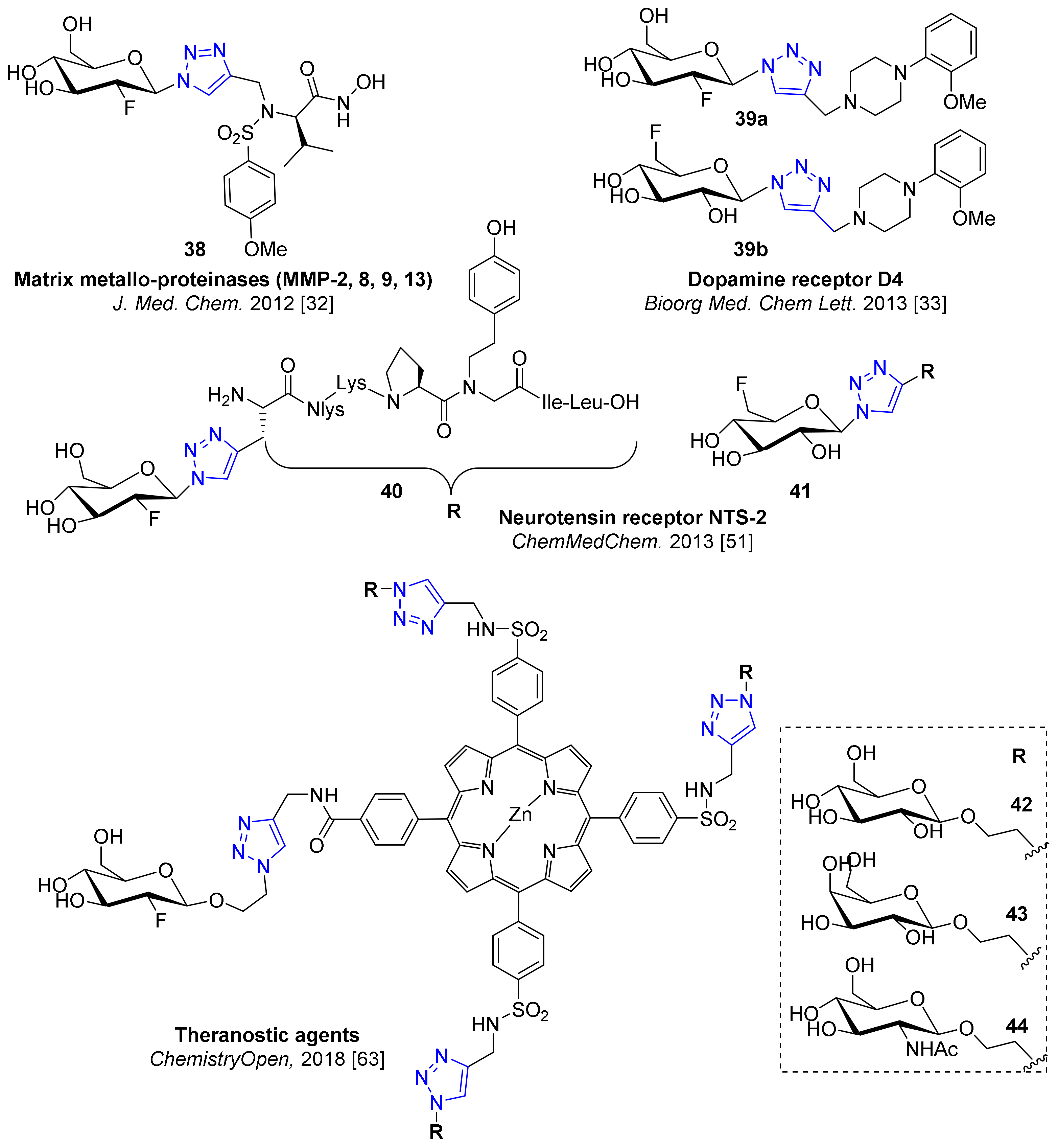
Some interesting studies on the effect of fluoroglycosylation on inhibitory potency or receptor affinity are of precedence in the literature (Figure 4). Without any doubt, it is of special importance to study such effects to further improve our knowledge of the effectiveness of 18F-fluoroglycosylated tracers for PET. For example, a series of triazolyl-linked inhibitors for the matrix metalloproteinases (MMPs) MMP-2, MMP-8 MMP-9 and MMP-13 as attractive targets for PET were developed by Hugenberg et al. [32]. The fluoroglycosylated compound 38, which was synthesized by a click chemistry-based method, displayed a logD7.4 of 0.58 and subnanomolar inhibition constant of 0.2–0.6 nM. However, the more lipophilic fluoroethyl-1,2,3-triazole analog (clogD7.4 = 1.53) revealed outstanding inhibition potencies of 0.006–0.13 nM, therefore rendering the 18F-glycoconjugate 38 to be a less suitable PET tracer candidate.
Furthermore, Banerjee et al. reported an example of fluoroglycosylation in their search for subtype selective dopamine D4 receptor radioligands [33], introducing the deoxyfluoroglucosyl compounds 39a and 39b (Figure 4). However, the affinities for the D4 receptor with 500 nM and 340 nM, respectively, were 100 to 66 times lower when compared to the fluoropropoxyphenyl compound (5.1 nM), rendering 18F-fluoroglycosylation not suitable for this type of ligands.
In addition, the aforementioned study of Held et al. in search of NTS2 selective PET ligands clearly revealed the difference between the introduction of the 2-deoxy-2-fluoroglycosyl and the 6-deoxy-6-fluoroglycosyl moiety to the NTS2-selective Pra-Nlys-Lys-Pro-N-homo-Tyr-Ile-Leu-OH peptide analog, when both 40 and 41 showed a dramatic loss of NTS2 affinity compared to the non-glycosylated compound (110–290 nM vs. 4 nM), while interestingly, 41 demonstrated superior subtype selectivity for NTS2 (350-fold) compared to 40 (11-fold) [51].
Arja et al. reported a fluoroglucosylated porphyrin derivative for the application in photodynamic therapy [63]. The synthesized 2-deoxy-2-fluoro-β-glucosylated porphyrins 42–44 showed fluorescent properties for optical imaging, generated singlet oxygen in vitro and were showed preferred uptake in melanoma cells. These glycoconjugated porphyrins could be promising radiotraces for combined photodynamic therapy and PET imaging studies, when radiolabeled by chemoselective 18F-fluoroglycosylation using [18F]FDG as prosthetic group.
6. [18F]FDG for Chemoselective 18F-Fluoroglycosylation by Oxime Linkage
The application of aldehyde click chemistry for oxime bond formation in radiopharmaceutical chemistry could be considered as a straightforward alternative method for CuAAC, owing to the favorable properties of oxime bond formation, such as chemoselectivity, high efficiency, and high biocompatibility due to the formation in aqueous solvents [64,65]. Scheme 3 shows the chemoselective oxime formation by click reaction between an aminooxy precursor and the aldehyde functionality of [18F]FDG in aqueous solution. [18F]FDG could in principle be readily applied to 18F-fluoroglycosylation through oxime formation, since in aqueous solution [18F]FDG undergoes mutarotation, that is isomerization between the α- and β-anomer via the intermediate acyclic aldehyde, which is favored at high temperatures (80–120 °C) and acidic pH (1.5–2.5).
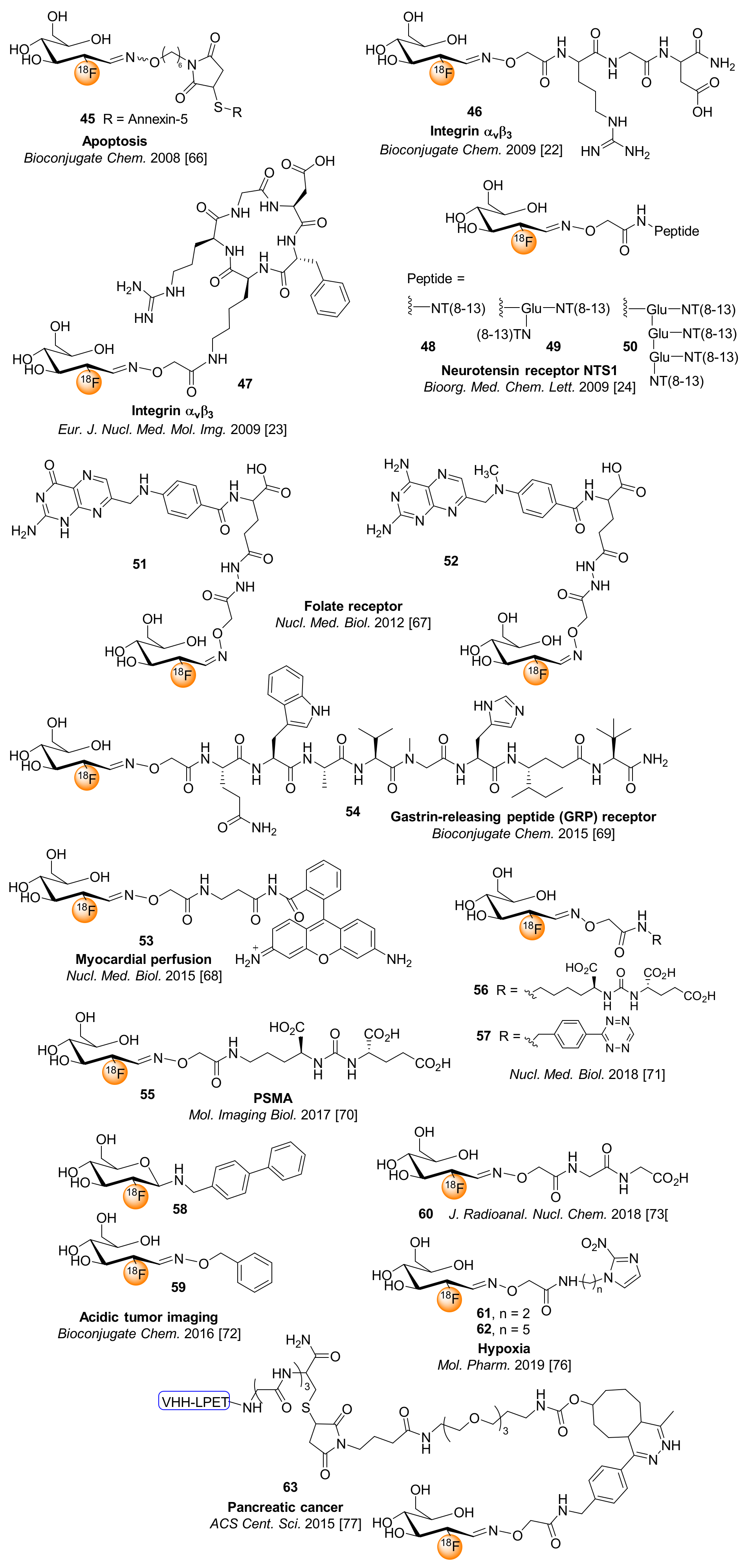
Alongside the indirect use of [18F]FDG for the 18F-fluoroglycosylation of thiol-containing peptides through the preparation of a [18F]FDG-maleimidehexyloxime prosthetic group ([18F]FDG-MHO) [66], the direct 18F-fluoroglycosylation of aminooxy-functionalized peptides was first published in 2009 using [18F]FDG as prosthetic group [23,24]. These approaches, including the [18F]FDG oxime-conjugation with the 36 kDa thiol-group-containing protein annexin-V (45) [66] and various aminooxy-functionalized peptides (46–50 [23,24,25], Figure 5) are discussed in detail in a previous review [21]. Notably, the clinically available [18F]FDG solution could not easily been applied for direct 18F-fluoroglycosylation of peptide precursors, since the concentration of approximately 0.2 g/mL glucose clearly hampers the 18F-fluoroglycosylation reaction, making HPLC purification of [18F]FDG prior to use indispensable. Another disadvantage of the 18F-fluoroglycosylation by the use of [18F]FDG is that high amounts of aminooxy-functionalized peptides (7.5–50 mM) are needed and these precursors often lack stability on storage. However, the direct 18F-fluoroglycosylation by oxime formation with [18F]FDG is a straightforward approach allowing RCY of up to 80–93% for peptide labeling, providing interesting 18F-glycopeptides for PET imaging studies (Figure 5).
Alongside peptides, [18F]FDG as a prosthetic group has been applied to the 18F-fluoroglycosylation of folate and methotrexate to give conjugates 51 and 52 [67]. The aminooxy-functionalized precursors (9 mM) were conjugated with [18F]FDG in DMSO/1% acetic acid/EtOH (1:1, pH∼4.5) at 60 °C for 10–15 min, achieving glycoconjugates 51 and 52 in overall RCY of at least 80%, within a total synthesis time of 20 min and in molar activities of >9 GBq/µmol. The 18F-glycoconjugates 51 and 52 displayed favorable binding affinities to folate receptor-positive KB cells when compared to aromatic conjugates and in vivo studies in KB tumor-bearing nude mice showed low uptake in intestine, liver and kidney, rapid clearance from the blood, and high specific uptake of 51 in the tumor, resulting in tumor-to-blood ratio of 11 [67].
More recently, [18F]FDG was conjugated to rhodamine by oxime coupling [68]. The radiosynthesis of [18F]FDG-rhodamine conjugate 53 was achieved in a simple and convenient way by a one-step process, affording high RCY and 98% radiochemical purity of the formulated tracer after 20 min total synthesis time. Biodistribution studies of 53 in rats revealed an uptake of 11%ID/g in the heart at 60 min p.i., rendering 53 suitable as an imaging agent for the PET evaluation of myocardial perfusion after translation into the clinic.
Richter et al. developed the [18F]FDG-conjugated bombesin analog QWAV-Sar-H-FA01010-Tle-NH2 ([18F]FDG-AOAc-BBN2, 54) for PET imaging of gastrin-releasing peptide (GRP) receptor-expressing prostate tumors by PET [69]. The bombesin-[18F]FDG conjugate 54 provided a favorable pharmacokinetic profile compared to BBN2 conjugated to other 18F-labeled prosthetic groups. The 18F-glycopeptide 54 revealed high tumor accumulation, fast renal excretion due to low lipophilicity, and high metabolic stability in mouse xenografts using small-animal PET, such that 54 was considered as favorable candidate for imaging GRP-positive prostate cancer by PET.
The Wuest group has described the synthesis and evaluation of PSMA inhibitors conjugated to various 18F-labeled prosthetic groups [70]. The 18F-fluoroglycosylation of a suitable PSMA derivative with lysins-urea-glutamate scaffold was achieved with [18F]FDG via oxime bond formation. The resulting 18F-glycoconjugate 55 was isolated by HPLC purification in a decay-corrected RCY of 69% and molar activity of 40 GBq/µmol. Glycoconjugate 55 showed an IC50 value of 62 nM for PSMA inhibitory potency, which was a factor of 10 worse than the corresponding fluorophenyl analog. In vivo tumor uptake of the glycoconjugate 55 was similarly inferior by a factor of 10 compared with the fluorophenyl analog, as demonstrated by dynamic PET studies in LNCaP tumor-bearing mice [70].
To avoid the HPLC purifications after 18F-fluoroglycosylation via oxime formation, Keinänen et al. developed solid-phase extraction and resin purification protocols for the synthesis of glycoconjugates 56 and 57 (Figure 5) [71]. The purification of the final 18F-glycoconjugates was achieved by removal of unreacted carbohydrate via derivatization with 4,4′-dimethoxytrityl chloride (DMT-Cl) and the removal of excess aminooxy precursors after 18F-fluoroglycoconjugation was achieved by the use of an aldehyde resin (AminoLink).
Flavell et al. developed an [18F]FDG amine prodrug targeting tissue regions with low interstitial pH [72]. The [18F]FDG-derived glycosylamine 58 was synthesized in one step from [18F]FDG by treatment with 4-phenylbenzylamine in acidic acid at 80 °C. The resulting 18F-glycosylamine 58 was isolated in 20% RAY and showed greater uptake in tumor tissue relative to benign tissue, revealing favorable and pH-dependent properties for tumor uptake when compared to the oxime-linked analog 59 (Figure 5). Therefore, the 18F-glycosylamine 58 could be a promising acid-responsive PET tracer for tumor imaging.
The very simple procedure for using clinically readily available FDG for ligation with peptides was demonstrated using the small peptide glycylglycine as an example [73]. Starting from the commercially available [18F]FDG solution and after cleavage of the BOC protecting group from the aminooxy-derivatized peptide, [18F]FDG-GlyGly (60) could be obtained in a RCY of 98% at 100 °C after 30 min.
The PET imaging of tumor hypoxia using 2-nitroimidazole tracers is well established in nuclear medicine practice [74]. By using an approach similar to that described by Patt et al. [75], Yang et al. synthesized the [18F]FDG-conjugated 2-nitroimidazole 61 and 62 via oxime bond formation, introducing ethyl and pentylaminecarbamate alkyl chains as spacers with different lengths between the glycosyl moiety and the nitroimidazole [76]. The biodistribution studies revealed that compound 61 (ethyl spacer) showed better in vivo properties than compound 62 (pentyl spacer), probably due to its lower lipophilicity.
Rashidian et al. developed the 18F-labeled single domain antibody fragment for PET imaging of pancreatic tumors in mice [77]. [18F]FDG was coupled to a tetrazine scaffold by oxime ligation and subsequently conjugated to the trans-cyclooctene (TCO)-functionalized peptide VHH-LPET via TCO-tetrazine sortase-mediated reaction, affording the 18F-glycosylated antibody fragment 63 (Figure 5) after 20 min of constant agitation at 25 °C. Conjugate 63 showed promising characteristics for the detection of the growth and regression of small pancreatic tumors by immune-PET imaging.
7. 5-[18F]fluoro-5-deoxyribose ([18F]FDR) for Chemoselective 18F-fluoroglycosylation by Oxime Linkage
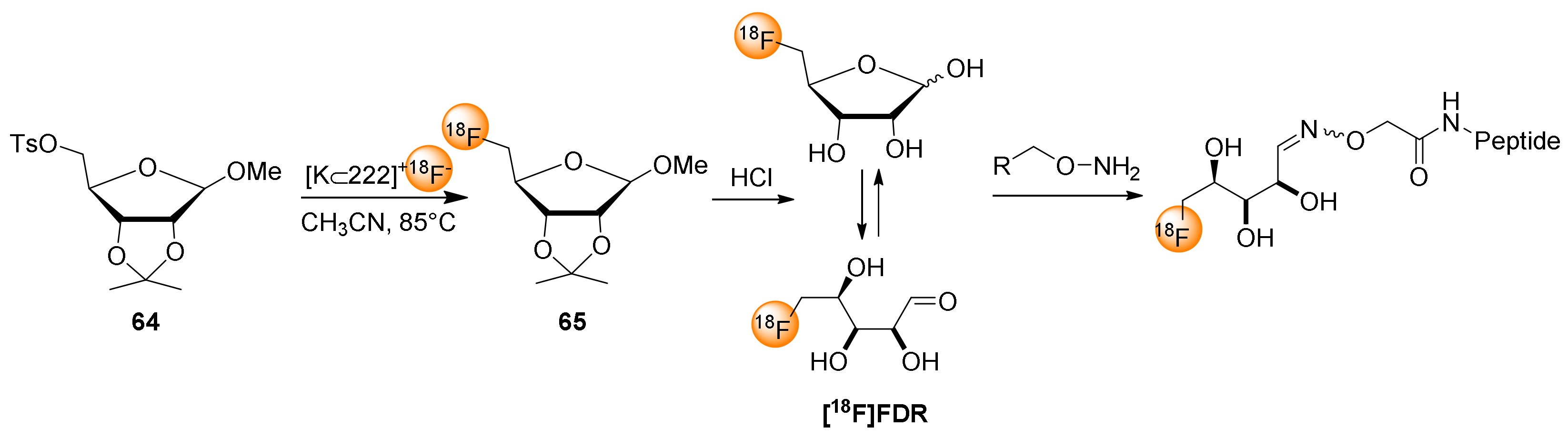
The use of [18F]FDG for direct 18F-fluoroglycosylation via oxime formation requires relatively harsh reaction conditions, namely high temperature and acidic pH, which is unfavorable for sensitive biomolecules. To overcome this limitation, 5-[18F]fluoro-5-deoxyribose ([18F]FDR) can be considered as an alternative prosthetic group for 18F-fluoroglycosylation, because the location of the fluorine at C-5 of the 5-deoxyribose ring facilitates the formation of the acyclic form of [18F]FDR making oxime bond formation possible at ambient temperature and pH of 4–5 [65,78]. Scheme 4 shows the aminopolyether-supported radiosynthesis of [18F]FDR starting from methyl 2,3-O-isopropylidene-5-O-(p-toluenesulfonyl)-β-d-ribofuranoside (64) and the subsequent oxime bond formation with aminooxy-functionalized peptides.
Noteworthily, HPLC separation of the intermediate methyl 2,3-O-isopropylidene-5-deoxy-5-[18F]fluororibofuranoside (65) from excess precursor 64 turned out to be essential for the efficient use of [18F]FDR in subsequent 18F-fluoroglycosylation reactions. After the acidic hydrolysis of 65 and solid phase extraction, [18F]FDR was obtained in average at 35% RCY with a total synthesis time of 85 min [27,78].
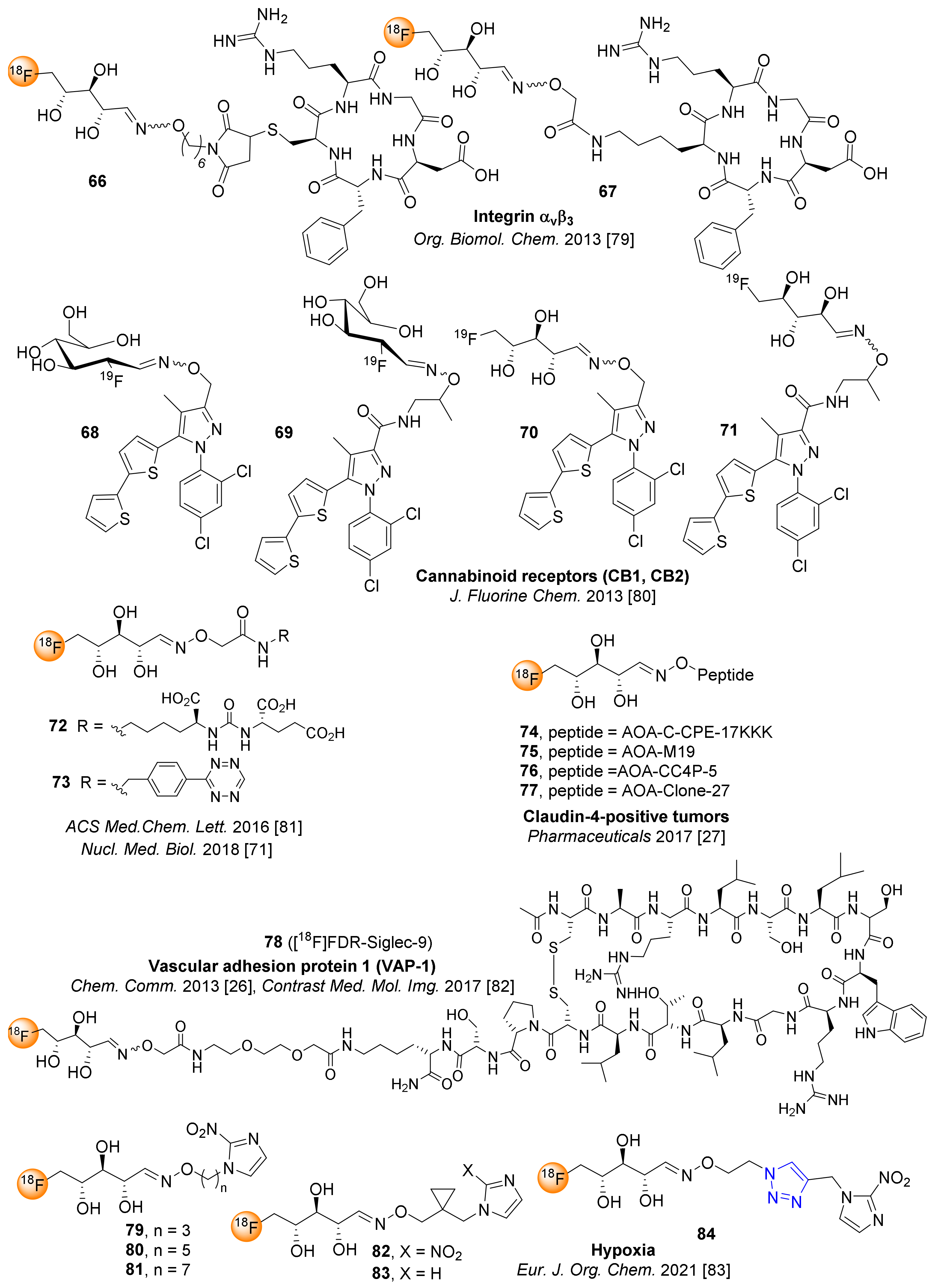
[18F]FDR was conjugated to the aminooxy-functionalized RGD peptides c(RGDfK) and c(RGDfC) at room temperature in sodium acetate buffer at pH 4.6, affording 65–92% RCY in 15 min [79]. The resulting 18F-glycopeptides 66 and 67 (Figure 6) were isolated by radio-HPLC and showed specific binding to αvβ3-expressing PC3 cells, demonstrating that [18F]FDR is an effective prosthetic group for 18F-fluoroglycosylation of bioactive RGD peptides [79].
In a comparative study of non-radioactive FDR and FDG for oxime formation with hydroxylamine-functionalized rimonabant-type pyrazoles, glycoconjugates 68–71 were synthesized as candidate PET ligands for cannabinoid receptors 1 (CB1) and 2 (CB2) [80]. As expected, FDR conjugation proved to be superior to FDG analogues, as the conjugation proceeded at room temperature in 20 min, whereas FDG conjugation required 100 °C (30 min). However, 68–71 showed only weak affinities to CB1 (540–720 nM) and CB2 (310–1400 nM), such that subsequent studies on 18F-glycosylation were not reasonable.
Alongside the [18F]FDG conjugates 56 and 57 (see Figure 5), Keinänen et al. reported the [18F]FDR-conjugated PSMA inhibitor 72 (Figure 6) and tetrazine analog 73 as a prosthetic group for inverse electron-demand Diels–Alder cycloaddition (IEDDA) reactions with trans-cyclooctene derivatives, being compatible for pretargeted in vivo PET imaging studies [81]. The 18F-glycosylated tetrazine 73 showed low lipophilicity and excellent stability in phosphate-buffered saline and in mouse plasma. The biodistribution study of 73 in mice demonstrated promising pharmacokinetics that could be suitable for in vivo bioorthogonal IEDDA reactions in future pre-targeted PET imaging studies. The reported solid-phase purification method applied for both [18F]FDG- and [18F]FDR-conjugated products, providing 56, 57 and 72, 73 in high radiochemical purity and molar activity (Figure 5 and 6) [71,81].
The Neumaier group synthesized and studied various 18F-labeled peptides for imaging of claudin-4 as candidate tracers for PET imaging of pancreatic tumors [28]. The various [18F]FDR-conjugated peptides 74–77 were synthesized via oxime ligation of claudin-derived peptides, applying [18F]FDR obtained by 18F-labeling of the naphthalene onium salt of 5-deoxyribose (Table 1). The 18F-glycosylated peptide 77 (Figure 6) was afforded in high radiochemical purity (>98%) and 15% RCY after a total synthesis time of 98 min, successfully introducing a “minimalist” protocol for 18F-synthesis by taking advantage of the onium salt precursor [28].
Li et al. demonstrated the 18F-fluoroglycosylation by the use of [18F]FDR using sialic acid-binding Ig-like lectin 9 (siglec-9) [26], a protein ligand for vascular adhesion protein 1 (VAP-1) which is upregulated in inflammation. Since siglec-9 is a rare temperature sensitive peptide, the authors optimized the 18F-fluoroglycosylation with [18F]FDR by the use of an anilinium buffer (pH 4.6) instead of sodium acetate, to allow oxime bond formation at a minimized peptide concentration of 0.3 mM, affording the desired 18F-glycopeptide with 50–60% RCY after ligation for 10 min at room temperature. [18F]FDR-Siglec-9 (78) was formulated within 120 min after the final HPLC purification with a RAY of 27% and a molar activity of 36–43 GBq/µmol. In vivo experiments clearly demonstrated that 78 could be successfully applied for the detection of inflammatory foci in rats [27]. Moreover, the glycoconjugate 78 was compared with 68Ga-DOTA-Siglec-9, revealing very similar tracer properties for the detection of inflammatory lesions in vivo [82]; however, since the radiosynthesis of 78 turned out to be more laborious and time-consuming, the 68Ga-DOTA-conjugated Siglec-9 analog was suggested as more advantageous for future clinical studies.
More recently, Musolino et al. reported the synthesis of radiotracers 79–84 for detection of hypoxia cells using PET [83]. The hypoxia-reactive 2-nitroimidazoles, bearing different alkyl chains or triazole moieties as spacers, were conjugated to [18F]FDR via oxime linkage. Interestingly, the introduction of the cyclopropyl ring in the spacer (82, Figure 6) showed superior uptake kinetics and selectivity for hypoxia cells.
8. Miscellaneous 18F-fluoroglycosylation Reactions
There are alternative 18F-glycosylation strategies present in the literature, consisting of the early works that apply tetraacetylated FDG, the intermediate of the FDG synthesis, as 18F-glycosyl donor in the presence of Lewis acids or Koenigs–Knorr conditions [75,84,85], alternative approaches toward thiol-selective 18F-fluoroglycosylation [66,83,86], the use of thiol-reactive FDG derivative for 18F-labeling of magnetic nanoparticles for combined PET/MR studies [87,88], and rarely studied enzymatic 18F-fluoroglycosylation reactions [89,90] or, more recently, the use of acidic reaction conditions for the direct ligation of [18F]FDG to 4-amino-phenylalanine in 79% RCY [91], providing a novel 18F-glycosylated amino acid PET tracer that could be valuable for the differentiation of tumor tissue from inflammatory lesions in future clinical studies.
9. Conclusions and Future Prospects
The number of literature examples for the application of 18F-fluoroglycosylation as a strategy for the successful development of PET tracers has increased further since 2014. Above all, the biocompatible methods of the mild chemoselective click chemistry conjugations are preferred in most cases. Almost every suitable 18F-glycosylated tracer described in the literature has high stability in vivo, and very good clearance properties in vivo, whereby the clearance through kidneys can be significantly influenced by the position of 18F-substitution in the carbohydrate ring.
In particular, lipophilic receptor ligands gain hydrophilicity and lose blood protein binding by 18F-fluoroglycosylation. Therefore, 18F-labeling and concomitant glycosylation for lipophilic ligands is a very suitable method for the development of PET ligands, provided that receptor binding is not affected by glycosylation. Furthermore, 18F-fluoroglycosylation is particularly well suited for the derivatization of peptides, since suitable alkyne-modified amino acids are commercially available for the targeted introduction to solid-phase peptide synthesis and the binding properties of resulting glycopeptides are thus very little or not affected at all.
The GMP-compliant automated two-step 18F-fluoroglycosylation has been established for promising 18F-fluoroglycosylated tracers and is being further improved to enable the translation of 18F-glycoconjugates into the clinic. However, the difficulty to be overcome with respect to the automation of 18F-fluoroglycosylation is mainly the two-step and thus laborious synthesis. In the future, it will be necessary to shorten and simplify the synthesis of 18F-glycoconjugates, for example by using solid-phase bound glycosyl precursors. In particular, for 18F-glycopeptide tracers or [18F]FGlc-FAPI (27), the improved availability could significantly promote and facilitate the use of these glyco-tracers in first-in-humans studies in the future.
Author Contributions
Conceptualization—S.S.S., S.M. and O.P.; writing—original draft preparation—S.S.S. and O.P.; writing—review and editing—S.S.S., S.M. and O.P. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the Alexander von Humboldt-Stiftung (ref. number 3.5-1134203-IND–HFST-E) and the Deutsche Forschungsgemeinschaft (DFG, grant MA 4295/2-1).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ametamey, S.M.; Honer, M.; Schubiger, P.A. Molecular imaging with PET. Chem. Rev. 2008, 108, 1501–1516. [Google Scholar] [CrossRef]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef] [PubMed]
- Coenen, H.H. Fluorine-18 Labeling Methods: Features and Possibilities of Basic Reactions. In PET Chemistry; Springer: Berlin/Heidelberg, Germany, 2007; pp. 15–50. [Google Scholar]
- Littich, R.; Scott, P.J.H. Novel Strategies for Fluorine-18 Radiochemistry. Angew. Chem. Int. Ed. 2012, 51, 1106–1109. [Google Scholar] [CrossRef] [Green Version]
- Shinde, S.S.; Patil, S.N. One molecule of ionic liquid and tert-alcohol on a polystyrene-support as catalysts for efficient nucleophilic substitution including fluorination. Org. Biomol. Chem. 2014, 12, 9264–9271. [Google Scholar] [CrossRef] [PubMed]
- Shinde, S.S.; Patil, S.N.; Ghatge, A.; Kumar, P. Nucleophilic fluorination using imidazolium based ionic liquid bearing tert-alcohol moiety. New J. Chem. 2015, 39, 4368–4374. [Google Scholar] [CrossRef]
- van der Born, D.; Pees, A.; Poot, A.J.; Orru, R.V.A.; Windhorst, A.D.; Vugts, D.J. Fluorine-18 labelled building blocks for PET tracer synthesis. Chem. Soc. Rev. 2017, 46, 4709–4773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamacher, K. Phase-transfer catalysed synthesis of 4-β-d-glucopyranosyl-4-thio-d-glucopyranose (thiocellobiose) and 2-β-d-glucopyranosyl-2-thio-d-glucopyranose (thiosophorose). Carbohydr. Res. 1984, 128, 291–295. [Google Scholar] [CrossRef]
- Hamacher, K.; Coenen, H.H.; Stöcklin, G. Efficient stereospecific synthesis of no-carrier-added 2-[18F]-fluoro-2-deoxy-D-glucose using aminopolyether supported nucleophilic substitution. J. Nucl. Med. 1986, 27, 235–238. [Google Scholar]
- Egleton, R.D.; Davis, T.P. Development of neuropeptide drugs that cross the blood-brain barrier. NeuroRx 2005, 2, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Haubner, R.; Kuhnast, B.; Mang, C.; Weber, W.A.; Kessler, H.; Wester, H.-J.; Schwaiger, M. [18F]Galacto-RGD: Synthesis, radiolabeling, metabolic stability, and radiation dose estimates. Bioconjug. Chem. 2004, 15, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Schottelius, M.; Rau, F.; Reubi, J.C.; Schwaiger, M.; Wester, H.-J. Modulation of pharmacokinetics of radioiodinated sugar-conjugated somatostatin analogues by variation of peptide net charge and carbohydration chemistry. Bioconjug. Chem. 2005, 16, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Haubner, R.; Wester, H.J.; Weber, W.A.; Mang, C.; Ziegler, S.I.; Goodman, S.L.; Senekowitsch-Schmidtke, R.; Kessler, H.; Schwaiger, M. Noninvasive imaging of αvβ3 integrin expression using 18F-labeled RGD-containing glycopeptide and positron emission tomography. Cancer Res. 2001, 61, 1781–1785. [Google Scholar] [PubMed]
- Schottelius, M.; Wester, H.-J.; Reubi, J.C.; Senekowitsch-Schmidtke, R.; Schwaiger, M. Improvement of pharmacokinetics of radioiodinated Tyr3-octreotide by conjugation with carbohydrates. Bioconjug. Chem. 2002, 13, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Wester, H.; Schottelius, M.; Scheidhauer, K.; Meisetschläger, G.; Herz, M.; Rau, F.; Reubi, J.; Schwaiger, M. PET imaging of somatostatin receptors: Design, synthesis and preclinical evaluation of a novel 18F-labelled, carbohydrated analogue of octreotide. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 117–122. [Google Scholar] [PubMed]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Witczak, Z.J.; Bielski, R. Click Chemistry in Glycoscience: New Developments and Strategies; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Maschauer, S.; Prante, O. A series of 2-O-trifluoromethylsulfonyl-D-mannopyranosides as precursors for concomitant 18F-labeling and glycosylation by click chemistry. Carbohydr. Res. 2009, 344, 753–761. [Google Scholar] [CrossRef]
- Maschauer, S.; Einsiedel, J.; Haubner, R.; Hocke, C.; Ocker, M.; Hübner, H.; Kuwert, T.; Gmeiner, P.; Prante, O. Labeling and Glycosylation of Peptides Using Click Chemistry: A General Approach to 18F-Glycopeptides as Effective Imaging Probes for Positron Emission Tomography. Angew. Chem. Int. Ed. 2010, 49, 976–979. [Google Scholar] [CrossRef] [PubMed]
- Maschauer, S.; Prante, O. Sweetening Pharmaceutical Radiochemistry by 18F-Fluoroglycosylation: A Short Review. Biomed. Res. Int. 2014, 2014, 214748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coenen, H.H.; Gee, A.D.; Adam, M.; Antoni, G.; Cutler, C.S.; Fujibayashi, Y.; Jeong, J.M.; Mach, R.H.; Mindt, T.L.; Pike, V.W.; et al. Consensus nomenclature rules for radiopharmaceutical chemistry-Setting the record straight. Nucl. Med. Biol. 2017, 55, v–xi. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namavari, M.; Cheng, Z.; Zhang, R.; De, A.; Levi, J.; Hoerner, J.K.; Yaghoubi, S.S.; Syud, F.A.; Gambhir, S.S. A Novel Method for Direct Site-Specific Radiolabeling of Peptides Using [18F]FDG. Bioconjug. Chem. 2009, 20, 432–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hultsch, C.; Schottelius, M.; Auernheimer, J.; Alke, A.; Wester, H.-J. 18F-Fluoroglucosylation of peptides, exemplified on cyclo(RGDfK). Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 1469–1474. [Google Scholar] [CrossRef] [PubMed]
- Wuest, F.; Hultsch, C.; Berndt, M.; Bergmann, R. Direct labelling of peptides with 2-[18F]fluoro-2-deoxy-D-glucose ([18F]FDG). Bioorg. Med. Chem. Lett. 2009, 19, 5426–5428. [Google Scholar] [CrossRef]
- Maschauer, S.; Haubner, R.; Kuwert, T.; Prante, O. 18F-Glyco-RGD Peptides for PET Imaging of Integrin Expression: Efficient Radiosynthesis by Click Chemistry and Modulation of Biodistribution by Glycosylation. Mol. Pharm. 2014, 11, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-G.; Autio, A.; Ahtinen, H.; Helariutta, K.; Liljenback, H.; Jalkanen, S.; Roivainen, A.; Airaksinen, A.J. Translating the concept of peptide labeling with 5-deoxy-5-[18F]fluororibose into preclinical practice: 18F-labeling of Siglec-9 peptide for PET imaging of inflammation. Chem. Commun. 2013, 49, 3682–3684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feni, L.; Omrane, M.A.; Fischer, M.; Zlatopolskiy, B.D.; Neumaier, B.; Neundorf, I. Convenient Preparation of 18F-Labeled Peptide Probes for Potential Claudin-4 PET Imaging. Pharmaceuticals 2017, 10, 99. [Google Scholar] [CrossRef] [Green Version]
- Mirfeizi, L.; Campbell-Verduyn, L.; Dierckx, R.A.; Feringa, B.L.; Elsinga, P.H. Application of Click Chemistry for PET. Curr. Org. Chem. 2013, 17, 2108–2118. [Google Scholar] [CrossRef]
- Fischer, C.R.; Muller, C.; Reber, J.; Muller, A.; Kramer, S.D.; Ametamey, S.M.; Schibli, R. [18F]Fluoro-Deoxy-Glucose Folate: A Novel PET Radiotracer with Improved in Vivo Properties for Folate Receptor Targeting. Bioconjug. Chem. 2012, 23, 805–813. [Google Scholar] [CrossRef]
- Fischer, C.R.; Groehn, V.; Reber, J.; Schibli, R.; Ametamey, S.M.; Muller, C. Improved PET imaging of tumors in mice using a novel 18F-folate conjugate with an albumin-binding entity. Mol. Imaging Biol. 2013, 15, 649–654. [Google Scholar] [CrossRef]
- Hugenberg, V.; Breyholz, H.-J.; Riemann, B.; Hermann, S.; Schober, O.; Schaefers, M.; Gangadharmath, U.; Mocharla, V.; Kolb, H.; Walsh, J.; et al. A New Class of Highly Potent Matrix Metalloproteinase Inhibitors Based on Triazole-Substituted Hydroxamates: (Radio)Synthesis and in Vitro and First In Vivo Evaluation. J. Med. Chem. 2012, 55, 4714–4727. [Google Scholar] [CrossRef]
- Banerjee, A.; Maschauer, S.; Hübner, H.; Gmeiner, P.; Prante, O. Click chemistry based synthesis of dopamine D4 selective receptor ligands for the selection of potential PET tracers. Bioorg. Med. Chem. Lett. 2013, 23, 6079–6082. [Google Scholar] [CrossRef] [PubMed]
- Maschauer, S.; Michel, K.; Tripal, P.; Büther, K.; Kuwert, T.; Schober, O.; Kopka, K.; Riemann, B.; Prante, O. Synthesis and In Vivo Evaluation of an 18F-Labeled Glycoconjugate of PD156707 for Imaging ETA Expression in Thyroid Carcinoma by Positron Emission Tomography. Am. J. Nucl. Med. Mol. Imaging 2013, 3, 425–436. [Google Scholar] [PubMed]
- Lang, C.; Maschauer, S.; Hübner, H.; Gmeiner, P.; Prante, O. Synthesis and Evaluation of a 18F-Labeled Diarylpyrazole Glycoconjugate for the Imaging of NTS1-Positive Tumors. J. Med. Chem. 2013, 56, 9361–9365. [Google Scholar] [CrossRef]
- Pisaneschi, F.; Slade, R.L.; Iddon, L.; George, G.P.; Nguyen, Q.D.; Spivey, A.C.; Aboagye, E.O. Synthesis of a new fluorine-18 glycosylated ‘click’ cyanoquinoline for the imaging of epidermal growth factor receptor. J. Labelled Compd. Radiopharm. 2014, 57, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Boutureira, O.; D’Hooge, F.; Fernandez-Gonzalez, M.; Bernardes, G.J.L.; Sanchez-Navarro, M.; Koeppe, J.R.; Davis, B.G. Fluoroglycoproteins: Ready chemical site-selective incorporation of fluorosugars into proteins. Chem. Commun. 2010, 46, 8142–8144. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, S.; Maschauer, S.; Kuwert, T.; Beck-Sickinger, A.G.; Prante, O. Synthesis and in Vitro and in Vivo Evaluation of an 18F-Labeled Neuropeptide Y Analogue for Imaging of Breast Cancer by PET. Mol. Pharm. 2015, 12, 1121–1130. [Google Scholar] [CrossRef] [PubMed]
- Boutureira, O.; Bernardes, G.J.; D’Hooge, F.; Davis, B.G. Direct radiolabelling of proteins at cysteine using [18F]fluorosugars. Chem. Commun. 2011, 47, 10010–10012. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Liu, Z.; Chen, K.; Yan, Y.; Watzlowik, P.; Wester, H.J.; Chin, F.T.; Chen, X. 18F-labeled galacto and PEGylated RGD dimers for PET imaging of αvβ3 integrin expression. Mol. Imaging Biol. 2010, 12, 530–538. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Lang, L.; Hu, S.; Guo, N.; Zhu, L.; Sun, Z.; Ma, Y.; Kiesewetter, D.O.; Niu, G.; Xie, Q. Comparison of three dimeric 18F-AlF-NOTA-RGD tracers. Mol. Imaging Biol. 2014, 16, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, Z.; Lozada, J.; Wong, M.Q.; Lin, K.-S.; Yapp, D.; Perrin, D.M. Single step 18F-labeling of dimeric cycloRGD for functional PET imaging of tumors in mice. Nucl. Med. Biol. 2013, 40, 959–966. [Google Scholar] [CrossRef]
- Maschauer, S.; Einsiedel, J.; Hübner, H.; Gmeiner, P.; Prante, O. 18F- and 68Ga-Labeled Neurotensin Peptides for PET Imaging of Neurotensin Receptor 1. J. Med. Chem. 2016, 59, 6480–6492. [Google Scholar] [CrossRef] [PubMed]
- Potemkin, R.; Strauch, B.; Kuwert, T.; Prante, O.; Maschauer, S. Development of 18F-Fluoroglycosylated PSMA-Ligands with Improved Renal Clearance Behavior. Mol. Pharm. 2020, 17, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Maschauer, S.; Greff, C.; Einsiedel, J.; Ott, J.; Tripal, P.; Hübner, H.; Gmeiner, P.; Prante, O. Improved radiosynthesis and preliminary in vivo evaluation of a 18F-labeled glycopeptide–peptoid hybrid for PET imaging of neurotensin receptor 2. Biorg. Med. Chem. 2015, 23, 4026–4033. [Google Scholar] [CrossRef]
- Swift, S.L.; Burns, J.E.; Maitland, N.J. Altered Expression of Neurotensin Receptors Is Associated with the Differentiation State of Prostate Cancer. Cancer Res. 2010, 70, 347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korner, M.; Waser, B.; Strobel, O.; Buchler, M.; Reubi, J.C. Neurotensin receptors in pancreatic ductal carcinomas. EJNMMI Res. 2015, 5, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, S.; Nikolaou, S.; Zhu, J.; Jeffery, P.; Goldin, R.; Kinross, J.; Alexander, J.L.; Rasheed, S.; Tekkis, P.; Kontovounisios, C. Characterisation of the Expression of Neurotensin and Its Receptors in Human Colorectal Cancer and Its Clinical Implications. Biomolecules 2020, 10, 1145. [Google Scholar] [CrossRef]
- Reubi, J.C.; Waser, B.; Friess, H.; Buchler, M.; Laissue, J. Neurotensin receptors: A new marker for human ductal pancreatic adenocarcinoma. Gut 1998, 42, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Souaze, F.; Dupouy, S.; Viardot-Foucault, V.; Bruyneel, E.; Attoub, S.; Gespach, C.; Gompel, A.; Forgez, P. Expression of neurotensin and NT1 receptor in human breast cancer: A potential role in tumor progression. Cancer Res. 2006, 66, 6243–6249. [Google Scholar] [CrossRef] [Green Version]
- Held, C.; Plomer, M.; Hübner, H.; Meltretter, J.; Pischetsrieder, M.; Gmeiner, P. Development of a Metabolically Stable Neurotensin Receptor 2 (NTS2) Ligand. Chem. Med. Chem. 2013, 8, 75–81. [Google Scholar] [CrossRef]
- Maschauer, S.; Heilmann, M.; Wängler, C.; Schirrmacher, R.; Prante, O. Radiosynthesis and Preclinical Evaluation of 18F-Fluoroglycosylated Octreotate for Somatostatin Receptor Imaging. Bioconjug. Chem. 2016, 27, 2707–2714. [Google Scholar] [CrossRef] [PubMed]
- Maschauer, S.; Ott, J.J.; Bernhardt, G.; Kuwert, T.; Keller, M.; Prante, O. 18F-labelled triazolyl-linked argininamides targeting the neuropeptide Y Y1R for PET imaging of mammary carcinoma. Sci. Rep. 2019, 9, 12990. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, E.M.; Roise, J.J.; Li, I.C.; Das, R.; Murthy, N. Advances in Imaging Reactive Oxygen Species. J. Nucl. Med. 2021, 62, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Toms, J.; Reshetnikov, V.; Maschauer, S.; Mokhir, A.; Prante, O. Radiosynthesis of an 18F-fluoroglycosylated aminoferrocene for in-vivo imaging of reactive oxygen species activity by PET. J. Label. Compd. Radiopharm. 2018, 61, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Daum, S.; Toms, J.; Reshetnikov, V.; Ozkan, H.G.; Hampel, F.; Maschauer, S.; Hakimioun, A.; Beierlein, F.; Sellner, L.; Schmitt, M.; et al. Identification of Boronic Acid Derivatives as an Active Form of N-Alkylaminoferrocene-Based Anticancer Prodrugs and Their Radiolabeling with 18F. Bioconjug. Chem. 2019, 30, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Kratochwil, C.; Flechsig, P.; Lindner, T.; Abderrahim, L.; Altmann, A.; Mier, W.; Adeberg, S.; Rathke, H.; Röhrich, M.; Winter, H.; et al. 68Ga-FAPI PET/CT: Tracer Uptake in 28 Different Kinds of Cancer. J. Nucl. Med. 2019, 60, 801. [Google Scholar] [CrossRef] [Green Version]
- Giesel, F.L.; Kratochwil, C.; Lindner, T.; Marschalek, M.M.; Loktev, A.; Lehnert, W.; Debus, J.; Jäger, D.; Flechsig, P.; Altmann, A.; et al. 68Ga-FAPI PET/CT: Biodistribution and Preliminary Dosimetry Estimate of 2 DOTA-Containing FAP-Targeting Agents in Patients with Various Cancers. J. Nucl. Med. 2019, 60, 386. [Google Scholar] [CrossRef] [Green Version]
- Toms, J.; Kogler, J.; Maschauer, S.; Daniel, C.; Schmidkonz, C.; Kuwert, T.; Prante, O. Targeting Fibroblast Activation Protein: Radiosynthesis and Preclinical Evaluation of an 18F-Labeled FAP Inhibitor. J. Nucl. Med. 2020, 61, 1806. [Google Scholar] [CrossRef]
- Elgland, M.; Nordeman, P.; Fyrner, T.; Antoni, G.; Nilsson, K.P.R.; Konradsson, P. β-Configured clickable [18F]FDGs as novel 18F-fluoroglycosylation tools for PET. New J. Chem. 2017, 41, 10231–10236. [Google Scholar] [CrossRef] [Green Version]
- Collet, C.; Vucko, T.; Ariztia, J.; Karcher, G.; Pellegrini-Moïse, N.; Lamandé-Langle, S. Fully automated radiosynthesis of [18F]fluoro-C-glyco-c(RGDfC): Exploiting all the abilities of the AllInOne synthesizer. React. Chem. Eng. 2019, 4, 2088–2098. [Google Scholar] [CrossRef]
- Collet, C.; Maskali, F.; Clément, A.; Chrétien, F.; Poussier, S.; Karcher, G.; Marie, P.-Y.; Chapleur, Y.; Lamandé-Langle, S. Development of 6-[18F]fluoro-carbohydrate-based prosthetic groups and their conjugation to peptides via click chemistry. J. Label. Compd. Radiopharm. 2016, 59, 54–62. [Google Scholar] [CrossRef]
- Arja, K.; Elgland, M.; Appelqvist, H.; Konradsson, P.; Lindgren, M.; Nilsson, K.P.R. Synthesis and Characterization of Novel Fluoro-glycosylated Porphyrins that can be Utilized as Theranostic Agents. ChemistryOpen 2018, 7, 495–503. [Google Scholar] [CrossRef]
- Ulrich, S.; Boturyn, D.; Marra, A.; Renaudet, O.; Dumy, P. Oxime ligation: A chemoselective click-type reaction for accessing multifunctional biomolecular constructs. Chemistry 2014, 20, 34–41. [Google Scholar] [CrossRef]
- Li, X.G.; Haaparanta, M.; Solin, O. Oxime formation for fluorine-18 labeling of peptides and proteins for positron emission tomography (PET) imaging: A review. J. Fluor. Chem. 2012, 143, 49–56. [Google Scholar] [CrossRef]
- Wuest, F.; Berndt, M.; Bergmann, R.; van, d.H.J.; Pietzsch, J. Synthesis and Application of [18F]FDG-Maleimidehexyloxime ([18F]FDG-MHO): A [18F]FDG-Based Prosthetic Group for the Chemoselective 18F-Labeling of Peptides and Proteins. Bioconjug. Chem. 2008, 19, 1202–1210. [Google Scholar] [CrossRef] [PubMed]
- Al Jammaz, I.; Al-Otaibi, B.; Amer, S.; Al-Hokbany, N.; Okarvi, S. Novel synthesis and preclinical evaluation of folic acid derivatives labeled with 18F-FDG for PET imaging of folate receptor-positive tumors. Nucl. Med. Biol. 2012, 39, 864–870. [Google Scholar] [CrossRef]
- AlJammaz, I.; Al-Otaibi, B.; AlHindas, H.; Okarvi, S.M. Novel synthesis and initial preclinical evaluation of 18F[FDG] labeled rhodamine: A potential PET myocardial perfusion imaging agent. Nucl. Med. Biol. 2015, 42, 804–808. [Google Scholar] [CrossRef] [PubMed]
- Richter, S.; Wuest, M.; Bergman, C.N.; Way, J.D.; Krieger, S.; Rogers, B.E.; Wuest, F. Rerouting the Metabolic Pathway of 18F-Labeled Peptides: The Influence of Prosthetic Groups. Bioconjug. Chem. 2015, 26, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Bouvet, V.; Wuest, M.; Bailey, J.J.; Bergman, C.; Janzen, N.; Valliant, J.F.; Wuest, F. Targeting Prostate-Specific Membrane Antigen (PSMA) with F-18-Labeled Compounds: The Influence of Prosthetic Groups on Tumor Uptake and Clearance Profile. Mol. Imaging Biol. 2017, 19, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Keinänen, O.; Partelová, D.; Alanen, O.; Antopolsky, M.; Sarparanta, M.; Airaksinen, A.J. Efficient cartridge purification for producing high molar activity [18F]fluoro-glycoconjugates via oxime formation. Nucl. Med. Biol. 2018, 67, 27–35. [Google Scholar] [CrossRef]
- Flavell, R.R.; Truillet, C.; Regan, M.K.; Ganguly, T.; Blecha, J.E.; Kurhanewicz, J.; VanBrocklin, H.F.; Keshari, K.R.; Chang, C.J.; Evans, M.J.; et al. Caged [18F]FDG Glycosylamines for Imaging Acidic Tumor Microenvironments Using Positron Emission Tomography. Bioconjug. Chem. 2016, 27, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Şenışık, A.M.; İçhedef, Ç.; Kılçar, A.Y.; Uçar, E.; Arı, K.; Göksoy, D.; Parlak, Y.; Sayıt Bilgin, B.E.; Teksöz, S. One-step conjugation of glycylglycine with [18F]FDG and a pilot PET imaging study. J. Radioanal. Nucl. Chem. 2018, 316, 457–463. [Google Scholar] [CrossRef]
- Lopci, E.; Grassi, I.; Chiti, A.; Nanni, C.; Cicoria, G.; Toschi, L.; Fonti, C.; Lodi, F.; Mattioli, S.; Fanti, S. PET radiopharmaceuticals for imaging of tumor hypoxia: A review of the evidence. Am. J. Nucl. Med. Mol. Imaging 2014, 4, 365–384. [Google Scholar] [PubMed]
- Patt, M.; Sorger, D.; Scheunemann, M.; Stöcklin, G. Adduct of 2-[18F]FDG and 2-nitroimidazole as a putative radiotracer for the detection of hypoxia with PET: Synthesis, in vitro- and in vivo-characterization. Appl. Radiat. Isot. 2002, 57, 705–712. [Google Scholar] [CrossRef]
- Yang, X.; Wang, F.; Zhu, H.; Yang, Z.; Chu, T. Synthesis and Bioevaluation of Novel [18F]FDG-Conjugated 2-Nitroimidazole Derivatives for Tumor Hypoxia Imaging. Mol. Pharm. 2019, 16, 2118–2128. [Google Scholar] [CrossRef]
- Rashidian, M.; Keliher, E.J.; Dougan, M.; Juras, P.K.; Cavallari, M.; Wojtkiewicz, G.R.; Jacobsen, J.T.; Edens, J.G.; Tas, J.M.J.; Victora, G.; et al. Use of 18F-2-Fluorodeoxyglucose to Label Antibody Fragments for Immuno-Positron Emission Tomography of Pancreatic Cancer. ACS Cent. Sci. 2015, 1, 142–147. [Google Scholar] [CrossRef] [Green Version]
- Li, X.G.; Dall’Angelo, S.; Schweiger, L.F.; Zanda, M.; O’Hagan, D. [18F]-5-Fluoro-5-deoxyribose, an efficient peptide bioconjugation ligand for positron emission tomography (PET) imaging. Chem. Commun. 2012, 48, 5247–5249. [Google Scholar] [CrossRef]
- Dall’Angelo, S.; Zhang, Q.; Fleming, I.N.; Piras, M.; Schweiger, L.F.; O’Hagan, D.; Zanda, M. Efficient bioconjugation of 5-fluoro-5-deoxy-ribose (FDR) to RGD peptides for positron emission tomography (PET) imaging of αvβ3 integrin receptor. Org. Biomol. Chem. 2013, 11, 4551–4558. [Google Scholar] [CrossRef] [PubMed]
- Frau, S.; Dall’Angelo, S.; Baillie, G.L.; Ross, R.A.; Pira, M.; Tseng, C.-C.; Lazzari, P.; Zanda, M. Pyrazole-type cannabinoid ligands conjugated with fluoro-deoxy-carbohydrates as potential PET-imaging agents: Synthesis and CB1/CB2 receptor affinity evaluation. J. Fluor. Chem. 2013, 152, 166–172. [Google Scholar] [CrossRef]
- Keinänen, O.; Li, X.-G.; Chenna, N.K.; Lumen, D.; Ott, J.; Molthoff, C.F.M.; Sarparanta, M.; Helariutta, K.; Vuorinen, T.; Windhorst, A.D.; et al. A New Highly Reactive and Low Lipophilicity Fluorine-18 Labeled Tetrazine Derivative for Pretargeted PET Imaging. ACS Med. Chem. Lett. 2016, 7, 62–66. [Google Scholar] [CrossRef] [Green Version]
- Virtanen, H.; Silvola, J.M.U.; Autio, A.; Li, X.-G.; Liljenbäck, H.; Hellberg, S.; Siitonen, R.; Ståhle, M.; Käkelä, M.; Airaksinen, A.J.; et al. Comparison of 68Ga-DOTA-Siglec-9 and 18F-Fluorodeoxyribose-Siglec-9: Inflammation Imaging and Radiation Dosimetry. Contrast Media Mol. Imaging 2017, 2017, 7645070. [Google Scholar] [CrossRef] [Green Version]
- Musolino, M.; Fleming, I.N.; Schweiger, L.F.; O’Hagan, D.; Dall’Angelo, S.; Zanda, M. Synthesis, Radiosynthesis, and in vitro Studies on Novel Hypoxia PET Tracers Incorporating [18F]FDR. Eur. J. Org. Chem. 2021, 2021, 1429–1439. [Google Scholar] [CrossRef]
- Maschauer, S.; Kuwert, T.; Prante, O. 18F-glycosylation using Koenigs-Knorr conditions: A comparative study. J. Label. Compd. Radiopharm. 2006, 49, 101–108. [Google Scholar] [CrossRef]
- Maschauer, S.; Pischetsrieder, M.; Kuwert, T.; Prante, O. Utility of 1,3,4,6-tetra-O-acetyl-2-deoxy-2-[18F]fluoroglucopyranoside for no-carrier-added 18F-glycosylation of amino acids. J. Label. Compd. Radiopharm. 2005, 48, 701–719. [Google Scholar] [CrossRef]
- Prante, O.; Einsiedel, J.; Haubner, R.; Gmeiner, P.; Wester, H.J.; Kuwert, T.; Maschauer, S. 3,4,6-tri-O-acetyl-2-deoxy-2-[18F]fluoroglucopyranosyl phenylthiosulfonate: A thiol-reactive agent for the chemoselective 18F-glycosylation of peptides. Bioconjug. Chem. 2007, 18, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Ozkaya, F.; Unak, P.; Medine, E.I.; Sakarya, S.; Unak, G.; Timur, S. 18FDG conjugated magnetic nanoparticle probes: Synthesis and in vitro investigations on MCF-7 breast cancer cells. J. Radioanal. Nucl. Chem. 2013, 295, 1789–1796. [Google Scholar] [CrossRef]
- Unak, G.; Ozkaya, F.; Ilker Medine, E.; Kozgus, O.; Sakarya, S.; Bekis, R.; Unak, P.; Timur, S. Gold nanoparticle probes: Design and in vitro applications in cancer cell culture. Colloids Surf. B. Biointerfaces 2012, 90, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Bormans, G.; Verbruggen, A. Enzymatic synthesis and biodistribution in mice of beta-O-D-galactopyranosyl-(1,4′)-2 ‘-[18F]fluoro-2′-deoxy-D-glucopyranose (2’-[18F]fluorodeoxylactose). J. Label. Compd. Radiopharm. 2001, 44, 417–423. [Google Scholar] [CrossRef]
- Prante, O.; Hamacher, K.; Coenen, H.H. Chemoenzymatic n.c.a synthesis of the coenzyme UDP-2-deoxy-2-[18F]fluoro-α-D-glucopyranose as substrate of glycosyltransferases. J. Label. Compd. Radiopharm. 2007, 50, 55–63. [Google Scholar] [CrossRef]
- Wu, Z.; Ma, J.; Brownell, A.-L.; Wang, H.; Li, C.; Meng, X.; Yuan, L.; Liu, H.; Li, S.; Xie, J. Synthesis and evaluation of an N-[18F]fluorodeoxyglycosyl amino acid for PET imaging of tumor metabolism. Nucl. Med. Biol. 2018, 66, 40–48. [Google Scholar] [CrossRef]
Scheme 1.
Two-step 18F-fluoroglycosylation by click cycloaddition using 2-deoxy-2-[18F]fluoroglucopyranosyl azide (2), starting from the 2-O-triflate precursor of triacetylated β-mannosyl azide (1).
Scheme 1.
Two-step 18F-fluoroglycosylation by click cycloaddition using 2-deoxy-2-[18F]fluoroglucopyranosyl azide (2), starting from the 2-O-triflate precursor of triacetylated β-mannosyl azide (1).

Figure 1.
Click chemistry-based 2-deoxy-2-[18F]fluoroglycosylated ligands and their target receptors.
Figure 1.
Click chemistry-based 2-deoxy-2-[18F]fluoroglycosylated ligands and their target receptors.

Scheme 2.
18F-fluoroglycosylation by CuAAC using 6-deoxy-6-[18F]fluoroglucopyranosyl azide 14 or 6´-deoxy-6´-[18F]fluoromaltosyl azide 17 with the cyclic peptide c(RGDfPra) alkyne (according to [26]).
Scheme 2.
18F-fluoroglycosylation by CuAAC using 6-deoxy-6-[18F]fluoroglucopyranosyl azide 14 or 6´-deoxy-6´-[18F]fluoromaltosyl azide 17 with the cyclic peptide c(RGDfPra) alkyne (according to [26]).

Figure 2.
Overview of PET tracers synthesized via CuAAC using the 18F-labeled prosthetic group 6-deoxy-6-[18F]fluoro-β-glucosyl azide (14). For a direct comparison (see text), the structures of 6′-deoxy-6′-[18F]fluoromaltosyl peptide 20 and 2-deoxy-2-[18F]fluoroglucosyl peptide 21 are included.
Figure 2.
Overview of PET tracers synthesized via CuAAC using the 18F-labeled prosthetic group 6-deoxy-6-[18F]fluoro-β-glucosyl azide (14). For a direct comparison (see text), the structures of 6′-deoxy-6′-[18F]fluoromaltosyl peptide 20 and 2-deoxy-2-[18F]fluoroglucosyl peptide 21 are included.

Figure 3.
Overview of triazolylalkyl-linked 18F-glycoconjugates achieved by click chemistry-based 18F-fluoroglycosylation.
Figure 3.
Overview of triazolylalkyl-linked 18F-glycoconjugates achieved by click chemistry-based 18F-fluoroglycosylation.

Figure 4.
Examples of fluoroglycosylated compounds that have been studied for their biological activity prior to 18F-fluoroglycosylation.
Figure 4.
Examples of fluoroglycosylated compounds that have been studied for their biological activity prior to 18F-fluoroglycosylation.

Scheme 3.
18F-fluoroglycosylation via oxime formation using [18F]FDG.

Figure 5.
18F-glycoconjugates synthesized by direct 18F-fluoroglycosylation through oxime formation with [18F]FDG.
Figure 5.
18F-glycoconjugates synthesized by direct 18F-fluoroglycosylation through oxime formation with [18F]FDG.

Scheme 4.
18F-fluoroglycosylation via oxime formation using [18F]FDR.

Figure 6.
18F-glycoconjugates synthesized by oxime formation with [18F]FDR.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Overview of a selection of 18F-labeled prosthetic groups for 18F-fluoroglycosylation reactions.
Table 1.
Overview of a selection of 18F-labeled prosthetic groups for 18F-fluoroglycosylation reactions.
| Labeling Precursor | Prosthetic Group | Reaction Conditions | Ref. |
|---|---|---|---|
 |  | 1. K222, K2CO3 2. NaOH | [23,24,25] |
 |  | 1. K222, K2CO3 2. NaOH | [19] |
 |  | 1. K222, K2CO3, KH2PO4 2. NaOH | [26] |
 |  | 1. K222, K2CO3, KH2PO4 2. NaOH | [26] |
 | 1. K222, K2CO3, 2. DMT-Cl, pyridine 3. HCl | [27] | |
 |  | 1. MeCN, 120 °C 2. 1M HCl, 110 °C | [28] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Shinde, S.S.; Maschauer, S.; Prante, O. Sweetening Pharmaceutical Radiochemistry by 18F-Fluoroglycosylation: Recent Progress and Future Prospects. Pharmaceuticals 2021, 14, 1175. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14111175
AMA Style
Shinde SS, Maschauer S, Prante O. Sweetening Pharmaceutical Radiochemistry by 18F-Fluoroglycosylation: Recent Progress and Future Prospects. Pharmaceuticals. 2021; 14(11):1175. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14111175
Chicago/Turabian StyleShinde, Sandip S., Simone Maschauer, and Olaf Prante. 2021. "Sweetening Pharmaceutical Radiochemistry by 18F-Fluoroglycosylation: Recent Progress and Future Prospects" Pharmaceuticals 14, no. 11: 1175. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14111175
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.