
Discovery and Development of Inhibitors of the Plasmodial FNT-Type Lactate Transporter as Novel Antimalarials


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
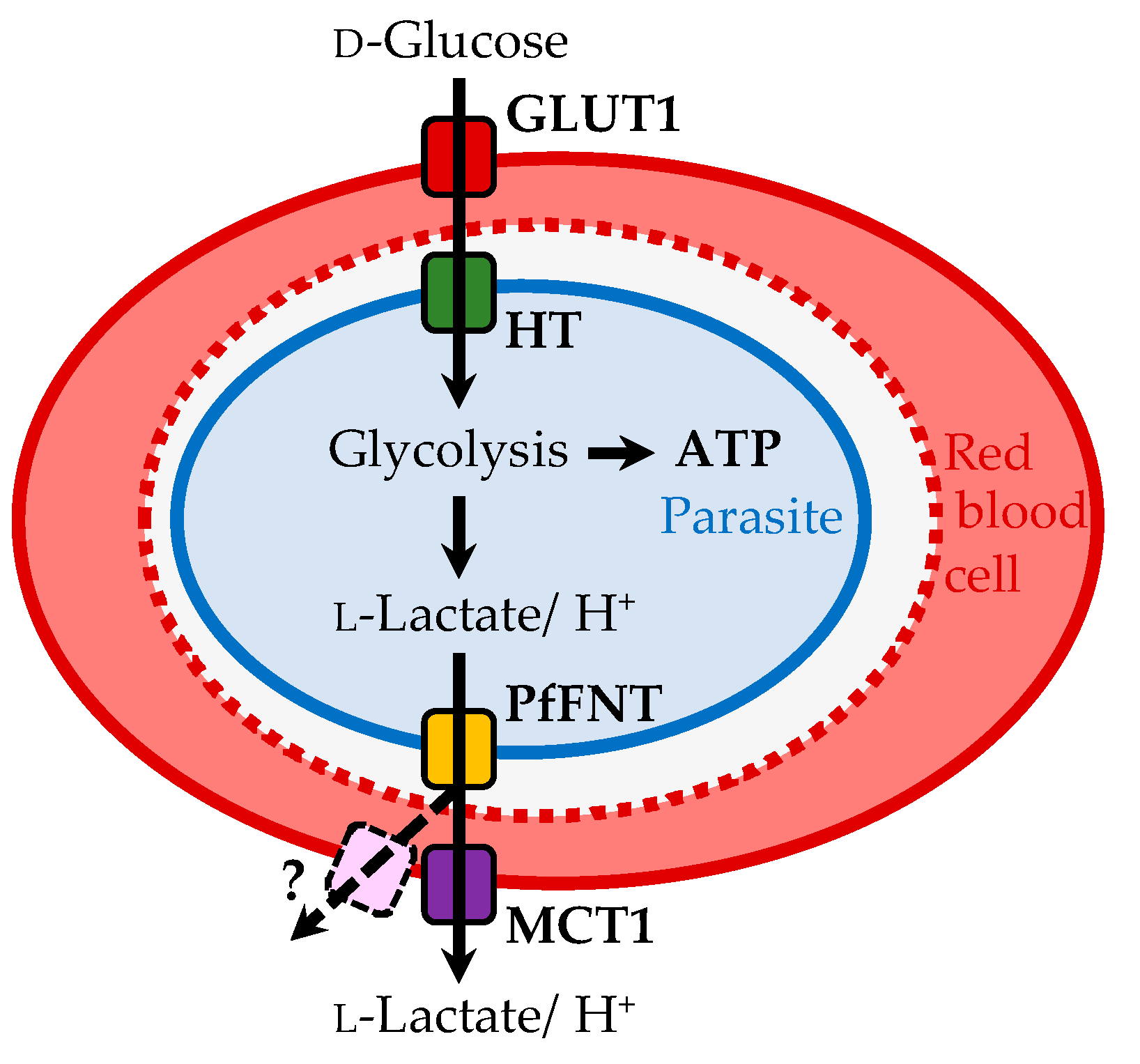
1. Introduction
2. Structures of Small Molecule PfFNT Inhibitors
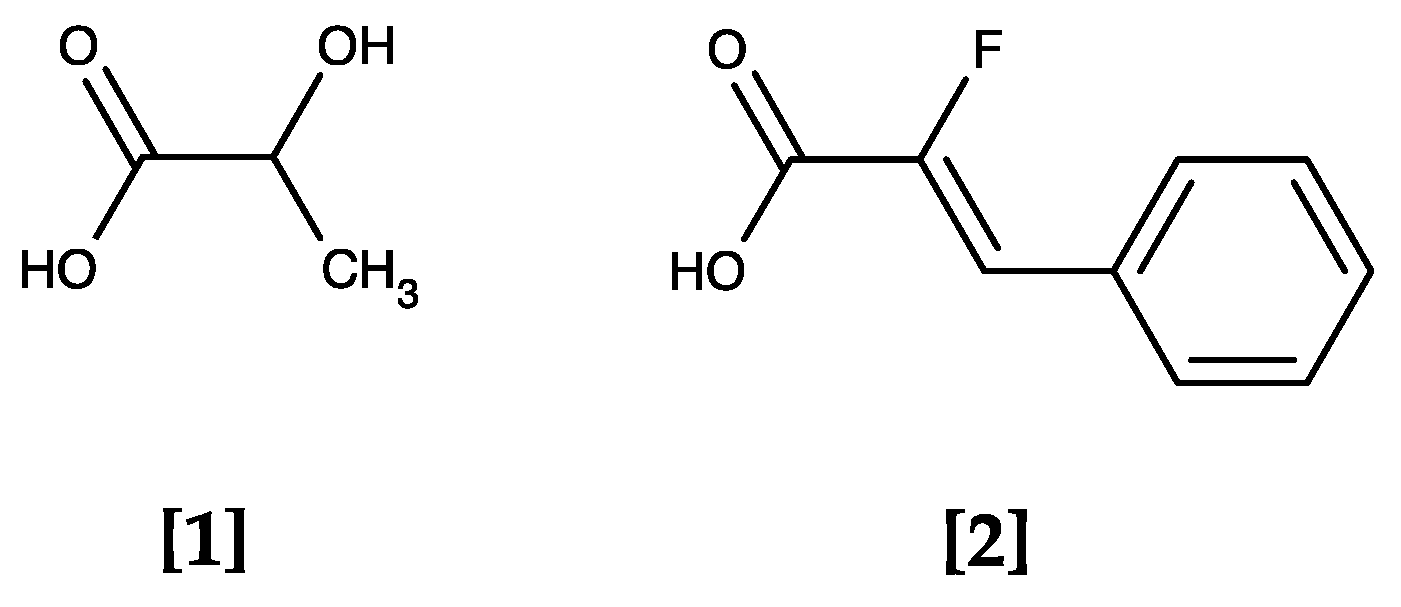
2.1. Initial Weak Inhibitors Hinted at the Therapeutic Potential of Targeting Plasmodial Lactate Transport
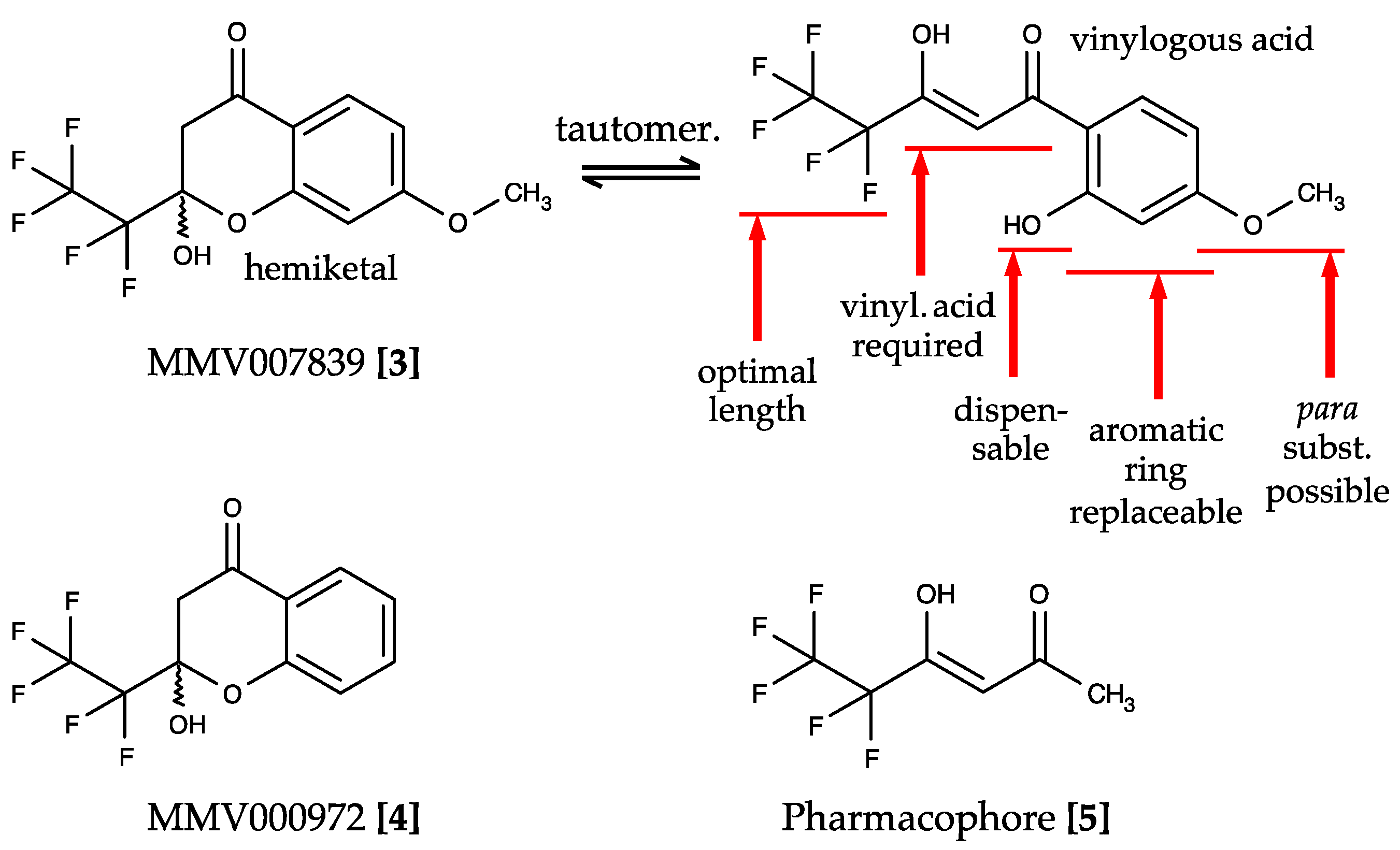
2.2. The MMV Malaria Box Contains Two Potent PfFNT Inhibitors
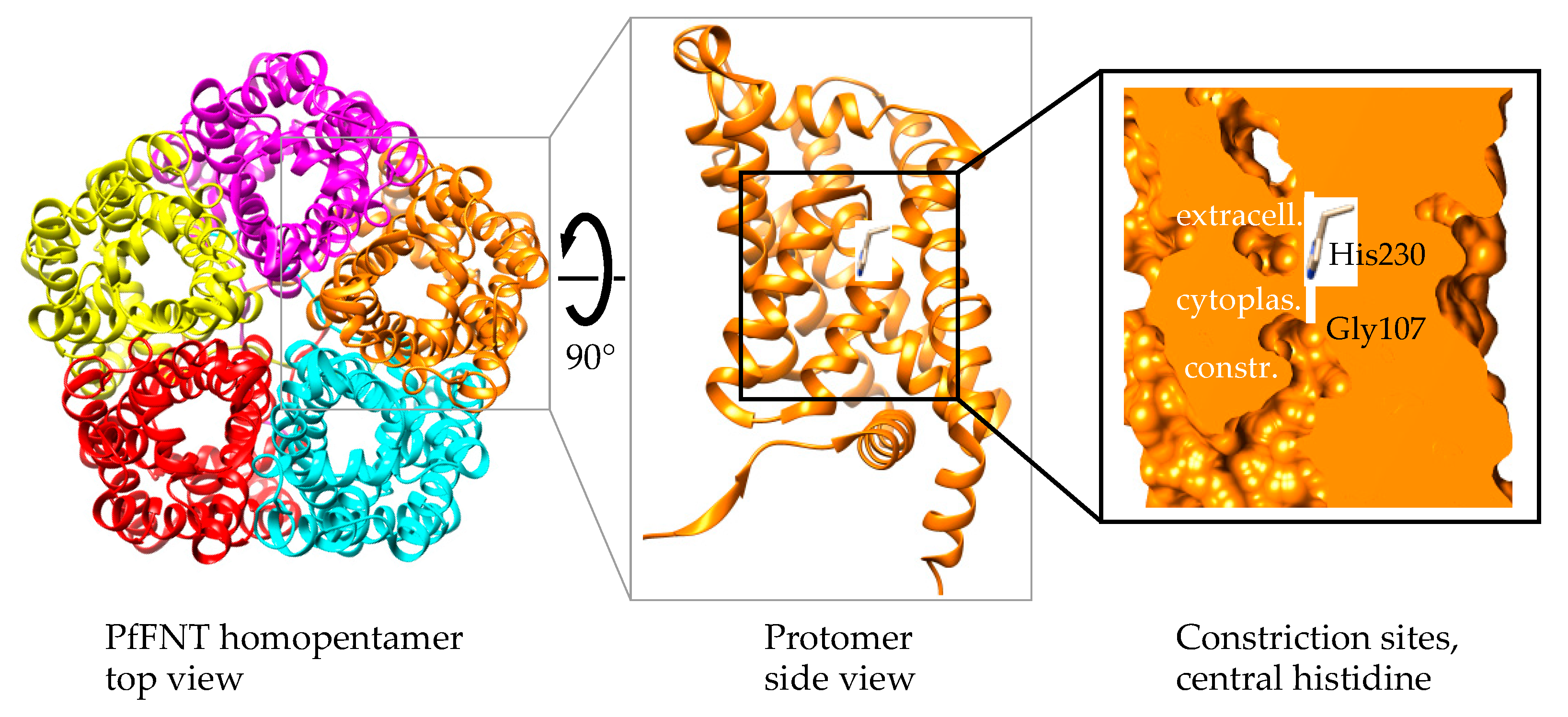
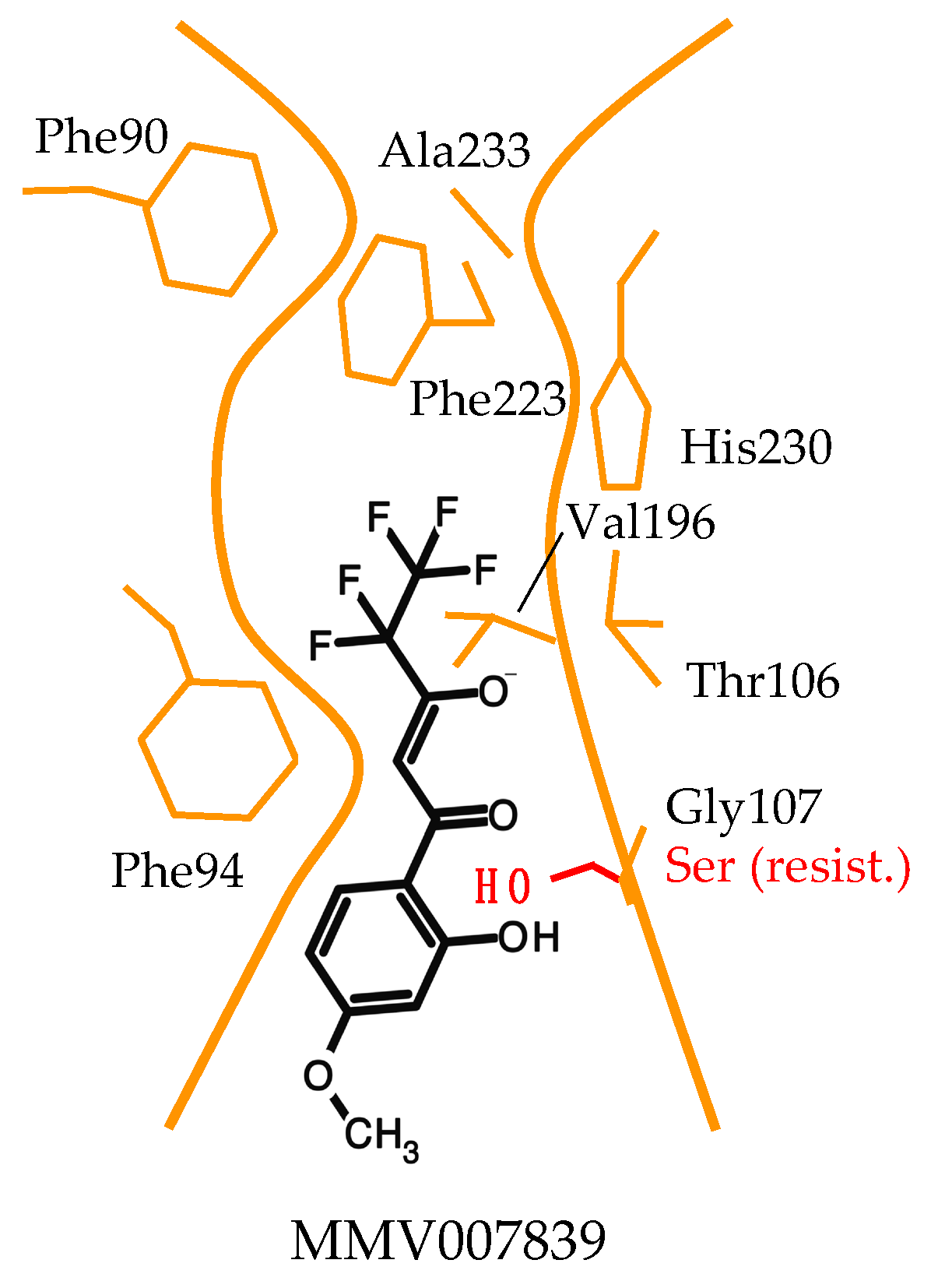
2.3. Forced Resistance Selection Revealed the Binding Site of PfFNT Inhibitors
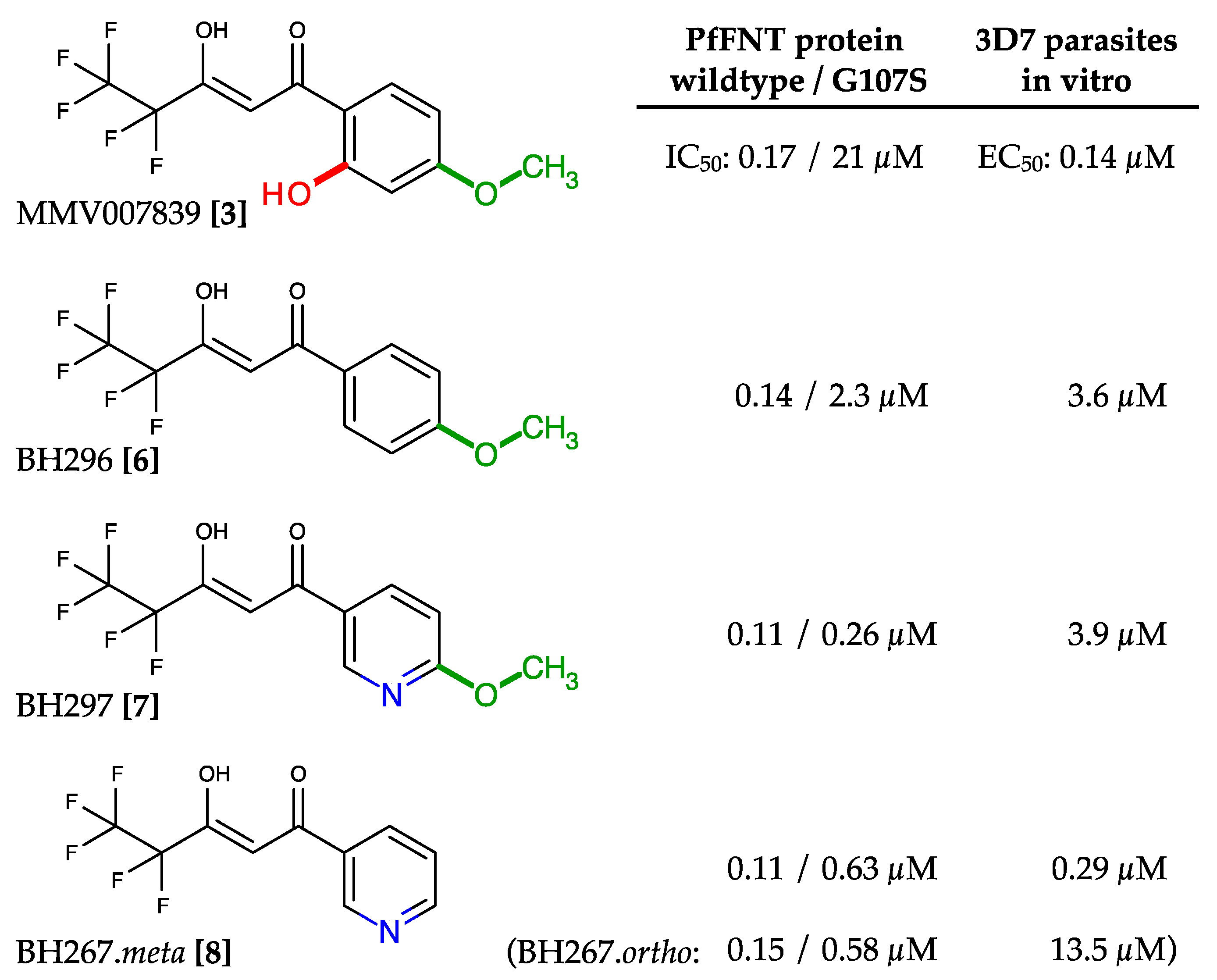
2.4. Circumvention of the PfFNT G107S Resistance Mutation by Introduction of Scaffold Nitrogen Atoms
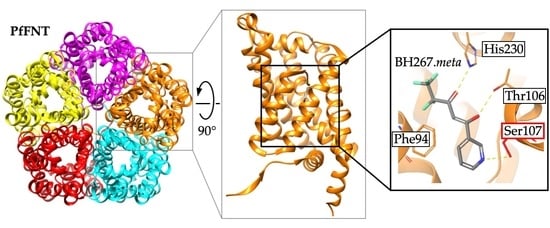
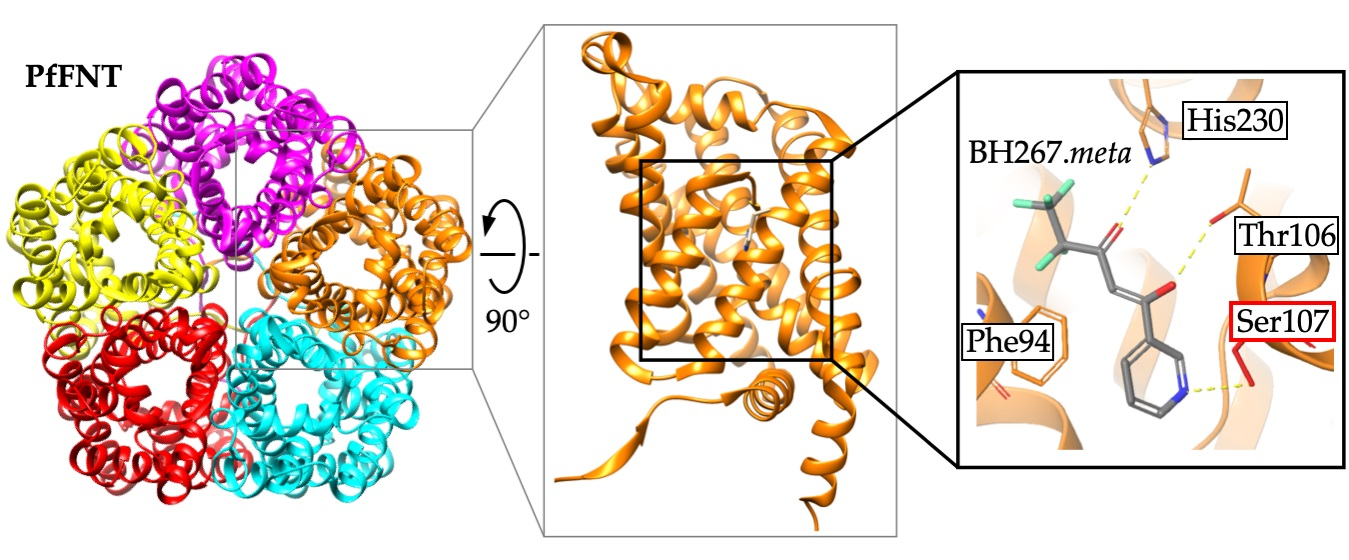
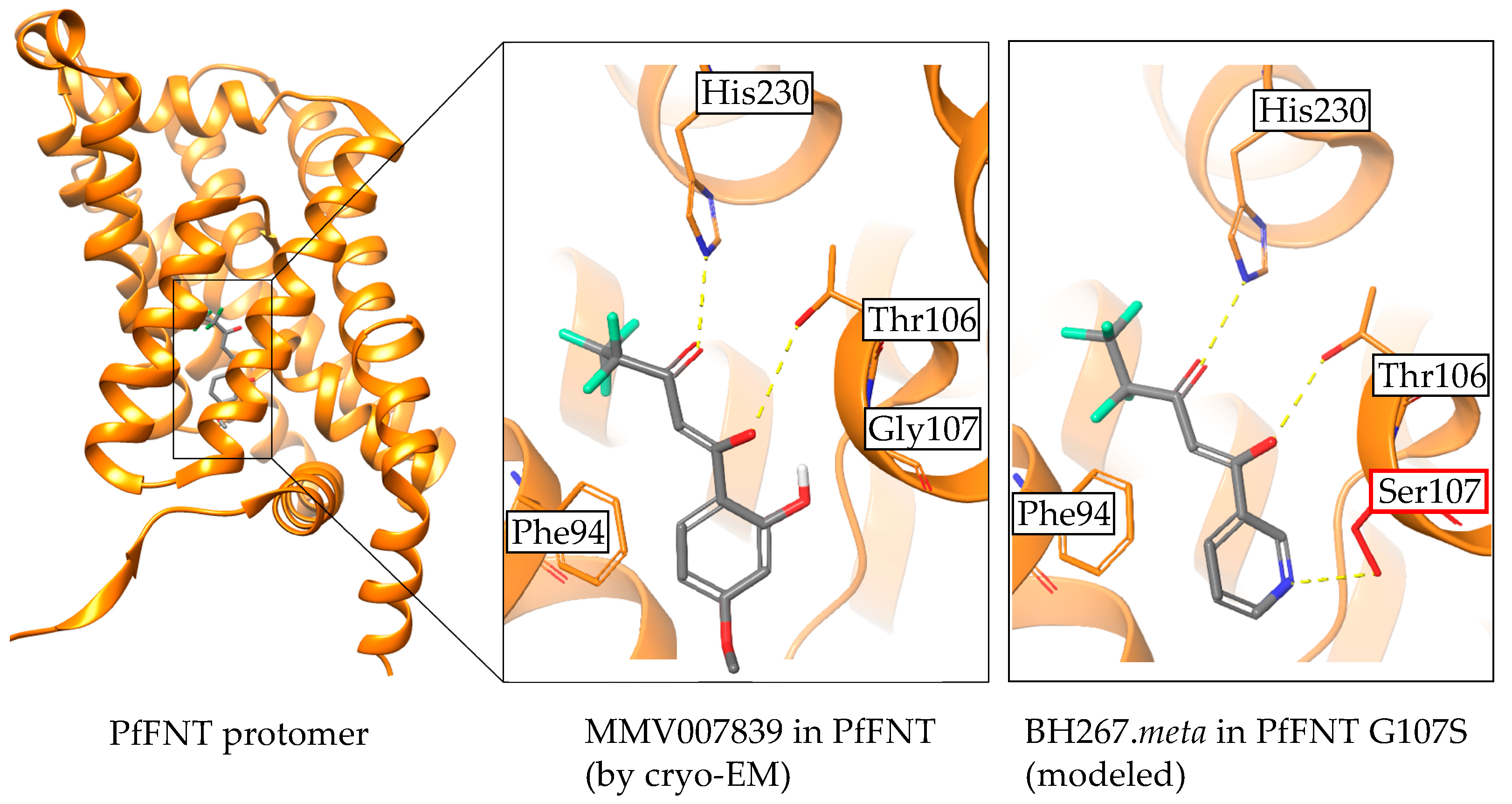
2.5. The PfFNT Cryo-Electron Microscopy Structure Confirms the Proposed Binding Mode
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Malaria Report 2020. Available online: https://www.who.int/publications/i/item/9789240015791 (accessed on 11 October 2021).
- Wells, T.N.C.; van Huijsduijnen, R.H.; van Voorhis, W.C. Malaria medicines: A glass half full? Nat. Rev. Drug Discov. 2015, 14, 424–442. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Mharakurwa, S.; Ndiaye, D.; Rathod, P.K.; Rosenthal, P.J. Antimalarial drug resistance: Literature review and activities and findings of the ICEMR network. Am. J. Trop. Med. Hyg. 2015, 93, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venugopal, K.; Hentzschel, F.; Valkiūnas, G.; Marti, M. Plasmodium asexual growth and sexual development in the haematopoietic niche of the host. Nat. Rev. Microbiol. 2020, 18, 177–189. [Google Scholar] [CrossRef]
- McKee, R.W.; Ormsbee, R.A.; Anfinsen, C.B.; Geiman, Q.M.; Ball, E.G. Studies on malarial parasites. J. Exp. Med. 1946, 84, 569–582. [Google Scholar] [CrossRef]
- MacRae, J.I.; Dixon, M.W.; Dearnley, M.K.; Chua, H.H.; Chambers, J.M.; Kenny, S.; Bottova, I.; Tilley, L.; McConville, M.J. Mitochondrial metabolism of sexual and asexual blood stages of the malaria parasite Plasmodium falciparum. BMC Biol. 2013, 13, 11–67. [Google Scholar] [CrossRef] [Green Version]
- Meier, A.; Erler, H.; Beitz, E. Targeting channels and transporters in protozoan parasite infections. Front. Chem. 2018, 6, 88. [Google Scholar] [CrossRef] [Green Version]
- Kasahara, M.; Hinkle, P.C. Reconstitution and purification of the D-glucose transporter from human erythrocytes. J. Biol. Chem. 1977, 252, 7384–7390. [Google Scholar] [CrossRef]
- Mehta, M.; Sonawat, H.M.; Sharma, S. Malaria parasite-infected erythrocytes inhibit glucose utilization in uninfected red cells. FEBS Lett. 2005, 579, 6151–6158. [Google Scholar] [CrossRef] [Green Version]
- Woodrow, C.J.; Penny, J.I.; Krishna, S. Intraerythrocytic Plasmodium falciparum expresses a high affinity facilitative hexose transporter. J. Biol. Chem. 1999, 274, 7272–7277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz, D.; Guiguemde, W.A.; Johnson, A.; Elya, C.; Anderson, J.; Clark, J.; Connelly, M.; Yang, L.; Min, J.; Sato, Y.; et al. Identification of selective inhibitors of the Plasmodium falciparum hexose transporter PfHT by screening focused libraries of anti-malarial compounds. PLoS ONE 2015, 10, e0123598. [Google Scholar] [CrossRef]
- Deng, D.; Xu, C.; Sun, P.; Wu, J.; Yan, C.; Hu, M.; Yan, N. Crystal structure of the human glucose transporter GLUT1. Nature 2014, 510, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Yuan, Y.; Zhao, N.; Pu, D.; Tang, Q.; Zhang, S.; Luo, S.; Yang, X.; Wang, N.; Xiao, Y.; et al. Orthosteric-allosteric dual inhibitors of PfHT1 as selective antimalarial agents. Proc. Natl. Acad. Sci. USA 2021, 118, e2017749118. [Google Scholar] [CrossRef]
- Jiang, X.; Yuan, Y.; Huang, J.; Zhang, S.; Luo, S.; Wang, N.; Pu, D.; Zhao, N.; Tang, Q.; Hirata, K.; et al. Structural basis for blocking sugar uptake into the malaria parasite Plasmodium falciparum. Cell 2020, 183, 258–268. [Google Scholar] [CrossRef]
- Davis, M.I.; Patrick, S.L.; Blanding, W.M.; Dwivedi, V.; Suryadi, J.; Golden, J.E.; Coussens, N.P.; Lee, O.W.; Shen, M.; Boxer, M.B.; et al. Identification of novel Plasmodium falciparum hexokinase inhibitors with antiparasitic activity. Antimicrob. Agents Chemother. 2016, 60, 6023–6033. [Google Scholar] [CrossRef] [Green Version]
- Wei, M.; Lu, L.; Sui, W.; Liu, Y.; Shi, X.; Lv, L. Inhibition of GLUTs by WZB117 mediates apoptosis in blood-stage Plasmodium parasites by breaking redox balance. Biochem. Biophys. Res. Commun. 2018, 503, 1154–1159. [Google Scholar] [CrossRef]
- Heitmeier, M.R.; Hresko, R.C.; Edwards, R.L.; Prinsen, M.J.; Ilagan, M.X.G.; Odom John, A.R.; Hruz, P.W. Identification of druggable small molecule antagonists of the Plasmodium falciparum hexose transporter PfHT and assessment of ligand access to the glucose permeation pathway via FLAG-mediated protein engineering. PLoS ONE 2019, 14, e0216457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, B.; Rambow, J.; Bock, S.; Holm-Bertelsen, J.; Wiechert, M.; Blancke Soares, A.; Spielmann, T.; Beitz, E. Identity of a Plasmodium lactate/H+ symporter structurally unrelated to human transporters. Nat. Commun. 2015, 6, 6284. [Google Scholar] [CrossRef] [PubMed]
- Kanaani, J.; Ginsburg, H. Effects of cinnamic acid derivatives on in vitro growth of Plasmodium falciparum and on the permeability of the membrane of malaria-infected erythrocytes. Antimicrob. Agents Chemother. 1992, 36, 1102–1108. [Google Scholar] [CrossRef] [Green Version]
- Cranmer, S.L.; Conant, A.R.; Gutteridge, W.E.; Halestrap, A.P. Characterization of the enhanced transport of L- and D-lactate into human red blood cells infected with Plasmodium falciparum suggests the presence of a novel saturable lactate proton cotransporter. J. Biol. Chem. 1995, 270, 15045–15052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, J.L.; Saliba, K.J.; Kirk, K. Transport of lactate and pyruvate in the intraerythrocytic malaria parasite, Plasmodium falciparum. Biochem. J. 2001, 355, 733–739. [Google Scholar] [CrossRef]
- Marchetti, R.V.; Lehane, A.M.; Shafik, S.H.; Winterberg, M.; Martin, R.E.; Kirk, K. A lactate and formate transporter in the intraerythrocytic malaria parasite, Plasmodium falciparum. Nat. Commun. 2015, 6, 6721. [Google Scholar] [CrossRef] [Green Version]
- Poole, R.C.; Halestrap, A.P. N-terminal protein sequence analysis of the rabbit erythrocyte lactate transporter suggests identity with the cloned monocarboxylate transport protein MCT1. Biochem. J. 1994, 303, 755–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walloch, P.; Hansen, C.; Priegann, T.; Schade, D.; Beitz, E. Pentafluoro-3-hydroxy-pent-2-en-1-ones potently inhibit FNT-type lactate transporters from all five human-pathogenic Plasmodium species. ChemMedChem 2021, 16, 1283–1289. [Google Scholar] [CrossRef]
- Erler, H.; Ren, B.; Gupta, N.; Beitz, E. The intracellular parasite Toxoplasma gondii harbors three druggable FNT-type formate and L-lactate transporters in the plasma membrane. J. Biol. Chem. 2018, 293, 17622–17630. [Google Scholar] [CrossRef] [Green Version]
- Helmstetter, F.; Arnold, P.; Höger, B.; Petersen, L.M.; Beitz, E. Formate-nitrite transporters carrying nonprotonatable amide amino acids instead of a central histidine maintain pH-dependent transport. J. Biol. Chem. 2019, 294, 623–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajek, P.; Bader, A.; Helmstetter, F.; Henke, B.; Arnold, P.; Beitz, E. Cell-free and yeast-based production of the malarial lactate transporter, PfFNT, delivers comparable yield and protein quality. Front. Pharmacol. 2019, 10, 375. [Google Scholar] [CrossRef]
- Lyu, M.; Su, C.; Kazura, J.W.; Yu, E.W. Structural basis of transport and inhibition of the Plasmodium falciparum transporter PfFNT. EMBO Rep. 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Wang, N.; Zhu, A.; Xu, H.; Li, J.; Zhou, Y.; Wang, C.; Xiao, Q.; Guo, L.; Liu, F.; et al. Structural characterization of the Plasmodium falciparum lactate transporter PfFNT alone and in complex with antimalarial compound MMV007839 reveals its inhibition mechanism. PLoS Biol. 2021, 19, e3001386. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, Y.; Wang, J.; Cheng, C.; Huang, W.; Lu, P.; Xu, Y.N.; Wang, P.; Yan, N.; Shi, Y. Structure of the formate transporter FocA reveals a pentameric aquaporin-like channel. Nature 2009, 462, 467–472. [Google Scholar] [CrossRef]
- Wiechert, M.; Beitz, E. Mechanism of formate–nitrite transporters by dielectric shift of substrate acidity. EMBO J. 2017, 36, 949–958. [Google Scholar] [CrossRef] [Green Version]
- Wiechert, M.; Beitz, E. Formate-nitrite transporters: Monoacids ride the dielectric slide. Channels 2017, 11, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Bader, A.; Beitz, E. Transmembrane facilitation of lactate/H+ instead of lactic acid is not a question of semantics but of cell viability. Membranes 2020, 10, 236. [Google Scholar] [CrossRef]
- Walloch, P.; Henke, B.; Häuer, S.; Bergmann, B.; Spielmann, T.; Beitz, E. Introduction of scaffold nitrogen atoms renders inhibitors of the malarial L-lactate transporter, PfFNT, effective against the Gly107Ser resistance mutation. J. Med. Chem. 2020, 63, 9731–9741. [Google Scholar] [CrossRef] [PubMed]
- Hapuarachchi, S.V.; Cobbold, S.A.; Shafik, S.H.; Dennis, A.S.M.; McConville, M.J.; Martin, R.E.; Kirk, K.; Lehane, A.M. The malaria parasite’s lactate transporter PfFNT is the target of antiplasmodial compounds identified in whole cell phenotypic screens. PLoS Pathog. 2017, 13, e1006180. [Google Scholar] [CrossRef]
- Spangenberg, T.; Burrows, J.N.; Kowalczyk, P.; McDonald, S.; Wells, T.N.C.; Willis, P. The open access malaria box: A drug discovery catalyst for neglected diseases. PLoS ONE 2013, 8, e62906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golldack, A.; Henke, B.; Bergmann, B.; Wiechert, M.; Erler, H.; Blancke Soares, A.; Spielmann, T.; Beitz, E. Substrate-analogous inhibitors exert antimalarial action by targeting the Plasmodium lactate transporter PfFNT at nanomolar scale. PLoS Pathog. 2017, 13, e1006172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakobowska, I.; Becker, F.; Minguzzi, S.; Hansen, K.; Henke, B.; Epalle, N.H.; Beitz, E.; Hannus, S. Fluorescence cross-correlation spectroscopy yields true affinity and binding kinetics of Plasmodium lactate transport inhibitors. Pharmaceuticals 2021, 14, 757. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nerlich, C.; Epalle, N.H.; Seick, P.; Beitz, E. Discovery and Development of Inhibitors of the Plasmodial FNT-Type Lactate Transporter as Novel Antimalarials. Pharmaceuticals 2021, 14, 1191. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14111191
Nerlich C, Epalle NH, Seick P, Beitz E. Discovery and Development of Inhibitors of the Plasmodial FNT-Type Lactate Transporter as Novel Antimalarials. Pharmaceuticals. 2021; 14(11):1191. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14111191
Chicago/Turabian StyleNerlich, Cornelius, Nathan H. Epalle, Philip Seick, and Eric Beitz. 2021. "Discovery and Development of Inhibitors of the Plasmodial FNT-Type Lactate Transporter as Novel Antimalarials" Pharmaceuticals 14, no. 11: 1191. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14111191