
Contribution of Mitochondrial Dysfunction Combined with NLRP3 Inflammasome Activation in Selected Neurodegenerative Diseases

 , ,
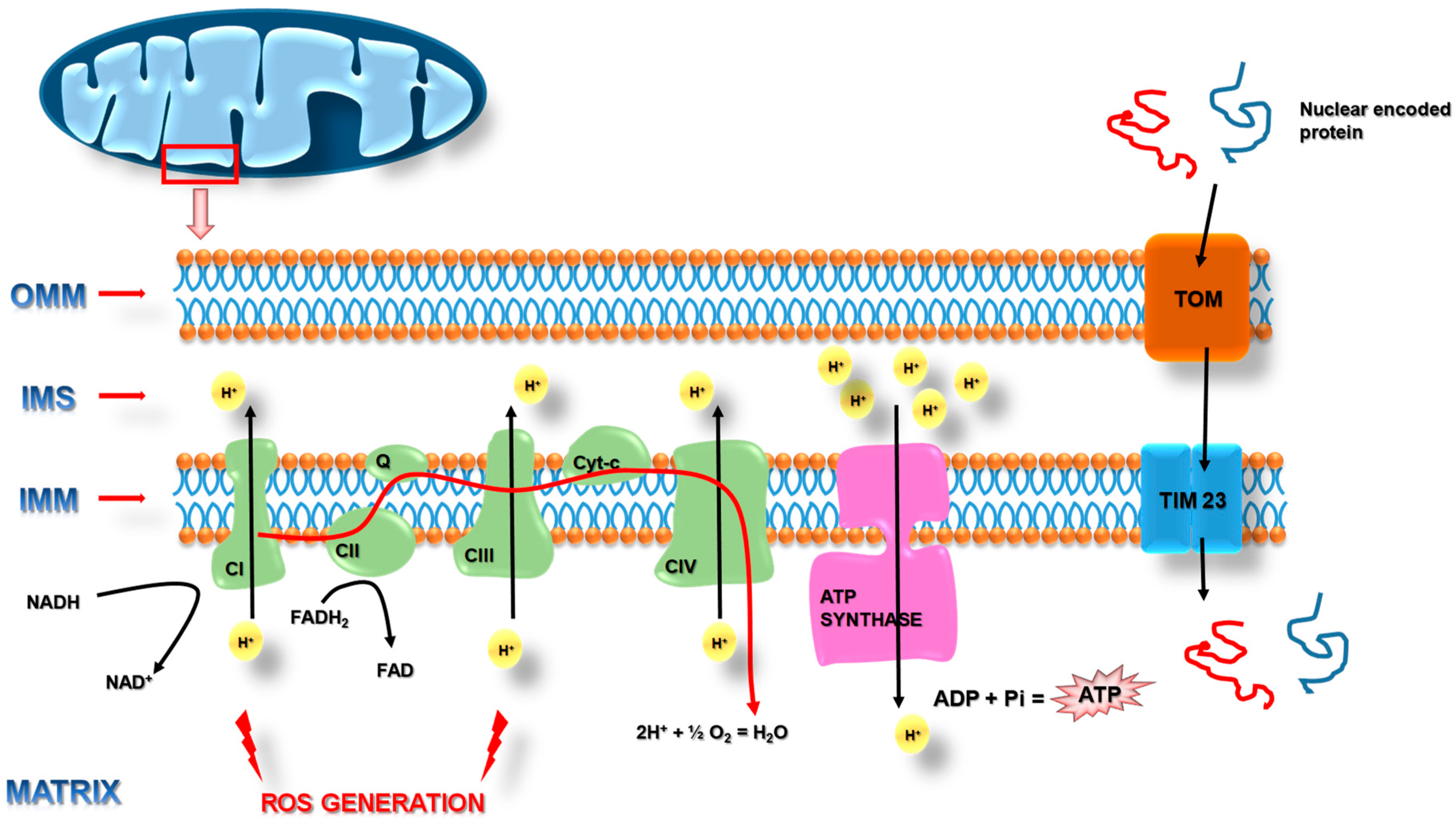
, , 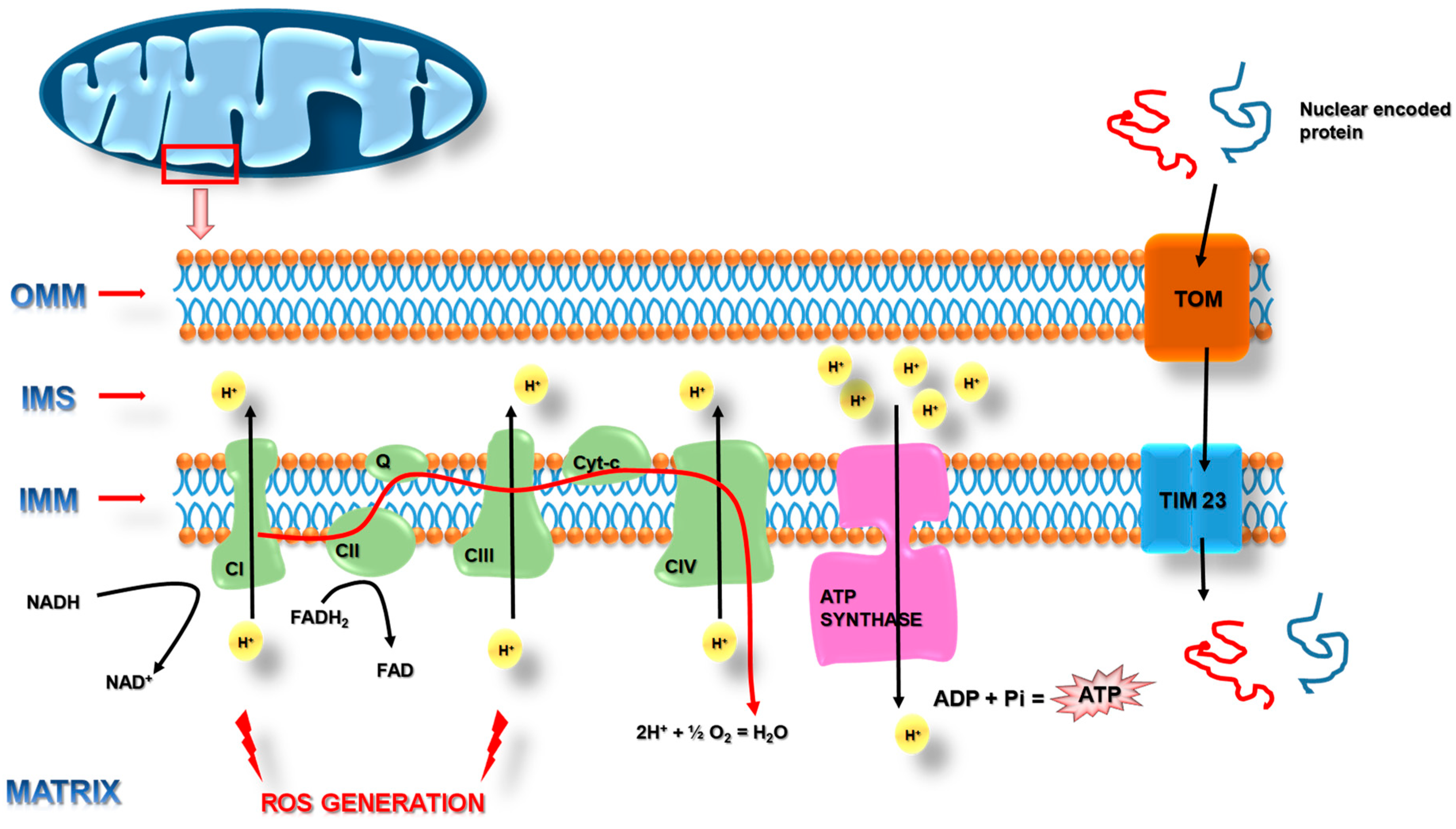{kind=link}
{kind=link}
{kind=link}
{kind=link}
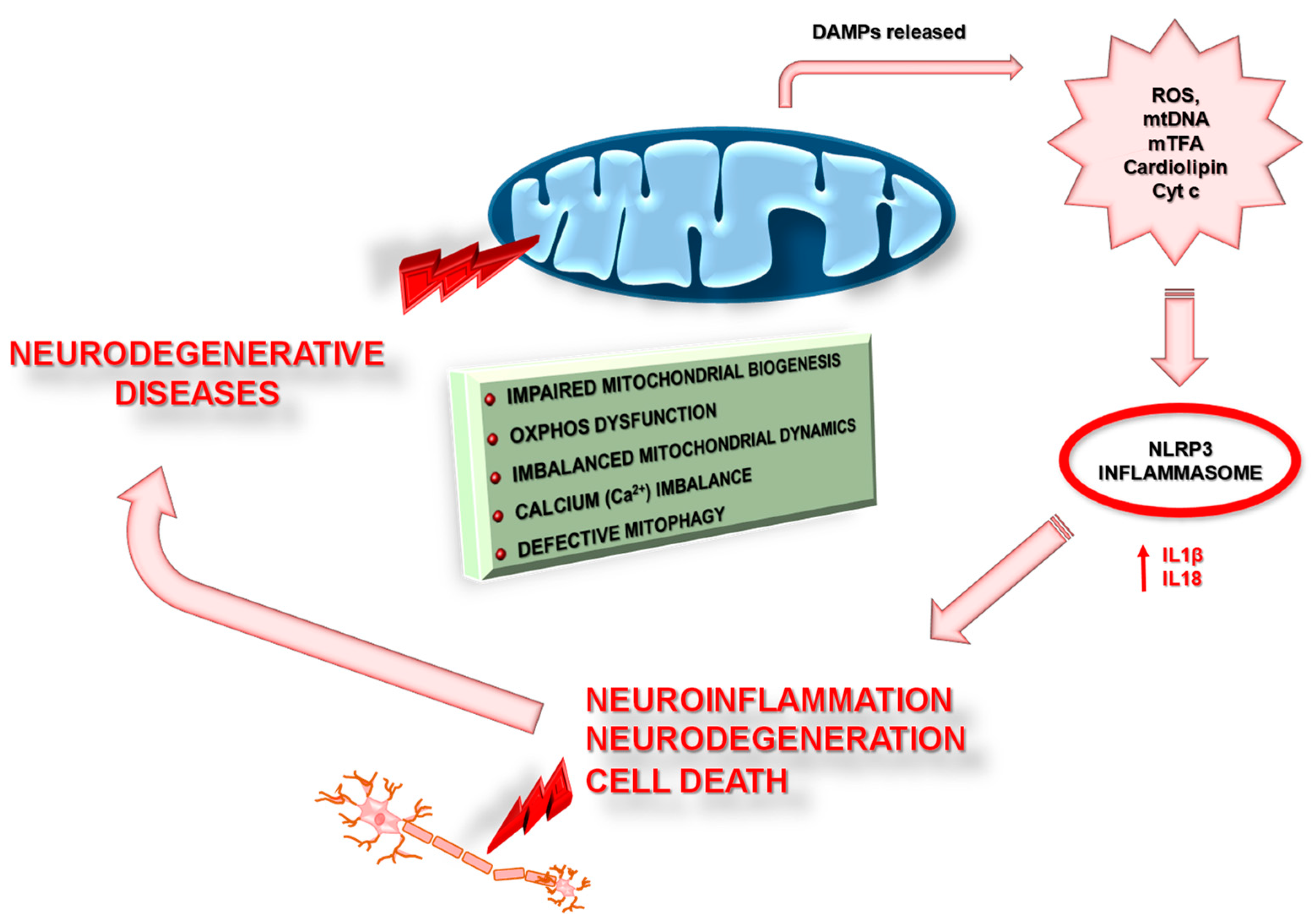{kind=link}
Abstract
:1. Introduction
2. Mitochondria
2.1. Function and Structure of Mitochondria
2.2. Mitochondrial Homeostasis
3. Inflammasomes
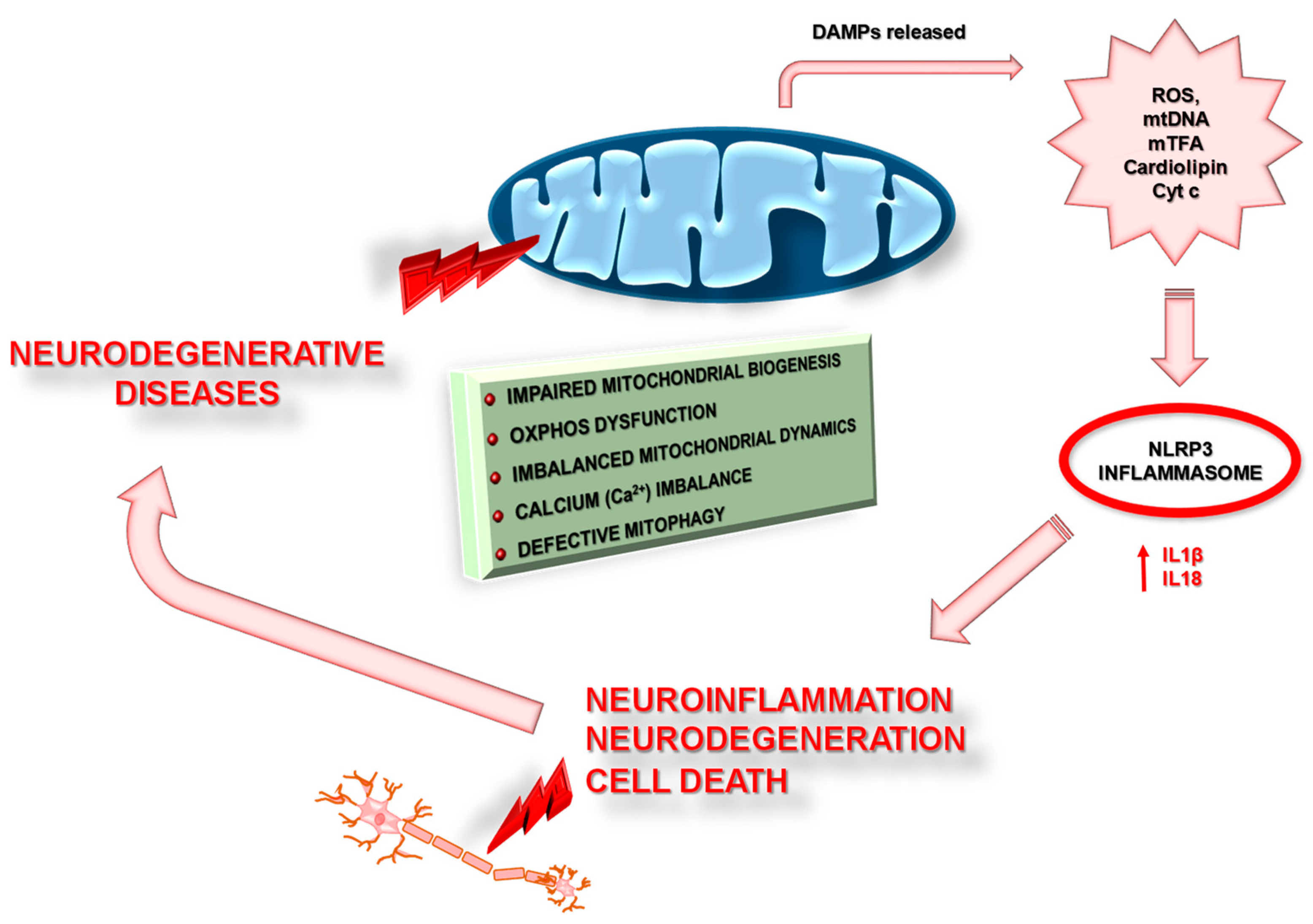
3.1. Inflammasomes and Mitochondria
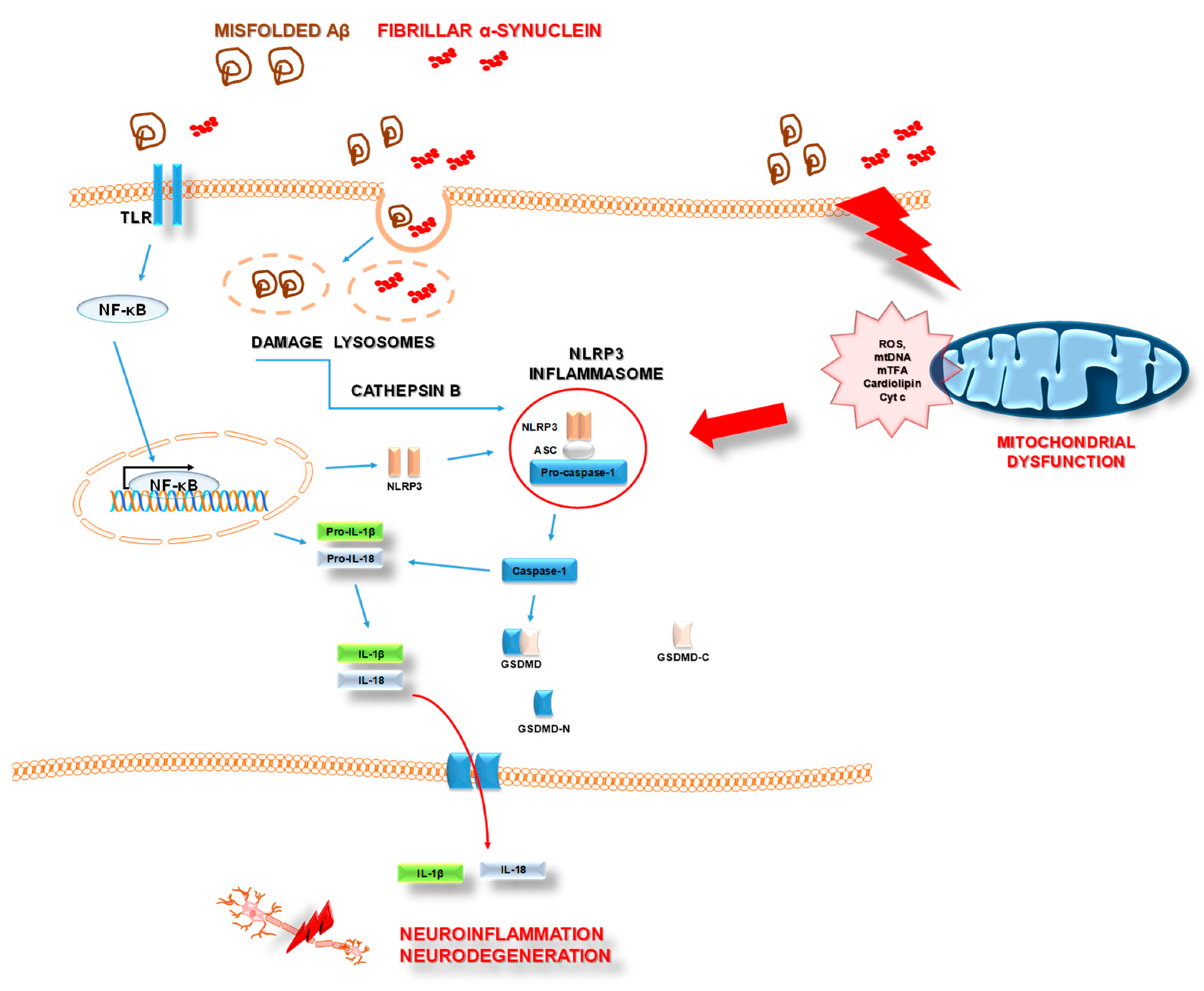
3.2. NLRP3 Inflammasome and AD
3.3. NLRP3 Inflammasome and PD
4. Mitochondrial Dysfunction in Neurodegenerative Disease
4.1. Mitochondrial Dysfunction in Alzheimer’s Disease
4.1.1. Deficiency in Mitochondrial Oxidative Phosphorylation in AD
4.1.2. Disruption of Mitochondrial Homeostasis in AD
4.1.3. Mitochondrial Dysfunction and Neuroinflammation in AD
4.2. Mitochondrial Dysfunction in Parkinson Disease
4.2.1. Deficiency in Mitochondrial Oxidative Phosphorylation in PD
4.2.2. Disruption of Mitochondrial Homeostasis in PD
4.2.3. Mitochondrial Dysfunction and Neuroinflammation in PD
5. Mitochondria as a Therapeutic Goal in Neurodegenerative Disease Treatment
5.1. Enhance Electron Transport Chain Activity
5.2. Regulation of Mitochondrial Homeostasis and Inflammation
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s Disease |
| AIM2 | Absent in Melanoma 2 |
| ALS | Amyotrophic Lateral Sclerosis |
| AMPK | 5′AMP-activated protein kinase |
| APOE4 | ApolipoproteinaE-allele4 |
| APP | Amyloid Precursor Protein |
| ASC | Adaptor Apoptosis Speck Protein |
| ATP | Adenosine Triphosphate |
| Aβ | β-amyloid |
| AβPP | Amyloid-β Protein Precursor |
| CL | Cardiolipin |
| CNS | Central Nervous System |
| COX | Cytochrome c oxidase |
| DA | Dopamine |
| DAMP | Damage-Associated Molecular Patterns |
| Drp1 | Dynamin-Related GTPase |
| ERR-β | Estrogen-related receptors -β |
| ERR-γ | Estrogen-related receptors -γ |
| ERR-α | Estrogen-related receptors -α |
| ETC | Electron Transport Chains |
| Fis1 | Mitochondrial fission 1 protein |
| HD | Huntington’s Disease |
| IL-18 | Interleukin 18 |
| IL-1β | Interleukin 1β |
| IL-6 | Interleukin 6 |
| IL-8 | Interleukin 8 |
| IMM | Inter Mitochondria Membrane |
| INF-γ | Interferon γ |
| LBs | Lewy bodies |
| LC3 | Microtubule Associated Proteins 1A/1B light chain 3B |
| LOAD | Late-onset AD |
| MAP-5 | Microtubule Associated Protein-5 |
| Mff | Mitochondrial Fission Factor |
| MFN1 | Mitofusin 1 |
| MFN2 | Mitofusin 2 |
| Mid49 | Mitochondrial Dynamics Proteins of 49 |
| Mid51 | Mitochondrial Dynamics Proteins of 51 |
| Miro1 | Mitochondrial Rho GTPase1 |
| MPP+ | 1-methyl-4-phenylpyridinium |
| mPTP | Mitochondrial Permeability Transition Pore |
| MS | Multiple Sclerosis |
| mtDNA | Mitochondria DNA |
| mTFA | Mitochondrial Transcription Factor A |
| mtΔΨ | Mitochondrial Membrane Potential |
| nDNA | Nuclear DNA |
| NDs | Neurodegenerative Diseases |
| NFTs | Neurofibrillary Tangles |
| NF-κβ | Nuclear Factor kappa-light-chain-enhancer of activated β cells |
| NLRC4 | CARD domain-containing protein 4/IPAF ICE-Protease Activating Factor |
| NLRP2 | NOD-like receptor protein 2 |
| NLRP3 | NOD-like receptor protein 3 |
| NRF1 | Nuclear Respiratory Factor 1 |
| NRF2 | Nuclear Respiratory Factor 2 |
| OMM | Outer Mitochondrial Membrane |
| OPA1 | Optic Dominant Atrophy 1 |
| OXPHOS | Mitochondrial Oxidative Phosphorylation System |
| PAMP | Pathogen-Associated Molecular Patterns |
| PD | Parkinson’s disease |
| PGC-1α | Peroxisome proliferator activated receptor gamma coactivator-1α |
| PINK1 | PTEN-induced putative kinase 1 |
| PRRs | Pattern Recognition Receptors |
| PS1 | Presenilin-1 |
| RNS | Reactive Nitrogen Species |
| ROS | Reactive Oxygen Species |
| rRNAs | Ribosomal RNAs |
| SN | Substantia Nigra |
| SNCA | α-synuclein |
| sPD | Sporadic Form of Parkinson’s Disease |
| SPs | Senile Plaques |
| TCA | Tricarboxylic Acid Cycle |
| TIM | Translocase of the Inner Membrane |
| TIM23 | Translocase of the Inner Mitochondrial Membrane 23 |
| TLRs | Toll-Like Receptors |
| TNF-α | Tumor Necrosis Factor α |
| TOM | Translocase of the Outer Membrane |
| TOM20 | Translocase of Outer Membrane 20 |
| Trem2 | Triggering receptor expressed on myeloid Cells 2 |
| TREM2 | Triggering Receptor Expressed on Myeloid Cells 2 |
| tRNAs | Transfer RNAs |
| VDAC1 | Voltage-Dependent Anion-Selective Channel 1 |
| αKGDH | Pyruvate Dehydrogenase Complex, α-Ketoglutarate Dehydrogenase |
References
- Holbrook, J.A.; Jarosz-Griffiths, H.H.; Caseley, E.; Lara-Reyna, S.; Poulter, J.A.; Williams-Gray, C.H.; Peckham, D.; McDermott, M.F. Neurodegenerative Disease and the NLRP3 Inflammasome. Front. Pharmacol. 2021, 12, 193. [Google Scholar] [CrossRef]
- Weller, J.; Budson, A. Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Research 2018, 7, 1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berchtold, N.; Cotman, C. Evolution in the Conceptualization of Dementia and Alzheimer’s Disease: Greco-Roman Period to the 1960s. Neurobiol. Aging 1998, 19, 173–189. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Cenini, G.; Voos, W. Mitochondria as Potential Targets in Alzheimer Disease Therapy: An Update. Front. Pharmacol. 2019, 10, 902. [Google Scholar] [CrossRef]
- Feigin, V.L.; Nichols, E.; Alam, T.; Bannick, M.S.; Beghi, E.; Blake, N.; Culpepper, W.J.; Dorsey, E.R.; Elbaz, A.; Ellenbogen, R.G.; et al. Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef] [Green Version]
- Forno, L.S. Neuropathology of Parkinson’s Disease. J. Neuropathol. Exp. Neurol. 1996, 55, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Noda, K.; Kitami, T.; Gai, W.P.; Chegini, F.; Jensen, P.H.; Fujimura, T.; Murayama, K.; Tanaka, K.; Mizuno, Y.; Hattori, N. Phosphorylated IκBα is a component of Lewy body of Parkinson’s disease. Biochem. Biophys. Res. Commun. 2005, 331, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Kim, Y.M.; Lee, G.; Junn, E.; Iwatsubo, T.; Mouradian, M.M. Aggresomes Formed by α-Synuclein and Synphilin-1 Are Cytoprotective. J. Biol. Chem. 2004, 279, 4625–4631. [Google Scholar] [CrossRef] [Green Version]
- Tapias, V. Editorial: Mitochondrial Dysfunction and Neurodegeneration. Front. Neurosci. 2019, 13, 1372. [Google Scholar] [CrossRef] [Green Version]
- Compagnoni, G.M.; Di Fonzo, A.; Corti, S.; Comi, G.P.; Bresolin, N.; Masliah, E. The Role of Mitochondria in Neurodegenerative Diseases: The Lesson from Alzheimer’s Disease and Parkinson’s Disease. Mol. Neurobiol. 2020, 57, 2959–2980. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, D.; Catania, A.; Viscomi, C.; Deleidi, M.; Bindoff, L.; Ghezzi, D.; Zeviani, M. Role of PITRM1 in Mitochondrial Dysfunction and Neurodegeneration. Biomedicines 2021, 9, 833. [Google Scholar] [CrossRef]
- Bahat, A.; MacVicar, T.; Langer, T. Metabolism and Innate Immunity Meet at the Mitochondria. Front. Cell Dev. Biol. 2021, 9, 2019. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef]
- Zhang, T.; Ding, S.; Wang, R. Research Progress of Mitochondrial Mechanism in NLRP3 Inflammasome Activation and Exercise Regulation of NLRP3 Inflammasome. Int. J. Mol. Sci. 2021, 22, 10866. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, A.; Scorrano, L. Mitochondria: From cell death executioners to regulators of cell differentiation. Trends Cell Biol. 2014, 24, 761–770. [Google Scholar] [CrossRef]
- Frey, T.G.; Mannella, C.A. The internal structure of mitochondria. Trends Biochem. Sci. 2000, 25, 319–324. [Google Scholar] [CrossRef]
- Picard, M.; Taivassalo, T.; Gouspillou, G.; Hepple, R.T. Mitochondria: Isolation, structure and function. J. Physiol. 2011, 589, 4413–4421. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; Schon, E.A. Mitochondrial Respiratory-Chain Diseases. N. Engl. J. Med. 2003, 348, 2656–2668. [Google Scholar] [CrossRef]
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial proteins: From biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Golpich, M.; Amini, E.; Mohamed, Z.; Ali, R.A.; Ibrahim, N.M.; Ahmadiani, A. Mitochondrial Dysfunction and Biogenesis in Neurodegenerative diseases: Pathogenesis and Treatment. CNS Neurosci. Ther. 2017, 23, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis and clearance: A balancing act. FEBS J. 2017, 284, 183–195. [Google Scholar] [CrossRef]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [CrossRef] [Green Version]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Balancing mitochondrial biogenesis and mitophagy to maintain energy metabolism homeostasis. Cell Death Differ. 2015, 22, 1399–1401. [Google Scholar] [CrossRef] [Green Version]
- Coppi, L.; Ligorio, S.; Mitro, N.; Caruso, D.; De Fabiani, E.; Crestani, M. PGC1s and Beyond: Disentangling the Complex Regulation of Mitochondrial and Cellular Metabolism. Int. J. Mol. Sci. 2021, 22, 6913. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.J.; Buckingham, J.A.; Brand, M.D. Dissociation of superoxide production by mitochondrial complex I from NAD(P)H redox state. FEBS Lett. 2008, 582, 1711–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinogradov, A.D.; Grivennikova, V.G. Oxidation of NADH and ROS production by respiratory complex I. Biochim. Biophys. Acta (BBA) Bioenerg. 2016, 1857, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Allio, R.; Donega, S.; Galtier, N.; Nabholz, B. Large Variation in the Ratio of Mitochondrial to Nuclear Mutation Rate across Animals: Implications for Genetic Diversity and the Use of Mitochondrial DNA as a Molecular Marker. Mol. Biol. Evol. 2017, 34, 2762–2772. [Google Scholar] [CrossRef] [Green Version]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef] [Green Version]
- Contis, A.; Mitrovic, S.; Lavie, J.; Douchet, I.; Lazaro, E.; Truchetet, M.-E.; Goizet, C.; Contin-Bordes, C.; Schaeverbeke, T.; Blanco, P.; et al. Neutrophil-derived mitochondrial DNA promotes receptor activator of nuclear factor κB and its ligand signalling in rheumatoid arthritis. Rheumatology 2017, 56, 1200–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Kroemer, G.; Kepp, O. Mitophagy: An Emerging Role in Aging and Age-Associated Diseases. Front. Cell Dev. Biol. 2020, 8, 200. [Google Scholar] [CrossRef] [Green Version]
- Meissner, C.; Lorenz, H.; Weihofen, A.; Selkoe, D.J.; Lemberg, M.K. The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J. Neurochem. 2011, 117, 856–867. [Google Scholar] [CrossRef]
- Deas, E.; Plun-Favreau, H.; Gandhi, S.; Desmond, H.; Kjaer, S.; Loh, S.H.; Renton, A.E.; Harvey, R.; Whitworth, A.; Martins, L.M.; et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 2011, 20, 867–879. [Google Scholar] [CrossRef]
- Yamano, K.; Youle, R.J. PINK1 is degraded through the N-end rule pathway. Autophagy 2013, 9, 1758–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, S.-M.; Jung, Y.-K. A Molecular Approach to Mitophagy and Mitochondrial Dynamics. Mol. Cells 2018, 41, 18–26. [Google Scholar] [PubMed]
- Bernardini, J.P.; Brouwer, J.M.; Tan, I.K.; Sandow, J.J.; Huang, S.; Stafford, C.A.; Bankovacki, A.; Riffkin, C.D.; Wardak, A.Z.; Czabotar, P.E.; et al. Parkin inhibits BAK and BAX apoptotic function by distinct mechanisms during mitophagy. EMBO J. 2019, 38, e99916. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Fusion and Fission: Interlinked Processes Critical for Mitochondrial Health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.; Ying, J.; Wang, X.; Zhao, T.; Yoon, S.; Fang, Y.; Zheng, Q.; Liu, X.; Yu, W.; Hua, F. Mitochondrial Dynamics: A Key Role in Neurodegeneration and a Potential Target for Neurodegenerative Disease. Front. Neurosci. 2021, 15, 359. [Google Scholar] [CrossRef] [PubMed]
- Kameoka, S.; Adachi, Y.; Okamoto, K.; Iijima, M.; Sesaki, H. Phosphatidic Acid and Cardiolipin Coordinate Mitochondrial Dynamics. Trends Cell Biol. 2018, 28, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, E.; Griparic, L.; Shurland, D.-L.; van der Bliek, A.M. Dynamin-related Protein Drp1 Is Required for Mitochondrial Division in Mammalian Cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhen, Y.; Zhang, H. NLRP3 Inflammasome and Inflammatory Bowel Disease. Front. Immunol. 2019, 10, 276. [Google Scholar] [CrossRef] [Green Version]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Groslambert, M.; Py, B.F. Spotlight on the NLRP3 inflammasome pathway. J. Inflamm. Res. 2018, 11, 359–374. [Google Scholar] [CrossRef] [Green Version]
- Ketelut-Carneiro, N.; Silva, G.K.; Rocha, F.A.; Milanezi, C.M.; Cavalcanti-Neto, F.F.; Zamboni, D.; Silva, J.S. IL-18 Triggered by the Nlrp3 Inflammasome Induces Host Innate Resistance in a Pulmonary Model of Fungal Infection. J. Immunol. 2015, 194, 4507–4517. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, C.; Hua, M.; Wang, R.; Zhong, C.; Yu, J.; Han, F.; He, N.; Zhao, Y.; Liu, G.; et al. NLRP3 inflammasome activation plays a carcinogenic role through effector cytokine IL-18 in lymphoma. Oncotarget 2017, 8, 108571–108583. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.-C.; Zhu, X.-X.; Xue, Y.; Zhang, L.-H.; Zou, H.-J.; Qiu, J.-H.; Liu, Q. Role of the NLRP3 inflammasome in the transient release of IL-1β induced by monosodium urate crystals in human fibroblast-like synoviocytes. J. Inflamm. 2015, 12, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, J.K.; MacPherson, M.B.; Beuschel, S.L.; Shukla, A. Asbestos-Induced Mesothelial to Fibroblastic Transition Is Modulated by the Inflammasome. Am. J. Pathol. 2017, 187, 665–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litwiniuk, A.; Domańska, A.; Chmielowska, M.; Martyńska, L.; Bik, W.; Kalisz, M. The Effects of Alpha-Linolenic Acid on the Secretory Activity of Astrocytes and β Amyloid-Associated Neurodegeneration in Differentiated SH-SY5Y Cells: Alpha-Linolenic Acid Protects the SH-SY5Y cells against β Amyloid Toxicity. Oxid. Med. Cell. Longev. 2020, 2020, 1–20. [Google Scholar] [CrossRef]
- Srivastava, S. The Mitochondrial Basis of Aging and Age-Related Disorders. Genes 2017, 8, 398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Elliott, E.I.; Miller, A.N.; Banoth, B.; Iyer, S.S.; Stotland, A.; Weiss, J.P.; Gottlieb, R.A.; Sutterwala, F.S.; Cassel, S.L. Cutting Edge: Mitochondrial Assembly of the NLRP3 Inflammasome Complex Is Initiated at Priming. J. Immunol. 2018, 200, 3047–3052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [Green Version]
- Piancone, F.; La Rosa, F.; Marventano, I.; Saresella, M.; Clerici, M. The Role of the Inflammasome in Neurodegenerative Diseases. Molecules 2021, 26, 953. [Google Scholar] [CrossRef]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [Green Version]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial Dysfunction and Defective β-Amyloid Clearance Pathways in Aging Alzheimer’s Disease Mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef]
- Lee, J.; Kim, H.; Kim, J.; Yook, T.; Kim, K.; Lee, J.; Yang, G. A Novel Treatment Strategy by Natural Products in NLRP3 Inflammasome-Mediated Neuroinflammation in Alzheimer’s and Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 1324. [Google Scholar] [CrossRef]
- Saresella, M.; La Rosa, F.; Piancone, F.; Zoppis, M.; Marventano, I.; Calabrese, E.; Rainone, V.; Nemni, R.; Mancuso, R.; Clerici, M. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xia, Y.; Yin, S.; Wan, F.; Hu, J.; Kou, L.; Sun, Y.; Wu, J.; Zhou, Q.; Huang, J.; et al. Targeting Microglial α-Synuclein/TLRs/NF-kappaB/NLRP3 Inflammasome Axis in Parkinson’s Disease. Front. Immunol. 2021, 12, 359. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 2018, 10, eaah4066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bifari, F.; Dolci, S.; Bottani, E.; Pino, A.; Di Chio, M.; Zorzin, S.; Ragni, M.; Zamfir, R.G.; Brunetti, D.; Bardelli, D.; et al. Complete neural stem cell (NSC) neuronal differentiation requires a branched chain amino acids-induced persistent metabolic shift towards energy metabolism. Pharmacol. Res. 2020, 158, 104863. [Google Scholar] [CrossRef]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain Energy Metabolism: Focus on Astrocyte-Neuron Metabolic Cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, E.J.; Rutter, G.A. Mitochondrial calcium as a key regulator of mitochondrial ATP production in mammalian cells. Biochim. Biophys. Acta (BBA) Bioenerg. 2009, 1787, 1324–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llorente-Folch, I.; Rueda, C.; Amigo, I.; del Arco, A.; Saheki, T.; Pardo, B.; Satrústegui, J. Calcium-Regulation of Mitochondrial Respiration Maintains ATP Homeostasis and Requires ARALAR/AGC1-Malate Aspartate Shuttle in Intact Cortical Neurons. J. Neurosci. 2013, 33, 13957–13971. [Google Scholar] [CrossRef]
- Huang, C.; Yan, S.; Zhang, Z. Maintaining the balance of TDP-43, mitochondria, and autophagy: A promising therapeutic strategy for neurodegenerative diseases. Transl. Neurodegener. 2020, 9, 1–16. [Google Scholar] [CrossRef]
- Sharma, C.; Kim, S.; Nam, Y.; Jung, U.; Kim, S. Mitochondrial Dysfunction as a Driver of Cognitive Impairment in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 4850. [Google Scholar] [CrossRef]
- Carinci, M.; Vezzani, B.; Patergnani, S.; Ludewig, P.; Lessmann, K.; Magnus, T.; Casetta, I.; Pugliatti, M.; Pinton, P.; Giorgi, C. Different Roles of Mitochondria in Cell Death and Inflammation: Focusing on Mitochondrial Quality Control in Ischemic Stroke and Reperfusion. Biomedicines 2021, 9, 169. [Google Scholar] [CrossRef]
- Mehta, A.R.; Gregory, J.M.; Dando, O.; Carter, R.N.; Burr, K.; Nanda, J.; Story, D.; McDade, K.; Smith, C.; Morton, N.M.; et al. Mitochondrial bioenergetic deficits in C9orf72 amyotrophic lateral sclerosis motor neurons cause dysfunctional axonal homeostasis. Acta Neuropathol. 2021, 141, 257–279. [Google Scholar] [CrossRef]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H. Amyloid beta, mitochondrial structural and functional dynamics in Alzheimer’s disease. Exp. Neurol. 2009, 218, 286–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonntag, K.-C.; Ryu, W.-I.; Amirault, K.M.; Healy, R.A.; Siegel, A.J.; McPhie, D.L.; Forester, B.; Cohen, B.M. Late-onset Alzheimer’s disease is associated with inherent changes in bioenergetics profiles. Sci. Rep. 2017, 7, 14038. [Google Scholar] [CrossRef] [Green Version]
- Maurer, I. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol. Aging 2000, 21, 455–462. [Google Scholar] [CrossRef]
- Gibson, G.E.; Shi, Q. A Mitocentric View of Alzheimer’s Disease Suggests Multi-Faceted Treatments. J. Alzheimer’s Dis. 2010, 20, S591–S607. [Google Scholar] [CrossRef] [Green Version]
- Wojsiat, J.; Prandelli, C.; Laskowska-Kaszub, K.; Martín-Requero, A.; Wojda, U. Oxidative Stress and Aberrant Cell Cycle in Alzheimer’s Disease Lymphocytes: Diagnostic Prospects. J. Alzheimer’s Dis. 2015, 46, 329–350. [Google Scholar] [CrossRef] [PubMed]
- Beck, S.J.; Guo, L.; Phensy, A.; Tian, J.; Wang, L.; Tandon, N.; Gauba, E.; Lu, L.; Pascual, J.M.; Kroener, S.; et al. Deregulation of mitochondrial F1FO-ATP synthase via OSCP in Alzheimer’s disease. Nat. Commun. 2016, 7, 11483. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, C.; Cardoso, S.; Correia, S.C.; Santos, R.X.; Santos, M.S.; Baldeiras, I.; Oliveira, C.R.; Moreira, P.I. Metabolic Alterations Induced by Sucrose Intake and Alzheimer’s Disease Promote Similar Brain Mitochondrial Abnormalities. Diabetes 2012, 61, 1234–1242. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Rissman, R.A.; Feng, J. Characterization of ATP Alternations in an Alzheimer’s Disease Transgenic Mouse Model. J. Alzheimer’s Dis. 2015, 44, 375–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of Aβ accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006, 15, 1437–1449. [Google Scholar] [CrossRef]
- Caspersen, C.; Wang, N.; Yao, J.; Sosunov, A.; Chen, X.; Lustbader, J.W.; Xu, H.W.; Stern, D.; McKhann, G.; Du Yan, S. Mitochondrial Aβ: A potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005, 19, 2040–2041. [Google Scholar] [CrossRef] [PubMed]
- Petersen, C.A.H.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamata, H.; Manfredi, G. Proteinopathies and OXPHOS dysfunction in neurodegenerative diseases. J. Cell Biol. 2017, 216, 3917–3929. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of Amyloid Precursor Protein in the Mitochondrial Import Channels of Human Alzheimer’s Disease Brain Is Associated with Mitochondrial Dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef] [Green Version]
- Gong, B.; Chen, F.; Pan, Y.; Arrieta-Cruz, I.; Yoshida, Y.; Haroutunian, V.; Pasinetti, G.M. SCFFbx2-E3-ligase-mediated degradation of BACE1 attenuates Alzheimer’s disease amyloidosis and improves synaptic function. Aging Cell 2010, 9, 1018–1031. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Guo, M.-N.; Liu, Z.-Z.; Ma, S.-F.; Liu, W.-J.; Qian, J.-J.; Zhang, W.-N. PGC-1α reduces Amyloid-β deposition in Alzheimer’s disease: Effect of increased VDR expression. Neurosci. Lett. 2021, 744, 135598. [Google Scholar] [CrossRef]
- Qin, W.; Haroutunian, V.; Katsel, P.; Cardozo, C.P.; Ho, L.; Buxbaum, J.; Pasinetti, G.M. PGC-1α Expression Decreases in the Alzheimer Disease Brain as a Function of Dementia. Arch. Neurol. 2009, 66, 352–361. [Google Scholar] [CrossRef]
- Oka, S.; Leon, J.; Sakumi, K.; Ide, T.; Kang, N.; LaFerla, F.M.; Nakabeppu, Y. Human mitochondrial transcriptional factor A breaks the mitochondria-mediated vicious cycle in Alzheimer’s disease. Sci. Rep. 2016, 6, 37889. [Google Scholar] [CrossRef] [Green Version]
- Song, C.; Li, M.; Xu, L.; Shen, Y.; Yang, H.; Ding, M.; Liu, X.; Xie, Z. Mitochondrial biogenesis mediated by melatonin in an APPswe/PS1dE9 transgenic mice model. NeuroReport 2018, 29, 1517–1524. [Google Scholar] [CrossRef]
- Reddy, P.H.; Reddy, T. Mitochondria as a Therapeutic Target for Aging and Neurodegenerative Diseases. Curr. Alzheimer Res. 2011, 8, 393–409. [Google Scholar] [CrossRef]
- Xu, L.-L.; Shen, Y.; Wang, X.; Wei, L.-F.; Wang, P.; Yang, H.; Wang, C.-F.; Xie, Z.-H.; Bi, J.-Z. Mitochondrial dynamics changes with age in an APPsw/PS1dE9 mouse model of Alzheimer’s disease. NeuroReport 2017, 28, 222–228. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Trushin, S.; Christensen, T.A.; Bachmeier, B.V.; Gateno, B.; Schroeder, A.; Yao, J.; Itoh, K.; Sesaki, H.; Poon, W.W.; et al. Altered brain energetics induces mitochondrial fission arrest in Alzheimer’s Disease. Sci. Rep. 2016, 6, 18725. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Su, B.; Lee, H.-G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired Balance of Mitochondrial Fission and Fusion in Alzheimer’s Disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef]
- Kandimalla, R.; Manczak, M.; Fry, D.; Suneetha, Y.; Sesaki, H.; Reddy, P.H. Reduced dynamin-related protein 1 protects against phosphorylated Tau-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum. Mol. Genet. 2016, 25, 4881–4897. [Google Scholar] [CrossRef] [Green Version]
- Manczak, M.; Kandimalla, R.; Fry, D.; Sesaki, H.; Reddy, P.H. Protective effects of reduced dynamin-related protein 1 against amyloid beta-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum. Mol. Genet. 2016, 25, 5148–5166. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Sun, X.; Starovoytov, V.; Cai, Q. Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer’s disease patient brains. Hum. Mol. Genet. 2015, 24, 2938–2951. [Google Scholar] [CrossRef] [PubMed]
- Martín-Maestro, P.; Gargini, R.; Perry, G.; Avila, J.; García-Escudero, V. PARK2 enhancement is able to compensate mitophagy alterations found in sporadic Alzheimer’s disease. Hum. Mol. Genet. 2015, 25, 792–806. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Yin, J.; Ma, X.; Zhao, F.; Siedlak, S.L.; Wang, Z.; Torres, S.; Fujioka, H.; Xu, Y.; Perry, G.; et al. Inhibition of mitochondrial fragmentation protects against Alzheimer’s disease in rodent model. Hum. Mol. Genet. 2017, 26, 4118–4131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Martín-Maestro, P.; Gargini, R.; García, E.; Simón, D.; Avila, J.; García-Escudero, V. Mitophagy Failure in APP and Tau Overexpression Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 70, 525–540. [Google Scholar] [CrossRef]
- Wilkins, H.; Koppel, S.J.; Weidling, I.W.; Roy, N.; Ryan, L.N.; Stanford, J.A.; Swerdlow, R.H. Extracellular Mitochondria and Mitochondrial Components Act as Damage-Associated Molecular Pattern Molecules in the Mouse Brain. J. Neuroimmune Pharmacol. 2016, 11, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Little, J.P.; Simtchouk, S.; Schindler, S.M.; Villanueva, E.B.; Gill, N.E.; Walker, D.G.; Wolthers, K.R.; Klegeris, A. Mitochondrial transcription factor A (Tfam) is a pro-inflammatory extracellular signaling molecule recognized by brain microglia. Mol. Cell. Neurosci. 2014, 60, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Schindler, S.M.; Frank, M.G.; Annis, J.L.; Maier, S.F.; Klegeris, A. Pattern recognition receptors mediate pro-inflammatory effects of extracellular mitochondrial transcription factor A (TFAM). Mol. Cell. Neurosci. 2018, 89, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, Y.; Yu, X.; Li, D.; Li, G. Mitochondria-mediated damage to dopaminergic neurons in Parkinson’s disease (Review). Int. J. Mol. Med. 2017, 41, 615–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasuhn, J.; Davis, R.L.; Kumar, K.R. Targeting Mitochondrial Impairment in Parkinson’s Disease: Challenges and Opportunities. Front. Cell Dev. Biol. 2021, 8, 1704. [Google Scholar] [CrossRef] [PubMed]
- Galkin, A.; Dröse, S.; Brandt, U. The proton pumping stoichiometry of purified mitochondrial complex I reconstituted into proteoliposomes. Biochim. Biophys. Acta (BBA) Bioenerg. 2006, 1757, 1575–1581. [Google Scholar] [CrossRef] [Green Version]
- Jackson-Lewis, V.; Przedborski, S. Protocol for the MPTP mouse model of Parkinson’s disease. Nat. Protoc. 2007, 2, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.R.; Greenamyre, J.T. Neurotoxic in vivo models of Parkinson’s disease. Prog. Brain Res. 2010, 184, 17–33. [Google Scholar] [CrossRef]
- Valdez, L.B.; Zaobornyj, T.; Bandez, M.J.; López-Cepero, J.M.; Boveris, A.; Navarro, A. Complex I syndrome in striatum and frontal cortex in a rat model of Parkinson disease. Free Radic. Biol. Med. 2019, 135, 274–282. [Google Scholar] [CrossRef]
- Perier, C.; Vila, M. Mitochondrial Biology and Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2011, 2, a009332. [Google Scholar] [CrossRef] [PubMed]
- Grünewald, A.; Rygiel, K.A.; Msc, P.D.H.; Morris, C.M.; Picard, M.; Turnbull, D.M. Mitochondrial DNA Depletion in Respiratory Chain–Deficient P arkinson Disease Neurons. Ann. Neurol. 2015, 79, 366–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barsoum, M.J.; Yuan, H.; Gerencser, A.A.; Liot, G.; Kushnareva, Y.; Gräber, S.; Kovacs, I.; Lee, W.D.; Waggoner, J.; Cui, J.; et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006, 25, 3900–3911. [Google Scholar] [CrossRef]
- Wang, X.; Becker, K.; Levine, N.; Zhang, M.; Lieberman, A.P.; Moore, D.; Ma, J. Pathogenic alpha-synuclein aggregates preferentially bind to mitochondria and affect cellular respiration. Acta Neuropathol. Commun. 2019, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Devoto, V.M.P.; Falzone, T.L. Mitochondrial dynamics in Parkinson’s disease: A role for α-synuclein? Dis. Models Mech. 2017, 10, 1075–1087. [Google Scholar] [CrossRef] [Green Version]
- Xie, W.; Chung, K.K.K. Alpha-synuclein impairs normal dynamics of mitochondria in cell and animal models of Parkinson’s disease. J. Neurochem. 2012, 122, 404–414. [Google Scholar] [CrossRef]
- Zilocchi, M.; Finzi, G.; Lualdi, M.; Sessa, F.; Fasano, M.; Alberio, T. Mitochondrial alterations in Parkinson’s disease human samples and cellular models. Neurochem. Int. 2018, 118, 61–72. [Google Scholar] [CrossRef]
- Houlden, H.; Singleton, A.B. The genetics and neuropathology of Parkinson’s disease. Acta Neuropathol. 2012, 124, 325–338. [Google Scholar] [CrossRef] [Green Version]
- Clark, I.E.; Dodson, M.W.; Jiang, C.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nat. Cell Biol. 2006, 441, 1162–1166. [Google Scholar] [CrossRef]
- Sang, T.-K.; Chang, H.-Y.; Lawless, G.M.; Ratnaparkhi, A.; Mee, L.; Ackerson, L.C.; Maidment, N.T.; Krantz, D.E.; Jackson, G.R. A Drosophila Model of Mutant Human Parkin-Induced Toxicity Demonstrates Selective Loss of Dopaminergic Neurons and Dependence on Cellular Dopamine. J. Neurosci. 2007, 27, 981–992. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, S.; Akamatsu, W.; Kisa, F.; Sone, T.; Ishikawa, K.-I.; Kuzumaki, N.; Katayama, H.; Miyawaki, A.; Hattori, N.; Okano, H. Efficient induction of dopaminergic neuron differentiation from induced pluripotent stem cells reveals impaired mitophagy in PARK2 neurons. Biochem. Biophys. Res. Commun. 2017, 483, 88–93. [Google Scholar] [CrossRef]
- Lei, Z.; Cao, G.; Wei, G. A30P mutant α-synuclein impairs autophagic flux by inactivating JNK signaling to enhance ZKSCAN3 activity in midbrain dopaminergic neurons. Cell Death Dis. 2019, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Mao, Z.; Liu, C.; Ji, S.; Yang, Q.; Ye, H.; Han, H.; Xue, Z. The NLRP3 Inflammasome is Involved in the Pathogenesis of Parkinson’s Disease in Rats. Neurochem. Res. 2017, 42, 1104–1115. [Google Scholar] [CrossRef] [PubMed]
- Martinez, E.M.; Young, A.L.; Patankar, Y.R.; Berwin, B.L.; Wang, L.; Von Herrmann, K.M.; Weier, J.M.; Havrda, M.C. Editor’s Highlight: Nlrp3 Is Required for Inflammatory Changes and Nigral Cell Loss Resulting From Chronic Intragastric Rotenone Exposure in Mice. Toxicol. Sci. 2017, 159, 64–75. [Google Scholar] [CrossRef]
- Lee, E.; Hwang, I.; Park, S.; Hong, S.; Hwang, B.; Cho, Y.; Son, J.; Yu, J.-W. MPTP-driven NLRP3 inflammasome activation in microglia plays a central role in dopaminergic neurodegeneration. Cell Death Differ. 2019, 26, 213–228. [Google Scholar] [CrossRef]
- Von Herrmann, K.M.; Salas, L.A.; Martinez, E.M.; Young, A.L.; Howard, J.M.; Feldman, M.S.; Christensen, B.C.; Wilkins, O.M.; Lee, S.L.; Hickey, W.F.; et al. NLRP3 expression in mesencephalic neurons and characterization of a rare NLRP3 polymorphism associated with decreased risk of Parkinson’s disease. NPJ Park. Dis. 2018, 4, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Codolo, G.; Plotegher, N.; Pozzobon, T.; Brucale, M.; Tessari, I.; Bubacco, L.; De Bernard, M. Triggering of Inflammasome by Aggregated α–Synuclein, an Inflammatory Response in Synucleinopathies. PLoS ONE 2013, 8, e55375. [Google Scholar] [CrossRef] [Green Version]
- Rakovic, A.; Ziegler, J.; Mårtensson, C.U.; Prasuhn, J.; Shurkewitsch, K.; König, P.; Paulson, H.L.; Klein, C. PINK1-dependent mitophagy is driven by the UPS and can occur independently of LC3 conversion. Cell Death Differ. 2019, 26, 1428–1441. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chi, J.; Huang, D.; Ding, L.; Zhao, X.; Jiang, L.; Yu, Y.; Gao, F. α-synuclein promotes progression of Parkinson’s disease by upregulating autophagy signaling pathway to activate NLRP3 inflammasome. Exp. Ther. Med. 2020, 19, 931–938. [Google Scholar] [CrossRef] [Green Version]
- Haque, E.; Akther, M.; Jakaria, M.; Kim, I.; Azam, S.; Choi, D. Targeting the Microglial NLRP3 Inflammasome and Its Role in Parkinson’s Disease. Mov. Disord. 2020, 35, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Oyewole, A.O.; Birch-Machin, M.A. Mitochondria-targeted antioxidants. FASEB J. 2015, 29, 4766–4771. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J.-H.; Koo, J.-H.; Yook, J.; Cho, J.-Y.; Kang, E.-B. Neuroprotective Benefits of Exercise and MitoQ on Memory Function, Mitochondrial Dynamics, Oxidative Stress, and Neuroinflammation in D-Galactose-Induced Aging Rats. Brain Sci. 2021, 11, 164. [Google Scholar] [CrossRef]
- Zhang, W.; Gu, G.-J.; Shen, X.; Zhang, Q.; Wang, G.-M.; Wang, P.-J. Neural stem cell transplantation enhances mitochondrial biogenesis in a transgenic mouse model of Alzheimer’s disease–like pathology. Neurobiol. Aging 2015, 36, 1282–1292. [Google Scholar] [CrossRef]
- Lehmann, S.; Loh, S.H.Y.; Martins, L.M. Enhancing NAD+ salvage metabolism is neuroprotective in a PINK1 model of Parkinson’s disease. Biol. Open 2017, 6, 141–147. [Google Scholar] [CrossRef] [Green Version]
- Shin, E.-J.; Nam, Y.; Lee, J.W.; Nguyen, P.-K.T.; Yoo, J.E.; Tran, T.-V.; Jeong, J.H.; Jang, C.-G.; Oh, Y.J.; Youdim, M.B.H.; et al. N-Methyl, N-propynyl-2-phenylethylamine (MPPE), a Selegiline Analog, Attenuates MPTP-induced Dopaminergic Toxicity with Guaranteed Behavioral Safety: Involvement of Inhibitions of Mitochondrial Oxidative Burdens and p53 Gene-elicited Pro-apoptotic Change. Mol. Neurobiol. 2016, 53, 6251–6269. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, Y.; Li, J.; Zhang, C. Resveratrol ameliorates spatial learning memory impairment induced by Aβ 1–42 in rats. Neuroscience 2017, 344, 39–47. [Google Scholar] [CrossRef]
- Cosín-Tomàs, M.; Senserrich, J.; Arumí-Planas, M.; Alquezar, C.; Pallàs, M.; Martín-Requero, Á.; Suñol, C.; Kaliman, P.; Sanfeliu, C. Role of Resveratrol and Selenium on Oxidative Stress and Expression of Antioxidant and Anti-Aging Genes in Immortalized Lymphocytes from Alzheimer’s Disease Patients. Nutrients 2019, 11, 1764. [Google Scholar] [CrossRef] [Green Version]
- Zeng, W.; Zhang, W.; Lu, F.; Gao, L.; Gao, G. Resveratrol attenuates MPP+-induced mitochondrial dysfunction and cell apoptosis via AKT/GSK-3β pathway in SN4741 cells. Neurosci. Lett. 2017, 637, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.-L.; Wang, P.-W.; Chuang, J.-H.; Lin, H.-Y.; Chen, S.-D.; Chuang, Y.-C.; Huang, S.-T.; Tiao, M.-M.; Chen, J.-B.; Huang, P.-H.; et al. Resveratrol provides neuroprotective effects through modulation of mitochondrial dynamics and ERK1/2 regulated autophagy. Free Radic. Res. 2018, 52, 1371–1386. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Dong, X.; Liu, Z.; Zhu, S.; Liu, H.; Fan, W.; Hu, Y.; Hu, T.; Yu, Y.; Li, Y.; et al. Resveratrol Suppresses Rotenone-induced Neurotoxicity Through Activation of SIRT1/Akt1 Signaling Pathway. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2018, 301, 1115–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, N.; Zhang, Y.; Chen, M.; Jin, H.; Nie, J.; Luo, Y.; Zhou, S.; Shi, J.; Jin, F. Resveratrol delays 6-hydroxydopamine-induced apoptosis by activating the PI3K/Akt signaling pathway. Exp. Gerontol. 2019, 124, 110653. [Google Scholar] [CrossRef]
- Guo, Y.-J.; Dong, S.-Y.; Cui, X.-X.; Feng, Y.; Liu, T.; Yin, M.; Kuo, S.-H.; Tan, E.-K.; Zhao, W.-J.; Wu, Y.-C. Resveratrol alleviates MPTP-induced motor impairments and pathological changes by autophagic degradation of α-synuclein via SIRT1-deacetylated LC3. Mol. Nutr. Food Res. 2016, 60, 2161–2175. [Google Scholar] [CrossRef] [PubMed]
- Palle, S.; Neerati, P. Improved neuroprotective effect of resveratrol nanoparticles as evinced by abrogation of rotenone-induced behavioral deficits and oxidative and mitochondrial dysfunctions in rat model of Parkinson’s disease. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2018, 391, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Spilman, P.; Podlutskaya, N.; Hart, M.J.; Debnath, J.; Gorostiza, O.; Bredesen, D.; Richardson, A.; Strong, R.; Galvan, V. Inhibition of mTOR by Rapamycin Abolishes Cognitive Deficits and Reduces Amyloid-β Levels in a Mouse Model of Alzheimer’s Disease. PLoS ONE 2010, 5, e9979. [Google Scholar] [CrossRef] [Green Version]
- Di Domenico, F.; Tramutola, A.; Barone, E.; Lanzillotta, C.; Defever, O.; Arena, A.; Zuliani, I.; Foppoli, C.; Iavarone, F.; Vincenzoni, F.; et al. Restoration of aberrant mTOR signaling by intranasal rapamycin reduces oxidative damage: Focus on HNE-modified proteins in a mouse model of down syndrome. Redox Biol. 2019, 23, 101162. [Google Scholar] [CrossRef]
- Vingtdeux, V.; Chandakkar, P.; Zhao, H.; D’Abramo, C.; Davies, P.; Marambsud, P. Novel synthetic small-molecule activators of AMPK as enhancers of autophagy and amyloid-β peptide degradation. FASEB J. 2010, 25, 219–231. [Google Scholar] [CrossRef] [Green Version]
- Su, S.-H.; Wu, Y.-F.; Lin, Q.; Wang, D.-P.; Hai, J. URB597 protects against NLRP3 inflammasome activation by inhibiting autophagy dysfunction in a rat model of chronic cerebral hypoperfusion. J. Neuroinflamm. 2019, 16, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Lonnemann, N.; Hosseini, S.; Marchetti, C.; Skouras, D.B.; Stefanoni, D.; D’Alessandro, A.; Dinarello, C.A.; Korte, M. The NLRP3 inflammasome inhibitor OLT1177 rescues cognitive impairment in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2020, 117, 32145–32154. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Litwiniuk, A.; Baranowska-Bik, A.; Domańska, A.; Kalisz, M.; Bik, W. Contribution of Mitochondrial Dysfunction Combined with NLRP3 Inflammasome Activation in Selected Neurodegenerative Diseases. Pharmaceuticals 2021, 14, 1221. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14121221
Litwiniuk A, Baranowska-Bik A, Domańska A, Kalisz M, Bik W. Contribution of Mitochondrial Dysfunction Combined with NLRP3 Inflammasome Activation in Selected Neurodegenerative Diseases. Pharmaceuticals. 2021; 14(12):1221. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14121221
Chicago/Turabian StyleLitwiniuk, Anna, Agnieszka Baranowska-Bik, Anita Domańska, Małgorzata Kalisz, and Wojciech Bik. 2021. "Contribution of Mitochondrial Dysfunction Combined with NLRP3 Inflammasome Activation in Selected Neurodegenerative Diseases" Pharmaceuticals 14, no. 12: 1221. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14121221