
Design, Synthesis, Anticancer Activity, and Solid Lipid Nanoparticle Formulation of Indole- and Benzimidazole-Based Compounds as Pro-Apoptotic Agents Targeting Bcl-2 Protein


 , , ,
, , ,  , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
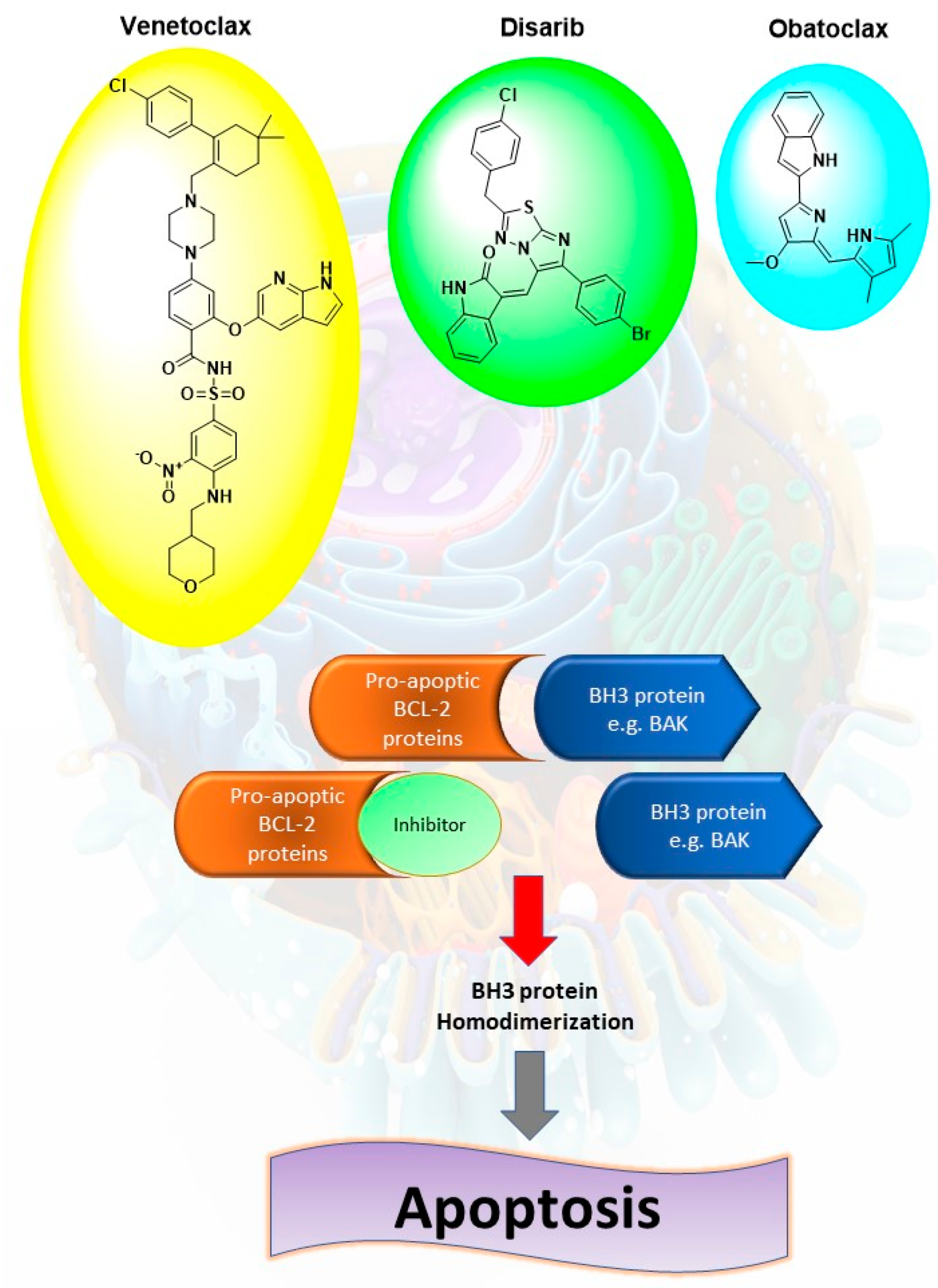
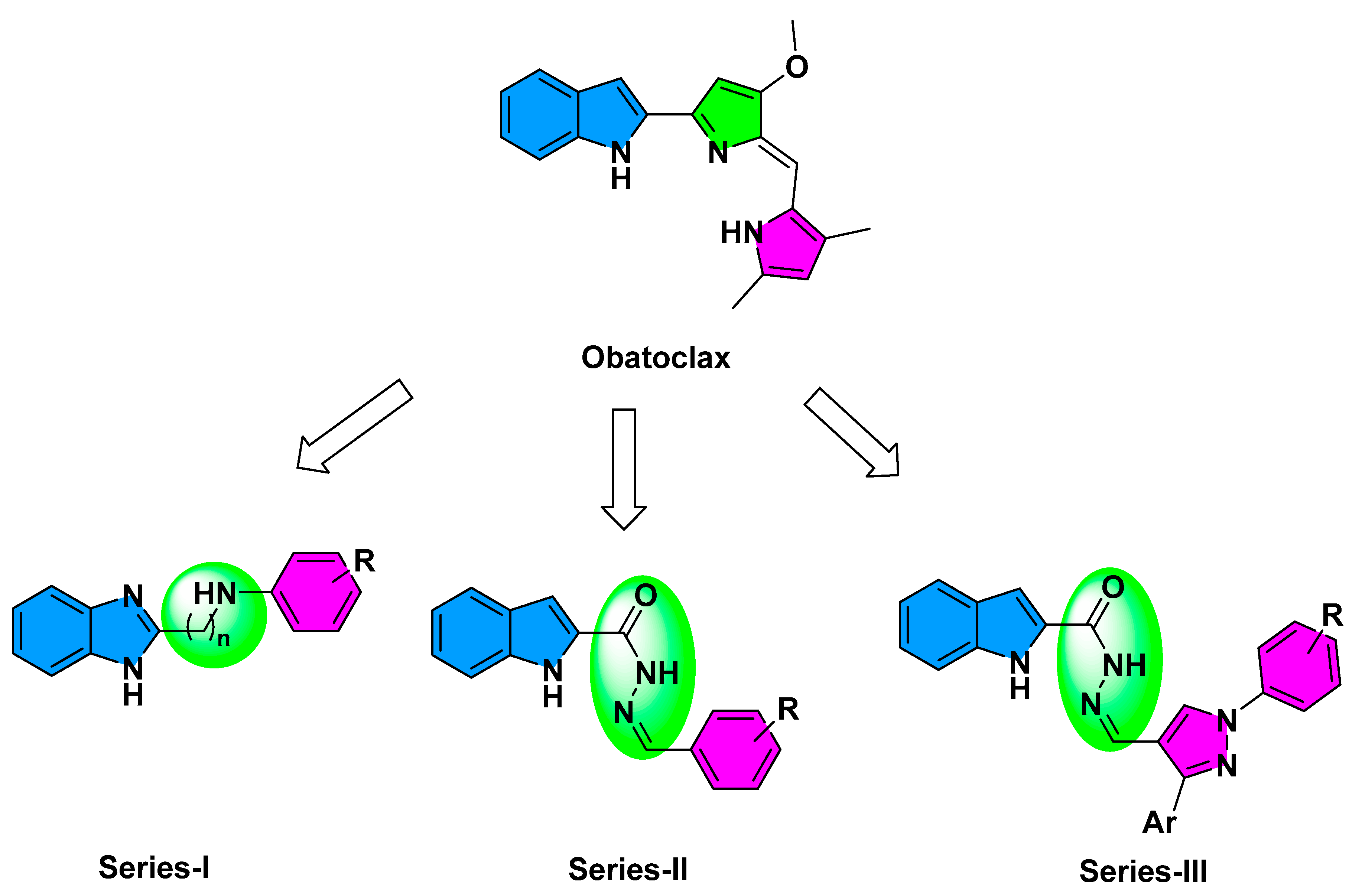
2.1. Compounds Design
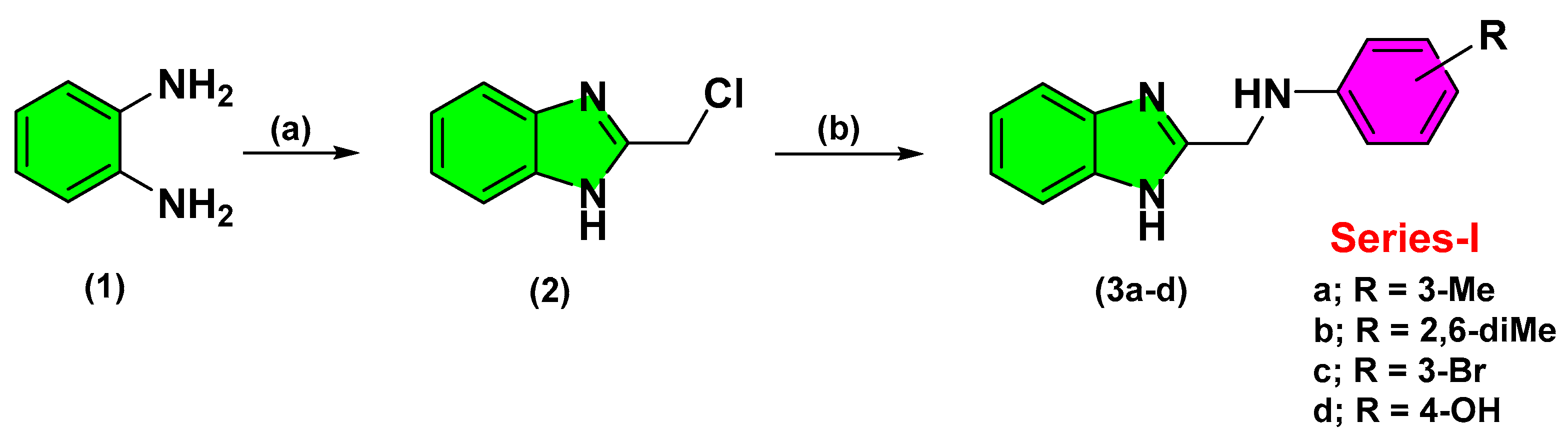
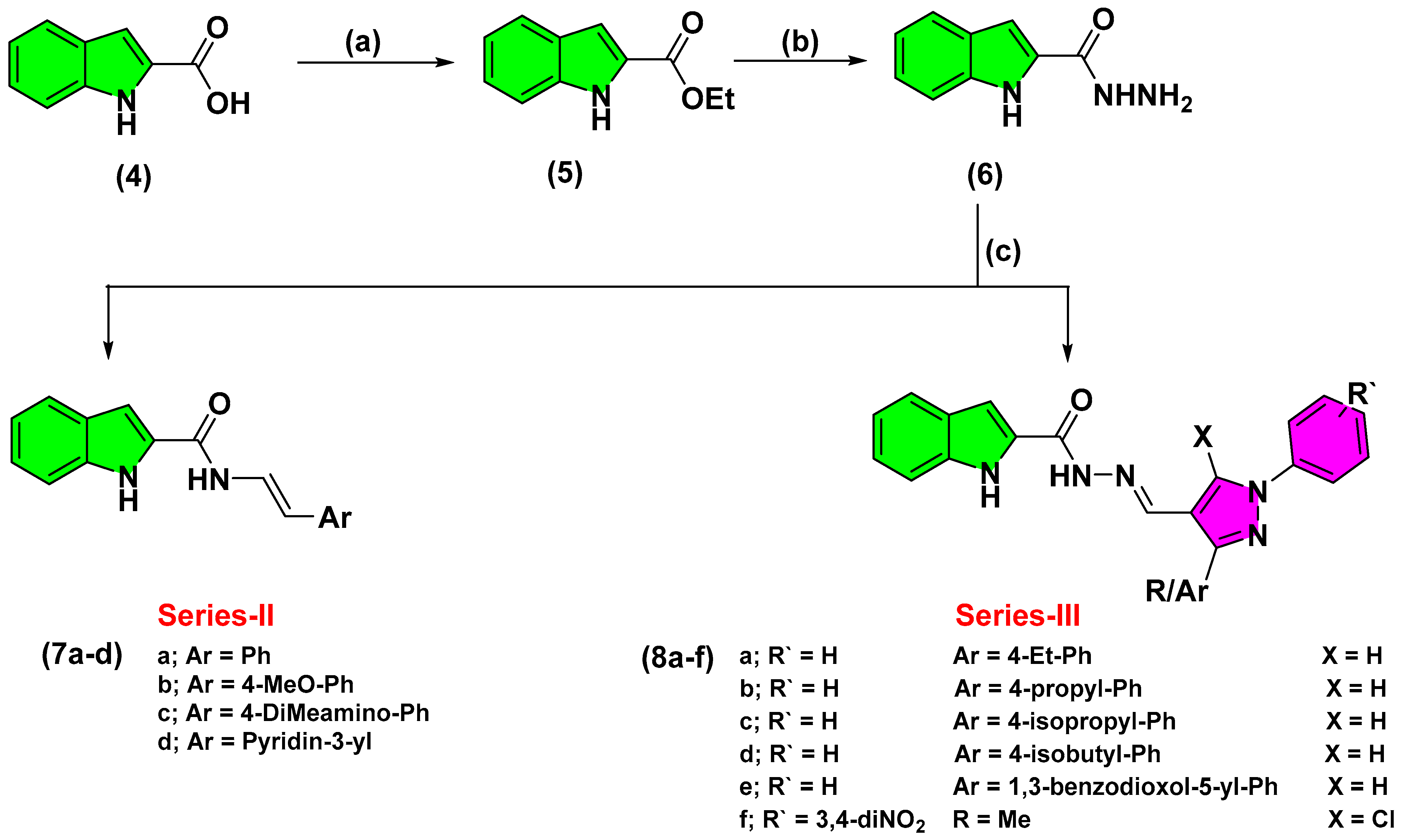
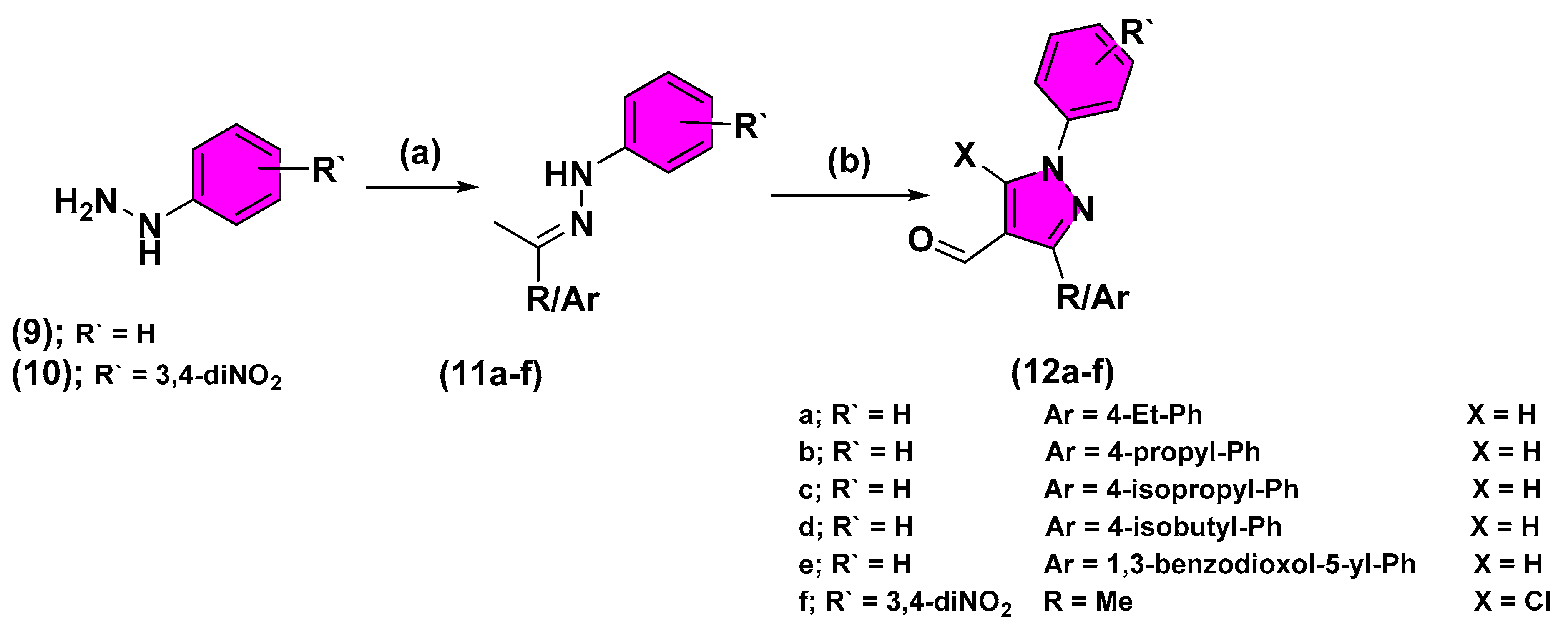
2.2. Chemistry
2.3. Biological Activity
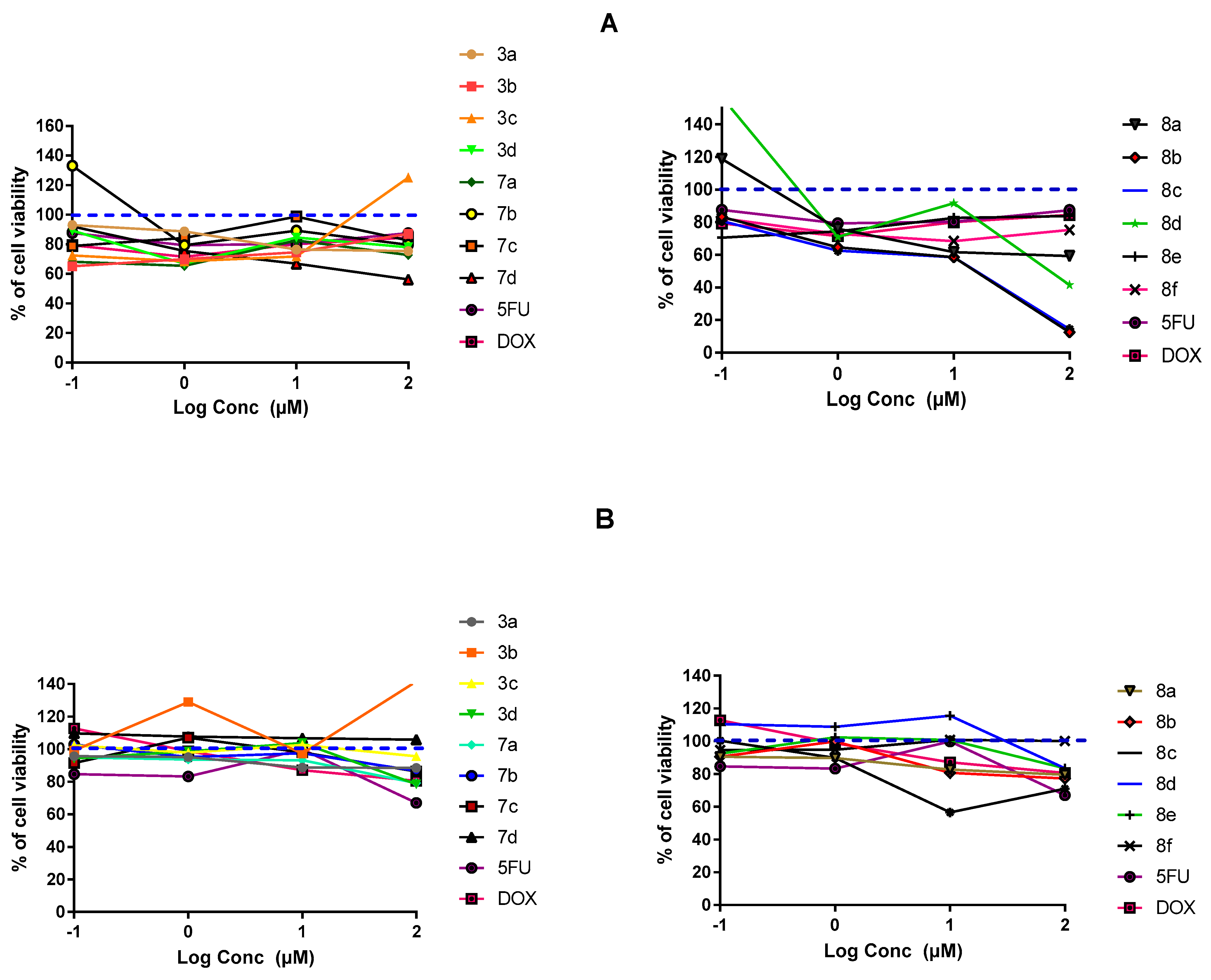
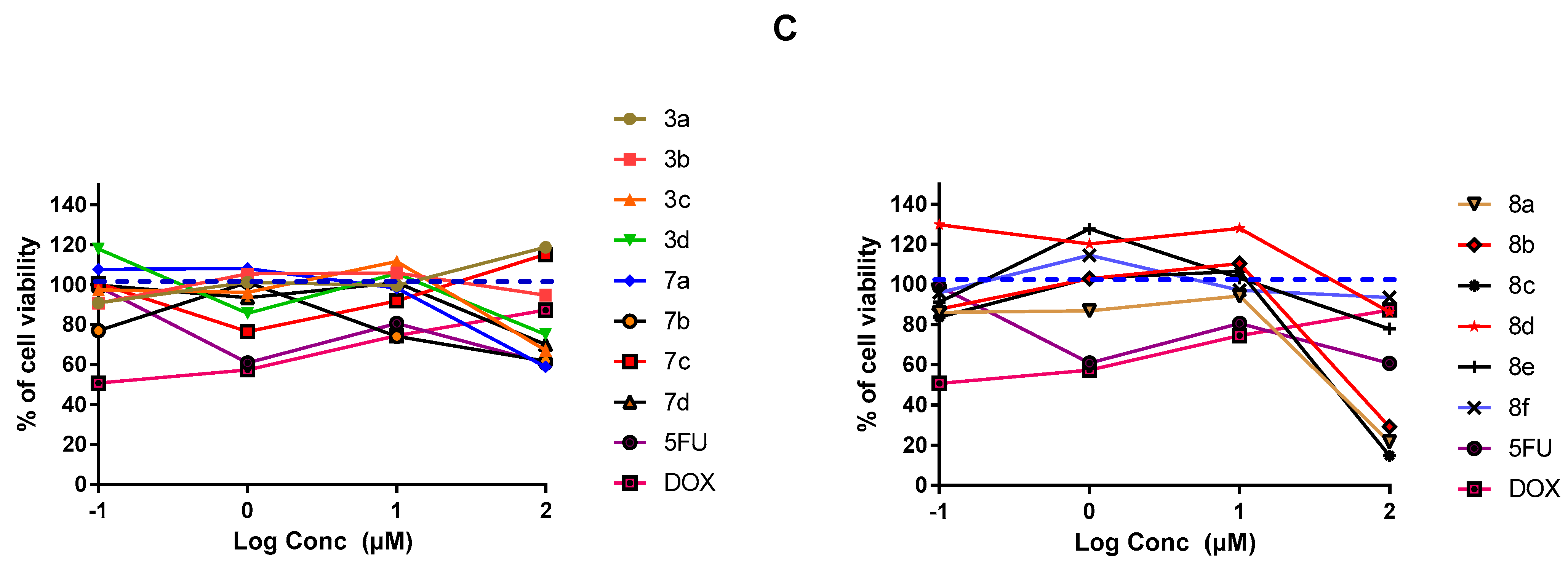
2.3.1. In-Vitro Cytotoxic Activity
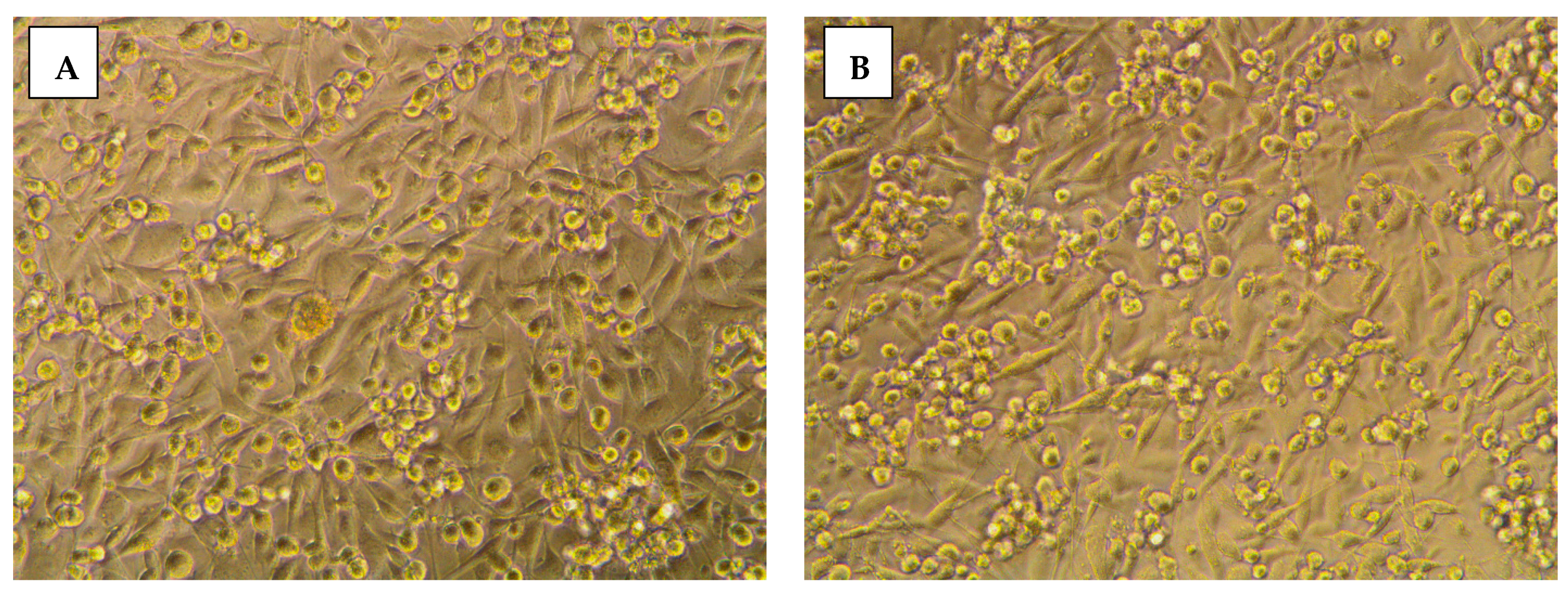
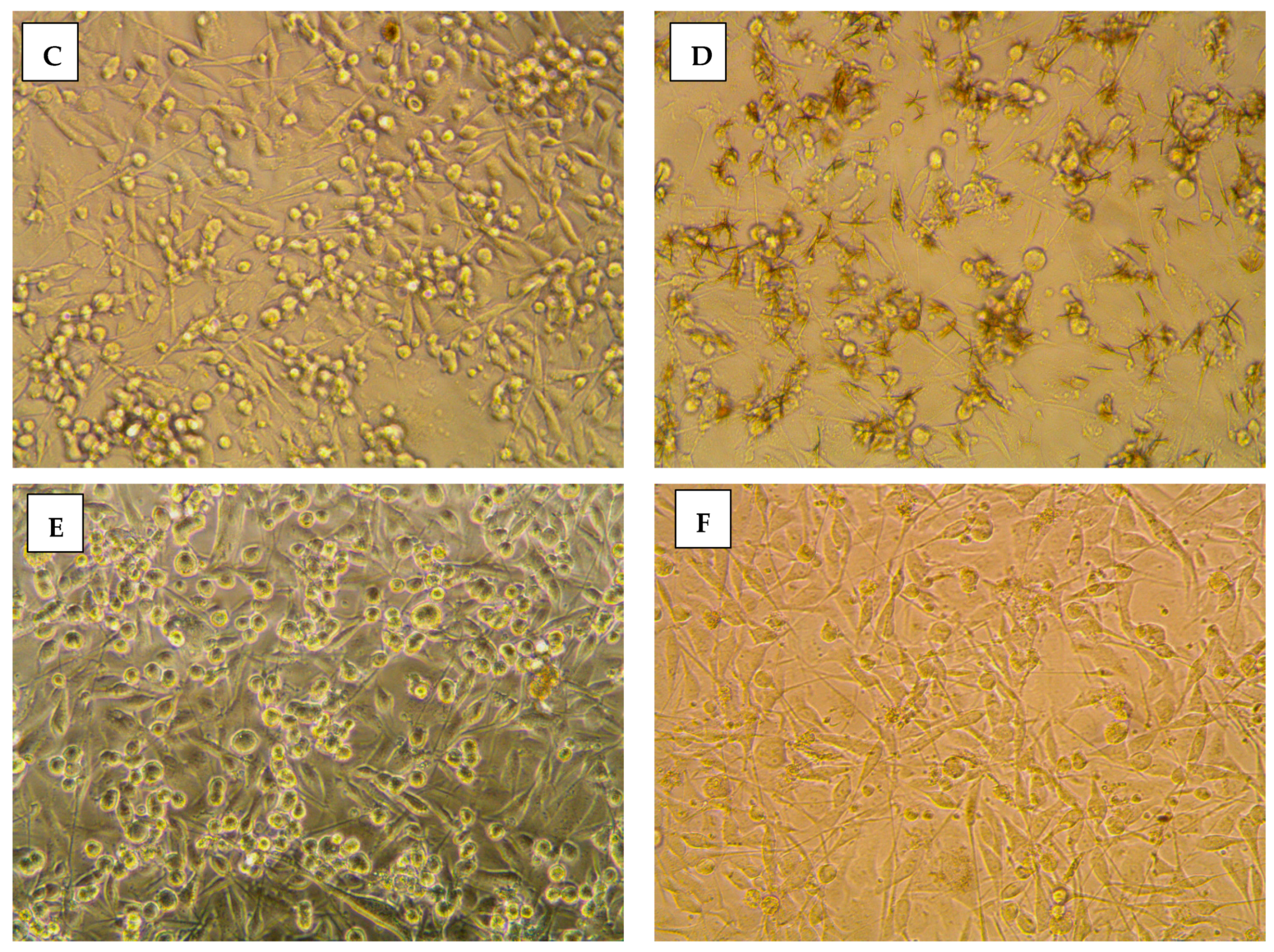
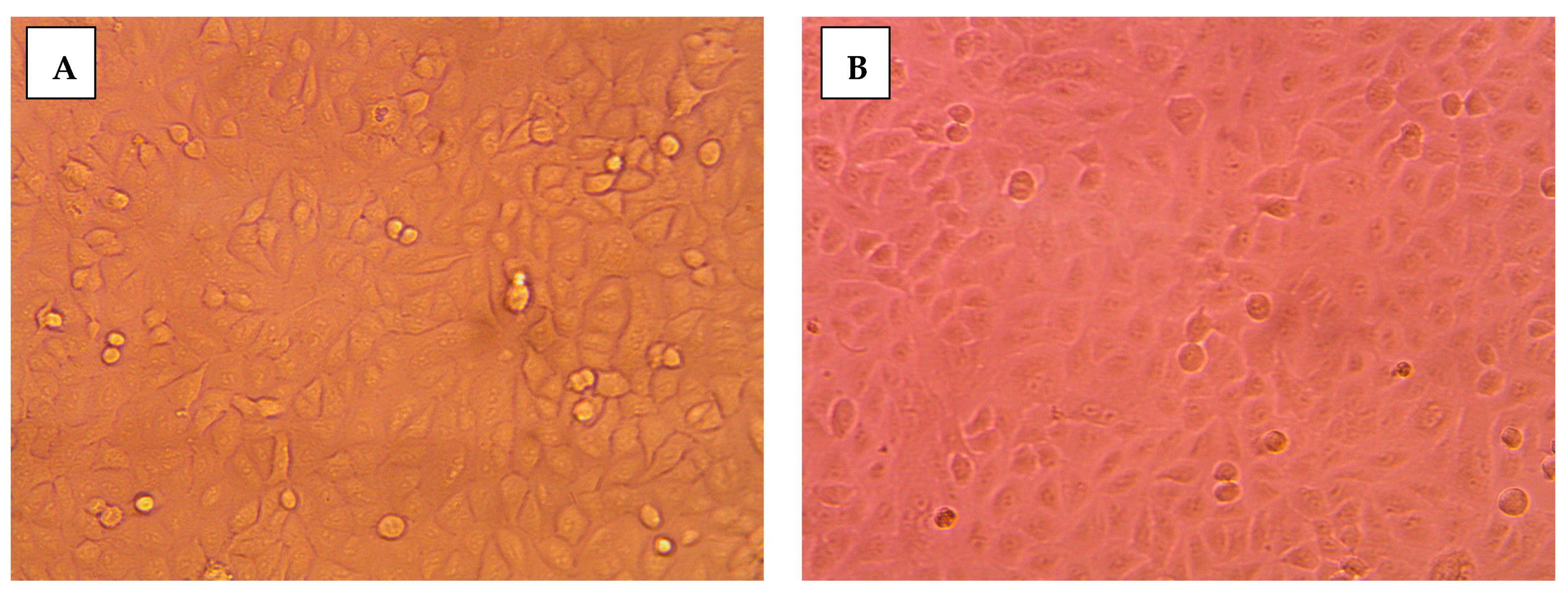
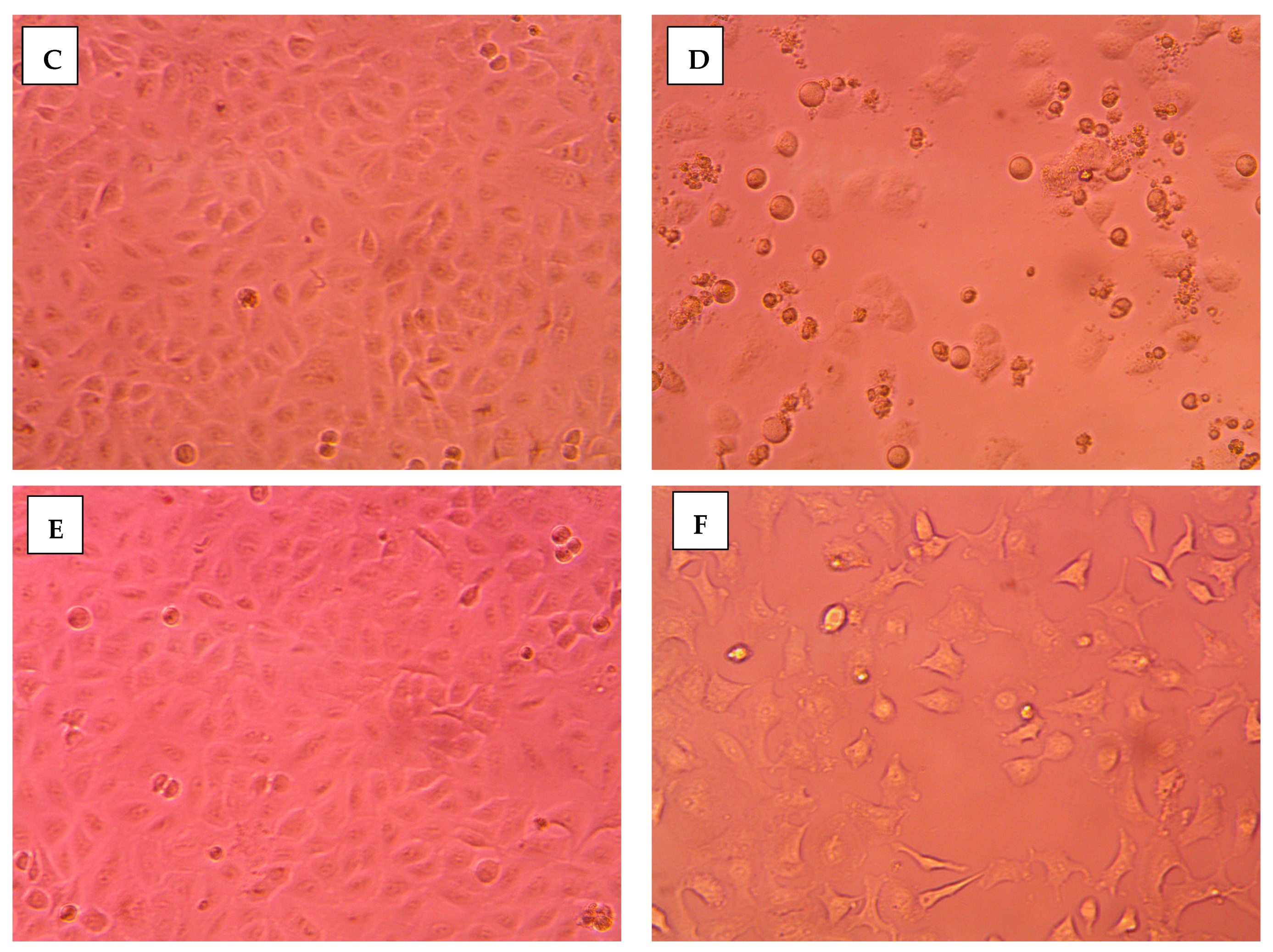
2.3.2. Morphological Assessment
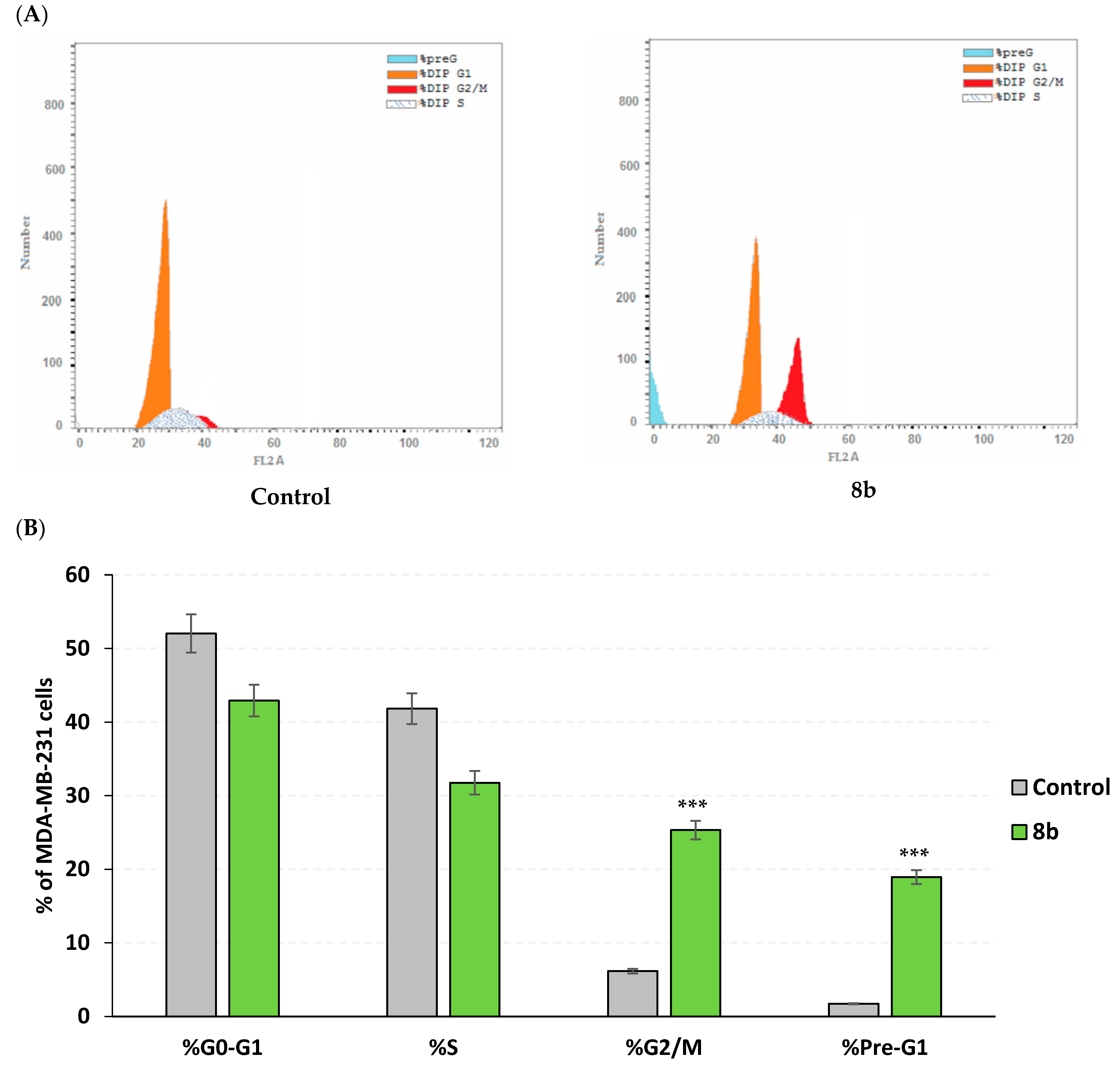
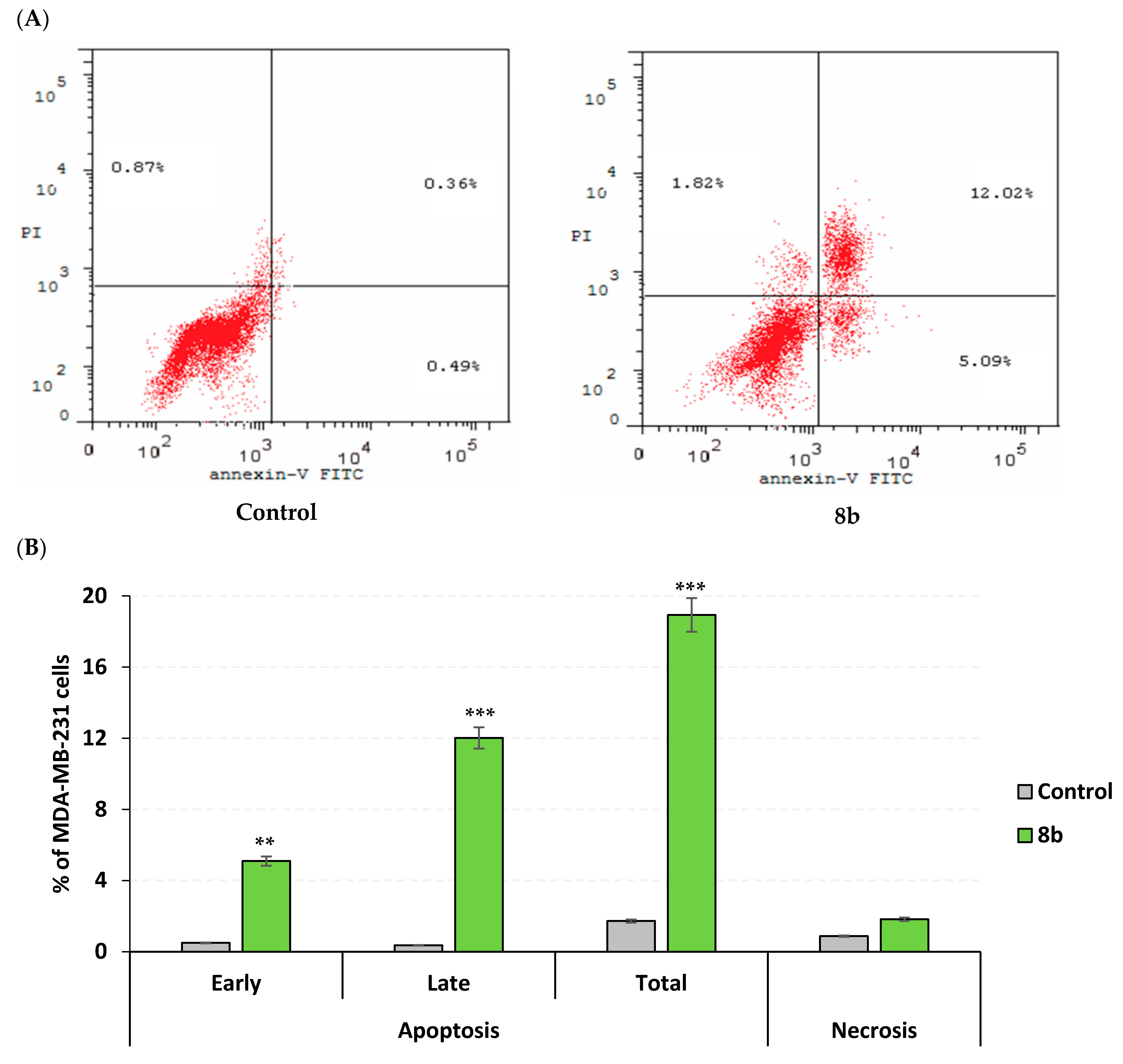
2.3.3. Apoptosis Rate and Cell Cycle Analysis
2.3.4. DNA Fragmentation Determination
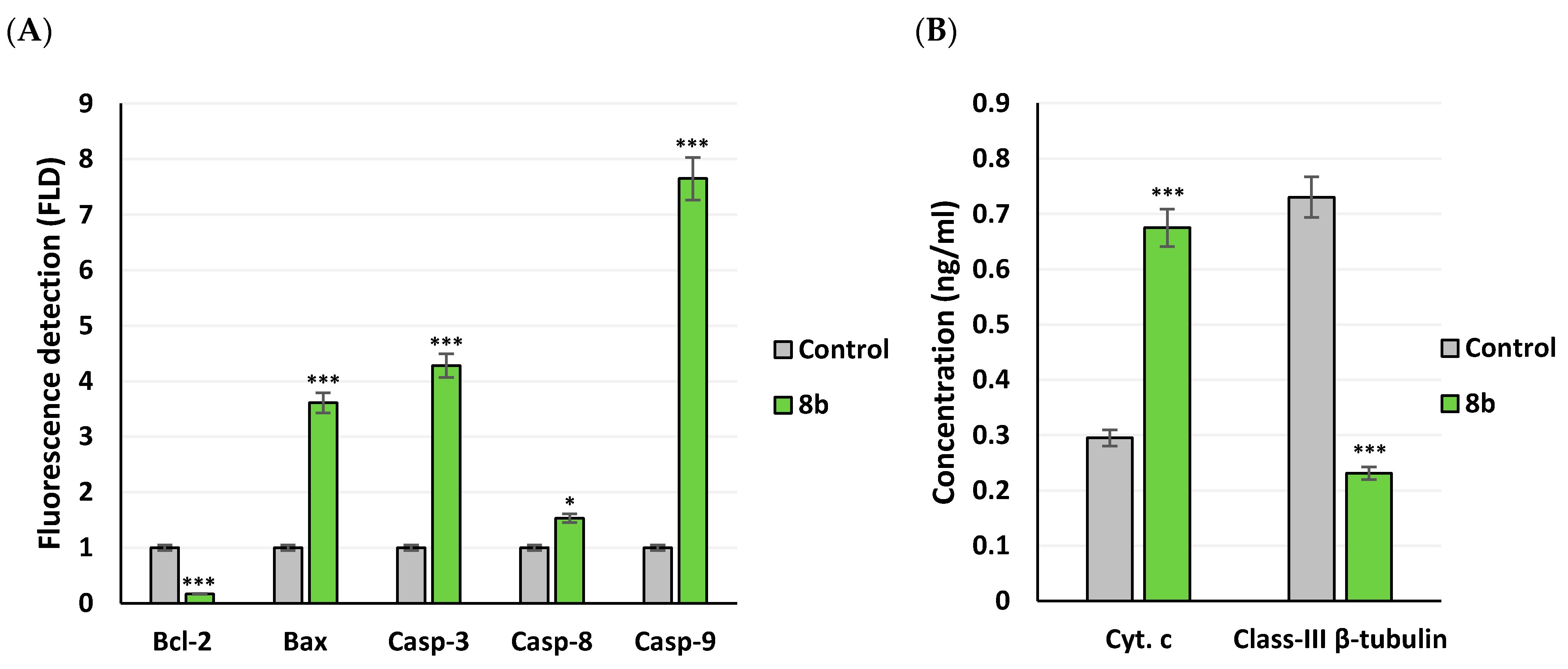
2.3.5. Apoptotic Gene Expression and Protein Level Analysis
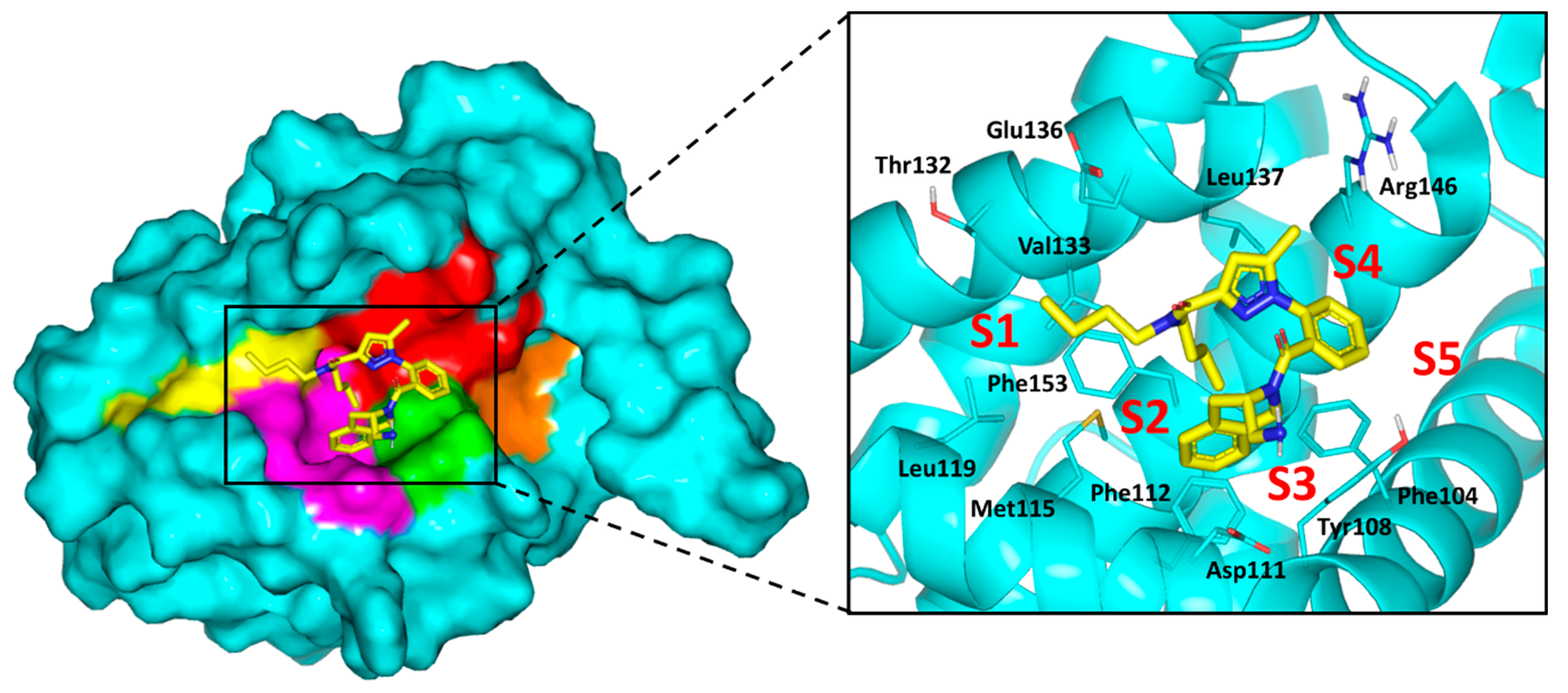
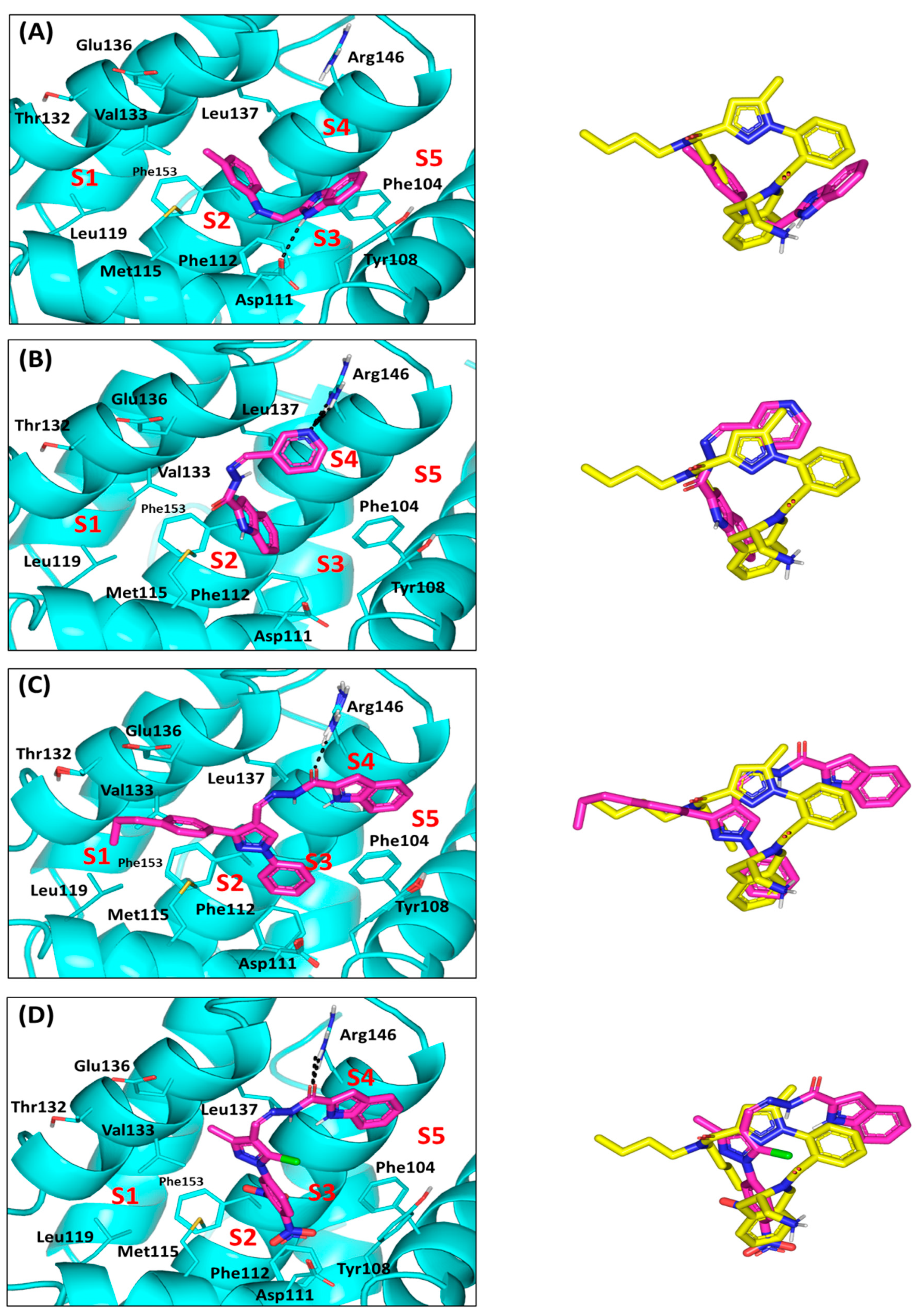
2.4. Computational Study
2.5. Solid Lipid Nanoparticle Formulation Studies
2.5.1. Design, Preparation and Optimization of Drug-SLN
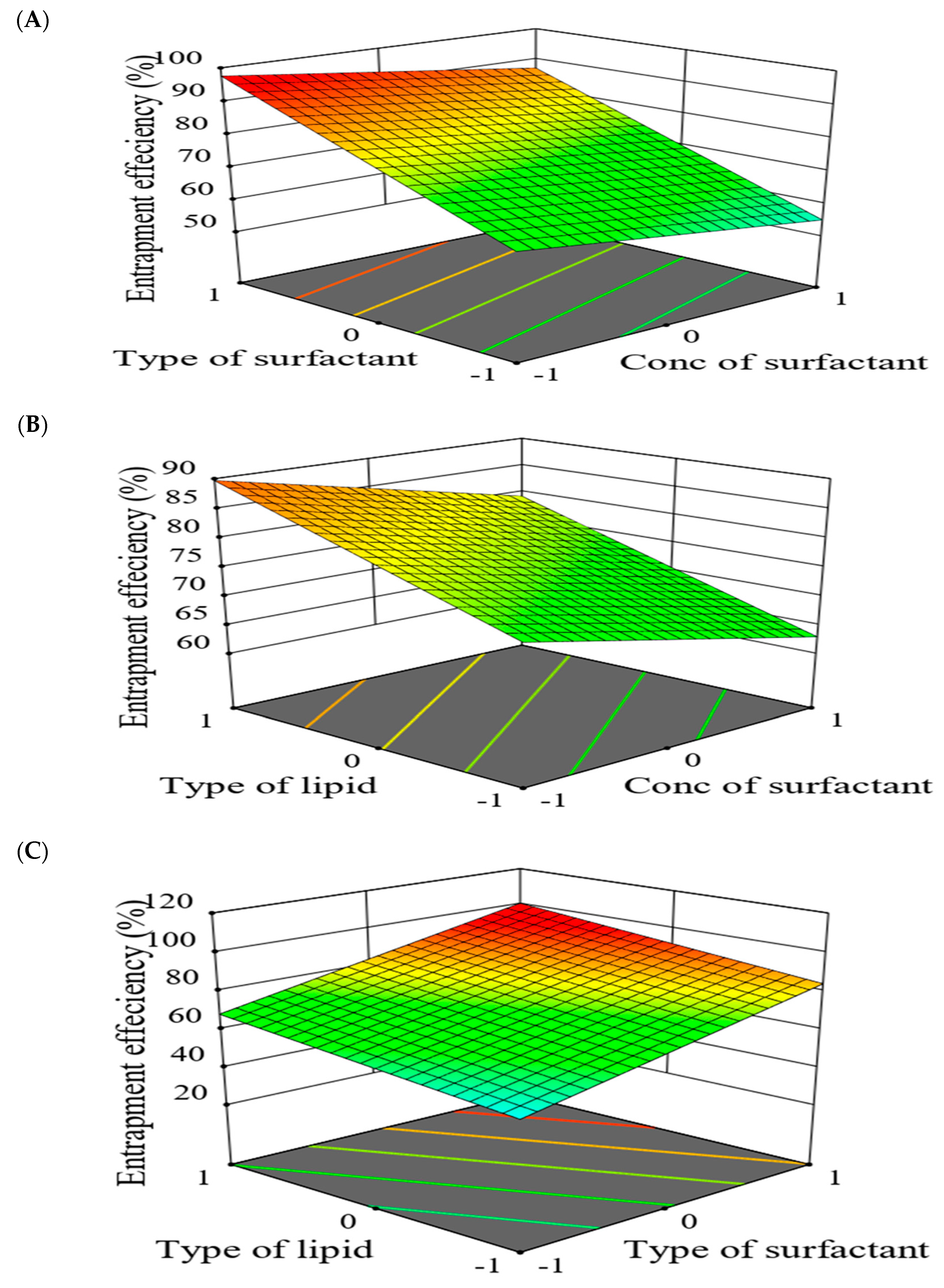
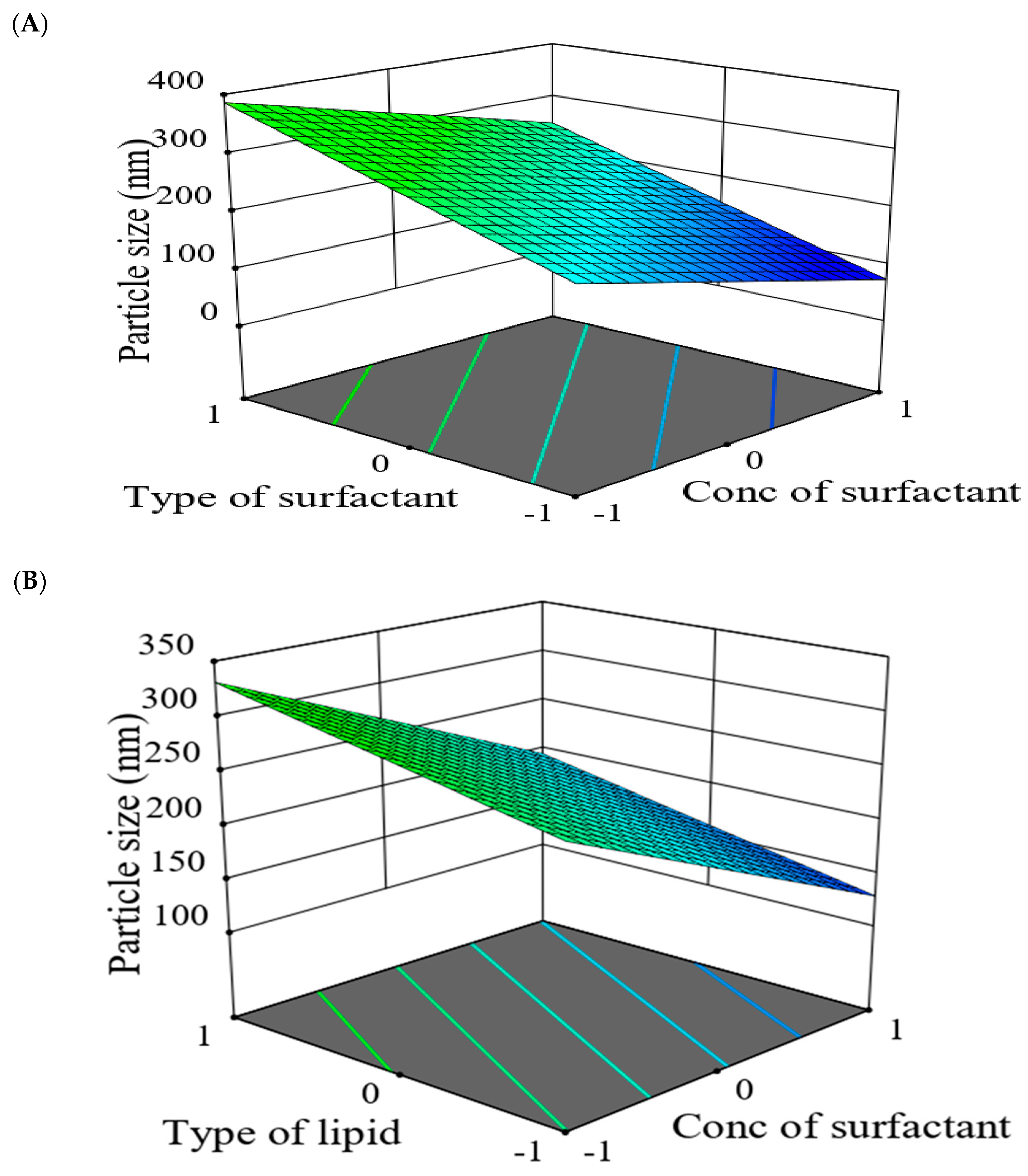
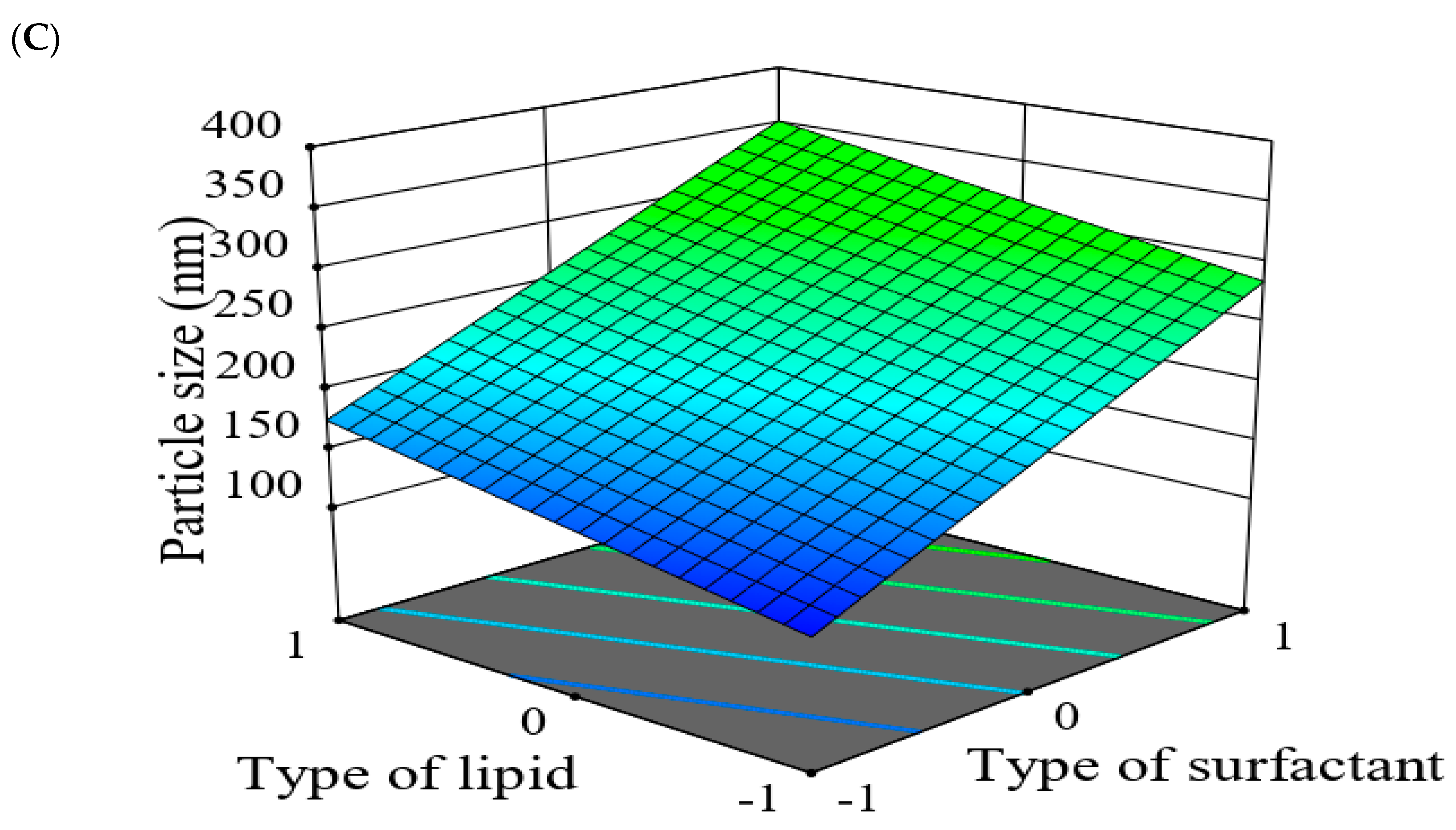
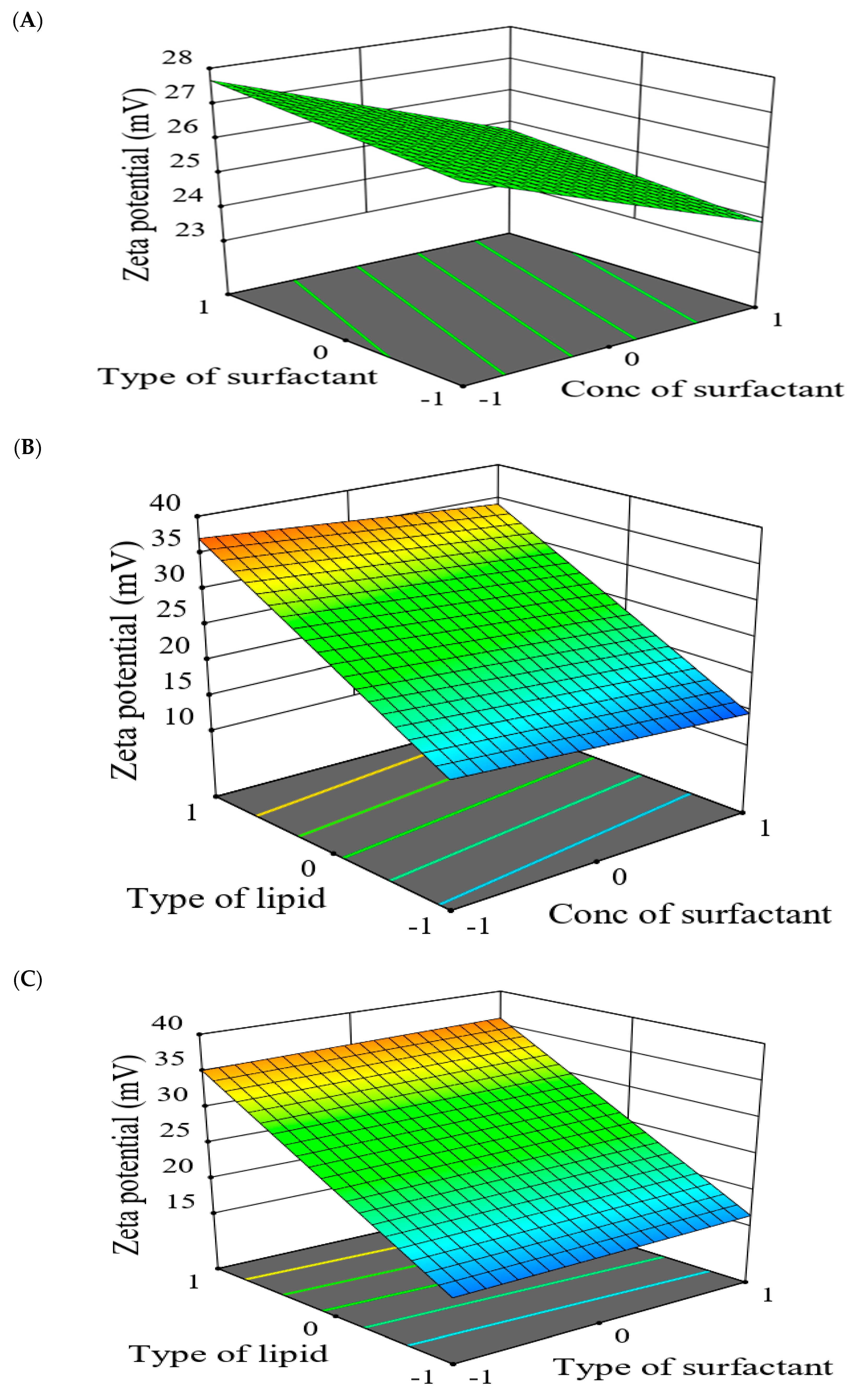
2.5.2. The Effect of Formulation Factors in the Responses
2.5.3. Optimization of Formulation Variables to Select the Best Formula
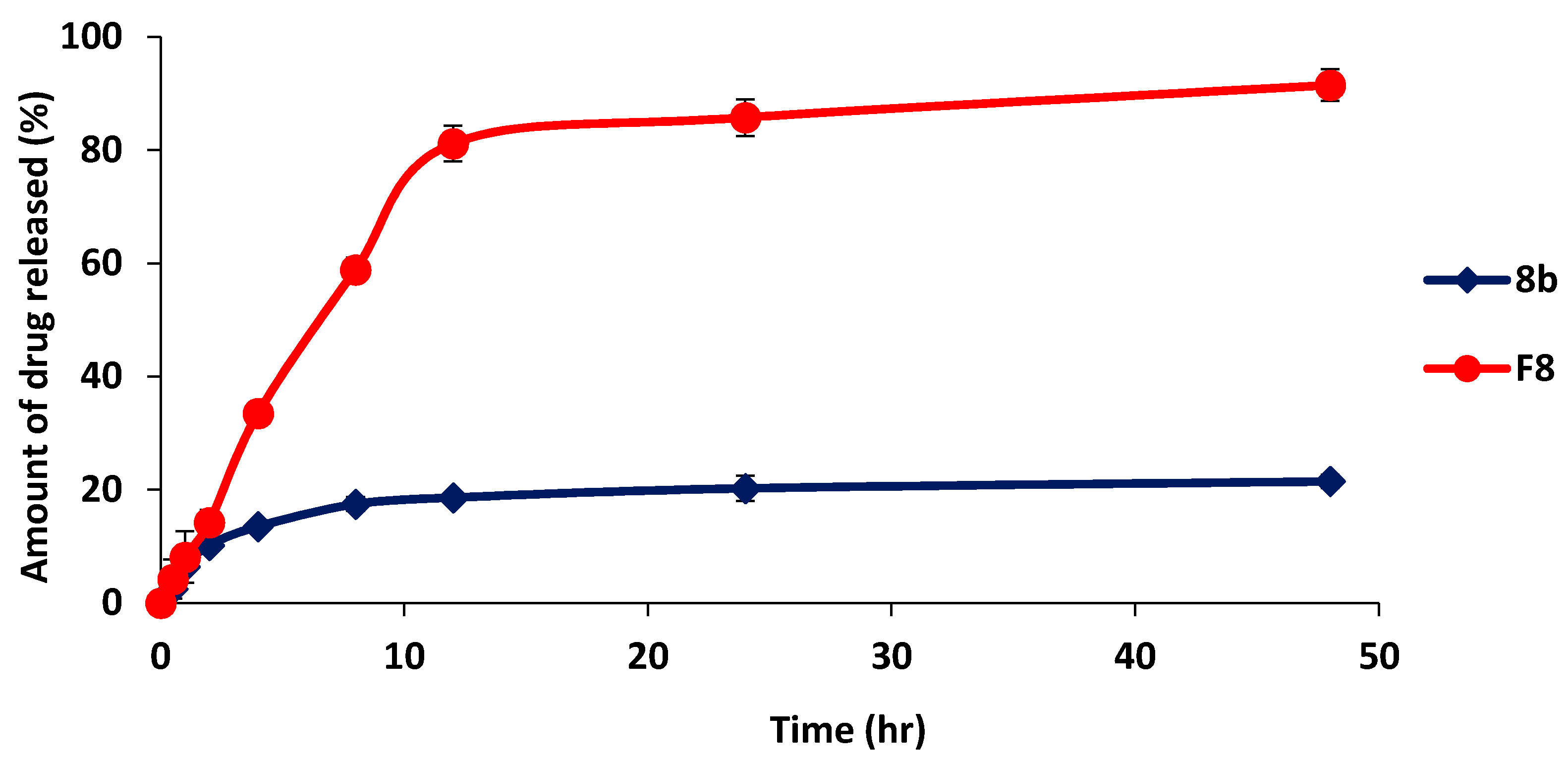
2.5.4. In-Vitro Release Study of Optimized Formulation (F8) in Comparison with 8b
2.5.5. Cytotoxicity Study of Optimized Formulation
3. Materials and Methods
3.1. General Experimental
3.2. Chemical Synthesis
3.2.1. Synthesis of 2-(chloromethyl)-1H-Benzimidazole (2)
3.2.2. Synthesis of 2-Aminomethyl-Benzimidazole Derivatives (3a–d)
3.2.3. Synthesis of Ethyl, 1H-Indole-2-Carboxylate (5)
3.2.4. Synthesis of 1H-Indole-2-Carbohydrazide (6)
3.2.5. Synthesis of Different Carbohydrazide Derivatives (7, 8)
3.3. MTT-Cell Proliferation Assay and Morphological Evaluation
3.4. SRB Cytotoxicity Assay
3.5. Morphological Evaluation
3.6. Flow Cytometer Analysis
3.7. DNA Fragmentation Assay
3.8. Real-Time PCR Analysis
3.9. ELISA Assay
3.10. Molecular Docking Studies
3.11. Preparation of Drug-Loaded SLNs
3.12. HPLC Analysis
3.13. Determination of the Drug-SLNs Parameters (Y1–3)
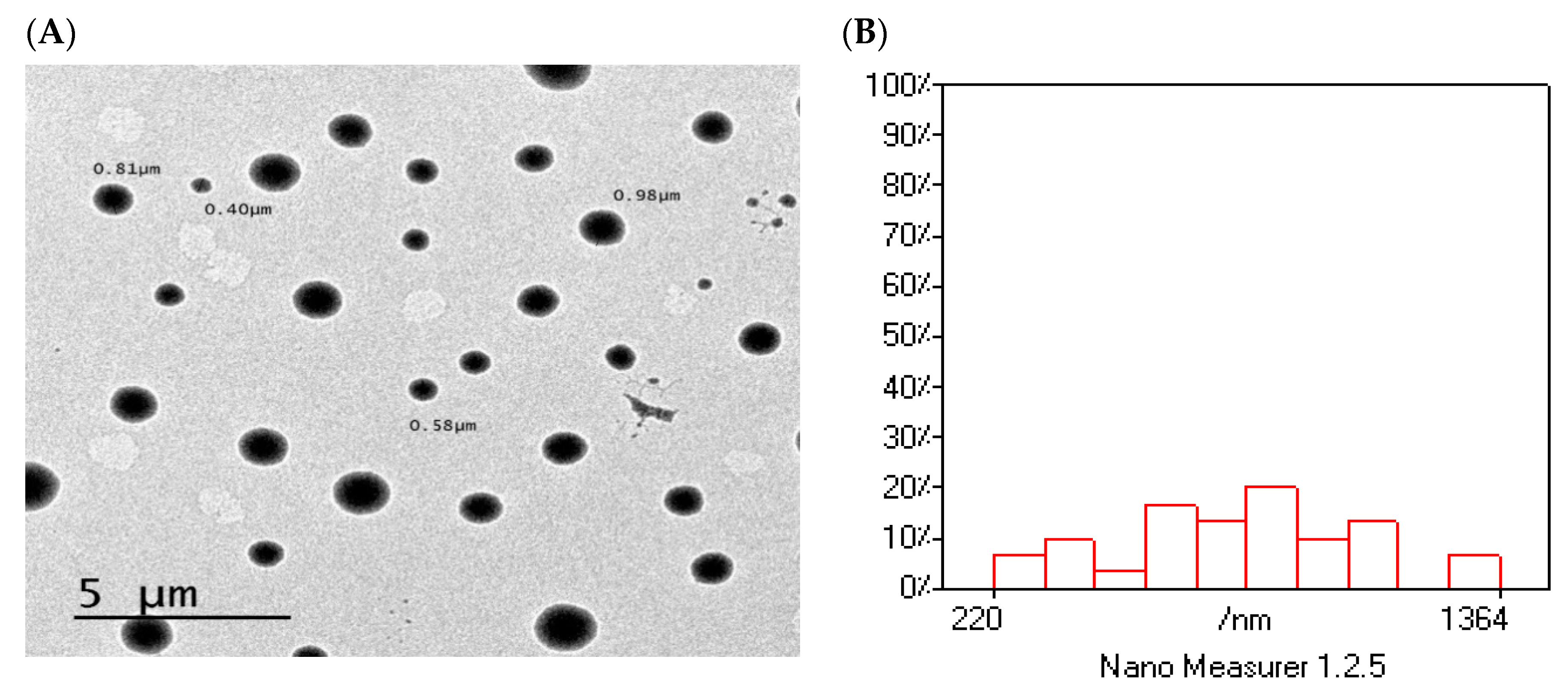
3.14. Surface Morphology of Optimized Formulation
3.15. In-Vitro Release Study of the Optimized Formulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Portt, L.; Norman, G.; Clapp, C.; Greenwood, M.; Greenwood, M.T. Anti-apoptosis and cell survival: A review. Biochim. Biophys. Acta 2011, 1813, 238–259. [Google Scholar] [CrossRef] [Green Version]
- Okada, H.; Mak, T.W. Pathways of apoptotic and non-apoptotic death in tumour cells. Nat. Rev. Cancer 2004, 4, 592–603. [Google Scholar] [CrossRef]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef]
- Cory, S.; Adams, J.M. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Garner, T.P.; Lopez, A.; Reyna, D.E.; Spitz, A.Z.; Gavathiotis, E. Progress in targeting the BCL-2 family of proteins. Curr. Opin. Chem. Biol. 2017, 39, 133–142. [Google Scholar] [CrossRef]
- Thomas, S.; Quinn, B.A.; Das, S.K.; Dash, R.; Emdad, L.; Dasgupta, S.; Wang, X.Y.; Dent, P.; Reed, J.C.; Pellecchia, M.; et al. Targeting the Bcl-2 family for cancer therapy. Expert Opin. Ther. Targets 2013, 17, 61–75. [Google Scholar] [CrossRef] [Green Version]
- Yip, K.W.; Reed, J.C. Bcl-2 family proteins and cancer. Oncogene 2008, 27, 6398–6406. [Google Scholar] [CrossRef] [Green Version]
- Birkinshaw, R.W.; Gong, J.-N.; Luo, C.S.; Lio, D.; White, C.A.; Anderson, M.A.; Blombery, P.; Lessene, G.; Majewski, I.J.; Thijssen, R.; et al. Structures of BCL-2 in complex with venetoclax reveal the molecular basis of resistance mutations. Nat. Commun. 2019, 10, 2385. [Google Scholar] [CrossRef]
- Besbes, S.; Mirshahi, M.; Pocard, M.; Billard, C. New dimension in therapeutic targeting of BCL-2 family proteins. Oncotarget 2015, 6, 12862–12871. [Google Scholar] [CrossRef] [Green Version]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Iyer, D.; Vartak, S.V.; Mishra, A.; Goldsmith, G.; Kumar, S.; Srivastava, M.; Hegde, M.; Gopalakrishnan, V.; Glenn, M.; Velusamy, M.; et al. Identification of a novel BCL2-specific inhibitor that binds predominantly to the BH1 domain. Febs J. 2016, 283, 3408–3437. [Google Scholar] [CrossRef] [PubMed]
- Vartak, S.V.; Hegde, M.; Iyer, D.; Gaikwad, S.; Gopalakrishnan, V.; Srivastava, M.; Karki, S.S.; Choudhary, B.; Ray, P.; Santhoshkumar, T.R.; et al. A novel inhibitor of BCL2, Disarib abrogates tumor growth while sparing platelets, by activating intrinsic pathway of apoptosis. Biochem. Pharmacol. 2016, 122, 10–22. [Google Scholar] [CrossRef]
- Kang, M.H.; Reynolds, C.P. Bcl-2 inhibitors: Targeting mitochondrial apoptotic pathways in cancer therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 1126–1132. [Google Scholar] [CrossRef] [Green Version]
- Paik, P.K.; Rudin, C.M.; Pietanza, M.C.; Brown, A.; Rizvi, N.A.; Takebe, N.; Travis, W.; James, L.; Ginsberg, M.S.; Juergens, R.; et al. A phase II study of obatoclax mesylate, a Bcl-2 antagonist, plus topotecan in relapsed small cell lung cancer. Lung Cancer 2011, 74, 481–485. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Viallet, J.; Haura, E.B. A small molecule pan-Bcl-2 family inhibitor, GX15–070, induces apoptosis and enhances cisplatin-induced apoptosis in non-small cell lung cancer cells. Cancer Chemother. Pharmacol. 2008, 61, 525–534. [Google Scholar] [CrossRef]
- Zhai, D.; Jin, C.; Satterthwait, A.C.; Reed, J.C. Comparison of chemical inhibitors of antiapoptotic Bcl-2-family proteins. Cell Death Differ. 2006, 13, 1419–1421. [Google Scholar] [CrossRef] [Green Version]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef] [Green Version]
- Koehler, B.C.; Jassowicz, A.; Scherr, A.L.; Lorenz, S.; Radhakrishnan, P.; Kautz, N.; Elssner, C.; Weiss, J.; Jaeger, D.; Schneider, M.; et al. Pan-Bcl-2 inhibitor Obatoclax is a potent late stage autophagy inhibitor in colorectal cancer cells independent of canonical autophagy signaling. BMC Cancer 2015, 15, 919. [Google Scholar] [CrossRef] [Green Version]
- Schwartz-Roberts, J.L.; Shajahan, A.N.; Cook, K.L.; Wärri, A.; Abu-Asab, M.; Clarke, R. GX15–070 (obatoclax) induces apoptosis and inhibits cathepsin D- and L-mediated autophagosomal lysis in antiestrogen-resistant breast cancer cells. Mol. Cancer Ther. 2013, 12, 448–459. [Google Scholar] [CrossRef] [Green Version]
- Stamelos, V.A.; Fisher, N.; Bamrah, H.; Voisey, C.; Price, J.C.; Farrell, W.E.; Redman, C.W.; Richardson, A. The BH3 mimetic obatoclax accumulates in lysosomes and causes their alkalinization. PLoS ONE 2016, 11, e0150696. [Google Scholar] [CrossRef] [PubMed]
- Basit, F.; Cristofanon, S.; Fulda, S. Obatoclax (GX15–070) triggers necroptosis by promoting the assembly of the necrosome on autophagosomal membranes. Cell Death Differ. 2013, 20, 1161–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, D.; Gu, C.; Shi, L.; Xun, T.; Li, X.; Liu, S.; Yu, L. Obatoclax induces G1/G0-phase arrest via p38/p21(waf1/Cip1) signaling pathway in human esophageal cancer cells. J. Cell Biochem. 2014, 115, 1624–1635. [Google Scholar] [CrossRef]
- Zacarías-Lara, O.J.; Correa-Basurto, J.; Bello, M. Exploring the conformational and binding properties of unphosphorylated/phosphorylated monomeric and trimeric Bcl-2 through docking and molecular dynamics simulations. Biopolymers 2016, 105, 393–413. [Google Scholar] [CrossRef]
- Cornils, B. Philips Reaction. In Catalysis from A to Z; Herrmann, W., Cornils, B., Zanthoff, H., Xu, J.-H., Eds.; Wiley & Sons: New York, NY, USA, 2020. [Google Scholar] [CrossRef]
- Achar, K.C.; Hosamani, K.M.; Seetharamareddy, H.R. In-vivo analgesic and anti-inflammatory activities of newly synthesized benzimidazole derivatives. Eur. J. Med. Chem. 2010, 45, 2048–2054. [Google Scholar] [CrossRef]
- Colella, M.; Degennaro, L.; Luisi, R. Continuous flow synthesis of heterocycles: A recent update on the flow synthesis of indoles. Molecules 2020, 25, 3242. [Google Scholar] [CrossRef]
- Kumar, S.; Kumar, P.; Sati, N. Synthesis and biological evaluation of Schiff bases and azetidinones of 1-naphthol. J. Pharm. Bioallied Sci. 2012, 4, 246–249. [Google Scholar] [CrossRef]
- Kishk, S.M.; McLean, K.J.; Sood, S.; Smith, D.; Evans, J.W.D.; Helal, M.A.; Gomaa, M.S.; Salama, I.; Mostafa, S.M.; de Carvalho, L.P.S.; et al. Design and synthesis of imidazole and triazole pyrazoles as mycobacterium tuberculosis CYP121A1 inhibitors. ChemistryOpen 2019, 8, 995–1011. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.V.; Herst, P.M.; Tan, A.S. Tetrazolium dyes as tools in cell biology: New insights into their cellular reduction. Biotechnol. Ann. Rev. 2005, 11, 127–152. [Google Scholar]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Van den Eijnde, S.M.; Luijsterburg, A.J.; Boshart, L.; De Zeeuw, C.I.; van Dierendonck, J.H.; Reutelingsperger, C.P.; Vermeij-Keers, C. In situ detection of apoptosis during embryogenesis with annexin V: From whole mount to ultrastructure. Cytometry 1997, 29, 313–320. [Google Scholar] [CrossRef]
- Darzynkiewicz, Z. Critical aspects in analysis of cellular DNA content. Curr. Protoc. Cytom. 2011, 56, 1–8. [Google Scholar] [CrossRef]
- Darzynkiewicz, Z.; Juan, G. DNA content measurement for DNA ploidy and cell cycle analysis. Curr. Protoc. Cytom. 2001, 1, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, K.C.; Gross, M.; Peirce, S.; Luyindula, D.; Liu, X.; Vu, A.; Sliozberg, M.; Guo, R.; Zhao, H.; Reynolds, C.P.; et al. Mitochondrial Bcl-2 family dynamics define therapy response and resistance in neuroblastoma. Cancer Res. 2012, 72, 2565–2577. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S. Regulation of caspase activation in apoptosis: Implications in pathogenesis and treatment of disease. Clin. Exp. Pharmacol. Physiol. 1999, 26, 295–303. [Google Scholar] [CrossRef]
- Ouyang, L.; Shi, Z.; Zhao, S.; Wang, F.T.; Zhou, T.T.; Liu, B.; Bao, J.K. Programmed cell death pathways in cancer: A review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012, 45, 487–498. [Google Scholar] [CrossRef]
- Ferreira, K.S.; Kreutz, C.; Macnelly, S.; Neubert, K.; Haber, A.; Bogyo, M.; Timmer, J.; Borner, C. Caspase-3 feeds back on caspase-8, Bid and XIAP in type I Fas signaling in primary mouse hepatocytes. Apoptosis 2012, 17, 503–515. [Google Scholar] [CrossRef]
- Mariani, M.; Karki, R.; Spennato, M.; Pandya, D.; He, S.; Andreoli, M.; Fiedler, P.; Ferlini, C. Class III β-tubulin in normal and cancer tissues. Gene 2015, 563, 109–114. [Google Scholar] [CrossRef]
- Sharifi, S.; Barar, J.; Hejazi, M.S.; Samadi, N. Roles of the Bcl-2/Bax ratio, caspase-8 and 9 in resistance of breast cancer cells to paclitaxel. Asian Pac. J. Cancer Prev. 2014, 15, 8617–8622. [Google Scholar] [CrossRef] [Green Version]
- Murray, J.B.; Davidson, J.; Chen, I.; Davis, B.; Dokurno, P.; Graham, C.J.; Harris, R.; Jordan, A.; Matassova, N.; Pedder, C.; et al. Establishing drug discovery and identification of hit series for the anti-apoptotic proteins, Bcl-2 and Mcl-1. ACS Omega 2019, 4, 8892–8906. [Google Scholar] [CrossRef]
- Kontoyianni, M.; McClellan, L.M.; Sokol, G.S. Evaluation of docking performance: Comparative data on docking algorithms. J. Med. Chem. 2004, 47, 558–565. [Google Scholar] [CrossRef]
- Severino, P.; Andreani, T.; Macedo, A.S.; Fangueiro, J.F.; Santana, M.H.A.; Silva, A.M.; Souto, E.B. Current state-of-art and new trends on lipid nanoparticles (SLN and NLC) for oral drug delivery. J. Drug Deliv. 2012, 2012, 750891. [Google Scholar] [CrossRef] [PubMed]
- Pardeshi, C.; Rajput, P.; Belgamwar, V.; Tekade, A.; Patil, G.; Chaudhary, K.; Sonje, A. Solid lipid based nanocarriers: An overview. Acta Pharm. 2012, 62, 433–472. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Ray, S.; Thakur, R.S. Solid lipid nanoparticles: A modern formulation approach in drug delivery system. Indian J. Pharm. Sci. 2009, 71, 349–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, R.H.; Mäder, K.; Gohla, S. Solid lipid nanoparticles (SLN) for controlled drug delivery—A review of the state of the art. Eur. J. Pharm. Biopharm. 2000, 50, 161–177. [Google Scholar] [CrossRef]
- Ekambaram, P.; Abdul, H.S.A. Formulation and evaluation of solid lipid nanoparticles of ramipril. J. Young Pharm. JYP 2011, 3, 216–220. [Google Scholar] [CrossRef] [Green Version]
- Qushawy, M.; Prabahar, K.; Abd-Alhaseeb, M.; Swidan, S.; Nasr, A. Preparation and evaluation of carbamazepine solid lipid nanoparticle for alleviating seizure activity in pentylenetetrazole-kindled mice. Molecules 2019, 24, 3971. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, R.S.; Silva, J.O.; Seabra, H.A.; Oliveira, M.S.; Carregal, V.M.; Vilela, J.M.C.; Andrade, M.S.; Townsend, D.M.; Colletti, P.M.; Leite, E.A.; et al. α-Tocopherol succinate loaded nano-structed lipid carriers improves antitumor activity of doxorubicin in breast cancer models in vivo. Biomed. Pharmacother. 2018, 103, 1348–1354. [Google Scholar] [CrossRef]
- Joseph, E.; Reddi, S.; Rinwa, V.; Balwani, G.; Saha, R. Design and in vivo evaluation of solid lipid nanoparticulate systems of Olanzapine for acute phase schizophrenia treatment: Investigations on antipsychotic potential and adverse effects. Eur. J. Pharm. Sci. 2017, 104, 315–325. [Google Scholar] [CrossRef]
- Sznitowska, M.; Wolska, E.; Baranska, H.; Cal, K.; Pietkiewicz, J. The effect of a lipid composition and a surfactant on the characteristics of the solid lipid microspheres and nanospheres (SLM and SLN). Eur. J. Pharm. Biopharm. 2017, 110, 24–30. [Google Scholar] [CrossRef]
- Priyanka, K.; Sathali, A.A. Preparation and evaluation of montelukast sodium loaded solid lipid nanoparticles. J. Young Pharm. 2012, 4, 129–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Housiny, S.; Shams Eldeen, M.A.; El-Attar, Y.A.; Salem, H.A.; Attia, D.; Bendas, E.R.; El-Nabarawi, M.A. Fluconazole-loaded solid lipid nanoparticles topical gel for treatment of pityriasis versicolor: Formulation and clinical study. Drug Deliv. 2018, 25, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Zielińska, A.; Ferreira, N.R.; Durazzo, A.; Lucarini, M.; Cicero, N.; Mamouni, S.E.; Silva, A.M.; Nowak, I.; Santini, A.; Souto, E.B. Development and optimization of alpha-pinene-loaded solid lipid nanoparticles (SLN) using experimental factorial design and dispersion analysis. Molecules 2019, 24, 2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardouh, A.R.; Attia, M.A.; Enan, E.T.; Elbahaie, A.M.; Fouad, R.A.; El-Shafey, M.; Youssef, A.M.; Alomar, S.Y.; Ali, Z.A.-E.; Zaitone, S.A.; et al. Synthesis and antitumor activity of doxycycline polymeric nanoparticles: Effect on tumor apoptosis in solid ehrlich carcinoma. Molecules 2020, 25, 3230. [Google Scholar] [CrossRef] [PubMed]
- Hong, I.K.; Kim, S.I.; Lee, S.B. Effects of HLB value on oil-in-water emulsions: Droplet size, rheological behavior, zeta-potential, and creaming index. J. Ind. Eng. Chem. 2018, 67, 123–131. [Google Scholar] [CrossRef]
- Schwarz, C.; Mehnert, W. Solid lipid nanoparticles (SLN) for controlled drug delivery. II. Drug incorporation and physicochemical characterization. J. Microencapsul. 1999, 16, 205–213. [Google Scholar] [CrossRef]
- Fasehee, H.; Dinarvand, R.; Ghavamzadeh, A.; Esfandyari-Manesh, M.; Moradian, H.; Faghihi, S.; Ghaffari, S.H. Delivery of disulfiram into breast cancer cells using folate-receptor-targeted PLGA-PEG nanoparticles: In vitro and in vivo investigations. J. Nanobiotechnol. 2016, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Agarwal, A.; Majumder, S.; Lariya, N.; Khaya, A.; Agrawal, H.; Majumdar, S.; Agrawal, G.P. Mannosylated solid lipid nanoparticles as vectors for site-specific delivery of an anti-cancer drug. J. Control. Release 2010, 148, 359–367. [Google Scholar] [CrossRef]
- Kalepu, S.; Nekkanti, V. Insoluble drug delivery strategies: Review of recent advances and business prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef] [Green Version]
- Swidan, S.; Ghonaim, H.; Samy, A.; Ghorab, M. Efficacy and in vitro cytotoxicity of nanostructured lipid carriers for paclitaxel delivery. J. Appl. Pharm. Sci. 2016, 6, 18–26. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhang, L.; Chen, T.; Guo, W.; Bao, X.; Wang, D.; Ren, B.; Wang, H.; Li, Y.; Wang, Y.; et al. Anticancer effects of resveratrol-loaded solid lipid nanoparticles on human breast cancer cells. Molecules 2017, 22, 1814. [Google Scholar] [CrossRef] [PubMed]
- Gutierréz-Hernández, A.; Galván-Ciprés, Y.; Domínguez-Mendoza, E.A.; Aguirre-Vidal, Y.; Estrada-Soto, S.; Almanza-Pérez, J.C.; Navarrete-Vázquez, G. Design, Synthesis, Antihyperglycemic studies, and docking simulations of benzimidazole-thiazolidinedione hybrids. J. Chem. 2019, 2019, 1650145. [Google Scholar] [CrossRef]
- Wang, W.; Pang, J.; Ha, E.H.; Zhou, M.; Li, Z.; Tian, S.; Li, H.; Hu, Q. Development of novel NLRP3-XOD dual inhibitors for the treatment of gout. Bioorg Med Chem Lett. 2020, 30, 126944. [Google Scholar] [CrossRef] [PubMed]
- Basceken, S.; Kaya, S.; Balci, M. Intramolecular gold-catalyzed and NaH-supported cyclization reactions of N-propargyl indole derivatives with pyrazole and pyrrole rings: Synthesis of pyrazolodiazepinoindole, pyrazolopyrazinoindole, and pyrrolopyrazinoindole. J. Org. Chem. 2015, 80, 12552–12561. [Google Scholar] [CrossRef]
- Demurtas, M.; Baldisserotto, A.; Lampronti, I.; Moi, D.; Balboni, G.; Pacifico, S.; Vertuani, S.; Manfredini, S.; Onnis, V. Indole derivatives as multifunctional drugs: Synthesis and evaluation of antioxidant, photoprotective and antiproliferative activity of indole hydrazones. Bioorganic Chem. 2019, 85, 568–576. [Google Scholar] [CrossRef]
- Mirfazli, S.S.; Kobarfard, F.; Firoozpour, L.; Asadipour, A.; Esfahanizadeh, M.; Tabib, K.; Shafiee, A.; Foroumadi, A. N-substituted indole carbohydrazide derivatives: Synthesis and evaluation of their antiplatelet aggregation activity. DARU 2014, 22, 65. [Google Scholar] [CrossRef] [Green Version]
- Boraei, A.T.; El Ashry, S.H.; Barakat, A.; Ghabbour, H.A. Synthesis of new functionalized indoles based on ethyl indol-2-carboxylate. Molecules 2016, 21, 333. [Google Scholar] [CrossRef] [Green Version]
- Al-Ebaisat, H.S.; Ababne, T.S.; Al Shboul, T.M.; Jazzazi, T.M. Synthesis, characterization and antifungal activity of some substituted 4-thiazolidinone derivatives. J. Pure Appl. Chem. Res. 2016, 5, 125–130. [Google Scholar] [CrossRef] [Green Version]
- Maurya, D.K.; Nandakumar, N.; Devasagayam, T.P.A. Anticancer property of gallic acid in A549, a human lung adenocarcinoma cell line, and possible mechanisms. J. Clin. Biochem. Nutr. 2011, 48, 85–90. [Google Scholar] [CrossRef] [Green Version]
- Ranganathan, S.; Halagowder, D.; Sivasithambaram, N.D. Quercetin suppresses twist to induce apoptosis in MCF-7 breast cancer cells. PLoS ONE 2015, 10, e0141370. [Google Scholar] [CrossRef] [Green Version]
- Koulaouzidou, E.A.; Papazisis, K.T.; Economides, N.A.; Beltes, P.; Kortsaris, A.H. Antiproliferative effect of mineral trioxide aggregate, zinc oxide-eugenol cement, and glass-ionomer cement against three fibroblastic cell lines. J. Endod. 2005, 31, 44–46. [Google Scholar] [CrossRef] [PubMed]
- Dey, N.S.; Mukherjee, B.; Maji, R.; Satapathy, B.S. Development of linker-conjugated nanosize lipid vesicles: A strategy for cell selective treatment in breast cancer. Curr. Cancer Drug Targets 2016, 16, 357–372. [Google Scholar] [PubMed]
- Wang, W.; Chen, T.; Xu, H.; Ren, B.; Cheng, X.; Qi, R.; Liu, H.; Wang, Y.; Yan, L.; Chen, S.; et al. Curcumin-loaded solid lipid nanoparticles enhanced anticancer efficiency in breast cancer. Molecules 2018, 23, 1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, B.; Shim, M.K.; Park, M.J.; Jang, E.H.; Yoon, H.Y.; Kim, K.; Kim, J.H. Hydrophobically modified polysaccharide-based on polysialic acid nanoparticles as carriers for anticancer drugs. Int. J. Pharm. 2017, 520, 111–118. [Google Scholar] [CrossRef]
- Hegazy, M.G.A.; Imam, A.M.; Abdelghany, B.E. Evaluation of cytotoxic and anticancer effect of Orobanche crenata methanolic extract on cancer cell lines. Tumor Biol. 2020, 42, 1010428320918685. [Google Scholar] [CrossRef]
- Tantawy, M.A.; Sroor, F.M.; Mohamed, M.F.; El-Naggar, M.E.; Saleh, F.M.; Hassaneen, H.M.; Abdelhamid, I.A. Molecular docking study, cytotoxicity, cell cycle arrest and apoptotic induction of novel chalcones incorporating thiadiazolyl isoquinoline in cervical cancer. Anticancer Agents Med. Chem. 2020, 20, 70–83. [Google Scholar] [CrossRef]
- Zhao, Y.; Xiang, S.; Dai, X.; Yang, K. A simplified diphenylamine colorimetric method for growth quantification. Appl. Microbiol. Biotechnol. 2013, 97, 5069–5077. [Google Scholar] [CrossRef]
- Preusse, M.; Tantawy, M.A.; Klawonn, F.; Schughart, K.; Pessler, F. Infection- and procedure-dependent effects on pulmonary gene expression in the early phase of influenza A virus infection in mice. BMC Microbiol. 2013, 13, 293. [Google Scholar] [CrossRef] [Green Version]
- Petersen, H.; Mostafa, A.; Tantawy, M.A.; Iqbal, A.A.; Hoffmann, D.; Tallam, A.; Selvakumar, B.; Pessler, F.; Beer, M.; Rautenschlein, S.; et al. NS segment of a 1918 influenza a virus-descendent enhances replication of H1N1pdm09 and virus-induced cellular immune response in mammalian and avian systems. Front. Microbiol. 2018, 9, 526. [Google Scholar] [CrossRef]
- El Raey, M.A.; El-Hagrassi, A.M.; Osman, A.F.; Darwish, K.M.; Emam, M. Acalypha wilkesiana flowers: Phenolic profiling, cytotoxic activity of their biosynthesized silver nanoparticles and molecular docking study for its constituents as Topoisomerase-I inhibitors. Biocatal. Agric. Biotechnol. 2019, 20, 101243. [Google Scholar] [CrossRef]
- Malebari, A.; Ibrahim, T.; Salem, I.; Salama, I.; Khayyat, A.; Mostafa, S.; El-Sabbagh, O.; Darwish, K. The anticancer activity for the bumetanide-based analogs via targeting the tumor-associated membrane bound human carbonic anhydrase-IX enzyme. Pharmaceuticals 2020, 13, 252. [Google Scholar] [CrossRef] [PubMed]
- Wadie, M.A.; Kishk, S.M.; Darwish, K.M.; Mostafa, S.M.; Elgawish, M.S. Simultaneous determination of losartan and rosuvastatin in rat plasma using liquid chromatography–tandem mass spectrometric technique for application into pharmacokinetic and drug–drug interaction studies. Chromatographia 2020, 83, 1477–1494. [Google Scholar] [CrossRef]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal chemistry and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System, 2.0.6; Schrödinger, LLC: New York, NY, USA, 2016.
- Souto, E.B.; Doktorovova, S.; Zielinska, A.; Silva, A.M. Key production parameters for the development of solid lipid nanoparticles by high shear homogenization. Pharm. Dev. Technol. 2019, 24, 1181–1185. [Google Scholar] [CrossRef] [PubMed]
- Da Rocha Lindner, G.; Khalil, N.M.; Mainardes, R.M. Resveratrol-loaded polymeric nanoparticles: Validation of an HPLC-PDA method to determine the drug entrapment and evaluation of its antioxidant activity. Sci. World J. 2013, 2013, 506083. [Google Scholar] [CrossRef] [Green Version]
- Nasr, A.M.; Qushawy, M.K.; Elkhoudary, M.M.; Gawish, A.Y.; Elhady, S.S.; Swidan, S.A. Quality by design for the development and analysis of enhanced in-situ forming vesicles for the improvement of the bioavailability of fexofenadine HCl in vitro and in vivo. Pharmaceutics 2020, 12, 409. [Google Scholar] [CrossRef]
- Aldawsari, H.M.; Singh, S. Rapid microwave-assisted cisplatin-loaded solid lipid nanoparticles: Synthesis, characterization and anticancer study. Nanomaterials 2020, 10, 510. [Google Scholar] [CrossRef] [Green Version]
- Mona, Q.; Ali, N. Solid lipid nanoparticles (SLNs) as nano drug delivery carriers: Preparation, characterization and application. Int. J. Appl. Pharm. 2019, 12, 1–9. [Google Scholar]

























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound b No. | MDA-MB-231 IC50 (µM) c | A549 IC50 (µM) c | MDCK IC50 (µM) c |
|---|---|---|---|
| 3a | 29.68 ± 4.03 | ND | ND |
| 7b | 43.77 ± 1.09 | ND | 28.92 ± 1.49 |
| 7d | 17.38 ± 3.23 | ND | ND |
| 8a | 12.69 ± 0.84 | ND | 73.86 ± 2.30 |
| 8b | 12.71 ± 2.48 | 23.05 ± 1.45 | 92.75 ± 0.43 |
| 8c | 12.83 ± 3.50 | 11.63 ± 2.57 | 87.29 ± 3.00 |
| 8d | 31.46 ± 4.66 | ND | ND |
| 8f | 21.64 ± 0.28 | ND | ND |
| Factors and Responses | Level Used | ||
| Factor | Name | Low (−1) | High (+1) |
| A: X1 | Type of lipid | Compritol 888 ATO (COMP) | Glyceryl Monostearate (GMS) |
| B: X2 | Type of surfactant | Cremophor RH40 | Poloxamer 188 |
| C: X3 | Surfactant conc. (% w/v) | 1% | 1.5% |
| Response | Name | Goal | |
| Y1 | EE (%) | Maximize | |
| Y2 | PS (nm) | Minimize | |
| Y3 | ZP (mV) | Maximize | |
| SNP Formulation No. | X1 | X2 | X3 |
|---|---|---|---|
| A: Lipid Type | B: Surfactant Lipid | C: Surfactant Conc. (% w/v) | |
| 1 | COMP | Cremophor RH40 | 1.5 |
| 2 | COMP | Cremophor RH40 | 1 |
| 3 | COMP | Poloxamer 188 | 1 |
| 4 | GMS | Cremophor RH40 | 1 |
| 5 | COMP | Poloxamer 188 | 1.5 |
| 6 | GMS | Cremophor RH40 | 1.5 |
| 7 | GMS | Poloxamer 188 | 1 |
| 8 | GMS | Poloxamer 188 | 1.5 |
| SNP Formulation No. | (Y1) EE% | (Y2) PS (nm) | (Y3) ZP (mV) | Polydispersity Index (PDI) |
|---|---|---|---|---|
| F1 | 37.1 ± 2.45 | 140.9 ± 3.2 | −12.3 ± 0.77 | 0.352 ± 0.03 |
| F2 | 53.4 ± 1.65 | 135.1 ± 1.0 | −13.6 ± 0.32 | 0.282 ± 0.01 |
| F3 | 95.3 ± 1.34 | 329.4 ± 6.1 | −21.7 ± 0.15 | 0.563 ± 0.03 |
| F4 | 82.8 ± 2.55 | 189.8 ± 3.3 | −36.1 ± 1.90 | 0.432 ± 0.04 |
| F5 | 86.8 ± 3.32 | 181.5 ± 2.1 | −16.4 ± 1.10 | 0.442 ± 0.02 |
| F6 | 67.7 ± 1.52 | 100.4 ± 0.4 | −39.4 ± 0.095 | 0.433 ± 0.01 |
| F7 | 94.6 ± 2.67 | 537.3 ± 10.4 | −37.5 ± 0.58 | 0.582 ± 0.04 |
| F8 | 92.8 ± 2.38 | 226.1 ±10.5 | −29.3 ± 1.17 | 0.639 ± 0.12 |
| Gene | Forward | Reverse |
|---|---|---|
| BAX | 5′-AGTGGCAGCTGACATGTTTT-3′ | 5′-GGAGGAAGTCCAATGTCCAG-3′ |
| Casp-3 | 5′-GGCCCTGAAATACGAAGTC-3′ | 5′-GGCAGTAGTCGACTCTGAAG-3′ |
| Casp-8 | 5′-GCCTCCCTCAAGTTCCT-3′ | 5′-CCTGGAGTCTCTGGAATAACA-3′ |
| Casp-9 | 5′-CGAACTAACAGGCAAGCAG-3′ | 5′-ACCTCACCAAATCCTCCAGAAC-3′ |
| BcL-2 | 5′-CCTGTGGATGACTGAGTACC-3′ | 5′-GAGACAGCCAGGAGAAATCA-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagy, M.I.; Darwish, K.M.; Kishk, S.M.; Tantawy, M.A.; Nasr, A.M.; Qushawy, M.; Swidan, S.A.; Mostafa, S.M.; Salama, I. Design, Synthesis, Anticancer Activity, and Solid Lipid Nanoparticle Formulation of Indole- and Benzimidazole-Based Compounds as Pro-Apoptotic Agents Targeting Bcl-2 Protein. Pharmaceuticals 2021, 14, 113. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14020113
Nagy MI, Darwish KM, Kishk SM, Tantawy MA, Nasr AM, Qushawy M, Swidan SA, Mostafa SM, Salama I. Design, Synthesis, Anticancer Activity, and Solid Lipid Nanoparticle Formulation of Indole- and Benzimidazole-Based Compounds as Pro-Apoptotic Agents Targeting Bcl-2 Protein. Pharmaceuticals. 2021; 14(2):113. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14020113
Chicago/Turabian StyleNagy, Manar I., Khaled M. Darwish, Safaa M. Kishk, Mohamed A. Tantawy, Ali M. Nasr, Mona Qushawy, Shady A. Swidan, Samia M. Mostafa, and Ismail Salama. 2021. "Design, Synthesis, Anticancer Activity, and Solid Lipid Nanoparticle Formulation of Indole- and Benzimidazole-Based Compounds as Pro-Apoptotic Agents Targeting Bcl-2 Protein" Pharmaceuticals 14, no. 2: 113. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14020113