
PARP Traps Rescue the Pro-Inflammatory Response of Human Macrophages in the In Vitro Model of LPS-Induced Tolerance


Abstract
:1. Introduction
2. Results
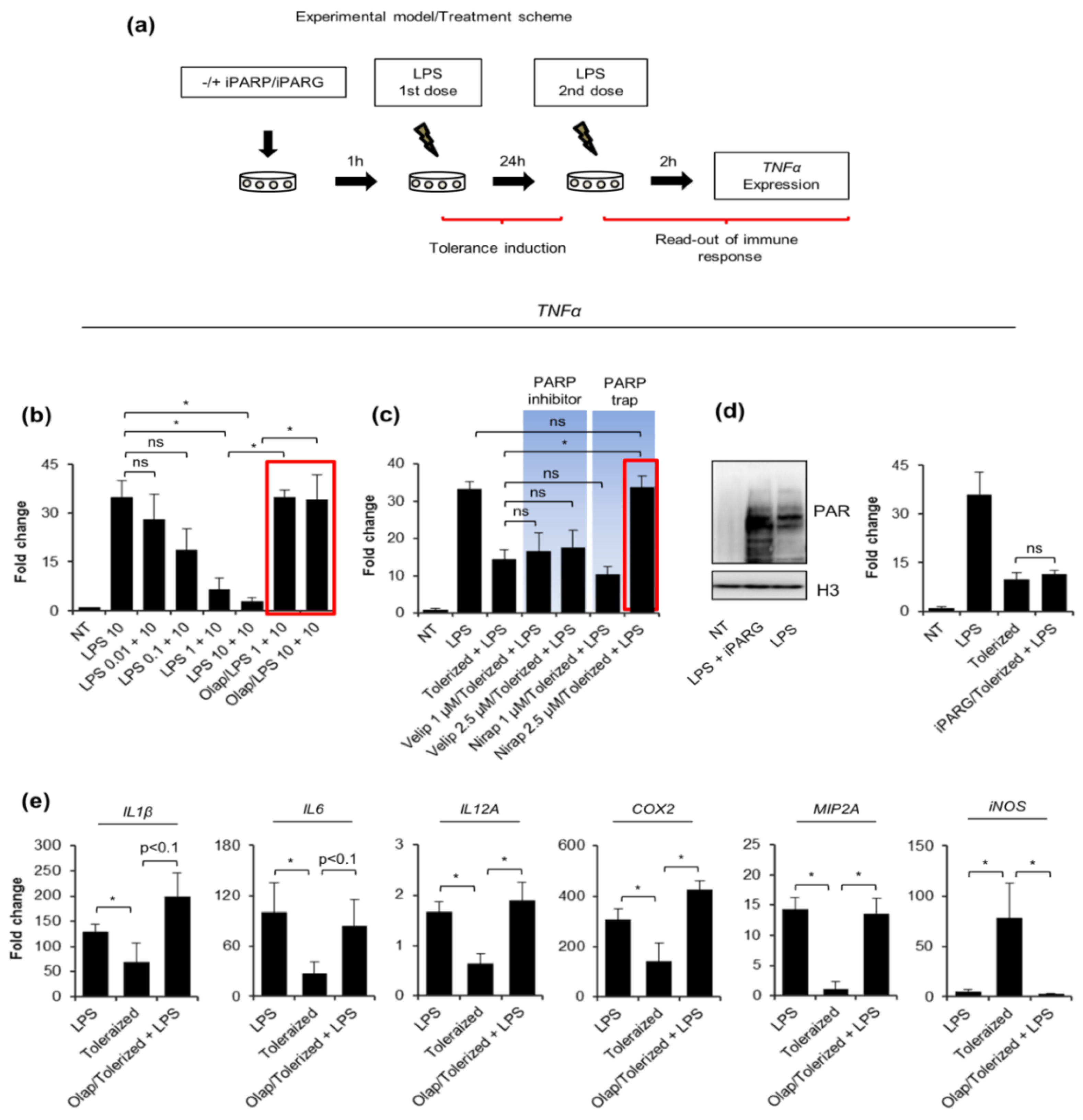
2.1. PARP Trapping Prior to Macrophage Activation Maintains Their Pro-Inflammatory Response
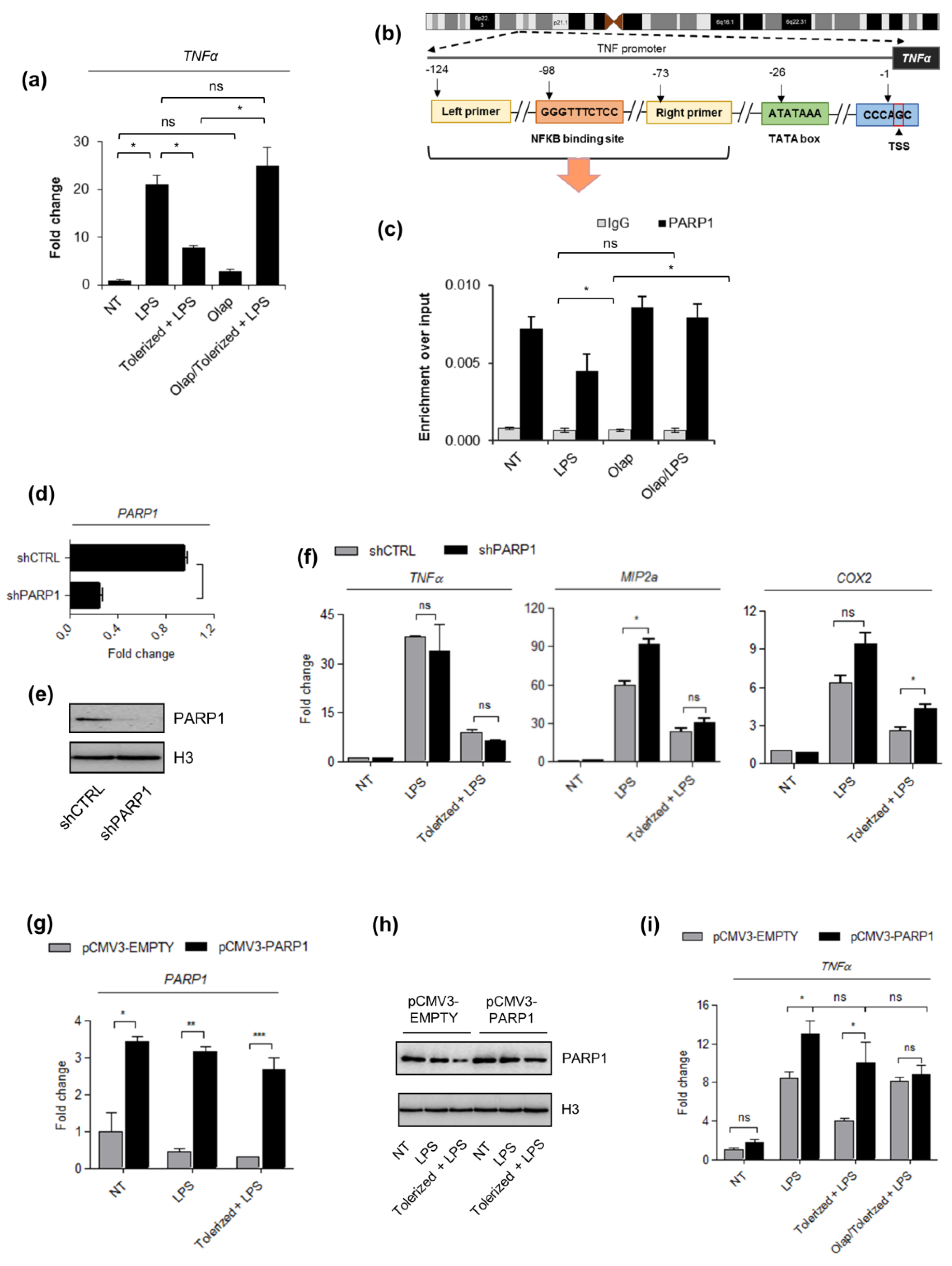
2.2. PARP1 Level Serves as a Key Determinant of Tolerance Development
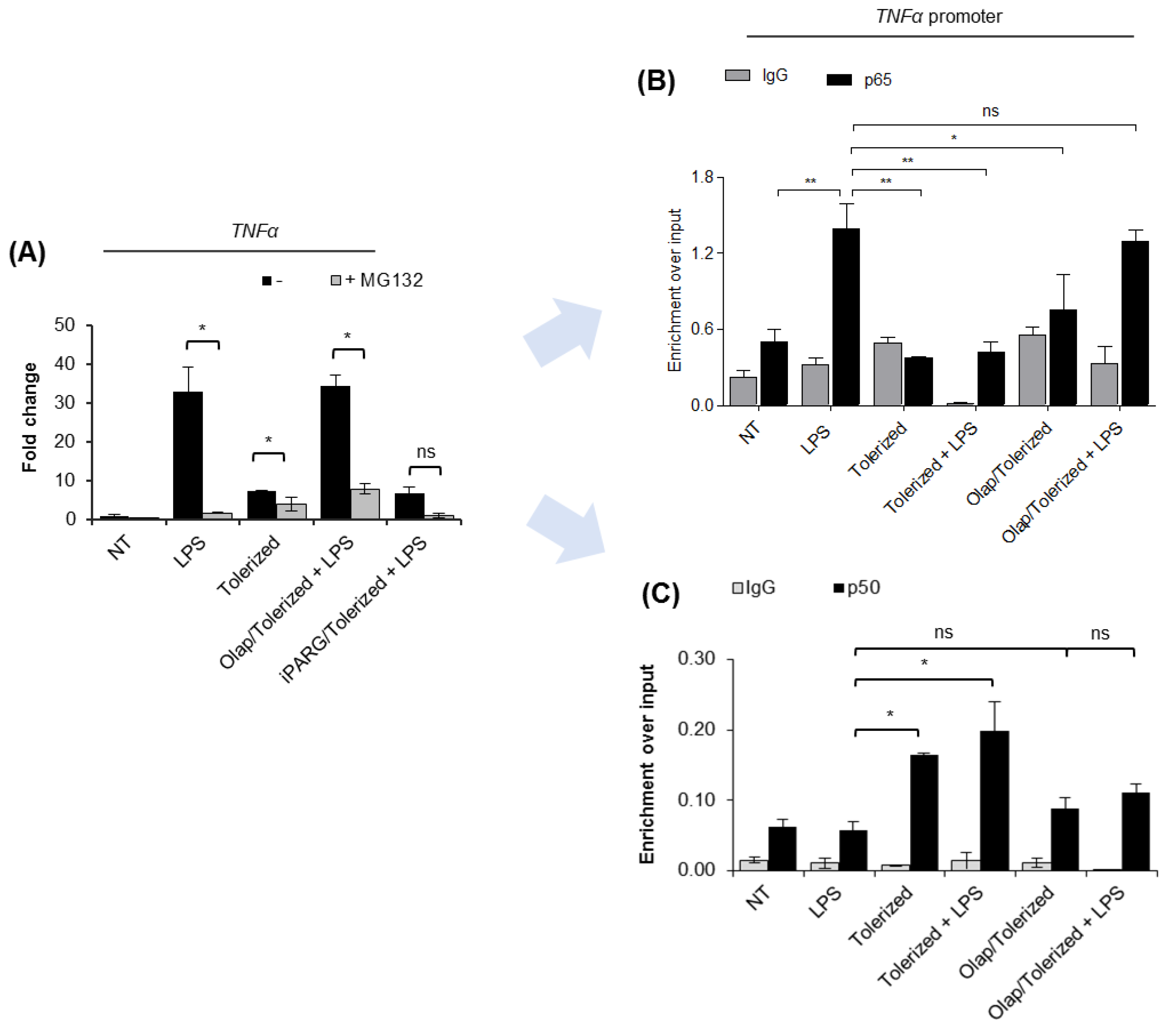
2.3. PARP1 Extrusion form TNF-α Promoter Hampers p65 Rebinding after Tolerance Induction
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Monocyte Isolation, Differentiation of Macrophages and Cell Culture
4.3. Induction of Immune Paralysis
4.4. Quantification of the Gene Expression
4.5. Western Blot
4.6. Chromatin Immunoprecipitation (CHIP) Assay
4.7. Permanent Gene Silencing and Transient Overexpression
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grondman, I.; Arts, R.; Koch, R.M.; Leijte, G.P.; Gerretsen, J.; Bruse, N.; Kempkes, R.; Ter Horst, R.; Kox, M.; Pickkers, P.; et al. Endotoxin-induced immunotolerance is associated with loss of monocyte metabolic plasticity and reduction of oxidative burst. J. Leukoc Biol. 2019, 5, 15–16. [Google Scholar] [CrossRef] [Green Version]
- Maddux, A.B.; Hiller, T.D.; Overdier, K.H.; Pyle, L.L.; Douglas, I.S. Innate immune function and organ failure recovery in adults with sepsis. J. Intensive Care Med. 2019, 34, 486–494. [Google Scholar] [CrossRef]
- Rhee, C.; Li, Z.; Wang, R.; Song, Y.; Kadri, S.S.; Septimus, E.J.; Chen, H.C.; Fram, D.; Jin, R.; Poland, R.; et al. Impact of risk adjustment using clinical vs. administrative data on hospital sepsis mortality comparisons. Open Forum Infect. Dis. 2020, 7, ofaa213. [Google Scholar] [CrossRef]
- Maddux, A.B.; Douglas, I.S. Is the developmentally immature immune response in paediatric sepsis a recapitulation of immune tolerance? Immunology 2015, 145, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, S.; Liu, B.; Wang, B.; He, S.; Zhang, R. Anti-inflammatory effects of adiponectin in cigarette smoke-activated alveolar macrophage through the COX-2/PGE2 and TLRs signaling pathway. Cytokine 2020, 133, 155148. [Google Scholar] [CrossRef]
- Zhang, D.; Chen, L.; Li, S.; Gu, Z.; Yan, J. Lipopolysaccharide (LPS) of Porphyromonas gingivalisinduces IL-1β, TNF-α and IL-6 production by THP-1 cells in a way different from that of Escherichia coli LPS. Innate Immunity 2008, 14, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Akhter, N.; Hasan, A.; Shenouda, S.; Wilson, A.; Kochumon, S.; Ali, S.; Tuomilehto, J.; Sindhu, S.; Ahmad, R. TLR4/MyD88-mediated CCL2 production by lipopolysaccharide (endotoxin): Implications for metabolic inflammation. J. Diabetes Metab. Disord. 2018, 17, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Sindhu, S.; Al-Roub, A.; Koshy, M.; Thomas, R.; Ahmad, R. Palmitate-Induced MMP-9 Expression in the human monocytic cells is mediated through the TLR4-MyD88 dependent mechanism. Cell Physiol. Biochem. 2016, 39, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Concetti, J.; Wilson, C.L. NFKB1 and cancer: Friend or foe? Cells 2018, 7, 133. [Google Scholar] [CrossRef] [Green Version]
- Eisemann, T.; Pascal, J.M. Poly(ADP-ribose) polymerase enzymes and the maintenance of genome integrity. Cell Mol. Life Sci. 2020, 77, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.; Kowluru, R.A. Role of PARP-1 as a novel transcriptional regulator of MMP-9 in diabetic retinopathy. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1761–1769. [Google Scholar] [CrossRef]
- Hassa, P.O.; Buerki, C.; Lombardi, C.; Imhof, R.; Hottiger, M.O. Transcriptional coactivation of nuclear factor-kappaB-dependent gene expression by p300 is regulated by poly(ADP)-ribose polymerase-1. J. Biol. Chem. 2003, 278, 45145–45153. [Google Scholar] [CrossRef] [Green Version]
- Ke, Y.; Wang, C.; Zhang, J.; Zhong, X.; Wang, R.; Zeng, X.; Ba, X. The role of PARPs in inflammation-and metabolic-related diseases: Molecular mechanisms and beyond. Cells 2019, 8, 1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobczak, M.; Zyma, M.; Robaszkiewicz, A. The role of PARP1 in monocyte and macrophage commitment and specification: Future perspectives and limitations for the treatment of monocyte and macrophage relevant diseases with PARP inhibitors. Cells 2020, 9, 2040. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- Pietrzak, J.; Płoszaj, T.; Pułaski, Ł.; Robaszkiewicz, A. EP300-HDAC1-SWI/SNF functional unit defines transcription of some DNA repair enzymes during differentiation of human macrophages. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Zhang, Y.; Morris, J.; Ji, J.; Takeda, S.; Doroshow, J.H.; Pommier, Y. Rationale for poly(ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibition. J. Pharmacol. Exp. Ther. 2014, 349, 408–416. [Google Scholar] [CrossRef] [Green Version]
- Leon-Ponte, M.; Kirchhof, M.G.; Sun, T.; Stephens, T.; Singh, B.; Sandhu, S.; Madrenas, J. Polycationic lipids inhibit the pro-inflammatory response to LPS. Immunol. Lett. 2005, 9, 73–83. [Google Scholar] [CrossRef]
- Regdon, Z.; Robaszkiewicz, A.; Kovács, K.; Rygielska, Ż.; Hegedűs, C.; Bodoor, K.; Szabó, É.; Virág, L. LPS protects macrophages from AIF-independent parthanatos by downregulation of PARP1 expression, induction of SOD2 expression, and a metabolic shift to aerobic glycolysis. Free Radic. Biol. Med. 2019, 131, 184–196. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Monneret, G.; Payen, D. Immunosuppression in sepsis: A novel understanding of the disorder and a new therapeutic approach. Lancet Infect. Dis. 2013, 13, 260–268. [Google Scholar] [CrossRef] [Green Version]
- Seeley, J.J.; Ghosh, S. Molecular mechanisms of innate memory and tolerance to LPS. J. Leukoc. Biol. 2017, 101, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Zamudio, R.; Ha, H.C. Histone ADP-ribosylation facilitates gene transcription by directly remodeling nucleosomes. Mol. Cell Biol. 2012, 32, 2490–2502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Günther, J.; Vogt, N.; Hampel, K.; Bikker, R.; Page, S.; Müller, B.; Kandemir, J.; Kracht, M.; Dittrich-Breiholz, O.; Huber, R.; et al. Identification of two forms of TNF tolerance in human monocytes: Differential inhibition of NF-κB/AP-1- and PP1-associated signaling. J. Immunol. 2014, 192, 3143–3155. [Google Scholar] [CrossRef]
- Haudek, S.B.; Bryant, D.D.; Giroir, B.P. Differential regulation of myocardial NF kappa B following acute or chronic TNF-alpha exposure. J. Mol. Cell Cardiol. 2001, 33, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.M.; Suarez-Alvarez, B.; Lopez-Larrea, C. Therapeutic epigenetic reprogramming of trained immunity in myeloid cells. Trends Immunol. 2019, 40, 66–80. [Google Scholar] [CrossRef]
- Yan, Q.; Carmody, R.J.; Qu, Z.; Ruan, Q.; Jager, J.; Mullican, S.E.; Lazar, M.A.; Chen, Y.H. Nuclear factor-κB binding motifs specify Toll-like receptor-induced gene repression through an inducible repressosome. Proc. Natl. Acad. Sci. USA 2012, 109, 14140–14145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; El Gazzar, M.; Yoza, B.K.; McCall, C.E. The NF-kappaB factor RelB and histone H3 lysine methyltransferase G9a directly interact to generate epigenetic silencing in endotoxin tolerance. J. Biol. Chem. 2009, 284, 27857–27865. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, D.; Ghosh, S. Regulation of the NF-κB-mediated transcription of inflammatory genes. Front. Immunol. 2014, 5, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novakovic, B.; Habibi, E.; Wang, S.Y.; Arts, R.J.W.; Davar, R.; Megchelenbrink, W.; Kim, B.; Kuznetsova, T.; Kox, M.; Zwaag, J.; et al. β-Glucan reverses the epigenetic state of LPS-induced immunological tolerance. Cell 2016, 167, 1354–1368. [Google Scholar] [CrossRef] [Green Version]
- Saeed, S.; Quintin, J.; Kerstens, H.H.; Rao, N.A.; Aghajanirefah, A.; Matarese, F.; Cheng, S.C.; Ratter, J.; Berentsen, K.; van der Ent, M.A.; et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 2014, 345, 1251086. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Zamudio, R.I.; Ha, H.C. PARP1 enhances inflammatory cytokine expression by alteration of promoter chromatin structure in microglia. Brain Behav. 2014, 4, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Rimoldi, M.; Raes, G.; Brys, L.; Ghezzi, P.; Di Liberto, D.; Dieli, F.; Ghisletti, S.; Natoli, G.; De Baetselier, P.; et al. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc. Natl. Acad. Sci. USA 2009, 106, 14978–14983. [Google Scholar] [CrossRef] [Green Version]
- Cartwright, T.N.; Worrell, J.C.; Marchetti, L.; Dowling, C.M.; Knox, A.; Kiely, P.; Mann, J.; Mann, D.A.; Wilson, C.L. HDAC1 interacts with the p50 NF-κB subunit via its nuclear localization sequence to constrain inflammatory gene expression. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 962–970. [Google Scholar] [CrossRef] [Green Version]
- Weisheit, C.K.; Klüners, A.; Wild, L.; Casalter, A.; Heilmann-Heimbach, S.; Sivalingam, S.; Kleiner, J.L.; Ehrentraut, S.F.; Hoeft, A.; Frede, S.; et al. Sustained immunoparalysis in endotoxin-tolerized monocytic cells. Mediators Inflamm. 2020, 2020, 8294342. [Google Scholar] [CrossRef]
- Cook, D.N.; Pisetsky, D.S.; Schwartz, D.A. Toll-like receptors in the pathogenesis of human disease. Nat. Immunol. 2004, 5, 975–979. [Google Scholar] [CrossRef] [PubMed]
- Möröy, T. The zinc finger transcription factor Growth factor independence 1 (Gfi1). Int. J. Biochem. Cell Biol. 2005, 37, 541–546. [Google Scholar] [CrossRef]
- Anwar, M.A.; Shah, M.; Kim, J.; Choi, S. Recent clinical trends in Toll-like receptor targeting therapeutics. Med. Res. Rev. 2019, 39, 1053–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharif-Askari, E.; Vassen, L.; Kosan, C.; Khandanpour, C.; Gaudreau, M.C.; Heyd, F.; Okayama, T.; Jin, J.; Rojas, M.E.; Grimes, H.L.; et al. Zinc finger protein Gfi1 controls the endotoxin-mediated Toll-like receptor inflammatory response by antagonizing NF-kappaB p65. Mol. Cell Biol. 2010, 30, 3929–3942. [Google Scholar] [CrossRef] [Green Version]
- Stilmann, M.; Hinz, M.; Arslan, S.C.; Zimmer, A.; Schreiber, V.; Scheidereit, C. A nuclear poly(ADP-ribose)-dependent signalosome confers DNA damage-induced IkappaB kinase activation. Mol. Cell. 2009, 36, 365–378. [Google Scholar] [CrossRef]
- Widdrington, J.D.; Gomez-Duran, A.; Pyle, A.; Ruchaud-Sparagano, M.H.; Scott, J.; Baudouin, S.V.; Rostron, A.J.; Lovat, P.E.; Chinnery, P.F.; Simpson, A.J. Exposure of monocytic cells to lipopolysaccharide induces coordinated endotoxin tolerance, mitochondrial biogenesis, mitophagy, and antioxidant defenses. Front. Immunol. 2018, 9, 2217. [Google Scholar] [CrossRef]
- Kox, M.; de Kleijn, S.; Pompe, J.C.; Ramakers, B.P.; Netea, M.G.; van der Hoeven, J.G.; Hoedemaekers, C.W.; Pickkers, P. Differential ex vivo and in vivo endotoxin tolerance kinetics following human endotoxemia. Crit. Care Med. 2011, 39, 1866–1870. [Google Scholar] [CrossRef]
- Genard, G.; Lucas, S.; Michiels, C. Reprogramming of tumor-associated macrophages with anticancer therapies: Radiotherapy versus chemo- and immunotherapies. Front. Immunol. 2017, 8, 828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: Analysis for the global burden of disease study. Lancet 2020, 395, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Polat, G.; Ugan, R.A.; Cadirci, E.; Halici, Z. Sepsis and septic shock: Current treatment strategies and new approaches. Eurasian J. Med. 2017, 49, 53–58. [Google Scholar] [CrossRef]
- Cheng, B.; Hoeft, A.H.; Book, M.; Shu, Q.; Pastores, S.M. Sepsis: Pathogenesis, biomarkers, and treatment. Biomed. Res. Int. 2015, 2015, 846935. [Google Scholar] [CrossRef] [PubMed]
- Reinhart, K.; Meier-Hellmann, A.; Beale, R.; Forst, H.; Boehm, D.; Willatts, S.; Rothe, K.F.; Adolph, M.; Hoffmann, J.E.; Boehme, M.; et al. Open randomized phase II trial of an extracorporeal endotoxin adsorber in suspected Gram-negative sepsis. Crit. Care Med. 2004, 32, 1662–1668. [Google Scholar] [CrossRef] [PubMed]
- McCann, K.E. Advances in the use of PARP inhibitors for BRCA1/2-associated breast cancer: Talazoparib. Future Oncol. 2019, 15, 1707–1715. [Google Scholar] [CrossRef]
- Boussios, S.; Abson, C.; Moschetta, M.; Rassy, E.; Karathanasi, A.; Bhat, T.; Ghumman, F.; Sheriff, M.; Pavlidis, N. Poly (ADP-Ribose) Polymerase Inhibitors: Talazoparib in ovarian cancer and beyond. Drugs R. D. 2020, 20, 55–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemzek, J.A.; Hugunin, K.M.; Opp, M.R. Modeling sepsis in the laboratory: Merging sound science with animal well-being. Comp. Med. 2008, 58, 120–128. [Google Scholar]
- Lewis, A.J.; Seymour, C.W.; Rosengart, M.R. Current murine models of sepsis. Surg. Infect. 2016, 17, 385–393. [Google Scholar] [CrossRef] [Green Version]
- Fink, M.P. Animal models of sepsis. Virulence 2014, 5, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Stanojcic, M.; Jeschke, M.G. Differences between murine and human sepsis. Surg. Clin. North. Am. 2014, 94, 1135–1149. [Google Scholar] [CrossRef] [PubMed]
- Wiśnik, E.; Płoszaj, T.; Robaszkiewicz, A. Downregulation of PARP1 transcription by promoter-associated E2F4-RBL2-HDAC1-BRM complex contributes to repression of pluripotency stem cell factors in human monocytes. Sci. Rep. 2017, 7, 9483. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| GENE | FORWARD | REVERSE |
|---|---|---|
| TNF-α | GGAGAAGGGTGACCGACTCA | TGCCCAGACTCGGCAAAG |
| ACTB | TGGCACCCAGCACAATGAA | CTAAGTCATAGTCCGCCTAGAAGCA |
| GAPDH | TTCTTTTGCGTCGCCAGCCGA | GTGACCAGGCGCCCAATACGA |
| COX2 | GAATCATTCACCAGGCAAATTG | TGGAAGCCTGTGATACTTTCTGTACT |
| MIP2a | CGCCCAAACCGAAGTCAT | GATTTGCCATTTTTCACATCTTT |
| PARP1 | AAGCCCTAAAGGCTCAGAACG | ACCATGCCATCAGCTACTCGGT |
| IL6 | GGCACTGGCAGAAAACAACC | GCAAGTCTCCTCATTGAATCC |
| IL12a | CTCCTGGACCACCTCAGTTTG | GGTGAAGGCATGGGAACATT |
| iNOS | GTTCTCAAGGCACAGGTCTC | GCAGGTCACTTATGTCACTTATC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pietrzak, J.; Gronkowska, K.; Robaszkiewicz, A. PARP Traps Rescue the Pro-Inflammatory Response of Human Macrophages in the In Vitro Model of LPS-Induced Tolerance. Pharmaceuticals 2021, 14, 170. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14020170
Pietrzak J, Gronkowska K, Robaszkiewicz A. PARP Traps Rescue the Pro-Inflammatory Response of Human Macrophages in the In Vitro Model of LPS-Induced Tolerance. Pharmaceuticals. 2021; 14(2):170. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14020170
Chicago/Turabian StylePietrzak, Julita, Karolina Gronkowska, and Agnieszka Robaszkiewicz. 2021. "PARP Traps Rescue the Pro-Inflammatory Response of Human Macrophages in the In Vitro Model of LPS-Induced Tolerance" Pharmaceuticals 14, no. 2: 170. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14020170