
Discovery of Substituted (2-Aminooxazol-4-yl)Isoxazole-3-carboxylic Acids as Inhibitors of Bacterial Serine Acetyltransferase in the Quest for Novel Potential Antibacterial Adjuvants

 , ,
, ,  , , , , and
, , , , and
Abstract
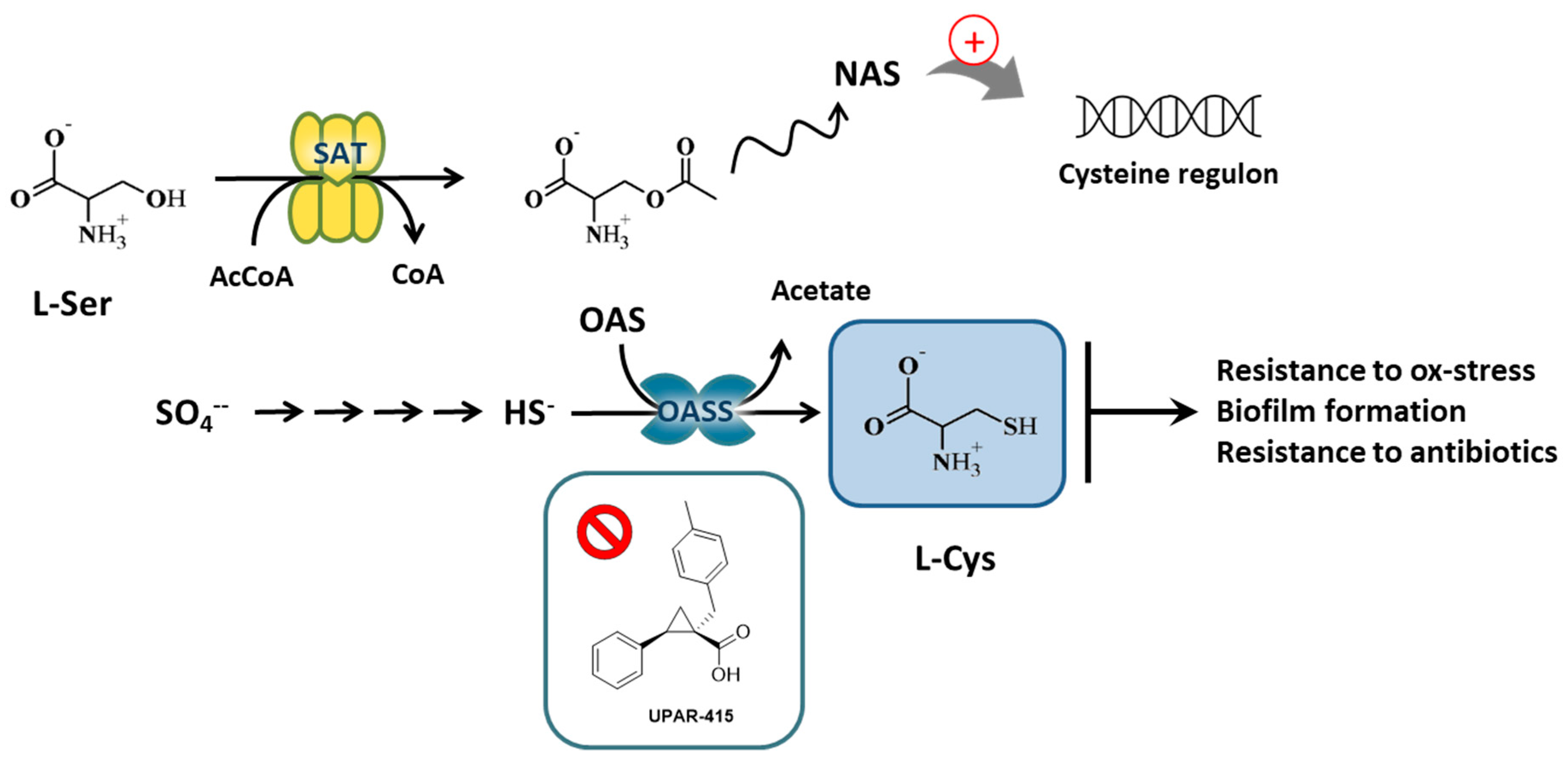
:1. Introduction
2. Results
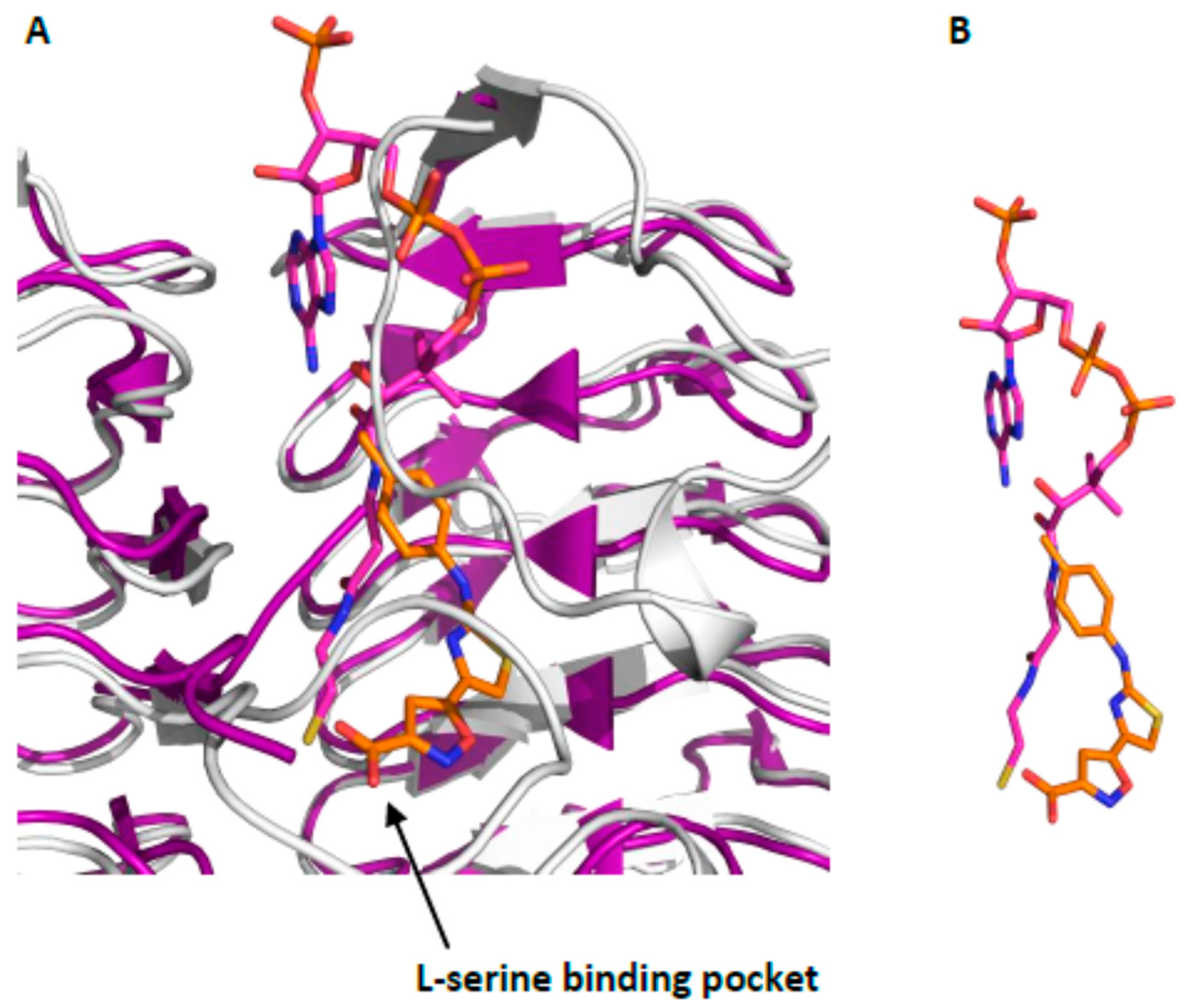
2.1. Hit Compounds from Virtual Screening
2.2. Hit Expansion
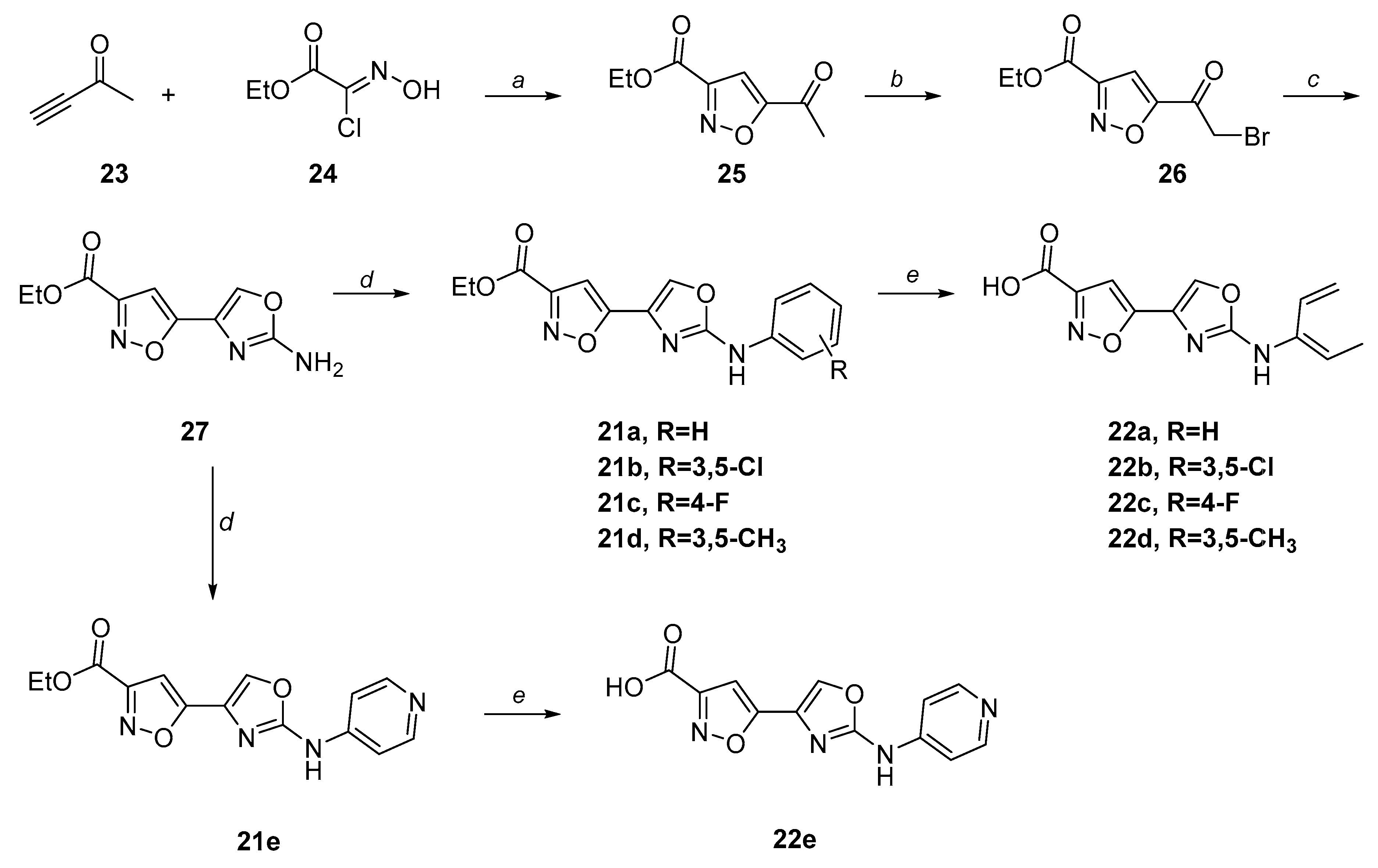
2.3. Chemistry
3. Discussion
3.1. Hints of Structure–Activity Relationships
3.2. Stability of the Isoxazole-Oxazole Nucleus
3.3. Antibacterial Activity
4. Materials and Methods
4.1. Enzyme Preparation and Activity Assay
4.2. Chemistry
4.3. Biology
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Becker, D.; Selbach, M.; Rollenhagen, C.; Ballmaier, M.; Meyer, T.F.; Mann, M.; Bumann, D. Robust Salmonella metabolism limits possibilities for new antimicrobials. Nature 2006, 440, 303–307. [Google Scholar] [CrossRef]
- Bhave, D.P.; Muse, W.B.; Carroll, K.S. Drug Targets in Mycobacterial Sulfur Metabolism. Infect. Disord. Drug Targets 2007, 7, 140–158. [Google Scholar] [CrossRef] [PubMed]
- Spyrakis, F.; Singh, R.; Cozzini, P.; Campanini, B.; Salsi, E.; Felici, P.; Raboni, S.; Benedetti, P.; Cruciani, G.; Kellogg, G.; et al. Isozyme-Specific Ligands for O-acetylserine sulfhydrylase, a Novel Antibiotic Target. PLoS ONE 2013, 8, e77558. [Google Scholar] [CrossRef] [Green Version]
- Spyrakis, F.; Felici, P.; Bayden, A.S.; Salsi, E.; Miggiano, R.; Kellogg, G.E.; Cozzini, P.; Cook, P.F.; Mozzarelli, A.; Campanini, B. Fine tuning of the active site modulates specificity in the interaction of O-acetylserine sulfhydrylase isozymes with serine acetyltransferase. Biochim. Biophys. Acta 2013, 1834, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Amori, L.; Katkevica, S.; Bruno, A.; Campanini, B.; Felici, P.; Mozzarelli, A.; Costantino, G. Design and synthesis of trans-2-substituted-cyclopropane-1-carboxylic acids as the first non-natural small molecule inhibitors of O-acetylserine sulfhydrylase. MedChemComm 2012, 3, 1111–1116. [Google Scholar] [CrossRef]
- Poyraz, O.; Jeankumar, V.U.; Saxena, S.; Schnell, R.; Haraldsson, M.; Yogeeswari, P.; Sriram, D.; Schneider, G. Structure-guided design of novel thiazolidine inhibitors of O-acetyl serine sulfhydrylase from Mycobacterium tuberculosis. J. Med. Chem. 2013, 56, 6457–6466. [Google Scholar] [CrossRef]
- Rabeh, W.M.; Cook, P.F. Structure and Mechanism of O-Acetylserine Sulfhydrylase. J. Biol. Chem. 2004, 279, 26803–26806. [Google Scholar] [CrossRef] [Green Version]
- Joshi, P.; Gupta, A.; Gupta, V. Insights into multifaceted activities of CysK for therapeutic interventions. 3 Biotech 2019, 9, 44. [Google Scholar] [CrossRef] [PubMed]
- Senaratne, R.H.; Silva, A.D.D.; Williams, S.J.; Mougous, J.D.; Reader, J.R.; Zhang, T.; Chan, S.; Sidders, B.; Lee, D.H.; Chan, J.; et al. 5′-Adenosinephosphosulphate reductase (CysH) protects Mycobacterium tuberculosis against free radicals during chronic infection phase in mice. Mol. Microbiol. 2006, 59, 1744–1753. [Google Scholar] [CrossRef]
- Lestrate, P.; Delrue, R.M.; Danese, I.; Didembourg, C.; Taminiau, B.; Mertens, P.; De Bolle, X.; Tibor, A.; Tang, C.M.; Letesson, J.J. Identification and characterization of in vivo attenuated mutants of Brucella melitensis. Mol. Microbiol. 2000, 38, 543–551. [Google Scholar] [CrossRef]
- Turnbull, A.L.; Surette, M.G. L-Cysteine is required for induced antibiotic resistance in actively swarming Salmonella enterica serovar Typhimurium. Microbiol. Read. Engl. 2008, 154, 3410–3419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connell, K.M.G.; Hodgkinson, J.T.; Sore, H.F.; Welch, M.; Salmond, G.P.C.; Spring, D.R. Combating multidrug-resistant bacteria: Current strategies for the discovery of novel antibacterials. Angew. Chem. Int. Ed Engl. 2013, 52, 10706–10733. [Google Scholar] [CrossRef]
- Blair, J.M.A.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J.V. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef]
- Antibiotics: Mode of Action and Mechanisms of Resistance: Art Science. Available online: https://www.scribd.com/document/202262261/Antibiotic-Action-and-Resistance (accessed on 3 September 2019).
- Taylor, P.L.; Wright, G.D. Novel approaches to discovery of antibacterial agents. Anim. Health Res. Rev. 2008, 9, 237–246. [Google Scholar] [CrossRef]
- Alekshun, M.N.; Levy, S.B. Targeting virulence to prevent infection: To kill or not to kill? Drug Discov. Today Ther. Strateg. 2004, 1, 483–489. [Google Scholar] [CrossRef]
- Shallcross, L.J.; Howard, S.J.; Fowler, T.; Davies, S.C. Tackling the threat of antimicrobial resistance: From policy to sustainable action. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2015, 370, 20140082. [Google Scholar] [CrossRef]
- Annunziato, G. Strategies to Overcome Antimicrobial Resistance (AMR) Making Use of Non-Essential Target Inhibitors: A Review. Int. J. Mol. Sci. 2019, 20, 5844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, G.D. Antibiotic Adjuvants: Rescuing Antibiotics from Resistance. Trends Microbiol. 2016, 24, 862–871. [Google Scholar] [CrossRef]
- Gill, E.E.; Franco, O.L.; Hancock, R.E.W. Antibiotic Adjuvants: Diverse Strategies for Controlling Drug-Resistant Pathogens. Chem. Biol. Drug Des. 2015, 85, 56–78. [Google Scholar] [CrossRef] [PubMed]
- Zaheer, Z.; Rahman, S.U.; Zaheer, I.; Younas, T.; Abbas, G.; Zahher, T. Antimicrobial adjuvant—An innovative strategy for handling antimicrobial resistance displayed by microbes. J. Bacteriol. Mycol. Open Access 2017, 5, 352–354. [Google Scholar] [CrossRef] [Green Version]
- Annunziato, G.; Pieroni, M.; Benoni, R.; Campanini, B.; Pertinhez, T.A.; Pecchini, C.; Bruno, A.; Magalhães, J.; Bettati, S.; Franko, N.; et al. Cyclopropane-1,2-dicarboxylic acids as new tools for the biophysical investigation of O-acetylserine sulfhydrylases by fluorimetric methods and saturation transfer difference (STD) NMR. J. Enzym. Inhib. Med. Chem. 2016, 31, 78–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magalhães, J.; Annunziato, G.; Franko, N.; Pieroni, M.; Campanini, B.; Bruno, A.; Costantino, G. Integration of Enhanced Sampling Methods with Saturation Transfer Difference Experiments to Identify Protein Druggable Pockets. J. Chem. Inf. Model. 2018, 58, 710–723. [Google Scholar] [CrossRef]
- Magalhães, J.; Franko, N.; Annunziato, G.; Welch, M.; Dolan, S.K.; Bruno, A.; Mozzarelli, A.; Armao, S.; Jirgensons, A.; Pieroni, M.; et al. Discovery of novel fragments inhibiting O-acetylserine sulphhydrylase by combining scaffold hopping and ligand-based drug design. J. Enzym. Inhib. Med. Chem. 2018, 33, 1444–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magalhães, J.; Franko, N.; Annunziato, G.; Pieroni, M.; Benoni, R.; Nikitjuka, A.; Mozzarelli, A.; Bettati, S.; Karawajczyk, A.; Jirgensons, A.; et al. Refining the structure-activity relationships of 2-phenylcyclopropane carboxylic acids as inhibitors of O-acetylserine sulfhydrylase isoforms. J. Enzym. Inhib. Med. Chem. 2019, 34, 31–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Yan, Q.; Tao, M.; Shi, H.; Han, X.; Jia, L.; Huang, Y.; Zhao, L.; Wang, C.; Ma, X.; et al. Characterization of serine acetyltransferase (CysE) from methicillin-resistant Staphylococcus aureus and inhibitory effect of two natural products on CysE. Microb. Pathog. 2019, 131, 218–226. [Google Scholar] [CrossRef]
- Agarwal, S.M.; Jain, R.; Bhattacharya, A.; Azam, A. Inhibitors of Escherichia coli serine acetyltransferase block proliferation of Entamoeba histolytica trophozoites. Int. J. Parasitol. 2008, 38, 137–141. [Google Scholar] [CrossRef]
- Palde, P.B.; Bhaskar, A.; Pedró Rosa, L.E.; Madoux, F.; Chase, P.; Gupta, V.; Spicer, T.; Scampavia, L.; Singh, A.; Carroll, K.S. First-in-Class Inhibitors of Sulfur Metabolism with Bactericidal Activity against Non-Replicating M. tuberculosis. ACS Chem. Biol. 2016, 11, 172–184. [Google Scholar] [CrossRef] [Green Version]
- Brunner, K.; Steiner, E.M.; Reshma, R.S.; Sriram, D.; Schnell, R.; Schneider, G. Profiling of in vitro activities of urea-based inhibitors against cysteine synthases from Mycobacterium tuberculosis. Bioorg. Med. Chem. Lett. 2017, 27, 4582–4587. [Google Scholar] [CrossRef]
- Pieroni, M.; Annunziato, G.; Beato, C.; Wouters, R.; Benoni, R.; Campanini, B.; Pertinhez, T.A.; Bettati, S.; Mozzarelli, A.; Costantino, G. Rational Design, Synthesis, and Preliminary Structure-Activity Relationships of α-Substituted-2-Phenylcyclopropane Carboxylic Acids as Inhibitors of Salmonella typhimurium O-Acetylserine Sulfhydrylase. J. Med. Chem. 2016, 59, 2567–2578. [Google Scholar] [CrossRef]
- Leu, L.S.; Cook, P.F. Kinetic mechanism of serine transacetylase from Salmonella typhimurium. Biochemistry 1994, 33, 2667–2671. [Google Scholar] [CrossRef]
- Johnson, C.M.; Huang, B.; Roderick, S.L.; Cook, P.F. Kinetic mechanism of the serine acetyltransferase from Haemophilus influenzae. Arch. Biochem. Biophys. 2004, 429, 115–122. [Google Scholar] [CrossRef]
- Inoue, K.; Noji, M.; Saito, K. Determination of the sites required for the allosteric inhibition of serine acetyltransferase by L-cysteine in plants. Eur. J. Biochem. 1999, 266, 220–227. [Google Scholar] [CrossRef] [Green Version]
- Tyrrell, R.; Verschueren, K.H.; Dodson, E.J.; Murshudov, G.N.; Addy, C.; Wilkinson, A.J. The structure of the cofactor-binding fragment of the LysR family member, CysB: A familiar fold with a surprising subunit arrangement. Structure 1997, 5, 1017–1032. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Raj, I.; Nagpal, I.; Subbarao, N.; Gourinath, S. Structural and Biochemical Studies of Serine Acetyltransferase Reveal Why the Parasite Entamoeba histolytica Cannot Form a Cysteine Synthase Complex. J. Biol. Chem. 2011, 286, 12533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pye, V.E.; Tingey, A.P.; Robson, R.L.; Moody, P.C.E. The structure and mechanism of serine acetyltransferase from Escherichia coli. J. Biol. Chem. 2004, 279, 40729–40736. Available online: https://pubmed.ncbi.nlm.nih.gov/15231846/ (accessed on 14 October 2020). [CrossRef] [Green Version]
- Magalhães, J.; Franko, N.; Raboni, S.; Annunziato, G.; Tammela, P.; Bruno, A.; Bettati, S.; Mozzarelli, A.; Pieroni, M.; Campanini, B.; et al. Inhibition of Nonessential Bacterial Targets: Discovery of a Novel Serine O-Acetyltransferase Inhibitor. ACS Med. Chem. Lett. 2020, 11, 790–797. [Google Scholar] [CrossRef]
- Denise, M.M.; Mueller, A.; Falkow, S. Persistent bacterial infections: The interface of the pathogen and the host immune system. Nat. Rev. Microbiol. 2004, 2, 747–765. Available online: https://pubmed.ncbi.nlm.nih.gov/15372085/ (accessed on 14 October 2020).
- Azzali, E.; Machado, D.; Kaushik, A.; Vacondio, F.; Flisi, S.; Cabassi, C.S.; Lamichhane, G.; Viveiros, M.; Costantino, G.; Pieroni, M. Substituted N-phenyl-5-(2-(phenylamino) thiazol-4-yl) isoxazole-3-carboxamides are valuable antitubercular candidates that evade innate efflux machinery. J. Med. Chem. 2017, 60, 7108–7122. [Google Scholar] [CrossRef] [PubMed]
- Azzali, E.; Girardini, M.; Annunziato, G.; Pavone, M.; Vacondio, F.; Mori, G.; Pasca, M.R.; Costantino, G.; Pieroni, M. 2-Aminooxazole as a Novel Privileged Scaffold in Antitubercular Medicinal Chemistry. ACS Med. Chem. Lett. 2020, 11, 1435–1441. [Google Scholar] [CrossRef] [PubMed]
- Turchi, I.J.; Dewar, M.J.S. Chemistry of oxazoles. Chem. Rev. 1975, 75, 389–437. [Google Scholar] [CrossRef]
- Balouiri, M.; Sadiki, M.; Ibnsouda, S.K. Methods for in vitro evaluating antimicrobial activity: A review. J. Pharm. Anal. 2016, 6, 71–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Copeland, R.A. Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Pieroni, M.; Wan, B.; Cho, S.; Franzblau, S.G.; Costantino, G. Design, synthesis and investigation on the structure–activity relationships of N-substituted 2-aminothiazole derivatives as antitubercular agents. Eur. J. Med. Chem. 2014, 72, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Clinical and Laboratory Standards Institute. M07-A11: Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, 10th ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2018. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd | % Inhibition at 1 mM | IC50 (μM) | Ki (μM) | Mechanism |
|---|---|---|---|---|
| 1 | 19.4 ± 1 | ND a | ND | |
| 2 | 34.9 ± 6 | ND | ND | |
| 3 | 31.4 ± 5 | ND | ND | |
| 4 | 18.7 ± 6 | ND | ND | |
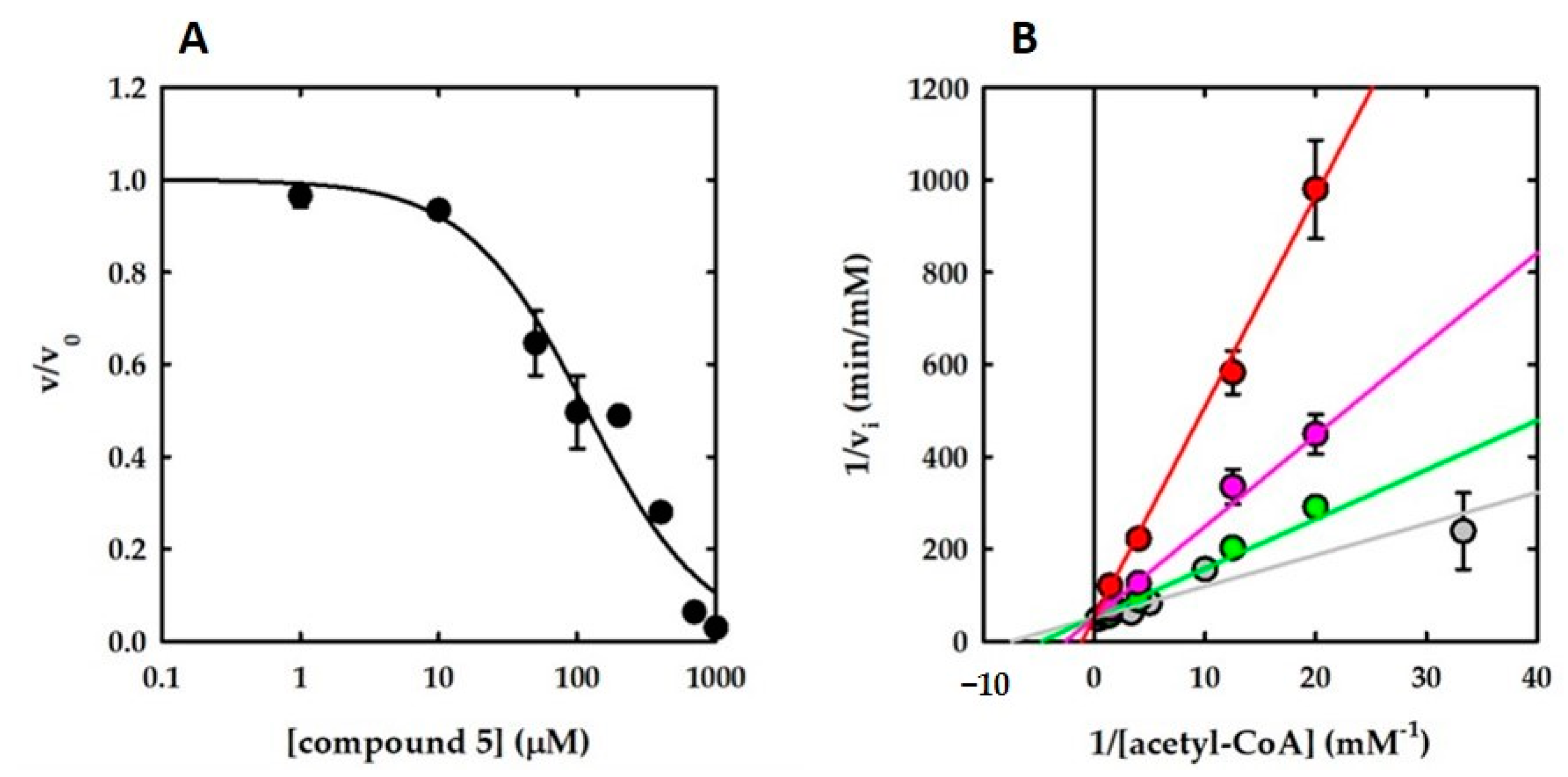
| 5 | 98.6 ± 0 | 110 ± 0 | 64 ± 12 | Competitive with AcCoA |
| 6 | 23.6 ± 0 | ND | ND | |
| 7 | 42 ± 5 | >2 mM | ND |
| Cpd | Structure | IC50 (μM) | Cpd | Structure | IC50 (μM) |
|---|---|---|---|---|---|
| 8 |  | >400 | 15 |  | 18 ± 2 |
| 9 |  | 12 ± 2 | 16 |  | 184 ± 13 |
| 10 |  | 9 ± 3 | 17 |  | 21 ± 5 |
| 11 |  | 26 ± 4 | 18 |  | 1.0 ± 0.2 |
| 12 |  | > 60 * | 19 |  | 7 ± 2 |
| 13 |  | 16 ± 3 | 20 |  | 11 ± 1 |
| 14 |  | ~10 * |
 |  | ||||
|---|---|---|---|---|---|
| Cmpd | R | StSAT IC50 (μM) | Cmpd | R | StSAT IC50 (μM) |
| 21a | Phenyl | 2.68 ± 0.27 | 22a | Phenyl | 1.54 ± 0.33 |
| 21b | 3,5-Dichlorophenyl | 2.52 ± 0.03 | 22b | 3,5-Dichlorophenyl | 8.03 ± 0.18 |
| 21c | 4-Fluorophenyl | 3.04 ± 0.37 | 22c | 4-Fluorophenyl | 2.51 ± 0.34 |
| 22d | 3,5-Dimethylphenyl | 4.24 ±0.11 | |||
| 21e | Pyridine | 3.95 ± 0.65 | 22e | Pyridine | 12.02 ± 1.25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magalhães, J.; Franko, N.; Raboni, S.; Annunziato, G.; Tammela, P.; Bruno, A.; Bettati, S.; Armao, S.; Spadini, C.; Cabassi, C.S.; et al. Discovery of Substituted (2-Aminooxazol-4-yl)Isoxazole-3-carboxylic Acids as Inhibitors of Bacterial Serine Acetyltransferase in the Quest for Novel Potential Antibacterial Adjuvants. Pharmaceuticals 2021, 14, 174. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14020174
Magalhães J, Franko N, Raboni S, Annunziato G, Tammela P, Bruno A, Bettati S, Armao S, Spadini C, Cabassi CS, et al. Discovery of Substituted (2-Aminooxazol-4-yl)Isoxazole-3-carboxylic Acids as Inhibitors of Bacterial Serine Acetyltransferase in the Quest for Novel Potential Antibacterial Adjuvants. Pharmaceuticals. 2021; 14(2):174. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14020174
Chicago/Turabian StyleMagalhães, Joana, Nina Franko, Samanta Raboni, Giannamaria Annunziato, Päivi Tammela, Agostino Bruno, Stefano Bettati, Stefano Armao, Costanza Spadini, Clotilde Silvia Cabassi, and et al. 2021. "Discovery of Substituted (2-Aminooxazol-4-yl)Isoxazole-3-carboxylic Acids as Inhibitors of Bacterial Serine Acetyltransferase in the Quest for Novel Potential Antibacterial Adjuvants" Pharmaceuticals 14, no. 2: 174. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14020174