
Ursolic Acid Inhibits Collective Cell Migration and Promotes JNK-Dependent Lysosomal Associated Cell Death in Glioblastoma Multiforme Cells

 ,
,  ,
,  , and
, and
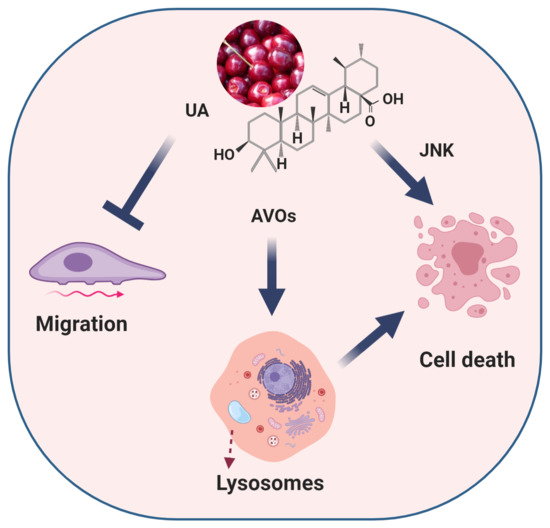
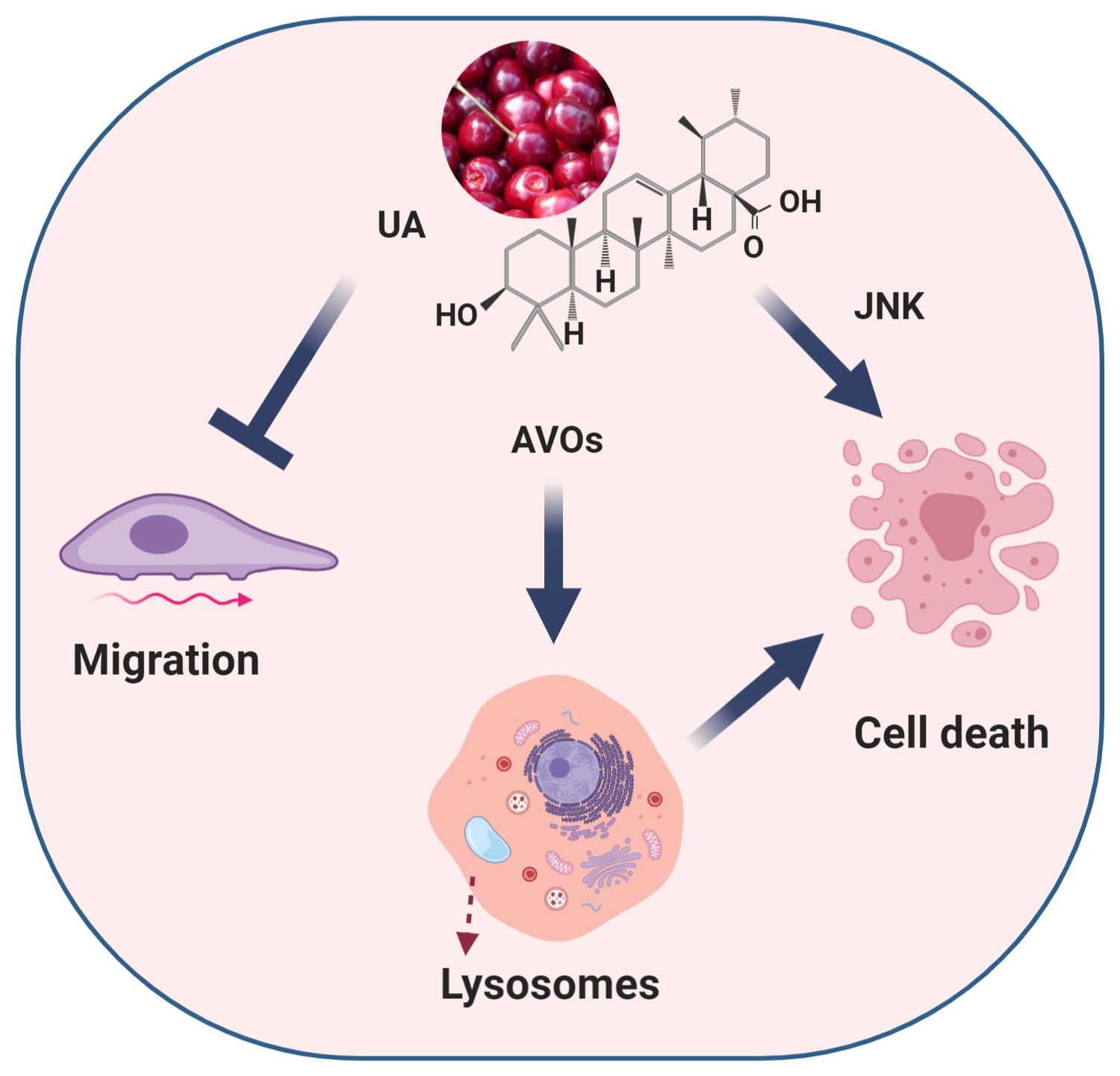
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
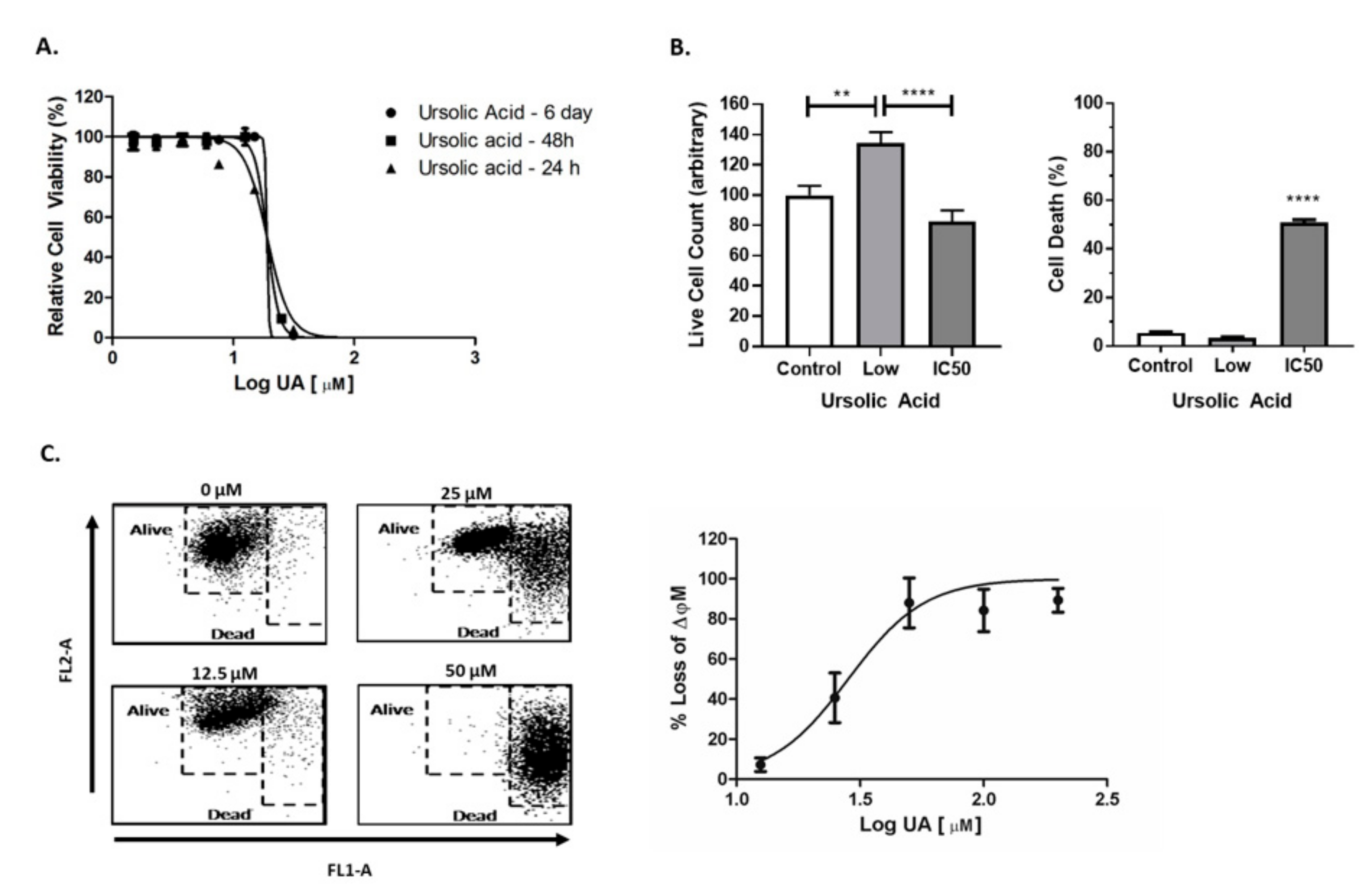
2.1. UA Induced Cytotoxicity Results in Rapid Mitochondrial Membrane Depolarisation
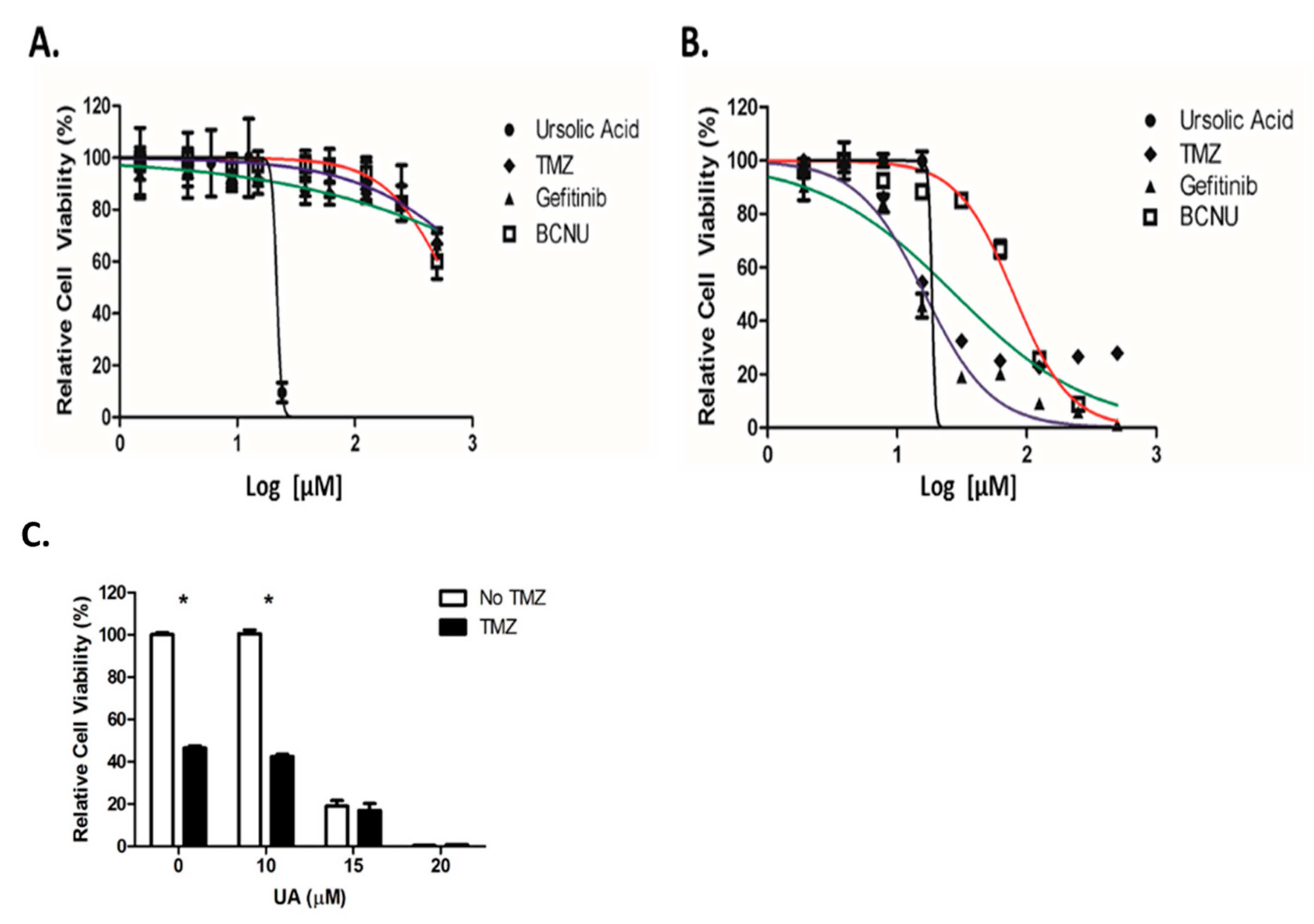
2.2. UA Demonstrates Enhanced Cytotoxicity Compared to Conventional Chemotherapeutic Drugs
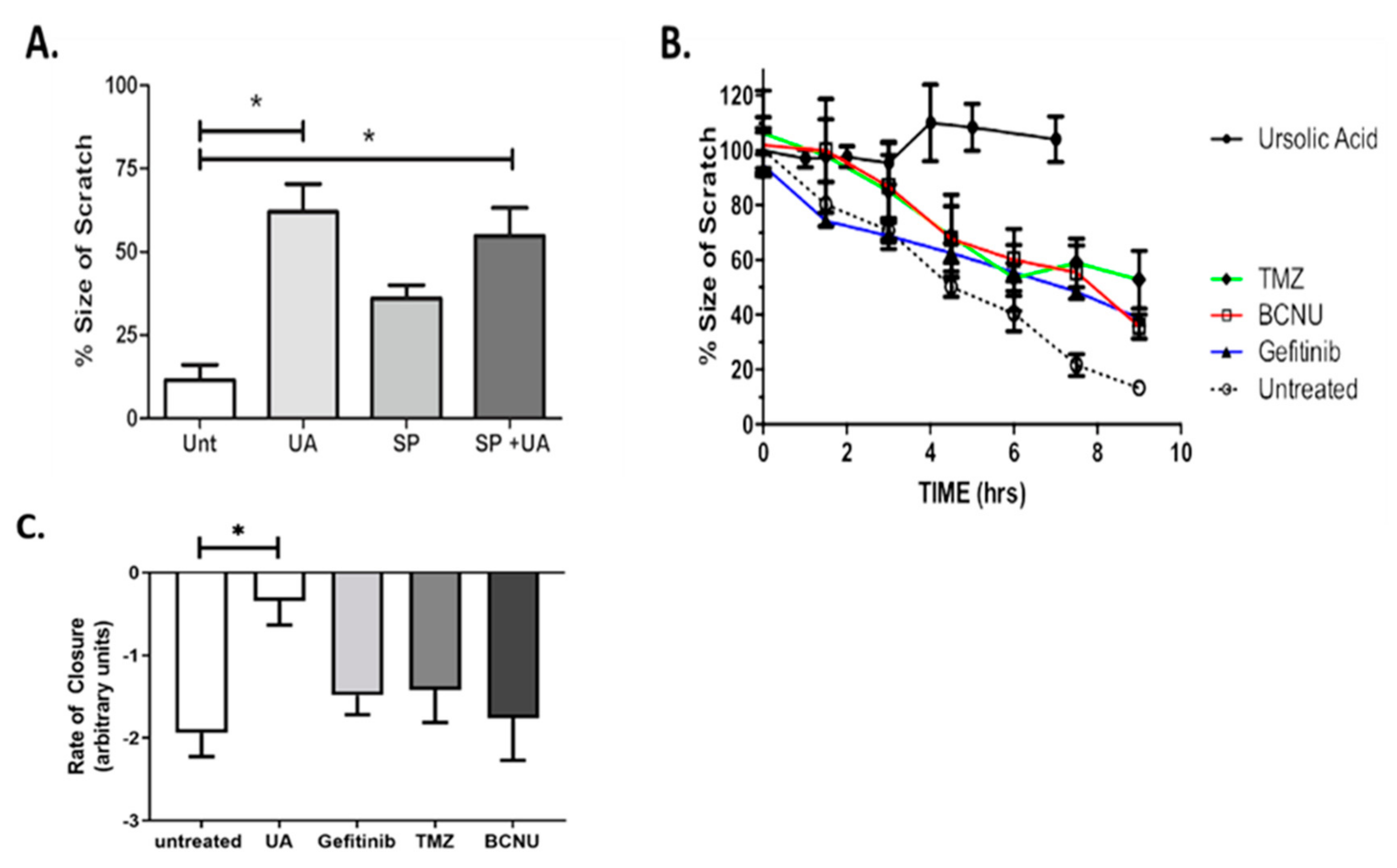
2.3. UA Inhibits JNK-Dependent GBM Collective Cell Migration
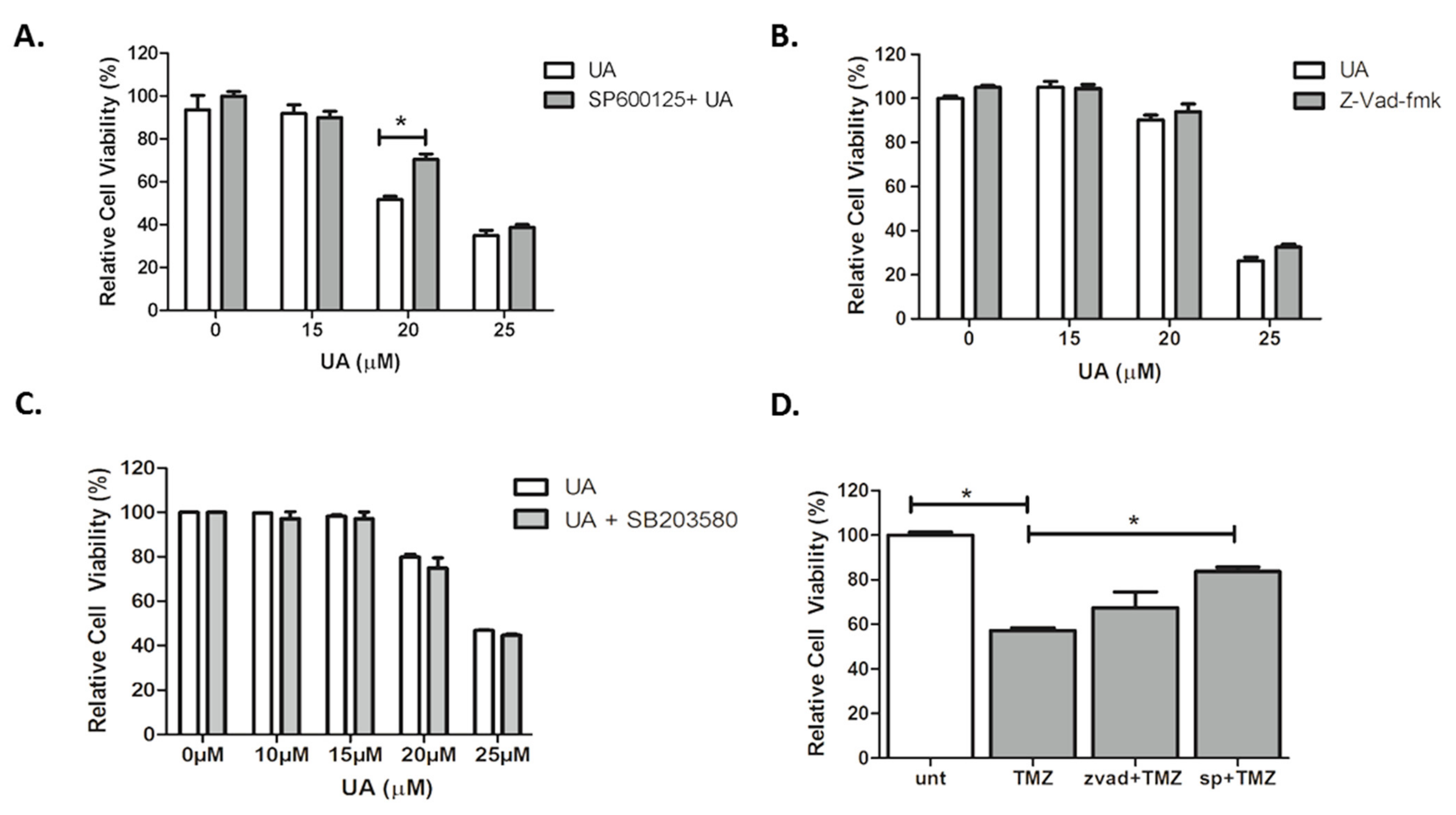
2.4. UA Induces Caspase-Independent, JNK-Dependent Cell Death
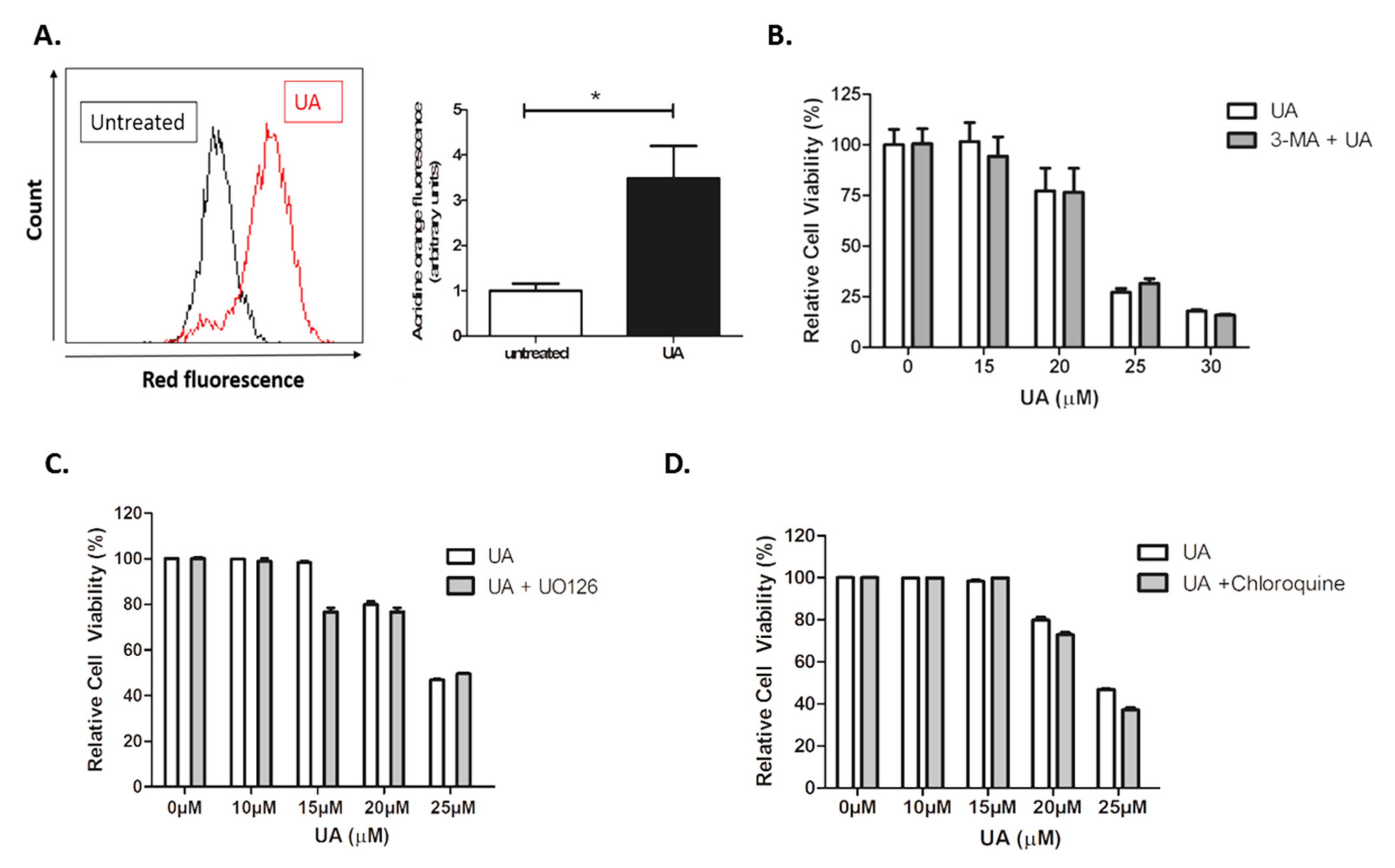
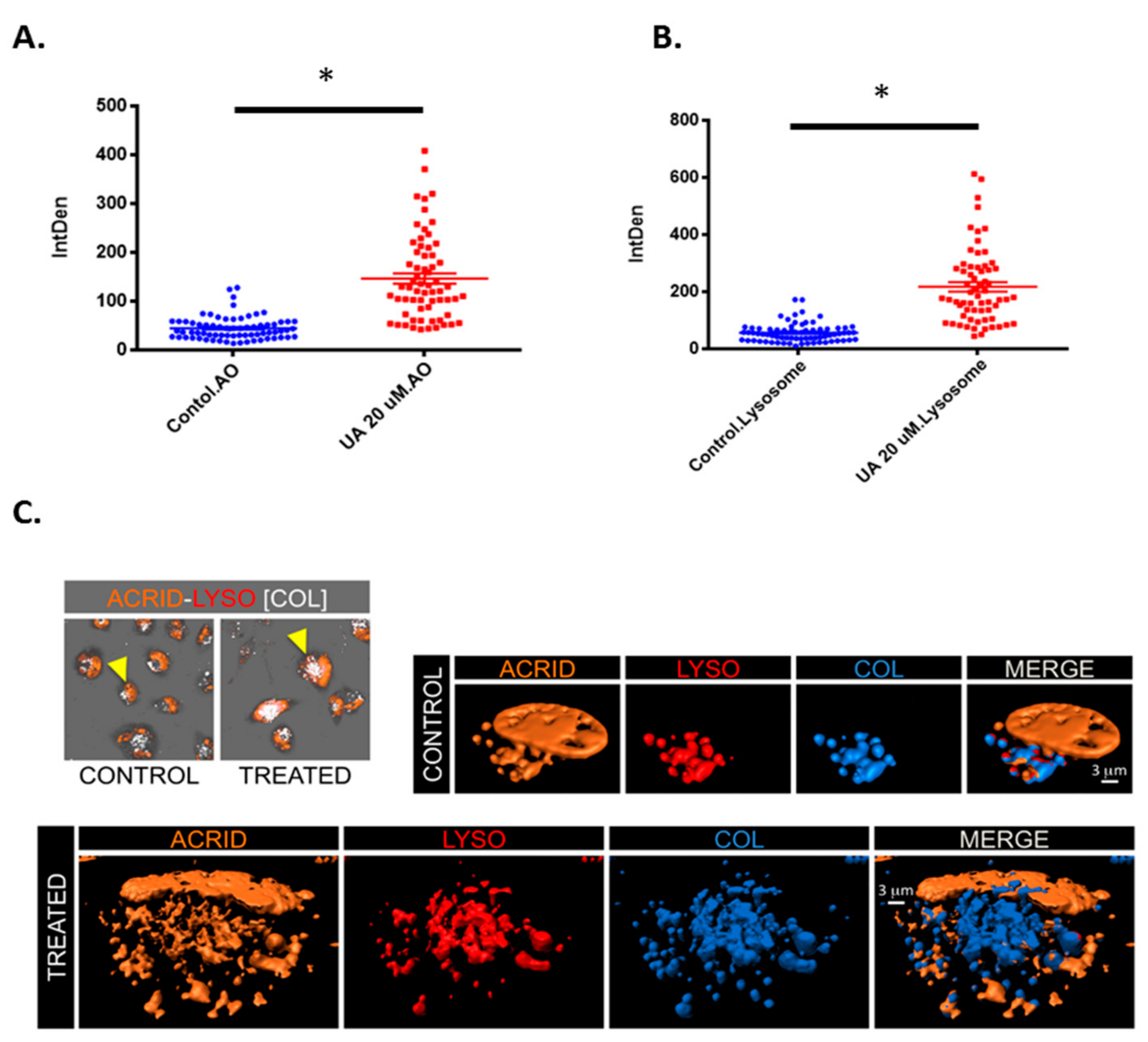
2.5. UA Triggers the Formation of Lysosomes
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cytotoxicity
4.3. Cell Viability Assays
4.4. Flow Cytometry
4.4.1. JC-1 Mitochondrial Membrane Potential Assay
4.4.2. Acridine Orange (AO)
4.5. Scratch Assay
4.6. Inhibitor Studies
4.7. Confocal Microscopy
4.8. Isosurface Rendering
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GBM | Glioblastoma multiforme |
| UA | Ursolic Acid |
| AVOs | Acidic vesicle organelles |
| TMZ | Temozolomide |
| NDBC | Naturally derived bioactive compound |
| NCCD | Nomenclature Committee on Cell Death |
| LDCD | Lysosome-dependant cell death |
| ADCD | Autophagy dependent cell death |
| AO | Acridine orange |
| 3-MA | 3-methyladenine |
References
- Philips, A.; Henshaw, D.L.; Lamburn, G.; O’Carroll, M.J. Brain tumours: Rise in glioblastoma multiforme incidence in England 1995–2015 suggests an adverse environmental or lifestyle factor. J. Environ. Public Health 2018, 2018. [Google Scholar] [CrossRef] [Green Version]
- Adamson, C.; Kanu, O.O.; Mehta, A.I.; Di, C.; Lin, N.; Mattox, A.K.; Bigner, D.D. Glioblastoma multiforme: A review of where we have been and where we are going. Expert Opin. Investig. Drugs 2009, 18, 1061–1083. [Google Scholar] [CrossRef]
- Candolfi, M.; Curtin, J.F.; Nichols, W.S.; Muhammad, A.G.; King, G.D.; Pluhar, G.E.; McNiel, E.A.; Ohlfest, J.R.; Freese, A.B.; Moore, P.F.; et al. Intracranial glioblastoma models in preclinical neuro-oncology: Neuropathological characterization and tumor progression. J. Neurooncol. 2007, 85, 133–148. [Google Scholar] [CrossRef] [Green Version]
- Nørøxe, D.S.; Poulsen, H.S.; Lassen, U. Hallmarks of glioblastoma: A systematic review. ESMO Open 2016, 1, 1. [Google Scholar] [CrossRef]
- Salim, S.; Snape, T.; Welsby, P.; Welsby, G. Phytochemicals—An alternative therapy for glioblastoma? Neuro. Oncol. 2018, 20, i5. [Google Scholar] [CrossRef] [Green Version]
- Øverby, A.; Zhao, C.-M.; Chen, D. Plant phytochemicals: Potential anticancer agents against gastric cancer. Curr. Opin. Pharmacol. 2014, 19, 6–10. [Google Scholar] [CrossRef]
- Koldaş, S.; Demirtas, I.; Ozen, T.; Demirci, M.A.; Behçet, L. Phytochemical screening, anticancer and antioxidant activities of Origanum vulgare L. ssp. viride (Boiss.) Hayek, a plant of traditional usage. J. Sci. Food Agric. 2015, 95, 786–798. [Google Scholar] [CrossRef]
- Neto, C.C. Cranberry and its phytochemicals: A review of in vitro anticancer studies. J. Nutr. 2007, 137, 186S–193S. [Google Scholar] [CrossRef] [Green Version]
- Sasidharan, S.; Chen, Y.; Saravanan, D.; Sundram, K.M.; Yoga-Latha, L. Extraction, isolation and characterization of bioactive compounds from plants’ extracts. Afr. J. Tradit. Complement. Altern. Med. 2011, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Khor, T.O.; Shu, L.; Su, Z.-Y.; Fuentes, F.; Lee, J.-H.; Kong, A.-N.T. Plants vs. cancer: A review on natural phytochemicals in preventing and treating cancers and their druggability. Anti Cancer Agents Med. Chem. 2012, 12, 1281–1305. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; Dai, X.; Kumar, A.P.; Tan, B.K.H.; Sethi, G.; Bishayee, A. Ursolic acid in cancer prevention and treatment: Molecular targets, pharmacokinetics and clinical studies. Biochem. Pharmacol. 2013, 85, 1579–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.-C.; Huang, B.-R.; Liao, P.-J.; Yen, G.-C. Ursolic acid triggers nonprogrammed death (necrosis) in human glioblastoma multiforme DBTRG-05MG cells through MPT pore opening and ATP decline. Mol. Nutr. Food Res. 2014, 58, 2146–2156. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Y.; Wang, X.; Jiang, C. Ursolic acid inhibits proliferation and induces apoptosis in human glioblastoma cell lines U251 by suppressing TGF-β1/miR-21/PDCD4 pathway. Basic Clin. Pharmacol. Toxicol. 2012, 111, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Bergamin, L.S.; Figueiró, F.; Dietrich, F.; de Manica, F.; Filippi-Chiela, E.C.; Mendes, F.B.; Jandrey, E.H.F.; Lopes, D.V.; Oliveira, F.H.; Nascimento, I.C.; et al. Interference of ursolic acid treatment with glioma growth: An in vitro and in vivo study. Eur. J. Pharmacol. 2017, 811. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.T.; Ho, C.T.; Yuan-Wang, Z.; Ferraro, T.; Lou, Y.R.; Stauber, K.; Ma, W.; Georgiadis, C.; Laskin, J.D.; Conney, A.H. Inhibition of skin tumorigenesis by rosemary and its constituents carnosol and ursolic acid. Cancer Res. 1994, 54, 701–708. [Google Scholar] [PubMed]
- Petiwala, S.M.; Puthenveetil, A.G.; Johnson, J.J. Polyphenols from the Mediterranean herb rosemary (Rosmarinus officinalis) for prostate cancer. Front. Pharmacol. 2013, 4, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navin, R.; Mi Kim, S. Therapeutic interventions using ursolic acid for cancer treatment. Med. Chem. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Woźniak, Ł.; Skąpska, S.; Marszałek, K. Ursolic acid-A pentacyclic triterpenoid with a wide spectrum of pharmacological activities. Molecules 2015, 20, 20614–20641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotecha, R.; Takami, A.; Espinoza, J.L. Dietary phytochemicals and cancer chemoprevention: A review of the clinical evidence. Oncotarget 2016, 7, 52517–52529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Du, S.; Ding, F.; Guo, S.; Ying, G.; Yan, Z. Ursolic acid attenuates temozolomide resistance in glioblastoma cells by downregulating O(6)-methylguanine-DNA methyltransferase (MGMT) expression. Am. J. Transl. Res. 2016, 8, 3299–3308. [Google Scholar] [PubMed]
- Bonaccorsi, I.; Altieri, F.; Sciamanna, I.; Oricchio, E.; Grillo, C.; Contartese, G.; Galati, E.M. Endogenous reverse transcriptase as a mediator of ursolic acid’s anti-proliferative and differentiating effects in human cancer cell lines. Cancer Lett. 2008, 263, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Kondo, M.; Mackinnon, S.L.; Craft, C.C.; Matchett, M.D.; Hurta, R.A.R.; Neto, C.C. Ursolic acid and its esters: Occurrence in cranberries and other Vaccinium fruit and effects on matrix metalloproteinase activity in DU145 prostate tumor cells. J. Sci. Food Agric. 2011, 91, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-C.; Huang, C.-Y.; Lin-Shiau, S.-Y.; Lin, J.-K. Ursolic acid inhibits IL-1beta or TNF-alpha-induced C6 glioma invasion through suppressing the association ZIP/p62 with PKC-zeta and downregulating the MMP-9 expression. Mol. Carcinog. 2009, 48, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Zhang, Y.; Zhang, R.; Tu, X.; Gong, X. Ursolic acid induces autophagy in U87MG cells via ROS-dependent endoplasmic reticulum stress. Chem. Biol. Interact. 2014, 218, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Salido, M.; Gonzalez, J.L.; Vilches, J. Loss of mitochondrial membrane potential is inhibited by bombesin in etoposide-induced apoptosis in PC-3 prostate carcinoma cells. Mol. Cancer Ther. 2007, 6, 1292–1299. [Google Scholar] [CrossRef] [Green Version]
- Pijuan, J.; Barceló, C.; Moreno, D.F.; Maiques, O.; Sisó, P.; Marti, R.M.; Macià, A.; Panosa, A. In vitro cell migration, invasion, and adhesion assays: From cell imaging to data analysis. Front. Cell Dev. Biol. 2019, 7, 107. [Google Scholar] [CrossRef] [Green Version]
- Portela, M.; Venkataramani, V.; Fahey-Lozano, N.; Seco, E.; Losada-Perez, M.; Winkler, F.; Casas-Tintó, S. Glioblastoma cells vampirize WNT from neurons and trigger a JNK/MMP signaling loop that enhances glioblastoma progression and neurodegeneration. PLoS Biol. 2019, 17, e3000545. [Google Scholar] [CrossRef]
- Zhou, X.; Hua, L.; Zhang, W.; Zhu, M.; Shi, Q.; Li, F.; Zhang, L.; Song, C.; Yu, R. FRK controls migration and invasion of human glioma cells by regulating JNK/c-Jun signaling. J. Neurooncol. 2012, 110, 9–19. [Google Scholar] [CrossRef]
- Murrow, L.; Debnath, J. Autophagy as a stress-response and quality-control mechanism: Implications for cell injury and human disease. Annu. Rev. Pathol. Mech. Dis. 2013, 8, 105–137. [Google Scholar] [CrossRef] [Green Version]
- Paglin, S.; Hollister, T.; Delohery, T.; Hackett, N.; McMahill, M.; Sphicas, E.; Domingo, D.; Yahalom, J. A Novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001, 61, 439–444. [Google Scholar] [PubMed]
- Shin, S.W.; Kim, S.Y.; Park, J.-W. Autophagy inhibition enhances ursolic acid-induced apoptosis in PC3 cells. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 451–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Conway, G.E.; He, Z.; Hutanu, A.L.; Cribaro, G.P.; Manaloto, E.; Casey, A.; Traynor, D.; Milosavljevic, V.; Howe, O.; Barcia, C.; et al. Cold atmospheric plasma induces accumulation of lysosomes and caspase-independent cell death in U373MG glioblastoma multiforme cells. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amin, A.; Gali-Muhtasib, H.; Ocker, M.; Schneider-Stock, R. Overview of major classes of plant-derived anticancer drugs. Int. J. Biomed. Sci. 2009, 5, 1–11. [Google Scholar]
- Weng, H.; Tan, Z.; Hu, Y.; Shu, Y.; Bao, R.; Jiang, L.; Wu, X. Ursolic acid induces cell cycle arrest and apoptosis of gallbladder carcinoma cells. Cancer Cell Int. 2014, 14, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhang, F.; Yang, L.; Mei, Y.; Long, H.; Zhang, X.; Zhang, J.; Qimuge-Suyila, S.; Su, X. Ursolic acid inhibits proliferation and induces apoptosis of cancer cells in vitro and in vivo. J. Biomed. Biotechnol. 2011, 2011, 419343. [Google Scholar] [CrossRef]
- Zhang, H.; Zheng, D.; Ding, J.; Xu, H.; Li, X.; Sun, W. Efficient delivery of ursolic acid by poly(N-vinylpyrrolidone)-block-poly (ε-caprolactone) nanoparticles for inhibiting the growth of hepatocellular carcinoma in vitro and in vivo. Int. J. Nanomed. 2015, 10, 1909–1920. [Google Scholar] [CrossRef] [Green Version]
- Yoon, Y.; Lim, J.W.; Kim, J.; Kim, Y.; Chun, K.H. Discovery of ursolic acid prodrug (NX-201): Pharmacokinetics and in vivo antitumor effects in PANC-1 pancreatic cancer. Bioorganic Med. Chem. Lett. 2016, 26, 5524–5527. [Google Scholar] [CrossRef]
- Hermisson, M.; Klumpp, A.; Wick, W.; Wischhusen, J.; Nagel, G.; Roos, W.; Kaina, B.; Weller, M. O6-methylguanine DNA methyltransferase and p53 status predict temozolomide sensitivity in human malignant glioma cells. J. Neurochem. 2006, 96, 766–776. [Google Scholar] [CrossRef]
- Cheung, K.J.; Ewald, A.J. A collective route to metastasis: Seeding by tumor cell clusters. Science 2016, 352, 167–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalil, A.A.; Ilina, O.; Gritsenko, P.G.; Bult, P.; Span, P.N.; Friedl, P. Collective invasion in ductal and lobular breast cancer associates with distant metastasis. Clin. Exp. Metastasis 2017, 34, 421–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volovetz, J.; Berezovsky, A.D.; Alban, T.; Chen, Y.; Lauko, A.; Aranjuez, G.F.; Burtscher, A.; Shibuya, K.; Silver, D.J.; Peterson, J.; et al. Identifying conserved molecular targets required for cell migration of glioblastoma cancer stem cells. Cell Death Dis. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alieva, M.; Leidgens, V.; Riemenschneider, M.J.; Klein, C.A.; Hau, P.; van Rheenen, J. Intravital imaging of glioma border morphology reveals distinctive cellular dynamics and contribution to tumor cell invasion. Sci. Rep. 2019, 9, 2054. [Google Scholar] [CrossRef]
- Kim, E.-S.E.; Moon, A. Ursolic acid inhibits the invasive phenotype of SNU-484 human gastric cancer cells. Oncol. Lett. 2015, 9, 897–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, C.-T.T.; Wu, C.-H.H.; Yen, G.-C.C. Ursolic acid, a naturally occurring triterpenoid, suppresses migration and invasion of human breast cancer cells by modulating c-Jun N-terminal kinase, Akt and mammalian target of rapamycin signaling. Mol. Nutr. Food Res. 2010, 54, 1285–1295. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Lin, C.-Y.; Tsai, C.-W.; Yin, M.-C. Inhibition of cell proliferation, invasion and migration by ursolic acid in human lung cancer cell lines. Toxicol. In Vitro 2011, 25, 1274–1280. [Google Scholar] [CrossRef] [PubMed]
- Ovesná, Z.; Kozics, K.; Slamenová, D. Protective effects of ursolic acid and oleanolic acid in leukemic cells. Mutat. Res. 2006, 600, 131–137. [Google Scholar] [CrossRef]
- Guo, G.; Yao, W.; Zhang, Q.; Bo, Y. Oleanolic acid suppresses migration and invasion of malignant glioma cells by inactivating MAPK/ERK signaling pathway. PLoS ONE 2013, 8, e72079. [Google Scholar] [CrossRef]
- Kassi, E.; Papoutsi, Z.; Pratsinis, H.; Aligiannis, N.; Manoussakis, M.; Moutsatsou, P. Ursolic acid, a naturally occurring triterpenoid, demonstrates anticancer activity on human prostate cancer cells. J. Cancer Res. Clin. Oncol. 2007, 133, 493–500. [Google Scholar] [CrossRef]
- Gao, N.; Cheng, S.; Budhraja, A.; Gao, Z.; Chen, J.; Liu, E.-H.; Huang, C.; Chen, D.; Yang, Z.; Liu, Q.; et al. Ursolic acid induces apoptosis in human leukaemia cells and exhibits anti-leukaemic activity in nude mice through the PKB pathway. Br. J. Pharmacol. 2012, 165, 1813–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Kong, C.; Zeng, Y.; Wang, L.; Li, Z.; Wang, H.; Xu, C.; Sun, Y. Ursolic acid induces PC-3 cell apoptosis via activation of JNK and inhibition of Akt pathways in vitro. Mol. Carcinog. 2010, 49, 374–385. [Google Scholar] [CrossRef]
- Xavier, C.P.R.; Lima, C.F.; Pedro, D.F.N.; Wilson, J.M.; Kristiansen, K.; Pereira-Wilson, C. Ursolic acid induces cell death and modulates autophagy through JNK pathway in apoptosis-resistant colorectal cancer cells. J. Nutr. Biochem. 2013, 24, 706–712. [Google Scholar] [CrossRef] [Green Version]
- Liang, X. Ursolic acid inhibits growth and induces apoptosis in gemcitabine-resistant human pancreatic cancer via the JNK and PI3K/Akt/NF-κB pathways. Oncol. Rep. 2012. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Q.; Li, P.; Jin, F.; Yao, C.; Zhang, G.; Zang, T.; Ai, X. Ursolic acid induces ER stress response to activate ASK1–JNK signaling and induce apoptosis in human bladder cancer T24 cells. Cell. Signal. 2013, 25, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Leng, S.; Hao, Y.; Du, D.; Xie, S.; Hong, L.; Gu, H.; Zhu, X.; Zhang, J.; Fan, D.; Kung, H. Ursolic acid promotes cancer cell death by inducing Atg5-dependent autophagy. Int. J. Cancer 2013, 133, 2781–2790. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Alnemri, E.S.; Altucci, L.; Andrews, D.; Annicchiarico-Petruzzelli, M.; et al. Essential versus accessory aspects of cell death: Recommendations of the NCCD 2015. Cell Death Differ. 2015, 22, 58–73. [Google Scholar] [CrossRef] [Green Version]
- Boya, P.; Wang, F.; Salvati, A.; Boya, P. Lysosome-dependent cell death and deregulated autophagy induced by amine-modified polystyrene nanoparticles. Open Biol. 2018, 4. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Gómez-Sintes, R.; Boya, P. Lysosomal membrane permeabilization and cell death. Traffic 2018, 19, 918–931. [Google Scholar] [CrossRef]
- Martins, W.K.; Costa, É.T.; Cruz, M.C.; Stolf, B.S.; Miotto, R.; Cordeiro, R.M.; Baptista, M.S. Parallel damage in mitochondrial and lysosomal compartments promotes efficient cell death with autophagy: The case of the pentacyclic triterpenoids. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH image to imageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conway, G.E.; Zizyte, D.; Mondala, J.R.M.; He, Z.; Lynam, L.; Lecourt, M.; Barcia, C.; Howe, O.; Curtin, J.F. Ursolic Acid Inhibits Collective Cell Migration and Promotes JNK-Dependent Lysosomal Associated Cell Death in Glioblastoma Multiforme Cells. Pharmaceuticals 2021, 14, 91. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14020091
Conway GE, Zizyte D, Mondala JRM, He Z, Lynam L, Lecourt M, Barcia C, Howe O, Curtin JF. Ursolic Acid Inhibits Collective Cell Migration and Promotes JNK-Dependent Lysosomal Associated Cell Death in Glioblastoma Multiforme Cells. Pharmaceuticals. 2021; 14(2):91. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14020091
Chicago/Turabian StyleConway, Gillian E., Deimante Zizyte, Julie Rose Mae Mondala, Zhonglei He, Lorna Lynam, Mathilde Lecourt, Carlos Barcia, Orla Howe, and James F. Curtin. 2021. "Ursolic Acid Inhibits Collective Cell Migration and Promotes JNK-Dependent Lysosomal Associated Cell Death in Glioblastoma Multiforme Cells" Pharmaceuticals 14, no. 2: 91. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14020091