
Repeated Administration of Clinically Relevant Doses of the Prescription Opioids Tramadol and Tapentadol Causes Lung, Cardiac, and Brain Toxicity in Wistar Rats

 ,
,  ,
, 
 and
and 
Abstract
:
1. Introduction
2. Results
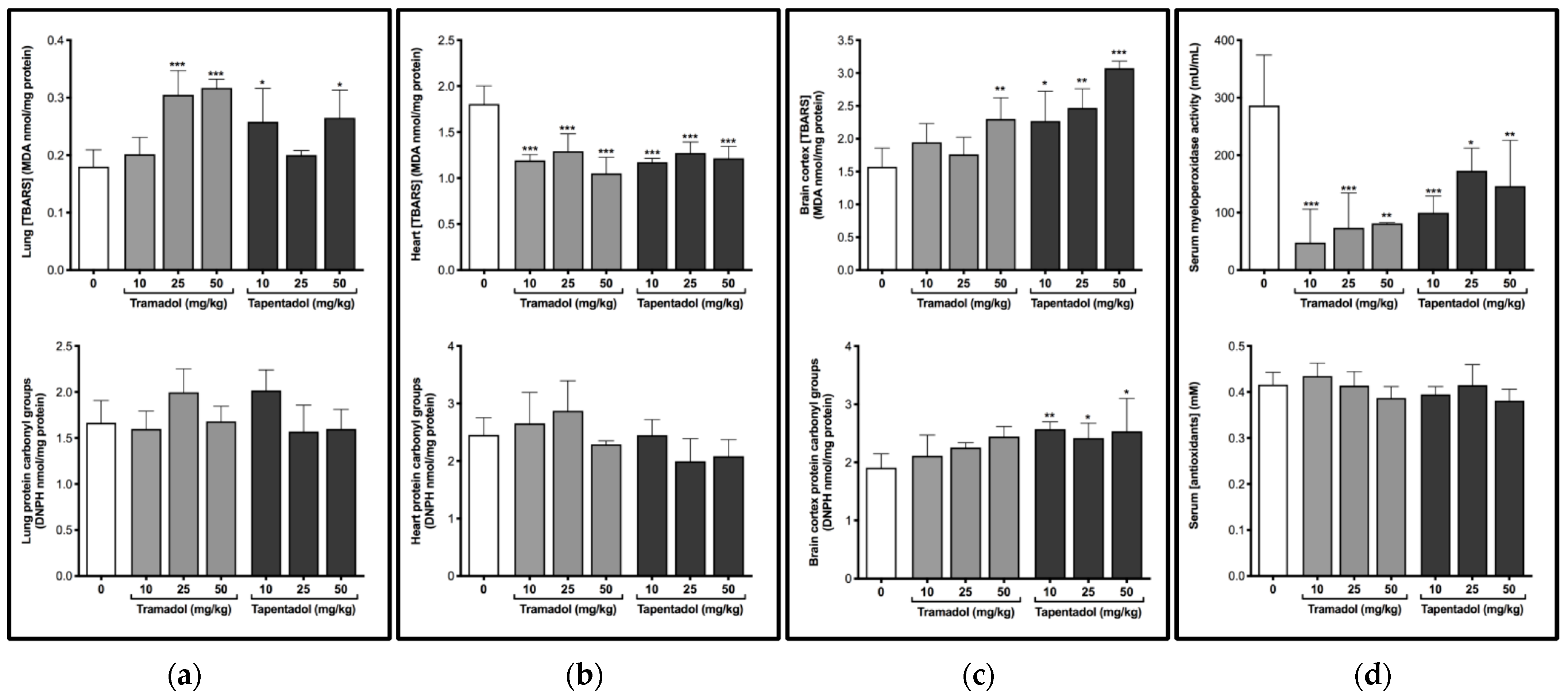
2.1. Repeated Exposure to Tramadol and Tapentadol Causes Oxidative Stress in Lung and Brain Cortex
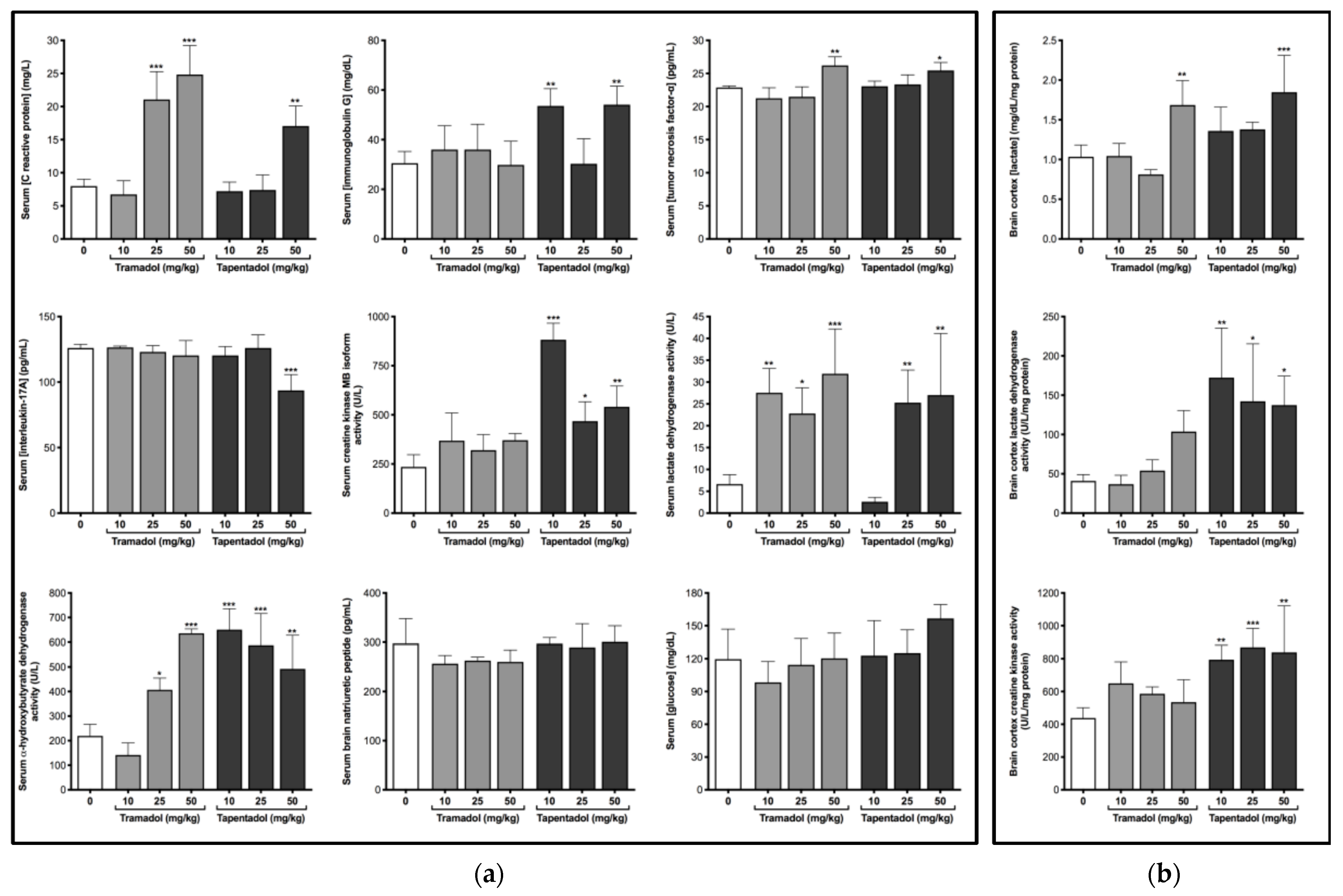
2.2. Repeated Exposure to Tramadol and Tapentadol Causes Alterations in Immunological and Inflammatory Biomarkers
2.3. Repeated Exposure to Tramadol and Tapentadol Compromises Cardiac Cell Integrity and Brain Cortex Metabolism
2.4. Repeated Exposure to Tramadol and Tapentadol Leads to Changes in the Expression of Lung, Heart and Brain Toxicity Biomarkers
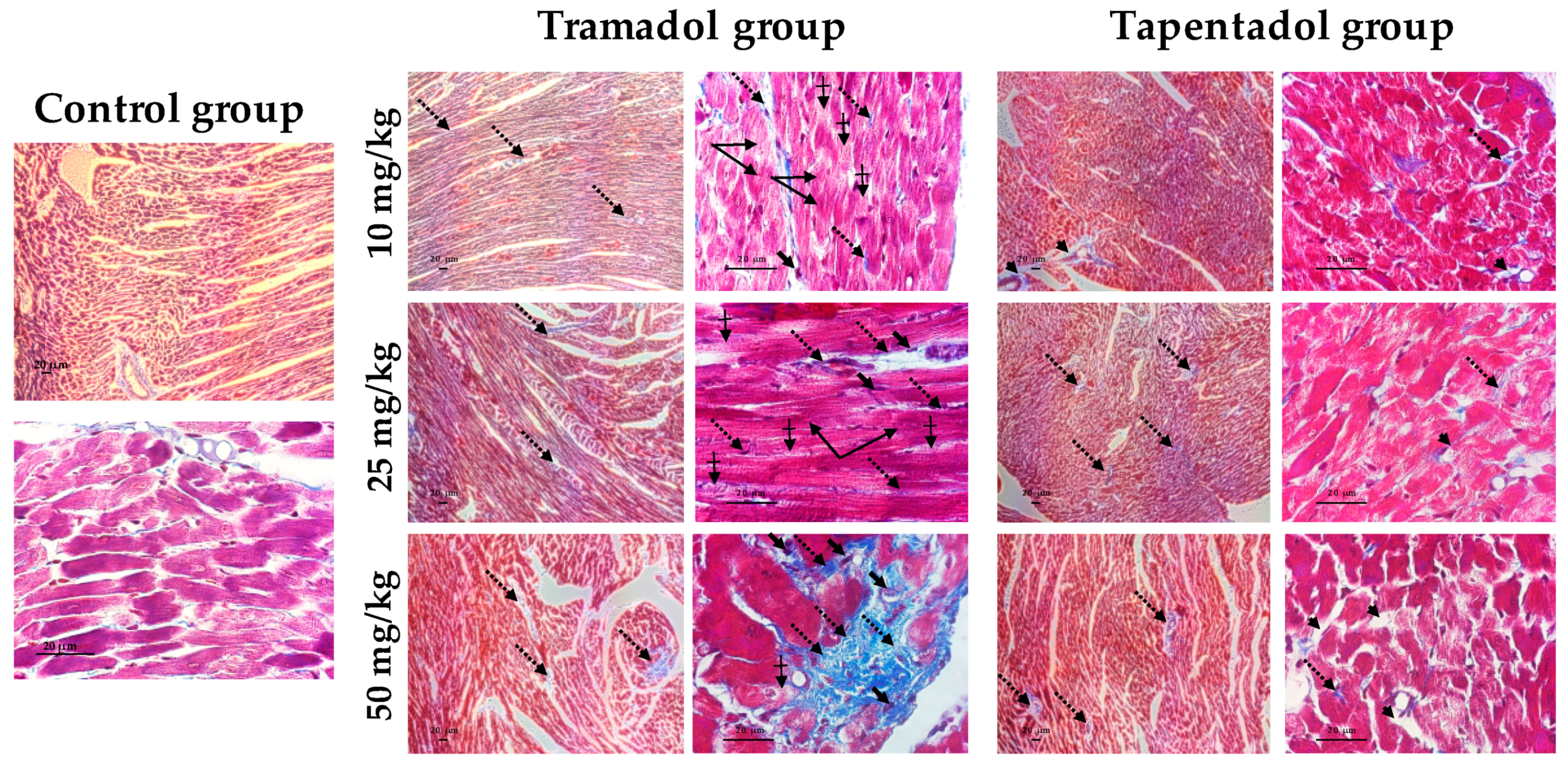
2.5. Repeated Exposure to Tramadol and Tapentadol Leads to Lung Alveolar Collapse, Cardiac Inflammation and Fibrosis and Neuronal Degeneration
3. Discussion
3.1. Repeated Administration of Tramadol and Tapentadol Leads Mainly to Lipid Peroxidation in Lung and Brain Cortex Tissues, but Has Seemingly a Protective Effect in Cardiac Tissue
3.2. Repeated Administration of Tramadol and Tapentadol Leads to Inflammation, with Possible Compensatory Recruitment of Anti-Inflammatory Pathways
3.3. Repeated Administration of Tramadol and Tapentadol Leads to Cardiac Muscle Cell Damage, though with No Impact on Ventricular Function
3.4. Repeated Administration of Tramadol and Tapentadol Modifies Brain Cortex Metabolism, with Tapentadol Causing a Higher Degree of Metabolic Modulation
3.5. Repeated Exposure to Tramadol and Tapentadol Alters the Expression of Lung, Heart and Brain Toxicity Biomarkers at the Gene and Protein Levels, Correlating with Oxidative Stress, Inflammation, Metabolic and Histological Parameters
3.6. Repeated Exposure to Tramadol and Tapentadol Leads to Histopathological Damage in Lung, Heart and Brain Cortex Tissues from the Lowest Therapeutic Dose
4. Materials and Methods
4.1. Chemicals
4.2. Experimental Models and Animal Handling
4.3. Experimental Design and Drug Treatment
4.4. Collection and Processing of Biological Samples
4.4.1. Quantification of Oxidative Stress Parameters
4.4.2. Quantification of Biochemical/Immunological Parameters in Serum Samples and in Brain Cortex Homogenates
4.4.3. Gene Expression Analysis Through qRT-PCR
4.4.4. Brain Cortex Protein Expression Analysis through Western Blotting
4.4.5. Lung, Heart, and Brain Cortex Histopathological Analysis
4.5. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Faria, J.; Barbosa, J.; Moreira, R.; Queiros, O.; Carvalho, F.; Dinis-Oliveira, R.J. Comparative pharmacology and toxicology of tramadol and tapentadol. Eur. J. Pain 2018, 22, 827–844. [Google Scholar] [CrossRef] [PubMed]
- Bravo, L.; Mico, J.A.; Berrocoso, E. Discovery and development of tramadol for the treatment of pain. Expert Opin. Drug Discov. 2017, 12, 1281–1291. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, S.; Chang, S.; Mehta, V. Tapentadol--the evidence so far. Anaesthesia 2015, 70, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, Y.; Kataoka, T.; Tasaki, Y.; Kondo, Y.; Sato, N.; Naiki, T.; Sakamoto, N.; Akechi, T.; Kimura, K. Efficacy of tapentadol for first-line opioid-resistant neuropathic pain in Japan. Jpn. J. Clin. Oncol. 2018, 48, 362–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommer, C.; Klose, P.; Welsch, P.; Petzke, F.; Hauser, W. Opioids for chronic non-cancer neuropathic pain. An updated systematic review and meta-analysis of efficacy, tolerability and safety in randomized placebo-controlled studies of at least 4 weeks duration. Eur. J. Pain 2020, 24, 3–18. [Google Scholar] [CrossRef]
- Caraci, F.; Merlo, S.; Drago, F.; Caruso, G.; Parenti, C.; Sortino, M.A. Rescue of Noradrenergic System as a Novel Pharmacological Strategy in the Treatment of Chronic Pain: Focus on Microglia Activation. Front. Pharmacol. 2019, 10, 1024. [Google Scholar] [CrossRef] [Green Version]
- Kress, H.G.; Koch, E.D.; Kosturski, H.; Steup, A.; Karcher, K.; Dogan, C.; Etropolski, M.; Eerdekens, M. Direct conversion from tramadol to tapentadol prolonged release for moderate to severe, chronic malignant tumour-related pain. Eur. J. Pain 2016, 20, 1513–1518. [Google Scholar] [CrossRef]
- Rensburg, R.; Reuter, H. An overview of analgesics: Opioids, tramadol, and tapentadol (Part 2). S. Afr. Fam. Pract. 2019, 61, 16–23. [Google Scholar] [CrossRef]
- Barbosa, J.; Faria, J.; Queiros, O.; Moreira, R.; Carvalho, F.; Dinis-Oliveira, R.J. Comparative metabolism of tramadol and tapentadol: A toxicological perspective. Drug Metab. Rev. 2016, 48, 577–592. [Google Scholar] [CrossRef] [PubMed]
- Matthiesen, T.; Wohrmann, T.; Coogan, T.P.; Uragg, H. The experimental toxicology of tramadol: An overview. Toxicol. Lett. 1998, 95, 63–71. [Google Scholar] [CrossRef]
- Vadivelu, N.; Chang, D.; Helander, E.M.; Bordelon, G.J.; Kai, A.; Kaye, A.D.; Hsu, D.; Bang, D.; Julka, I. Ketorolac, Oxymorphone, Tapentadol, and Tramadol: A Comprehensive Review. Anesthesiol. Clin. 2017, 35, e1–e20. [Google Scholar] [CrossRef] [PubMed]
- Pergolizzi, J.V., Jr.; Breve, F.; Taylor, R., Jr.; Raffa, R.B.; Strasburger, S.E.; LeQuang, J.A. Considering tapentadol as a first-line analgesic: 14 questions. Pain Manag. 2017, 7, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Grond, S.; Sablotzki, A. Clinical pharmacology of tramadol. Clin. Pharmacokinet 2004, 43, 879–923. [Google Scholar] [CrossRef] [PubMed]
- Raffa, R.B.; Buschmann, H.; Christoph, T.; Eichenbaum, G.; Englberger, W.; Flores, C.M.; Hertrampf, T.; Kogel, B.; Schiene, K.; Strassburger, W.; et al. Mechanistic and functional differentiation of tapentadol and tramadol. Expert Opin. Pharmacother. 2012, 13, 1437–1449. [Google Scholar] [CrossRef]
- Raffa, R.B. Basic pharmacology relevant to drug abuse assessment: Tramadol as example. J. Clin. Pharm. Ther. 2008, 33, 101–108. [Google Scholar] [CrossRef]
- Leppert, W. CYP2D6 in the metabolism of opioids for mild to moderate pain. Pharmacology 2011, 87, 274–285. [Google Scholar] [CrossRef]
- Giorgi, M.; Meizler, A.; Mills, P.C. Pharmacokinetics of the novel atypical opioid tapentadol following oral and intravenous administration in dogs. Vet. J. 2012, 194, 309–313. [Google Scholar] [CrossRef]
- Tzschentke, T.M.; Christoph, T.; Kogel, B.Y. The mu-opioid receptor agonist/noradrenaline reuptake inhibition (MOR-NRI) concept in analgesia: The case of tapentadol. CNS Drugs 2014, 28, 319–329. [Google Scholar] [CrossRef]
- Hartrick, C.T.; Rozek, R.J. Tapentadol in pain management: A mu-opioid receptor agonist and noradrenaline reuptake inhibitor. CNS Drugs 2011, 25, 359–370. [Google Scholar] [CrossRef]
- Meske, D.S.; Xie, J.Y.; Oyarzo, J.; Badghisi, H.; Ossipov, M.H.; Porreca, F. Opioid and noradrenergic contributions of tapentadol in experimental neuropathic pain. Neurosci. Lett. 2014, 562, 91–96. [Google Scholar] [CrossRef] [Green Version]
- Steigerwald, I.; Schenk, M.; Lahne, U.; Gebuhr, P.; Falke, D.; Hoggart, B. Effectiveness and tolerability of tapentadol prolonged release compared with prior opioid therapy for the management of severe, chronic osteoarthritis pain. Clin. Drug Investig. 2013, 33, 607–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsutaoka, B.T.; Ho, R.Y.; Fung, S.M.; Kearney, T.E. Comparative Toxicity of Tapentadol and Tramadol Utilizing Data Reported to the National Poison Data System. Ann. Pharmacother. 2015, 49, 1311–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzschentke, T.M.; Christoph, T.; Kogel, B.; Schiene, K.; Hennies, H.H.; Englberger, W.; Haurand, M.; Jahnel, U.; Cremers, T.I.; Friderichs, E.; et al. (-)-(1R,2R)-3-(3-dimethylamino-1-ethyl-2-methyl-propyl)-phenol hydrochloride (tapentadol HCl): A novel mu-opioid receptor agonist/norepinephrine reuptake inhibitor with broad-spectrum analgesic properties. J. Pharmacol. Exp. Ther. 2007, 323, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Bortolotto, V.; Grilli, M. Opiate Analgesics as Negative Modulators of Adult Hippocampal Neurogenesis: Potential Implications in Clinical Practice. Front. Pharmacol. 2017, 8, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, G.; Williams, D. The discovery and development of analgesics: New mechanisms, new modalities. J. Clin. Investig. 2010, 120, 3753–3759. [Google Scholar] [CrossRef] [Green Version]
- Langford, R.M.; Knaggs, R.; Farquhar-Smith, P.; Dickenson, A.H. Is tapentadol different from classical opioids? A review of the evidence. Br. J. Pain 2016, 10, 217–221. [Google Scholar] [CrossRef]
- Meneghini, V.; Cuccurazzu, B.; Bortolotto, V.; Ramazzotti, V.; Ubezio, F.; Tzschentke, T.M.; Canonico, P.L.; Grilli, M. The noradrenergic component in tapentadol action counteracts mu-opioid receptor-mediated adverse effects on adult neurogenesis. Mol. Pharmacol. 2014, 85, 658–670. [Google Scholar] [CrossRef] [Green Version]
- Cantrell, F.L.; Mallett, P.; Aldridge, L.; Verilhac, K.; McIntyre, I.M. A tapentadol related fatality: Case report with postmortem concentrations. Forensic Sci. Int. 2016, 266, e1–e3. [Google Scholar] [CrossRef]
- Channell, J.S.; Schug, S. Toxicity of tapentadol: A systematic review. Pain Manag. 2018, 8, 327–339. [Google Scholar] [CrossRef]
- Costa, I.; Oliveira, A.; Guedes de Pinho, P.; Teixeira, H.M.; Moreira, R.; Carvalho, F.; Dinis-Oliveira, R.J. Postmortem redistribution of tramadol and O-desmethyltramadol. J. Anal. Toxicol. 2013, 37, 670–675. [Google Scholar] [CrossRef]
- Franco, D.M.; Ali, Z.; Levine, B.; Middleberg, R.A.; Fowler, D.R. Case report of a fatal intoxication by Nucynta. Am. J. Forensic Med. Pathol. 2014, 35, 234–236. [Google Scholar] [CrossRef] [PubMed]
- Hawton, K.; Ferrey, A.; Casey, D.; Wells, C.; Fuller, A.; Bankhead, C.; Clements, C.; Ness, J.; Gunnell, D.; Kapur, N.; et al. Relative toxicity of analgesics commonly used for intentional self-poisoning: A study of case fatality based on fatal and non-fatal overdoses. J. Affect. Disord. 2019, 246, 814–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemp, W.; Schlueter, S.; Smalley, E. Death due to apparent intravenous injection of tapentadol. J. Forensic Sci. 2013, 58, 288–291. [Google Scholar] [CrossRef] [PubMed]
- Khaja, M.; Lominadze, G.; Millerman, K. Cardiac Arrest Following Drug Abuse with Intravenous Tapentadol: Case Report and Literature Review. Am. J. Case Rep. 2017, 18, 817–821. [Google Scholar] [CrossRef] [Green Version]
- Larson, S.J.; Pestaner, J.; Prashar, S.K.; Bayard, C.; Zarwell, L.W.; Pierre-Louis, M. Postmortem distribution of tapentadol and N-desmethyltapentadol. J. Anal. Toxicol. 2012, 36, 440–443. [Google Scholar] [CrossRef] [Green Version]
- Loughrey, M.B.; Loughrey, C.M.; Johnston, S.; O’Rourke, D. Fatal hepatic failure following accidental tramadol overdose. Forensic Sci. Int. 2003, 134, 232–233. [Google Scholar] [CrossRef]
- Murphy, D.L.; Lebin, J.A.; Severtson, S.G.; Olsen, H.A.; Dasgupta, N.; Dart, R.C. Comparative Rates of Mortality and Serious Adverse Effects Among Commonly Prescribed Opioid Analgesics. Drug Saf. 2018, 41, 787–795. [Google Scholar] [CrossRef]
- Partridge, E.; Teoh, E.; Nash, C.; Scott, T.; Charlwood, C.; Kostakis, C. The Increasing Use and Abuse of Tapentadol and Its Incorporation Into a Validated Quantitative Method. J. Anal. Toxicol. 2018, 42, 485–490. [Google Scholar] [CrossRef] [Green Version]
- Pilgrim, J.L.; Gerostamoulos, D.; Drummer, O.H. Deaths involving contraindicated and inappropriate combinations of serotonergic drugs. Int. J. Leg. Med. 2011, 125, 803–815. [Google Scholar] [CrossRef]
- Pilgrim, J.L.; Gerostamoulos, D.; Drummer, O.H. Deaths involving serotonergic drugs. Forensic Sci. Int. 2010, 198, 110–117. [Google Scholar] [CrossRef]
- Tjaderborn, M.; Jonsson, A.K.; Hagg, S.; Ahlner, J. Fatal unintentional intoxications with tramadol during 1995–2005. Forensic Sci. Int. 2007, 173, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.; Tchoe, H.J.; Li, J.; Shin, J.Y. All-Cause Mortality Associated with Tramadol Use: A Case-Crossover Study. Drug Saf. 2019, 42, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Barbera, N.; Fisichella, M.; Bosco, A.; Indorato, F.; Spadaro, G.; Romano, G. A suicidal poisoning due to tramadol. A metabolic approach to death investigation. J. Forensic Leg. Med. 2013, 20, 555–558. [Google Scholar] [CrossRef] [PubMed]
- De Backer, B.; Renardy, F.; Denooz, R.; Charlier, C. Quantification in postmortem blood and identification in urine of tramadol and its two main metabolites in two cases of lethal tramadol intoxication. J. Anal. Toxicol. 2010, 34, 599–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musshoff, F.; Madea, B. Fatality due to ingestion of tramadol alone. Forensic Sci. Int. 2001, 116, 197–199. [Google Scholar] [CrossRef]
- Lusthof, K.J.; Zweipfenning, P.G. Suicide by tramadol overdose. J. Anal. Toxicol. 1998, 22, 260. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.A.; Cina, S.J.; Jones, R.; Selby, D.M.; Levine, B.; Smith, M.L. Tissue distribution of tramadol and metabolites in an overdose fatality. Am. J. Forensic Med. Pathol. 1999, 20, 98–100. [Google Scholar] [CrossRef]
- Wang, S.Q.; Li, C.S.; Song, Y.G. Multiply organ dysfunction syndrome due to tramadol intoxication alone. Am. J. Emerg. Med. 2009, 27, 903.E5–903.E7. [Google Scholar] [CrossRef]
- Borys, D.; Stanton, M.; Gummin, D.; Drott, T. Tapentadol toxicity in children. Pediatrics 2015, 135, e392–e396. [Google Scholar] [CrossRef] [Green Version]
- Hedenmalm, K.; Slattery, J.; Skibicka-Stepien, I.; Kurz, X.; Morales, D. Prescribing patterns of tramadol in adults in IMS(R) primary care databases in France and Germany between 1 January 2006 and 30 June 2016. Eur. J. Clin. Pharmacol. 2019, 75, 707–716. [Google Scholar] [CrossRef] [Green Version]
- Karila, L.; Marillier, M.; Chaumette, B.; Billieux, J.; Franchitto, N.; Benyamina, A. New synthetic opioids: Part of a new addiction landscape. Neurosci. Biobehav. Rev. 2019, 106, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Pinho, S.; Oliveira, A.; Costa, I.; Gouveia, C.A.; Carvalho, F.; Moreira, R.F.; Dinis-Oliveira, R.J. Simultaneous quantification of tramadol and O-desmethyltramadol in hair samples by gas chromatography-electron impact/mass spectrometry. Biomed. Chromatogr. 2013, 27, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Suga, Y.; Uchida, M.; Suzuki, S.; Sugawara, H.; Torigoe, K.; Futamura, A.; Uesawa, Y.; Nakagawa, T.; Takase, H. Current Status of Adverse Events Related with Opioid Analgesics in Japan: Assessment Based on Japanese Adverse Drug Event Report Database. Biol. Pharm. Bull. 2019, 42, 801–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faria, J.; Barbosa, J.; Queiros, O.; Moreira, R.; Carvalho, F.; Dinis-Oliveira, R.J. Comparative study of the neurotoxicological effects of tramadol and tapentadol in SH-SY5Y cells. Toxicology 2016, 359–360, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Faria, J.; Barbosa, J.; Leal, S.; Afonso, L.P.; Lobo, J.; Moreira, R.; Queiros, O.; Carvalho, F.; Dinis-Oliveira, R.J. Effective analgesic doses of tramadol or tapentadol induce brain, lung and heart toxicity in Wistar rats. Toxicology 2017, 385, 38–47. [Google Scholar] [CrossRef]
- Barbosa, J.; Faria, J.; Leal, S.; Afonso, L.P.; Lobo, J.; Queiros, O.; Moreira, R.; Carvalho, F.; Dinis-Oliveira, R.J. Acute administration of tramadol and tapentadol at effective analgesic and maximum tolerated doses causes hepato- and nephrotoxic effects in Wistar rats. Toxicology 2017, 389, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, J.; Faria, J.; Garcez, F.; Leal, S.; Afonso, L.P.; Nascimento, A.V.; Moreira, R.; Queiros, O.; Carvalho, F.; Dinis-Oliveira, R.J. Repeated Administration of Clinical Doses of Tramadol and Tapentadol Causes Hepato- and Nephrotoxic Effects in Wistar Rats. Pharmaceuticals 2020, 13, 149. [Google Scholar] [CrossRef]
- Samaka, R.M.; Girgis, N.F.; Shams, T.M. Acute toxicity and dependence of tramadol in albino rats: Relationship of Nestin and Notch 1 as stem cell markers. J. Am. Sci. 2012, 8, 313–327. [Google Scholar]
- Awadalla, E.A.; Salah-Eldin, A.E. Molecular and histological changes in cerebral cortex and lung tissues under the effect of tramadol treatment. Biomed. Pharmacother. 2016, 82, 269–280. [Google Scholar] [CrossRef]
- Ghoneim, F.M.; Khalaf, H.A.; Elsamanoudy, A.Z.; Helaly, A.N. Effect of chronic usage of tramadol on motor cerebral cortex and testicular tissues of adult male albino rats and the effect of its withdrawal: Histological, immunohistochemical and biochemical study. Int. J. Clin. Exp. Pathol. 2014, 7, 7323–7341. [Google Scholar]
- Khodeary, M.F.; Sharaf El-Din, A.A.I.; El Kholy, S.M.S. A histopathological and immunohistochemical study of adult rats’ brain after long-term exposure to Amadol (tramadol hydrochloride). Mansoura J. Forensic Med. Clin. Toxicol. 2010, 18, 1–24. [Google Scholar] [CrossRef]
- Nafea, O.E.; ElKhishin, I.A.; Awad, O.A.; Mohamed, D.A. A study of the neurotoxic effects of tramadol and cannabis in adolescent male albino rats. Int. J. Sci. Rep. 2016, 2, 143–154. [Google Scholar] [CrossRef] [Green Version]
- Mehdizadeh, H.; Pourahmad, J.; Taghizadeh, G.; Vousooghi, N.; Yoonessi, A.; Naserzadeh, P.; Behzadfar, L.; Rouini, M.R.; Sharifzadeh, M. Mitochondrial impairments contribute to spatial learning and memory dysfunction induced by chronic tramadol administration in rat: Protective effect of physical exercise. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 79, 426–433. [Google Scholar] [CrossRef]
- Doostmohammadi, M.; Rahimi, H.R. ADME and toxicity considerations for tramadol: From basic research to clinical implications. Expert Opin. Drug Metab. Toxicol. 2020, 16, 627–640. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, T.; Christrup, L.L.; Drewes, A.M.; Fallon, M.T.; Kress, H.G.; McQuay, H.J.; Mikus, G.; Morlion, B.J.; Perez-Cajaraville, J.; Pogatzki-Zahn, E.; et al. European Pain Federation position paper on appropriate opioid use in chronic pain management. Eur. J. Pain 2017, 21, 3–19. [Google Scholar] [CrossRef]
- Lukas, G.; Brindle, S.D.; Greengard, P. The route of absorption of intraperitoneally administered compounds. J. Pharmacol. Exp. Ther. 1971, 178, 562–564. [Google Scholar] [PubMed]
- Abdel-Zaher, A.O.; Abdel-Rahman, M.S.; Elwasei, F.M. Protective effect of Nigella sativa oil against tramadol-induced tolerance and dependence in mice: Role of nitric oxide and oxidative stress. Neurotoxicology 2011, 32, 725–733. [Google Scholar] [CrossRef]
- Hussein, S.A.; Abdel Aal, S.A.; Ismail, H.K. Effect of tramadol drug on some biochemical and immunological parameters in albino male rats; evaluation of possible reversal following its withdrawal. Benhav. Vet. Med. J. 2017, 33, 418–429. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, H.M.; Mahmoud, A.M. Chronic exposure to the opioid tramadol induces oxidative damage, inflammation and apoptosis, and alters cerebral monoamine neurotransmitters in rats. Biomed. Pharmacother. 2019, 110, 239–247. [Google Scholar] [CrossRef]
- Ali, H.A.; Afifi, M.; Saber, T.M.; Makki, A.A.; Keshta, A.T.; Baeshen, M.; Al-Farga, A. Neurotoxic, Hepatotoxic and Nephrotoxic Effects of Tramadol Administration in Rats. J. Mol. Neurosci. 2020, 70, 1934–1942. [Google Scholar] [CrossRef]
- Xia, W.; Liu, G.; Shao, Z.; Xu, E.; Yuan, H.; Liu, J.; Gao, L. Toxicology of tramadol following chronic exposure based on metabolomics of the cerebrum in mice. Sci. Rep. 2020, 10, 11130. [Google Scholar] [CrossRef] [PubMed]
- Takhtfooladi, H.A.; Asl, A.H.; Shahzamani, M.; Takhtfooladi, M.A.; Allahverdi, A.; Khansari, M. Tramadol alleviates myocardial injury induced by acute hindlimb ischemia reperfusion in rats. Arq. Bras. Cardiol. 2015, 105, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Ndrepepa, G. Myeloperoxidase—A bridge linking inflammation and oxidative stress with cardiovascular disease. Clin. Chim Acta 2019, 493, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Takhtfooladi, M.A.; Jahanshahi, A.; Sotoudeh, A.; Jahanshahi, G.; Takhtfooladi, H.A.; Aslani, K. Effect of tramadol on lung injury induced by skeletal muscle ischemia-reperfusion: An experimental study. J. Bras. Pneumol. 2013, 39, 434–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyssen, P.; Mouithys-Mickalad, A.; Minguet, G.; Sauvage, E.; Wouters, J.; Franck, T.; Hoebeke, M. Morphine, a potential inhibitor of myeloperoxidase activity. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2236–2244. [Google Scholar] [CrossRef]
- Kitamura, A.; Higuchi, K.; Okura, T.; Deguchi, Y. Transport characteristics of tramadol in the blood-brain barrier. J. Pharm. Sci. 2014, 103, 3335–3341. [Google Scholar] [CrossRef]
- Pergolizzi, J.; Alegre, C.; Blake, D.; Alen, J.C.; Caporali, R.; Casser, H.R.; Correa-Illanes, G.; Fernandes, P.; Galilea, E.; Jany, R.; et al. Current considerations for the treatment of severe chronic pain: The potential for tapentadol. Pain Pract. 2012, 12, 290–306. [Google Scholar] [CrossRef]
- Rubio, C.P.; Hernandez-Ruiz, J.; Martinez-Subiela, S.; Tvarijonaviciute, A.; Ceron, J.J. Spectrophotometric assays for total antioxidant capacity (TAC) in dog serum: An update. BMC Vet. Res. 2016, 12, 166. [Google Scholar] [CrossRef] [Green Version]
- Young, I.S. Measurement of total antioxidant capacity. J. Clin. Pathol. 2001, 54, 339. [Google Scholar] [CrossRef] [Green Version]
- Tsai, Y.C.; Won, S.J. Effects of tramadol on T lymphocyte proliferation and natural killer cell activity in rats with sciatic constriction injury. Pain 2001, 92, 63–69. [Google Scholar] [CrossRef]
- El-Sharrawy, E.A.; El-Hakim, I.E.; Sameeh, E. Attenuation of C-reactive protein increases after exodontia by tramadol and ibuprofen. Anesth. Prog. 2006, 53, 78–82. [Google Scholar] [CrossRef]
- Aschermann, S.; Lux, A.; Baerenwaldt, A.; Biburger, M.; Nimmerjahn, F. The other side of immunoglobulin G: Suppressor of inflammation. Clin. Exp. Immunol. 2010, 160, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, H.Y. Immunoglobulin G and its function in the human respiratory tract. Mayo Clin. Proc. 1988, 63, 161–174. [Google Scholar] [CrossRef]
- Rabei, H.M. The immunological and histopathological changes of tramadol, tramadol/acetaminophen and acetaminophen in male albino rats—comparative study. Egypt J. Hosp. Med. 2011, 45, 477–503. [Google Scholar]
- Liu, Y.M.; Zhu, S.M.; Wang, K.R.; Feng, Z.Y.; Chen, Q.L. Effect of tramadol on immune responses and nociceptive thresholds in a rat model of incisional pain. J. Zhejiang Univ. Sci. B 2008, 9, 895–902. [Google Scholar] [CrossRef]
- Liu, Z.; Gao, F.; Tian, Y. Effects of morphine, fentanyl and tramadol on human immune response. J. Huazhong Univ. Sci. Technol. Med. Sci. 2006, 26, 478–481. [Google Scholar] [CrossRef]
- Sayed, J.A.; Abd Elshafy, S.K.; Kamel, E.Z.; Fathy Riad, M.A.; Mahmoud, A.A.; Khalaf, G.S. The impact of caudally administrated tramadol on immune response and analgesic efficacy for pediatric patients: A comparative randomized clinical trial. Korean J. Pain 2018, 31, 206–214. [Google Scholar] [CrossRef]
- Mannocchi, G.; Napoleoni, F.; Napoletano, S.; Pantano, F.; Santoni, M.; Tittarelli, R.; Arbarello, P. Fatal self administration of tramadol and propofol: A case report. J. Forensic Leg. Med. 2013, 20, 715–719. [Google Scholar] [CrossRef]
- Cole, J.B.; Sattiraju, S.; Bilden, E.F.; Asinger, R.W.; Bertog, S.C. Isolated tramadol overdose associated with Brugada ECG pattern. Pacing Clin. Electrophysiol. 2012, 35, e219–e221. [Google Scholar] [CrossRef]
- Hafez, E.; Issa, S.; Rahman, S.A. Parenchymatous toxicity of tramadol: Histopathological and biochemical study. J. Alcohol. Drug Depend. 2015, 3. [Google Scholar]
- Zhang, Y.; Lin, W.; Shen, S.; Wang, H.; Feng, X.; Sun, J. Randomized comparison of sevoflurane versus propofol-remifentanil on the cardioprotective effects in elderly patients with coronary heart disease. BMC Anesthesiol. 2017, 17, 104. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.J.; McDonagh, T.A.; Harkness, A.; McKay, S.E.; Dargie, H.J. Morphine for the relief of breathlessness in patients with chronic heart failure—A pilot study. Eur. J. Heart Fail. 2002, 4, 753–756. [Google Scholar] [CrossRef]
- Preus, M.; Bhargava, A.S.; Khater, A.E.; Gunzel, P. Diagnostic value of serum creatine kinase and lactate dehydrogenase isoenzyme determinations for monitoring early cardiac damage in rats. Toxicol. Lett. 1988, 42, 225–233. [Google Scholar] [CrossRef]
- Li, S.W.; Sun, X.; He, Y.; Guo, Y.; Zhao, H.J.; Hou, Z.J.; Xing, M.W. Assessment of arsenic trioxide in the heart of Gallus gallus: Alterations of oxidative damage parameters, inflammatory cytokines, and cardiac enzymes. Environ. Sci. Pollut. Res. Int. 2017, 24, 5781–5790. [Google Scholar] [CrossRef]
- Perez-Alvarez, S.; Cuenca-Lopez, M.D.; de Mera, R.M.; Puerta, E.; Karachitos, A.; Bednarczyk, P.; Kmita, H.; Aguirre, N.; Galindo, M.F.; Jordan, J. Methadone induces necrotic-like cell death in SH-SY5Y cells by an impairment of mitochondrial ATP synthesis. Biochim. Biophys. Acta 2010, 1802, 1036–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuo, H.Q.; Huang, L.; Huang, H.Q.; Cai, Z. Effects of chronic tramadol exposure on the zebrafish brain: A proteomic study. J. Proteom. 2012, 75, 3351–3364. [Google Scholar] [CrossRef]
- Mohamed, T.M.; Ghaffar, H.M.; El Husseiny, R.M. Effects of tramadol, clonazepam, and their combination on brain mitochondrial complexes. Toxicol. Ind. Health 2015, 31, 1325–1333. [Google Scholar] [CrossRef]
- Sharma, S.K.; Yashpal, K.; Fundytus, M.E.; Sauriol, F.; Henry, J.L.; Coderre, T.J. Alterations in brain metabolism induced by chronic morphine treatment: NMR studies in rat CNS. Neurochem. Res. 2003, 28, 1369–1373. [Google Scholar] [CrossRef]
- Veenith, T.; Menon, D.K. The cerebral circulation. In Core Topics in Neuroanaesthesia and Neurointensive Care; Matta, B.F., Menon, D.K., Smith, M., Eds.; Cambridge University Press: New York, NY, USA, 2011; pp. 21–22. [Google Scholar]
- Schlattner, U.; Klaus, A.; Ramirez Rios, S.; Guzun, R.; Kay, L.; Tokarska-Schlattner, M. Cellular compartmentation of energy metabolism: Creatine kinase microcompartments and recruitment of B-type creatine kinase to specific subcellular sites. Amino Acids 2016, 48, 1751–1774. [Google Scholar] [CrossRef]
- Hermans, C.; Bernard, A. Clara cell protein (CC16): Characteristics and potential applications as biomarker of lung toxicity. Biomarkers 1996, 1, 3–8. [Google Scholar] [CrossRef]
- Pang, M.; Liu, H.Y.; Li, T.; Wang, D.; Hu, X.Y.; Zhang, X.R.; Yu, B.F.; Guo, R.; Wang, H.L. Recombinant club cell protein 16 (CC16) ameliorates cigarette smokeinduced lung inflammation in a murine disease model of COPD. Mol. Med. Rep. 2018, 18, 2198–2206. [Google Scholar] [CrossRef] [PubMed]
- Laucho-Contreras, M.E.; Polverino, F.; Gupta, K.; Taylor, K.L.; Kelly, E.; Pinto-Plata, V.; Divo, M.; Ashfaq, N.; Petersen, H.; Stripp, B.; et al. Protective role for club cell secretory protein-16 (CC16) in the development of COPD. Eur. Respir. J. 2015, 45, 1544–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hukkanen, J.; Pelkonen, O.; Hakkola, J.; Raunio, H. Expression and regulation of xenobiotic-metabolizing cytochrome P450 (CYP) enzymes in human lung. Crit. Rev. Toxicol. 2002, 32, 391–411. [Google Scholar] [CrossRef] [PubMed]
- Zhai, J.; Insel, M.; Addison, K.J.; Stern, D.A.; Pederson, W.; Dy, A.; Rojas-Quintero, J.; Owen, C.A.; Sherrill, D.L.; Morgan, W.; et al. Club Cell Secretory Protein Deficiency Leads to Altered Lung Function. Am. J. Respir. Crit. Care Med. 2019, 199, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Lierova, A.; Jelicova, M.; Nemcova, M.; Proksova, M.; Pejchal, J.; Zarybnicka, L.; Sinkorova, Z. Cytokines and radiation-induced pulmonary injuries. J. Radiat. Res. 2018, 59, 709–753. [Google Scholar] [CrossRef]
- Siva, S.; MacManus, M.; Kron, T.; Best, N.; Smith, J.; Lobachevsky, P.; Ball, D.; Martin, O. A pattern of early radiation-induced inflammatory cytokine expression is associated with lung toxicity in patients with non-small cell lung cancer. PLoS ONE 2014, 9, e109560. [Google Scholar] [CrossRef]
- Shimada, A.L.; Ribeiro, A.L.; Bolonheis, S.M.; Ferraz-de-Paula, V.; Hebeda, C.B.; Farsky, S.H. In vivo hydroquinone exposure impairs MCP-1 secretion and monocyte recruitment into the inflamed lung. Toxicology 2012, 296, 20–26. [Google Scholar] [CrossRef]
- Rizzo, M.D.; Crawford, R.B.; Bach, A.; Sermet, S.; Amalfitano, A.; Kaminski, N.E. Delta(9)-Tetrahydrocannabinol Suppresses Monocyte-Mediated Astrocyte Production of Monocyte Chemoattractant Protein 1 and Interleukin-6 in a Toll-Like Receptor 7-Stimulated Human Coculture. J. Pharmacol. Exp. Ther. 2019, 371, 191–201. [Google Scholar] [CrossRef]
- Weiss, J.M.; Cuff, C.A.; Berman, J.W. TGF-beta downmodulates cytokine-induced monocyte chemoattractant protein (MCP)-1 expression in human endothelial cells. A putative role for TGF-beta in the modulation of TNF receptor expression. Endothelium 1999, 6, 291–302. [Google Scholar] [CrossRef]
- Zhong, Y.; Liu, T.; Lai, W.; Tan, Y.; Tian, D.; Guo, Z. Heme oxygenase-1-mediated reactive oxygen species reduction is involved in the inhibitory effect of curcumin on lipopolysaccharide-induced monocyte chemoattractant protein-1 production in RAW264.7 macrophages. Mol. Med. Rep. 2013, 7, 242–246. [Google Scholar] [CrossRef]
- Tan, R.J.; Fattman, C.L.; Niehouse, L.M.; Tobolewski, J.M.; Hanford, L.E.; Li, Q.; Monzon, F.A.; Parks, W.C.; Oury, T.D. Matrix metalloproteinases promote inflammation and fibrosis in asbestos-induced lung injury in mice. Am. J. Respir. Cell Mol. Biol. 2006, 35, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Oggionni, T.; Morbini, P.; Inghilleri, S.; Palladini, G.; Tozzi, R.; Vitulo, P.; Fenoglio, C.; Perlini, S.; Pozzi, E. Time course of matrix metalloproteases and tissue inhibitors in bleomycin-induced pulmonary fibrosis. Eur. J. Histochem. 2006, 50, 317–325. [Google Scholar]
- Morais, A.; Beltrao, M.; Sokhatska, O.; Costa, D.; Melo, N.; Mota, P.; Marques, A.; Delgado, L. Serum metalloproteinases 1 and 7 in the diagnosis of idiopathic pulmonary fibrosis and other interstitial pneumonias. Respir. Med. 2015, 109, 1063–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, Y.; White, E.S.; de Bernard, S.; Cornelisse, P.; Leconte, I.; Morganti, A.; Roux, S.; Nayler, O. MMP-7 is a predictive biomarker of disease progression in patients with idiopathic pulmonary fibrosis. ERJ Open Res. 2017, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil, H.W.; Oh, M.H.; Woo, K.M.; Lee, E.Y.; Oh, M.H.; Hong, S.Y. Relationship between pulmonary surfactant protein and lipid peroxidation in lung injury due to paraquat intoxication in rats. Korean J. Intern. Med. 2007, 22, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Ju, Q.; Cao, J.; Tang, W.; Zhang, J. Impact of serum SP-A and SP-D levels on comparison and prognosis of idiopathic pulmonary fibrosis: A systematic review and meta-analysis. Medicine 2017, 96, e7083. [Google Scholar] [CrossRef] [PubMed]
- Johansson, J.; Curstedt, T.; Robertson, B. The proteins of the surfactant system. Eur. Respir J. 1994, 7, 372–391. [Google Scholar] [CrossRef] [Green Version]
- Kuroki, Y.; Takahashi, H.; Chiba, H.; Akino, T. Surfactant proteins A and D: Disease markers. Biochim. Biophys. Acta 1998, 1408, 334–345. [Google Scholar] [CrossRef] [Green Version]
- Sorensen, G.L. Surfactant Protein D in Respiratory and Non-Respiratory Diseases. Front. Med. 2018, 5, 18. [Google Scholar] [CrossRef]
- Fontes, J.A.; Rose, N.R.; Cihakova, D. The varying faces of IL-6: From cardiac protection to cardiac failure. Cytokine 2015, 74, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Zhang, Y.; Ye, J. IL-6: A Potential Role in Cardiac Metabolic Homeostasis. Int. J. Mol. Sci. 2018, 19, 2474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriwaki, H.; Stempien-Otero, A.; Kremen, M.; Cozen, A.E.; Dichek, D.A. Overexpression of urokinase by macrophages or deficiency of plasminogen activator inhibitor type 1 causes cardiac fibrosis in mice. Circ. Res. 2004, 95, 637–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohensinner, P.J.; Takacs, N.; Kaun, C.; Thaler, B.; Krychtiuk, K.A.; Pfaffenberger, S.; Aliabadi, A.; Zuckermann, A.; Huber, K.; Wojta, J. Urokinase plasminogen activator protects cardiac myocytes from oxidative damage and apoptosis via hOGG1 induction. Apoptosis 2017, 22, 1048–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Jackson-Weaver, O.; Xu, J. The TGFbeta superfamily in cardiac dysfunction. Acta Biochim. Biophys. Sin. 2018, 50, 323–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, A.; Frangogiannis, N.G. The Role of the TGF-beta Superfamily in Myocardial Infarction. Front. Cardiovasc. Med. 2019, 6, 140. [Google Scholar] [CrossRef]
- Euler, G. Good and bad sides of TGFbeta-signaling in myocardial infarction. Front. Physiol. 2015, 6, 66. [Google Scholar] [CrossRef] [Green Version]
- Takawale, A.; Zhang, P.; Patel, V.B.; Wang, X.; Oudit, G.; Kassiri, Z. Tissue Inhibitor of Matrix Metalloproteinase-1 Promotes Myocardial Fibrosis by Mediating CD63-Integrin beta1 Interaction. Hypertension 2017, 69, 1092–1103. [Google Scholar] [CrossRef]
- Heymans, S.; Schroen, B.; Vermeersch, P.; Milting, H.; Gao, F.; Kassner, A.; Gillijns, H.; Herijgers, P.; Flameng, W.; Carmeliet, P.; et al. Increased cardiac expression of tissue inhibitor of metalloproteinase-1 and tissue inhibitor of metalloproteinase-2 is related to cardiac fibrosis and dysfunction in the chronic pressure-overloaded human heart. Circulation 2005, 112, 1136–1144. [Google Scholar] [CrossRef] [Green Version]
- Barton, P.J.; Birks, E.J.; Felkin, L.E.; Cullen, M.E.; Koban, M.U.; Yacoub, M.H. Increased expression of extracellular matrix regulators TIMP1 and MMP1 in deteriorating heart failure. J. Heart Lung Transpl. 2003, 22, 738–744. [Google Scholar] [CrossRef]
- Villar-Pique, A.; Lopes da Fonseca, T.; Outeiro, T.F. Structure, function and toxicity of alpha-synuclein: The Bermuda triangle in synucleinopathies. J. Neurochem. 2016, 139 (Suppl. 1), 240–255. [Google Scholar] [CrossRef]
- Ziolkowska, B.; Gieryk, A.; Bilecki, W.; Wawrzczak-Bargiela, A.; Wedzony, K.; Chocyk, A.; Danielson, P.E.; Thomas, E.A.; Hilbush, B.S.; Sutcliffe, J.G.; et al. Regulation of alpha-synuclein expression in limbic and motor brain regions of morphine-treated mice. J. Neurosci. 2005, 25, 4996–5003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dursteler-MacFarland, K.M.; Brugger, I.; Bonsch, D.; Schmid, O.; Kornhuber, J.; Bleich, S.; Wiesbeck, G.A. Alpha-synuclein and heroin craving in opiate-dependent patients on injectable heroin maintenance. Addict. Biol. 2012, 17, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Horvath, M.C.; Kovacs, G.G.; Kovari, V.; Majtenyi, K.; Hurd, Y.L.; Keller, E. Heroin abuse is characterized by discrete mesolimbic dopamine and opioid abnormalities and exaggerated nuclear receptor-related 1 transcriptional decline with age. J. Neurosci. 2007, 27, 13371–13375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suliman, N.A.; Mohd Moklas, M.A.; Mat Taib, C.N.; Adenan, M.I.; Hidayat Baharuldin, M.T.; Basir, R.; Amom, Z. Morphine Antidependence of Erythroxylum cuneatum (Miq.) Kurz in Neurotransmission Processes In Vitro. Evid. Based Complement. Alternat. Med. 2016, 2016, 3517209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ujcikova, H.; Vosahlikova, M.; Roubalova, L.; Svoboda, P. Proteomic analysis of protein composition of rat forebrain cortex exposed to morphine for 10days; comparison with animals exposed to morphine and subsequently nurtured for 20days in the absence of this drug. J. Proteom. 2016, 145, 11–23. [Google Scholar] [CrossRef]
- Ujcikova, H.; Cechova, K.; Jagr, M.; Roubalova, L.; Vosahlikova, M.; Svoboda, P. Proteomic analysis of protein composition of rat hippocampus exposed to morphine for 10 days; comparison with animals after 20 days of morphine withdrawal. PLoS ONE 2020, 15, e0231721. [Google Scholar] [CrossRef] [Green Version]
- Khoshdel, Z.; Ahmadpour Jirandeh, S.; Takhshid, M.A.; Jalali Mashayekhi, F.; Shojaei, S.; Owji, A.A. The BDNF Protein and its Cognate mRNAs in the Rat Spinal Cord during Amylin-induced Reversal of Morphine Tolerance. Neuroscience 2019, 422, 54–64. [Google Scholar] [CrossRef]
- Faron-Gorecka, A.; Kusmider, M.; Inan, S.Y.; Siwanowicz, J.; Piwowarczyk, T.; Dziedzicka-Wasylewska, M. Long-term exposure of rats to tramadol alters brain dopamine and alpha 1-adrenoceptor function that may be related to antidepressant potency. Eur. J. Pharmacol. 2004, 501, 103–110. [Google Scholar] [CrossRef]
- Tsai, M.C.; Huang, T.L. Brain-derived neurotrophic factor (BDNF) and oxidative stress in heroin-dependent male patients undergoing methadone maintenance treatment. Psychiatry Res. 2017, 249, 46–50. [Google Scholar] [CrossRef]
- Rouhani, F.; Khodarahmi, P.; Naseh, V. NGF, BDNF and Arc mRNA Expression in the Hippocampus of Rats After Administration of Morphine. Neurochem. Res. 2019, 44, 2139–2146. [Google Scholar] [CrossRef]
- Yang, C.; Li, X.; Wang, N.; Xu, S.; Yang, J.; Zhou, Z. Tramadol reinforces antidepressant effects of ketamine with increased levels of brain-derived neurotrophic factor and tropomyosin-related kinase B in rat hippocampus. Front. Med. 2012, 6, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Michot, B.; Bourgoin, S.; Kayser, V.; Hamon, M. Effects of tapentadol on mechanical hypersensitivity in rats with ligatures of the infraorbital nerve versus the sciatic nerve. Eur. J. Pain 2013, 17, 867–880. [Google Scholar] [CrossRef] [PubMed]
- Hussein, O.A.; Abdel Mola, A.F.; Rateb, A. Tramadol administration induced hippocampal cells apoptosis, astrogliosis, and microgliosis in juvenile and adult male mice, histological and immunohistochemical study. Ultrastruct. Pathol. 2020, 44, 81–102. [Google Scholar] [CrossRef] [PubMed]
- Torkzadeh-Mahani, S.; Esmaeili-Mahani, S.; Nasri, S.; Darvishzadeh, F.; Naderi, R. Ginger Extract Reduces Chronic Morphine-Induced Neuroinflammation and Glial Activation in Nucleus Accumbens of Rats. Addict. Health 2019, 11, 66–72. [Google Scholar] [CrossRef]
- Rose, C.F.; Verkhratsky, A.; Parpura, V. Astrocyte glutamine synthetase: Pivotal in health and disease. Biochem. Soc. Trans. 2013, 41, 1518–1524. [Google Scholar] [CrossRef]
- Wu, G.J.; Wen, Z.H.; Chen, W.F.; Chang, Y.C.; Cherng, C.H.; Wong, C.S. The effect of dexamethasone on spinal glutamine synthetase and glutamate dehydrogenase expression in morphine-tolerant rats. Anesth. Analg. 2007, 104, 726–730. [Google Scholar] [CrossRef]
- Bodzon-Kulakowska, A.; Suder, P.; Drabik, A.; Kotlinska, J.H.; Silberring, J. Constant activity of glutamine synthetase after morphine administration versus proteomic results. Anal. Bioanal. Chem. 2010, 398, 2939–2942. [Google Scholar] [CrossRef] [Green Version]
- Muscoli, C.; Cuzzocrea, S.; Ndengele, M.M.; Mollace, V.; Porreca, F.; Fabrizi, F.; Esposito, E.; Masini, E.; Matuschak, G.M.; Salvemini, D. Therapeutic manipulation of peroxynitrite attenuates the development of opiate-induced antinociceptive tolerance in mice. J. Clin. Investig. 2007, 117, 3530–3539. [Google Scholar] [CrossRef]
- Chetan, P.S.; Sangeetha, R.; Mohan, P.M.; Rajendra, W. Alterations in glutamate metabolism in rat brain by tramadol analgesia during non-induction of pain. Saudi J. Med. Pharm. Sci. 2015, 1, 26–36. [Google Scholar]
- Kozlova, M.; Kentroti, S.; Vernadakis, A. Maintenance of glial plasticity with aging in C-6 glial cells and normal astrocytes in culture: Responsiveness to opioid peptides. J. Neurosci. Res. 1993, 36, 570–579. [Google Scholar] [CrossRef]
- Tsai, R.Y.; Jang, F.L.; Tai, Y.H.; Lin, S.L.; Shen, C.H.; Wong, C.S. Ultra-low-dose naloxone restores the antinociceptive effect of morphine and suppresses spinal neuroinflammation in PTX-treated rats. Neuropsychopharmacology 2008, 33, 2772–2782. [Google Scholar] [CrossRef] [PubMed]
- Amri, J.; Sadegh, M.; Moulaei, N.; Palizvan, M.R. Transgenerational modification of hippocampus TNF-alpha and S100B levels in the offspring of rats chronically exposed to morphine during adolescence. Am. J. Drug Alcohol Abuse 2018, 44, 95–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suárez, P.M.; Santotoribio, J.D.; Ramos, V.R.; García, M.A.G.; Ramos, S.P.; Huertas, D.P.; Hoyos, A.M. Brain damage after general anesthesia. Med. Clin. 2016, 146, 384–388. [Google Scholar] [CrossRef]
- Zhi, X.L.; Li, C.Y.; Xue, M.; Hu, Y.; Ji, Y. Changes in cognitive function due to combined propofol and remifentanil treatment are associated with phosphorylation of Tau in the hippocampus, abnormal total water and calcium contents of the brain, and elevated serum S100beta levels. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 2156–2162. [Google Scholar] [PubMed]
- Kuklin, V.; Akhatov, N.; Kondratiev, T.; Konkayev, A.; Baigenzhin, A.; Konkayeva, M.; Karibekov, T.; Barlow, N.; Tveita, T.; Dahl, V. The influences of morphine or ketamine pre-treatment on hemodynamic, acid-base status, biochemical markers of brain damage and early survival in rats after asphyxial cardiac arrest. BMC Anesthesiol. 2019, 19, 214. [Google Scholar] [CrossRef] [Green Version]
- Marie-Claire, C.; Courtin, C.; Roques, B.P.; Noble, F. Cytoskeletal genes regulation by chronic morphine treatment in rat striatum. Neuropsychopharmacology 2004, 29, 2208–2215. [Google Scholar] [CrossRef] [Green Version]
- Song, P.; Zhao, Z.Q. The involvement of glial cells in the development of morphine tolerance. Neurosci. Res. 2001, 39, 281–286. [Google Scholar] [CrossRef]
- Clarkson, J.E.; Lacy, J.M.; Fligner, C.L.; Thiersch, N.; Howard, J.; Harruff, R.C.; Logan, B.K. Tramadol (Ultram) concentrations in death investigation and impaired driving cases and their significance. J. Forensic Sci. 2004, 49, 1101–1105. [Google Scholar] [CrossRef]
- Kronstrand, R.; Roman, M.; Thelander, G.; Eriksson, A. Unintentional fatal intoxications with mitragynine and O-desmethyltramadol from the herbal blend Krypton. J. Anal. Toxicol. 2011, 35, 242–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milroy, C.M.; Parai, J.L. The histopathology of drugs of abuse. Histopathology 2011, 59, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Dettmeyer, R.; Friedrich, K.; Schmidt, P.; Madea, B. Heroin-associated myocardial damages--conventional and immunohistochemical investigations. Forensic Sci. Int. 2009, 187, 42–46. [Google Scholar] [CrossRef]
- Najafipour, H.; Joukar, S.; Malekpour-Afshar, R.; Mirzaeipour, F.; Nasri, H.R. Passive opium smoking does not have beneficial effect on plasma lipids and cardiovascular indices in hypercholesterolemic rabbits with ischemic and non-ischemic hearts. J. Ethnopharmacol. 2010, 127, 257–263. [Google Scholar] [CrossRef]
- El Fatoh, M.F.; Farag, M.R.; Sayed, S.A.E.; Kamel, M.A.; Abdel-Hamid, N.E.; Hussein, M.A.; Salem, G.A. Some biochemical, neurochemical, pharmacotoxicological and histopathological alterations induced by long-term administration of tramadol in male rats. Int. J. Pharm. Sci. 2014, 4, 565–571. [Google Scholar]
- Baghishani, F.; Mohammadipour, A.; Hosseinzadeh, H.; Hosseini, M.; Ebrahimzadeh-Bideskan, A. The effects of tramadol administration on hippocampal cell apoptosis, learning and memory in adult rats and neuroprotective effects of crocin. Metab. Brain Dis. 2018, 33, 907–916. [Google Scholar] [CrossRef] [Green Version]
- Ragab, I.K.; Mohamed, H.Z.E. Histological changes of the adult albino rats entorhinal cortex under the effect of tramadol administration: Histological and morphometric study. Alexandria J. Med. 2017, 53, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.; McNeill, J.H. To scale or not to scale: The principles of dose extrapolation. Br. J. Pharmacol. 2009, 157, 907–921. [Google Scholar] [CrossRef] [Green Version]
- Buege, J.A.; Aust, S.D. Microsomal lipid peroxidation. Methods Enzymol. 1978, 52, 302–310. [Google Scholar] [CrossRef]
- Levine, R.L.; Williams, J.A.; Stadtman, E.R.; Shacter, E. Carbonyl assays for determination of oxidatively modified proteins. Methods Enzymol. 1994, 233, 346–357. [Google Scholar] [CrossRef]
- Costa, I.; Carvalho, F.; Magalhaes, T.; Guedes de Pinho, P.; Silvestre, R.; Dinis-Oliveira, R.J. Promising blood-derived biomarkers for estimation of the postmortem interval. Toxicol. Res. 2015, 4, 1443–1452. [Google Scholar] [CrossRef] [Green Version]
- Xiao, C.; Li, S.; Zhou, W.; Shang, D.; Zhao, S.; Zhu, X.; Chen, K.; Wang, R. The effect of air pollutants on the microecology of the respiratory tract of rats. Environ. Toxicol. Pharmacol. 2013, 36, 588–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; He, T.; Xing, Y.; Cao, T. Effects of quercetin on the expression of MCP-1, MMP-9 and VEGF in rats with diabetic retinopathy. Exp. Ther. Med. 2017, 14, 6022–6026. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, Y.; Maruta, R.; Takaki, K.; Kotani, E.; Kato, Y.; Yoshimura, R.; Endo, Y.; Whitty, C.; Pernstich, C.; Gandhi, R.; et al. Sustained Neurotrophin Release from Protein Nanoparticles Mediated by Matrix Metalloproteinases Induces the Alignment and Differentiation of Nerve Cells. Biomolecules 2019, 9, 510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ter Horst, S.A.; Fijlstra, M.; Sengupta, S.; Walther, F.J.; Wagenaar, G.T. Spatial and temporal expression of surfactant proteins in hyperoxia-induced neonatal rat lung injury. BMC Pulm Med. 2006, 6, 8. [Google Scholar] [CrossRef] [Green Version]
- Peinnequin, A.; Mouret, C.; Birot, O.; Alonso, A.; Mathieu, J.; Clarencon, D.; Agay, D.; Chancerelle, Y.; Multon, E. Rat pro-inflammatory cytokine and cytokine related mRNA quantification by real-time polymerase chain reaction using SYBR green. BMC Immunol. 2004, 5, 3. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.H.; Zhao, H.; Tian, Y.H.; Li, H.G.; Ding, X.F.; Xiong, C.L. Gene expression changes of urokinase plasminogen activator and urokinase receptor in rat testes at postnatal stages. Asian J. Androl. 2007, 9, 679–683. [Google Scholar] [CrossRef]
- Shynlova, O.; Tsui, P.; Dorogin, A.; Langille, B.L.; Lye, S.J. The expression of transforming growth factor beta in pregnant rat myometrium is hormone and stretch dependent. Reproduction 2007, 134, 503–511. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Yi, F.; Xia, M.; Boini, K.M.; Zhu, Q.; Laperle, L.A.; Abais, J.M.; Brimson, C.A.; Li, P.L. NMDA receptor-mediated activation of NADPH oxidase and glomerulosclerosis in hyperhomocysteinemic rats. Antioxid. Redox Signal. 2010, 13, 975–986. [Google Scholar] [CrossRef] [Green Version]
- Mishra, N.; Kumar, P.; Singh, R.; Sharma, D. Response of α-Synuclein Expression to Amyloid β40 and Amyloid β42 administration into Rat Brain. J. Alzheimers Dis. Parkinsonism 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Baj, G.; Del Turco, D.; Schlaudraff, J.; Torelli, L.; Deller, T.; Tongiorgi, E. Regulation of the spatial code for BDNF mRNA isoforms in the rat hippocampus following pilocarpine-treatment: A systematic analysis using laser microdissection and quantitative real-time PCR. Hippocampus 2013, 23, 413–423. [Google Scholar] [CrossRef]
- Ishikawa, M.; Yoshitomi, T.; Zorumski, C.F.; Izumi, Y. Downregulation of glutamine synthetase via GLAST suppression induces retinal axonal swelling in a rat ex vivo hydrostatic pressure model. Investig. Ophthalmol. Vis. Sci. 2011, 52, 6604–6616. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, G.Q.; Qu, J.; Che, C.Y.; Lin, J.; Jiang, N.; Zhao, H.; Wang, X.J. Expression of S100B during the innate immune of corneal epithelium against fungi invasion. Int. J. Ophthalmol. 2016, 9, 191–197. [Google Scholar] [CrossRef]
- Che, R.; Zhu, C.; Ding, G.; Zhao, M.; Bai, M.; Jia, Z.; Zhang, A.; Huang, S. Huaier Cream Protects against Adriamycin-Induced Nephropathy by Restoring Mitochondrial Function via PGC-1alpha Upregulation. PPAR Res. 2015, 2015, 720383. [Google Scholar] [CrossRef]
- Dinis-Oliveira, R.J.; Duarte, J.A.; Remiao, F.; Sanchez-Navarro, A.; Bastos, M.L.; Carvalho, F. Single high dose dexamethasone treatment decreases the pathological score and increases the survival rate of paraquat-intoxicated rats. Toxicology 2006, 227, 73–85. [Google Scholar] [CrossRef]
- Dinis-Oliveira, R.J.; Remiao, F.; Duarte, J.A.; Ferreira, R.; Sanchez Navarro, A.; Bastos, M.L.; Carvalho, F. P-glycoprotein induction: An antidotal pathway for paraquat-induced lung toxicity. Free Radic. Biol. Med. 2006, 41, 1213–1224. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5′→3′) | Reverse Primer (5′→3′) | Annealing Temperature (°C) | No. of Amplification Cycles | Reference |
|---|---|---|---|---|---|
| CC16 (Clara cell protein-16) | CATCAGCCCACATCTACAGAC | GGGCTTTAGCGTAGAATATCT | 55 | 35 | [173] |
| MCP-1 (Monocyte chemoattractant protein-1) | CCCACTCACCTGCTGCTACTC | AGAAGTGCTTGAGGTGGTTGTG | 55 | 40 | [174] |
| MMP-7 (Matrix metalloproteinase-7) | TCGGCGGAGATGCTCACT | TGGCAACAAACAGGAAGTTCAC | 50 | 40 | [175] |
| SP-A (Pulmonary surfactant protein A) | TACCAGAGCAGGAGGCAACA | CAATACTTGCAATGGCCTCGTT | 55 | 35 | [176] |
| SP-D (Pulmonary surfactant protein D) | AAATCTTCAGGGCGGCAAA | GGCCTGCCTGCACATCTC | 55 | 40 | [176] |
| IL-6 (Interleukin-6) | TCCTACCCCAACTTCCAATGCTC | TTGGATGGTCTTGGTCCTTAGCC | 55 | 40 | [177] |
| Plau/UPA (Plasminogen activator, urokinase) | TCACTGGCTTCGGACAAGAGA | CCAATGTGGGACTGAATCCAG | 55 | 40 | [178] |
| TGF-β2 (Transforming growth factor-β2) | TTCAGAATCGTCCGCTTCGAT | TTGTTCAGCCACTCTGGCCTT | 50 | 41 | [179] |
| TIMP-1 (Tissue inhibitor of metalloproteinase-1) | TCTGGCATCCTCTTGTTGCTAT | CCACAGCGTCGAATCCTT | 50 | 41 | [180] |
| SNCA (α-Synuclein) | TGCTGTGGATATTGTTGTGG | AGGTGCGTAGTCTCATGCTC | 55 | 35 | [181] |
| BDNF (Brain-derived neurotrophic factor) | AAACGTCCACGGACAAGGCA | TTCTGGTCCTCATCCAGCAGC | 55 | 37 | [182] |
| GS (Glutamine synthetase) | CCACTGTCCCTGGGCTTAGTTTA | AGTGACATGCTAGTCCCACCAA | 55 | 37 | [183] |
| S100β (S100 calcium binding protein B) | GGGTGACAAGCACAAGCTGAA | AGCGTCTCCATCACTTTGTCCA | 55 | 35 | [184] |
| 18S rRNA (18S ribosomal RNA) | TTCGGAACTGAGGCCATGATT | TTTCGCTCTGGTCCGTCTTG | In line with that of the target gene | [185] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbosa, J.; Faria, J.; Garcez, F.; Leal, S.; Afonso, L.P.; Nascimento, A.V.; Moreira, R.; Pereira, F.C.; Queirós, O.; Carvalho, F.; et al. Repeated Administration of Clinically Relevant Doses of the Prescription Opioids Tramadol and Tapentadol Causes Lung, Cardiac, and Brain Toxicity in Wistar Rats. Pharmaceuticals 2021, 14, 97. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14020097
Barbosa J, Faria J, Garcez F, Leal S, Afonso LP, Nascimento AV, Moreira R, Pereira FC, Queirós O, Carvalho F, et al. Repeated Administration of Clinically Relevant Doses of the Prescription Opioids Tramadol and Tapentadol Causes Lung, Cardiac, and Brain Toxicity in Wistar Rats. Pharmaceuticals. 2021; 14(2):97. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14020097
Chicago/Turabian StyleBarbosa, Joana, Juliana Faria, Fernanda Garcez, Sandra Leal, Luís Pedro Afonso, Ana Vanessa Nascimento, Roxana Moreira, Frederico C. Pereira, Odília Queirós, Félix Carvalho, and et al. 2021. "Repeated Administration of Clinically Relevant Doses of the Prescription Opioids Tramadol and Tapentadol Causes Lung, Cardiac, and Brain Toxicity in Wistar Rats" Pharmaceuticals 14, no. 2: 97. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14020097