
Deciphering the H-Bonding Preference on Nucleoside Molecular Recognition through Model Copper(II) Compounds

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
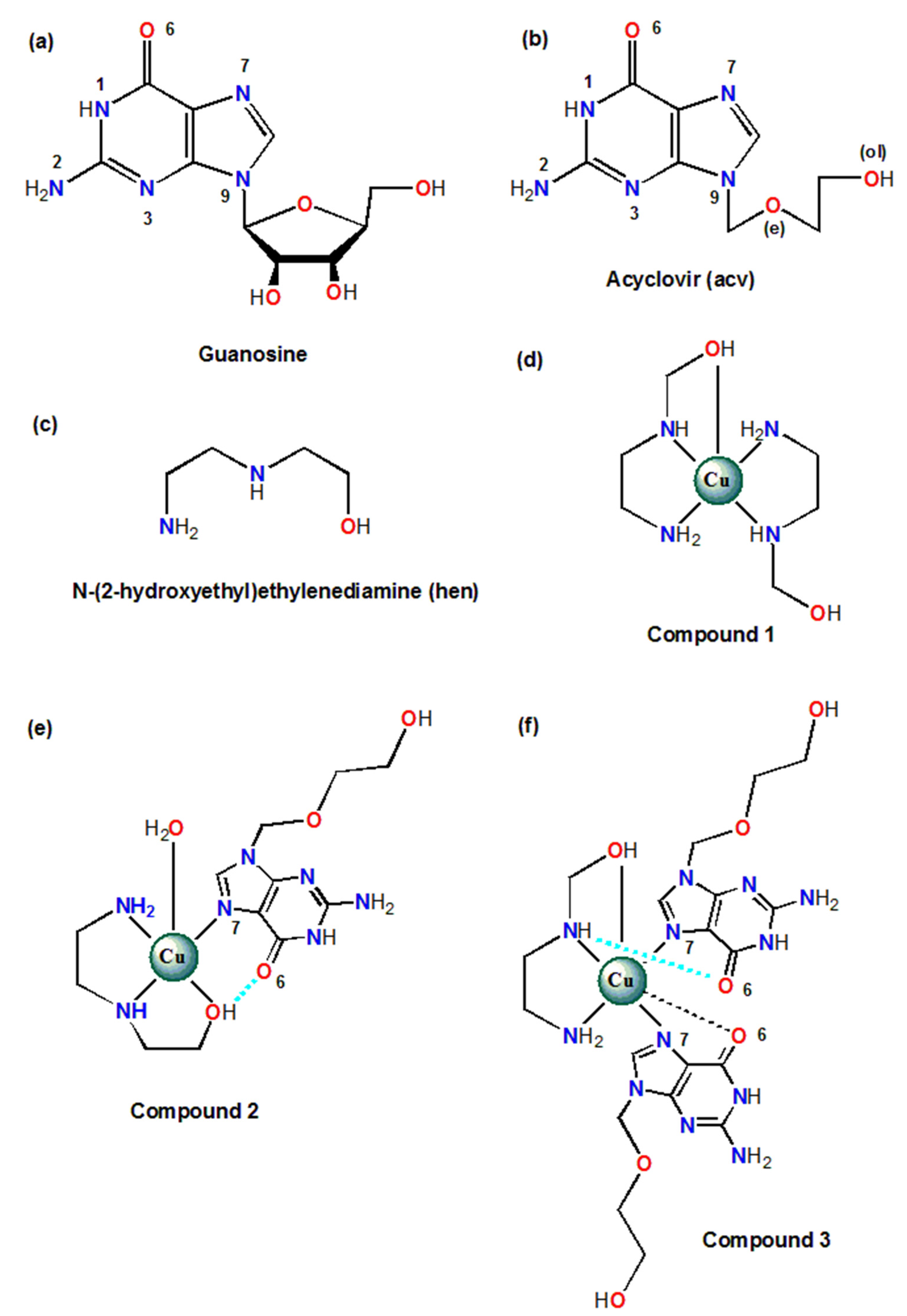
2.1. Analysis of the Structural and Theoretical Studies of Compound [Cu(hen)2]SO4 (1)
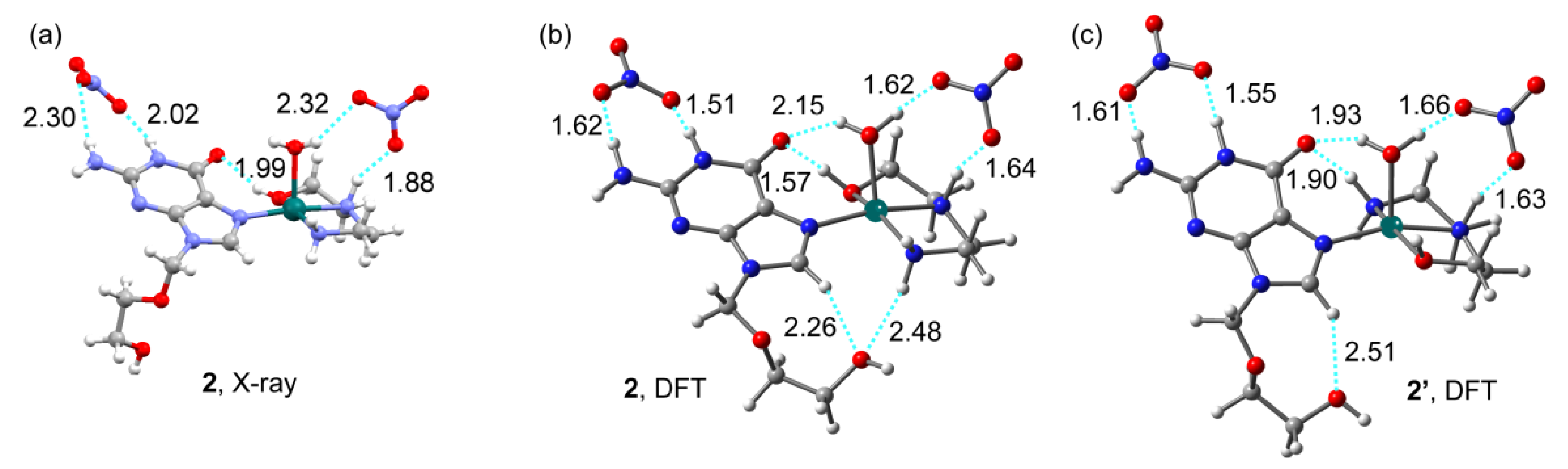
2.2. Analysis of the Structural and Theoretical Studies of Compound [Cu(hen)(acv)(H2O)](NO3)2 (2)
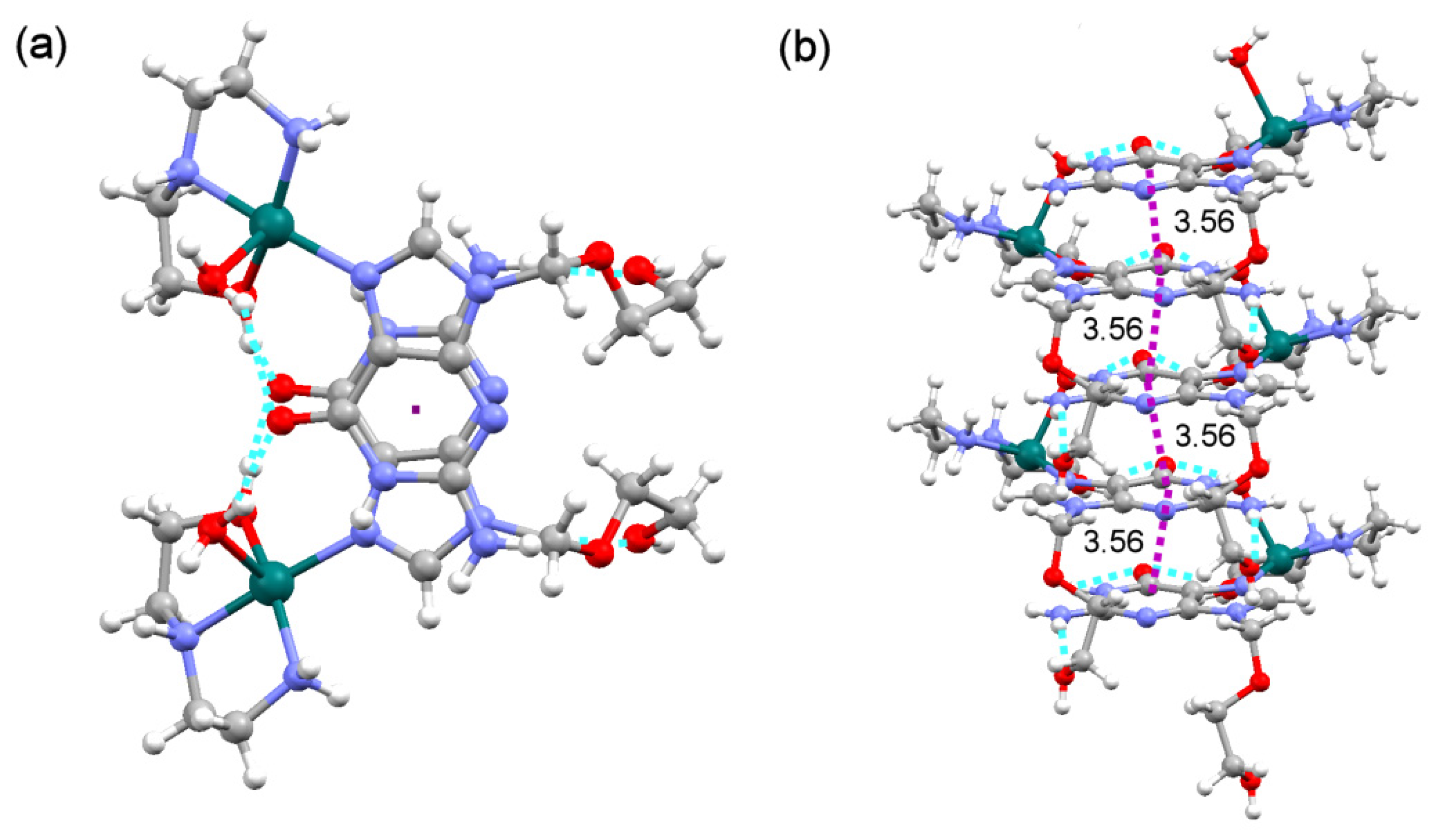
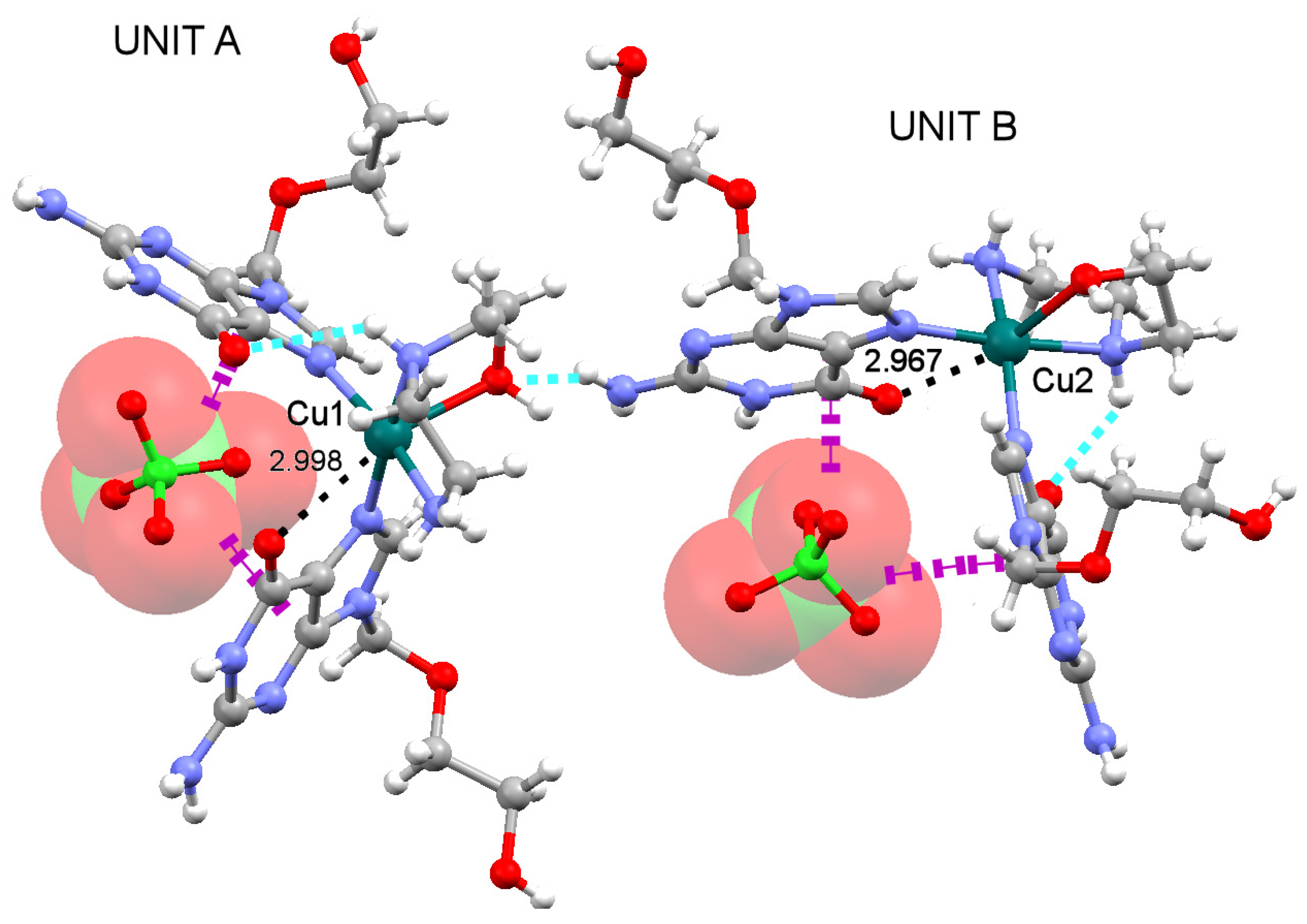
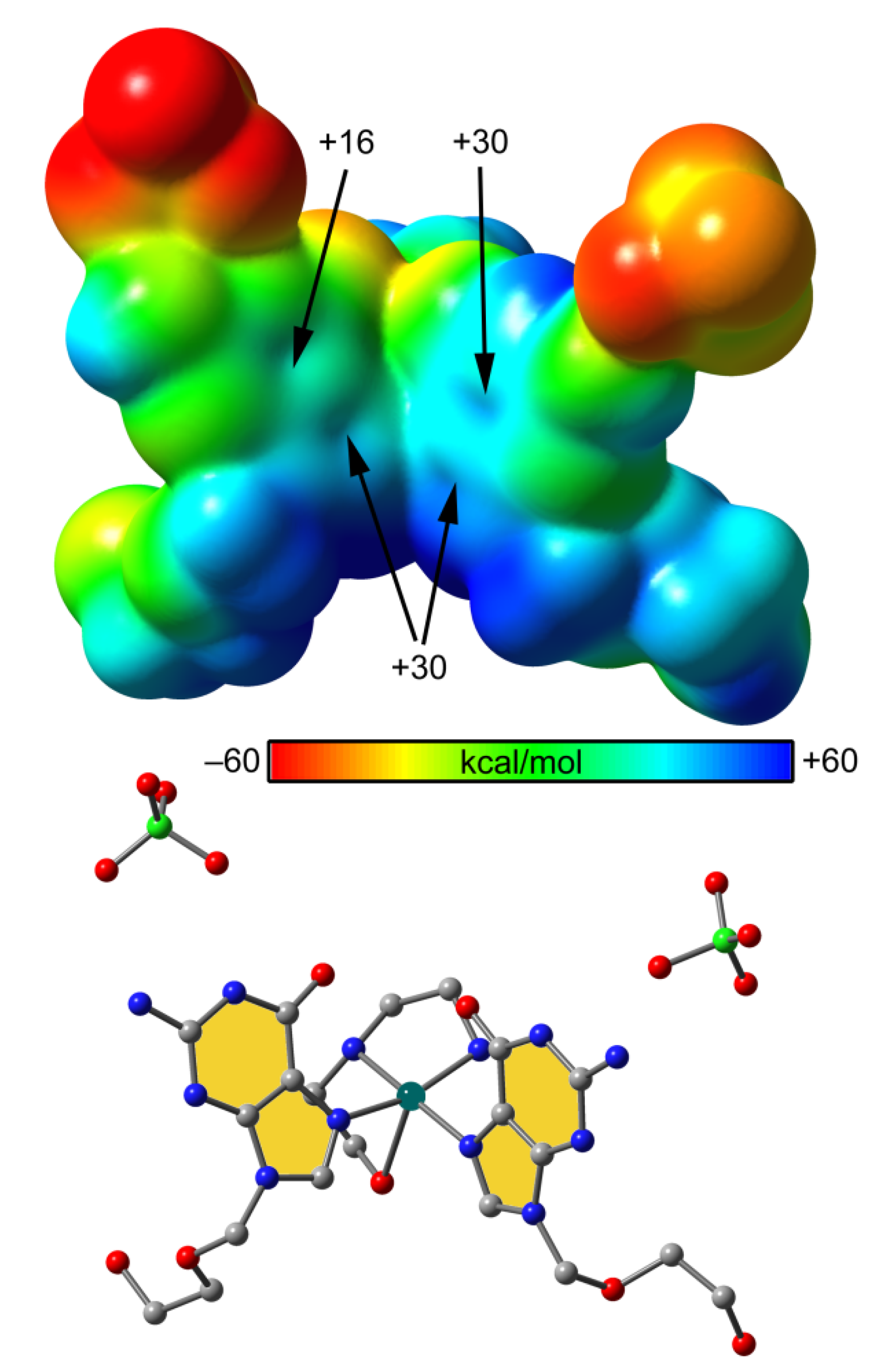
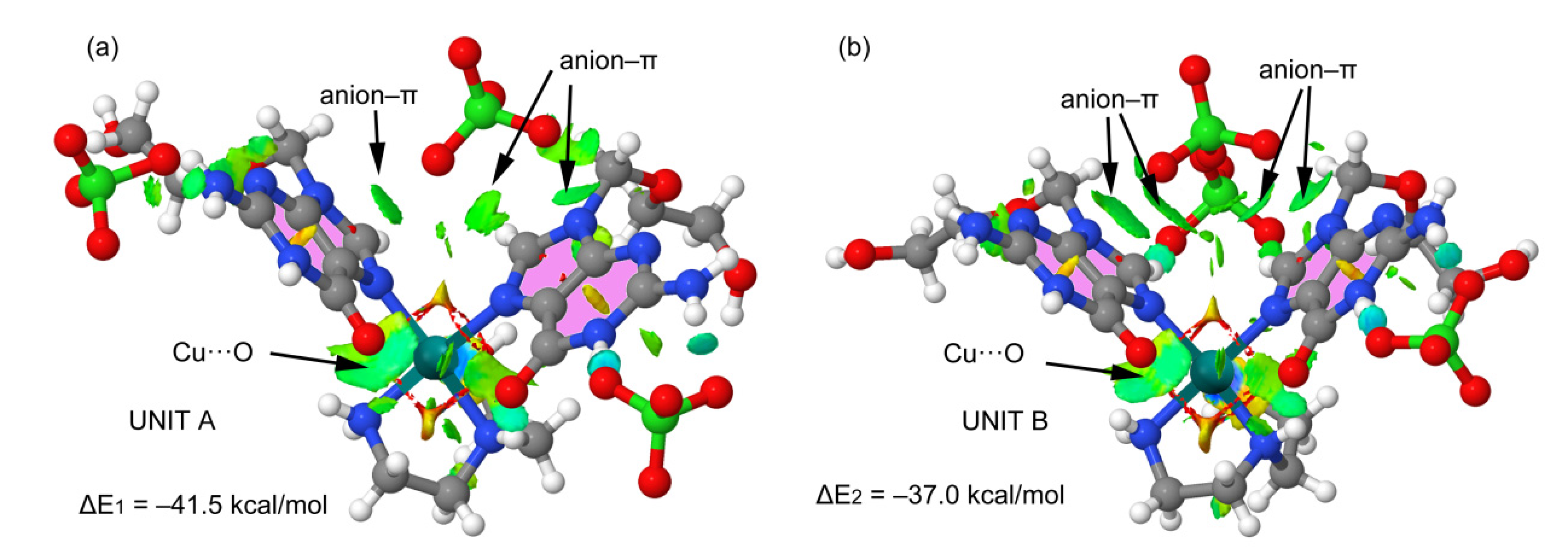
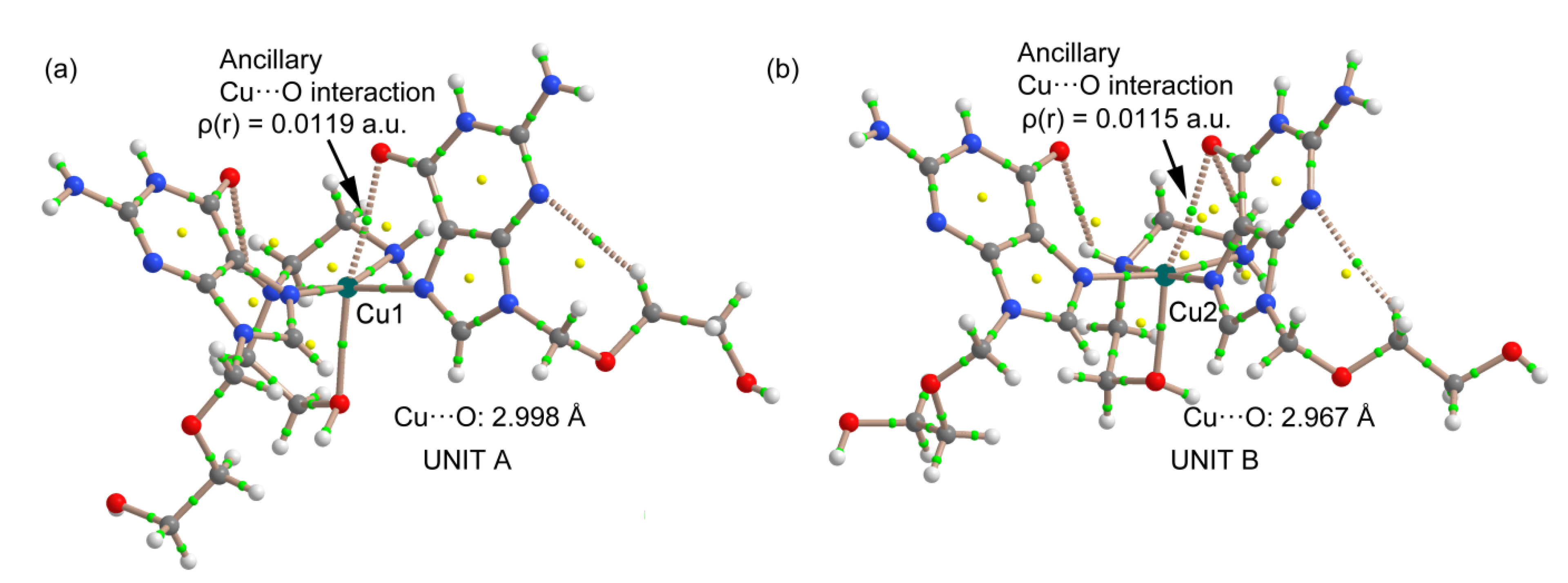
2.3. Analysis of the Structural and Theoretical Studies of Compound [Cu(hen)(acv)2](ClO4)2 (3)
3. Materials and Methods
3.1. Reagents
3.2. Synthesis
3.2.1. [Cu(hen)2]SO4 (1)
3.2.2. [Cu(hen)(acv)(H2O)](NO3)2 (2)
3.2.3. [Cu(hen)(acv)2](ClO4)2 (3)
3.3. Single-Crystal X-ray Diffraction
3.4. Other Physical Measurements
3.5. Theoretical Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kandimalla, E.R.; Bhagat, L.; Wang, D.; Yu, D.; Zhu, F.; Tang, J.; Wang, H.; Huang, P.; Zhang, R.; Agrawal, S. Divergent synthetic nucleotide motif recognition pattern: Design and development of potent immunomodulatory oligodeoxyribonucleotide agents with distinct cytokine induction profiles. Nucleic Acids Res. 2003, 31, 2393–2400. [Google Scholar] [CrossRef] [Green Version]
- Jordheim, L.P.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 2013, 12, 447. [Google Scholar] [CrossRef] [PubMed]
- Abraham, J.N.; Kedracki, D.; Prado, E.; Gourmel, C.; Maroni, P.; Nardin, C. Effect of the Interaction of the Amyloid β (1–42) Peptide with Short Single-Stranded Synthetic Nucleotide Sequences: Morphological Characterization of the Inhibition of Fibrils Formation and Fibrils Disassembly. Biomacromolecules 2014, 15, 3253–3258. [Google Scholar] [CrossRef]
- Kent, T.; Rusanov, T.D.; Hoang, T.M.; Velema, W.A.; Krueger, A.T.; Copeland, W.C.; Kool, E.T.; Pomerantz, R.T. DNA polymerase θ specializes in incorporating synthetic expanded-size (xDNA) nucleotides. Nucleic Acids Res. 2016, 44, 9381–9392. [Google Scholar] [CrossRef] [Green Version]
- Blindauer, C.A.; Holý, A.; Operschall, B.P.; Sigel, A.; Song, B.; Sigel, H. Metal Ion-Coordinating Properties in Aqueous Solutions of the Antivirally Active Nucleotide Analogue (S)-9-[3-Hydroxy-2-(phosphonomethoxy)propyl]adenine (HPMPA)—Quantification of Complex Isomeric Equilibria. Eur. J. Inorg. Chem. 2019, 2019, 3892–3903. [Google Scholar] [CrossRef]
- De Clercq, E.; Li, G. Approved Antiviral Drugs over the Past 50 Years. Clin. Microbiol. Rev. 2016, 29, 695–747. [Google Scholar] [CrossRef] [Green Version]
- Elion, G.B. The biochemistry and mechanism of action of acyclovir. J. Antimicrob. Chemother. 1983, 12, 9–17. [Google Scholar] [CrossRef]
- Blažič, B.; Turel, I.; Bukovec, N.; Bukovec, P.; Lazarini, F. Synthesis and structure of diaquadichlorobis {9-[(2-hydroxyethoxy)methyl]guanine} copper(II). J. Inorg. Biochem. 1993, 51, 737–744. [Google Scholar] [CrossRef]
- Sinur, A.; Grabner, S. A Platinum(II) Diammine Complex: Cis-[Pt(C8H11N5O3)2(NH3)2]Cl2.2H2O. Acta Crystallogr. Sect. C 1995, 51, 1769–1772. [Google Scholar] [CrossRef] [Green Version]
- Turel, I.; Bukovec, N.; Goodgame, M.; Williams, D.J. Synthesis and characterization of copper(II) coordination compounds with acyclovir: Crystal structure of triaquabis [9-{(2-hydroxyethoxy)methyl>guanine] copper(II) nitrate (V) hydrate. Polyhedron 1997, 16, 1701–1706. [Google Scholar] [CrossRef]
- Grabner, S.; Plavec, J.; Bukovec, N.; Di Leo, D.; Cini, R.; Natile, G. Synthesis and structural characterization of platinum(II)-acyclovir complexes. J. Chem. Soc. Dalt. Trans. 1998, 1447–1452. [Google Scholar] [CrossRef]
- Turel, I.; Anderson, B.; Sletten, E.; White, A.J.P.; Williams, D.J. New studies in the copper(II) acyclovir (acv) system. NMR relaxation studies and the X-ray crystal structure of [Cu(acv)2(H2O)2](NO3)2. Polyhedron 1998, 17, 4195–4201. [Google Scholar] [CrossRef]
- García-Raso, Á.J.; Fiol, J.; Bádenas, F.; Cons, R.; Terrón, Á.; Quirós, M. Synthesis and structural characteristics of metal–acyclovir (ACV) complexes: [Ni(or Co)(ACV)2(H2O)4]Cl2·2ACV, [Zn(ACV)Cl2(H2O)], [Cd(ACV)Cl2]·H2O and [{Hg(ACV)Cl2}]. Recognition of acyclovir by Ni–ACV. J. Chem. Soc. Dalt. Trans. 1999, 167–174. [Google Scholar] [CrossRef]
- Cini, R.; Grabner, S.; Bukovec, N.; Cerasino, L.; Natile, G. Synthesis and Structural Characterisation of a New Form of Bis(acyclovir)(ethylenediamine)platinum(II)—Correlation between the Puckering of the Carrier Ligand and the Canting of the Nucleobases. Eur. J. Inorg. Chem. 2000, 2000, 1601–1607. [Google Scholar] [CrossRef]
- Turel, I.; Pečanac, M.; Golobič, A.; Alessio, E.; Serli, B. Novel RuIII-DMSO Complexes of the Antiherpes Drug Acyclovir. Eur. J. Inorg. Chem. 2002, 2002, 1928–1931. [Google Scholar] [CrossRef]
- Barceló-Oliver, M.; Terrón, A.; García-Raso, A.; Fiol, J.J.; Molins, E.; Miravitlles, C. Ternary complexes metal [Co(II), Ni(II), Cu(II) and Zn(II)]—ortho-iodohippurate (I-hip)—acyclovir. X-ray characterization of isostructural [(Co, Ni or Zn)(I-hip)2(ACV)(H2O)3] with stacking as a recognition factor. J. Inorg. Biochem. 2004, 98, 1703–1711. [Google Scholar] [CrossRef] [PubMed]
- Turel, I.; Pečanac, M.; Golobič, A.; Alessio, E.; Serli, B.; Bergamo, A.; Sava, G. Solution, solid state and biological characterization of ruthenium(III)-DMSO complexes with purine base derivatives. J. Inorg. Biochem. 2004, 98, 393–401. [Google Scholar] [CrossRef]
- Del Brandi-Blanco, M.P.; Choquesillo-Lazarte, D.; Domínguez-Martín, A.; González-Pérez, J.M.; Castiñeiras, A.; Niclós-Gutiérrez, J. Metal ion binding patterns of acyclovir: Molecular recognition between this antiviral agent and copper(II) chelates with iminodiacetate or glycylglycinate. J. Inorg. Biochem. 2011, 105, 616–623. [Google Scholar] [CrossRef]
- Pérez-Toro, I.; Domínguez-Martín, A.; Choquesillo-Lazarte, D.; Vílchez-Rodríguez, E.; González-Pérez, J.M.; Castiñeiras, A.; Niclós-Gutiérrez, J. Lights and shadows in the challenge of binding acyclovir, a synthetic purine-like nucleoside with antiviral activity, at an apical–distal coordination site in copper(II)-polyamine chelates. J. Inorg. Biochem. 2015, 148, 84–92. [Google Scholar] [CrossRef]
- González-Pérez, J.M.; Choquesillo-Lazarte, D.; Domínguez-Martín, A.; Vílchez-Rodríguez, E.; Pérez-Toro, I.; Castiñeiras, A.; Arriortua, O.K.; García-Rubiño, M.E.; Matilla-Hernández, A.; Niclós-Gutiérrez, J. Metal binding pattern of acyclovir in ternary copper(II) complexes having an S-thioether or S-disulfide NO2S-tripodal tetradentate chelator. Inorganica Chim. Acta 2016, 452, 258–267. [Google Scholar] [CrossRef]
- Pérez-Toro, I.; Domínguez-Martín, A.; Choquesillo-Lazarte, D.; García-Rubiño, M.E.; González-Pérez, J.M.; Castiñeiras, A.; Bauzá, A.; Frontera, A.; Niclós-Gutiérrez, J. Copper(II) polyamine chelates as efficient receptors for acyclovir: Syntheses, crystal structures and dft study. Polyhedron 2018, 145, 218–226. [Google Scholar] [CrossRef]
- Pérez-Toro, I.; Domínguez-Martín, A.; Choquesillo-Lazarte, D.M.; González-Pérez, J.; Castiñeiras, A.; Niclós-Gutiérrez, J. Highest Reported Denticity of a Synthetic Nucleoside in the Unprecedented Tetradentate Mode of Acyclovir. Cryst. Growth Des. 2018, 18, 4282–4286. [Google Scholar] [CrossRef]
- Frieden, E. Non-covalent interactions: Key to biological flexibility and specificity. J. Chem. Educ. 1975, 52, 754. [Google Scholar] [CrossRef]
- Hathaway, B.J.; Billing, D.E. The electronic properties and stereochemistry of mono-nuclear complexes of the copper(II) ion. Coord. Chem. Rev. 1970, 5, 143–207. [Google Scholar] [CrossRef]
- Chastain, R.V.; Dominick, T.L. Crystal structure of bis[N-(2-hydroxyethyl)ethylenediamine]copper(II) perchlorate, [Cu(C4H12N2O)2](ClO4)2. Inorg. Chem. 1973, 12, 2621–2625. [Google Scholar] [CrossRef]
- Azadbakht, R.; Amiri Rudbari, H.; Bruno, G. Bis(2-{(2-aminoethyl)amino}ethan-1-ol)copper(II) dinitrate. Acta Crystallogr. Sect. E 2011, 67, m1203. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Sharma, R.P.; Venugopalan, P.; Aree, T. Effect of differently substituted methoxybenzoates on the supramolecular assemblies of three [Cu(N-hyden)2](o-/m-/p-methoxybenzoate)2 complexes: Synthesis, spectroscopic characterization and single crystal structure determination. Polyhedron 2017, 133, 213–221. [Google Scholar] [CrossRef]
- Kivelson, D.; Neiman, R. ESR Studies on the Bonding in Copper Complexes. J. Chem. Phys. 1961, 35, 149–155. [Google Scholar] [CrossRef]
- Crick, F.H.C.; Watson, J.D.; Bragg, W.L. The complementary structure of deoxyribonucleic acid. Proc. R. Soc. Lond. Ser. A Math. Phys. Sci. 1954, 223, 80–96. [Google Scholar] [CrossRef]
- Fichtinger-Schepman, A.M.J.; Veer, J.L.v.d.; Lohman, P.H.M.; Reedijk, J. A simple method for the inactivation of monofunctionally DNA-bound cis- diamminedichloroplatinum (II). J. Inorg. Biochem. 1984, 21, 103–111. [Google Scholar] [CrossRef]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.P.; Beratan, D.N.; Yang, W. NCIPLOT: A program for plotting noncovalent interaction regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Path, A.B. A Universal Indicator of Bonded Interactions. J. Phys. Chem. A 1998, 102, 7314–7323. [Google Scholar] [CrossRef]
- Bruker APEX3. APEX3 V2019.1, 2019.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—new features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian16 (Revision A.03); Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Richard, F.W.; Bader, A. Quantum Theory of Molecular Structure and Its Applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar]
- Keith, T.A. AIMALL (Version 19.10.12). In TK Gristmill Software; Overland Park, KS, USA, 2019. [Google Scholar]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Velo-Gala, I.; Barceló-Oliver, M.; Gil, D.M.; González-Pérez, J.M.; Castiñeiras, A.; Domínguez-Martín, A. Deciphering the H-Bonding Preference on Nucleoside Molecular Recognition through Model Copper(II) Compounds. Pharmaceuticals 2021, 14, 244. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14030244
Velo-Gala I, Barceló-Oliver M, Gil DM, González-Pérez JM, Castiñeiras A, Domínguez-Martín A. Deciphering the H-Bonding Preference on Nucleoside Molecular Recognition through Model Copper(II) Compounds. Pharmaceuticals. 2021; 14(3):244. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14030244
Chicago/Turabian StyleVelo-Gala, Inmaculada, Miquel Barceló-Oliver, Diego M. Gil, Josefa M. González-Pérez, Alfonso Castiñeiras, and Alicia Domínguez-Martín. 2021. "Deciphering the H-Bonding Preference on Nucleoside Molecular Recognition through Model Copper(II) Compounds" Pharmaceuticals 14, no. 3: 244. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14030244