
Exatecan Antibody Drug Conjugates Based on a Hydrophilic Polysarcosine Drug-Linker Platform

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
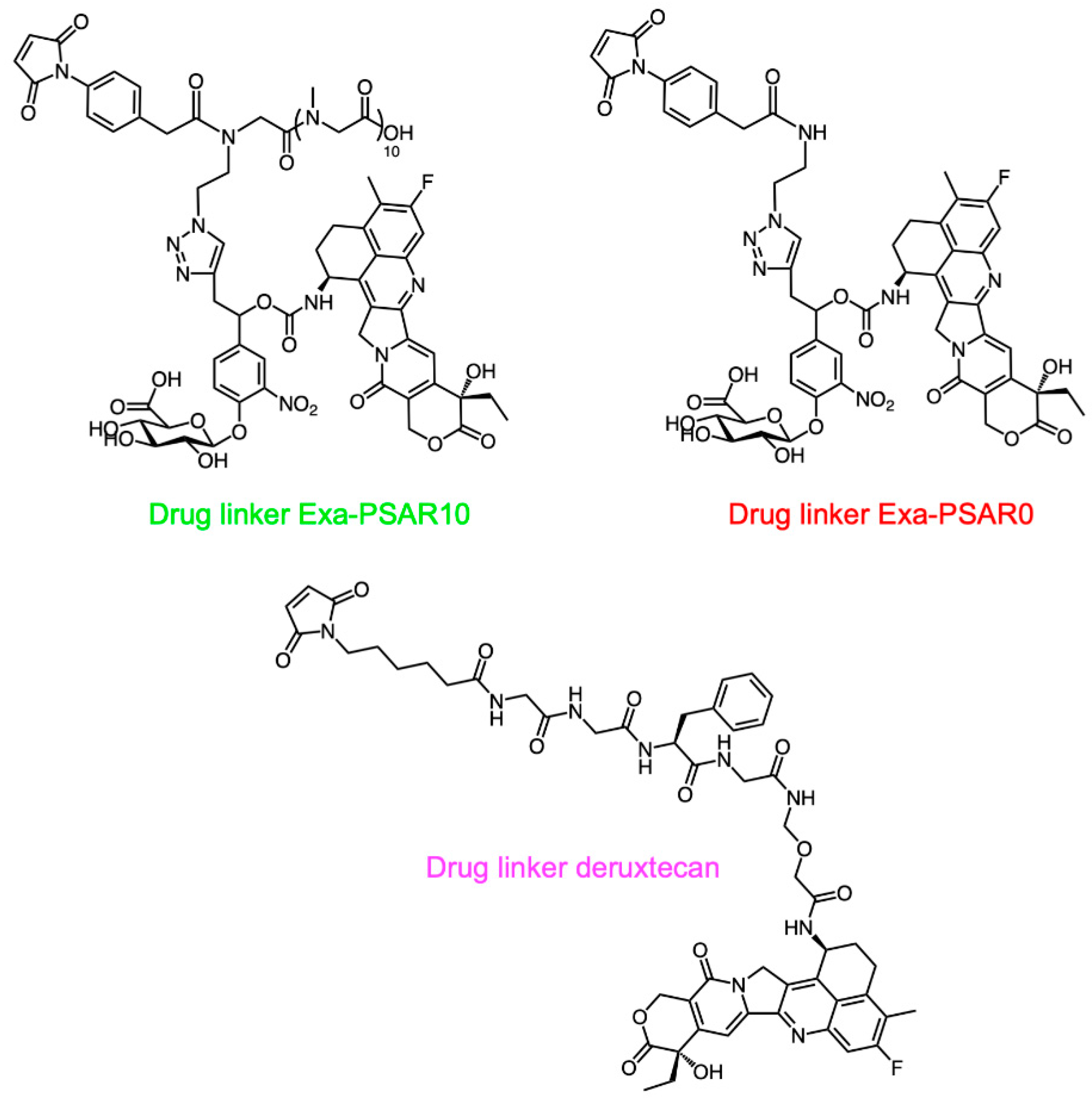
2.1. Drug-Linker Conception, Bioconjugation, and Physicochemical Characterization of ADCs
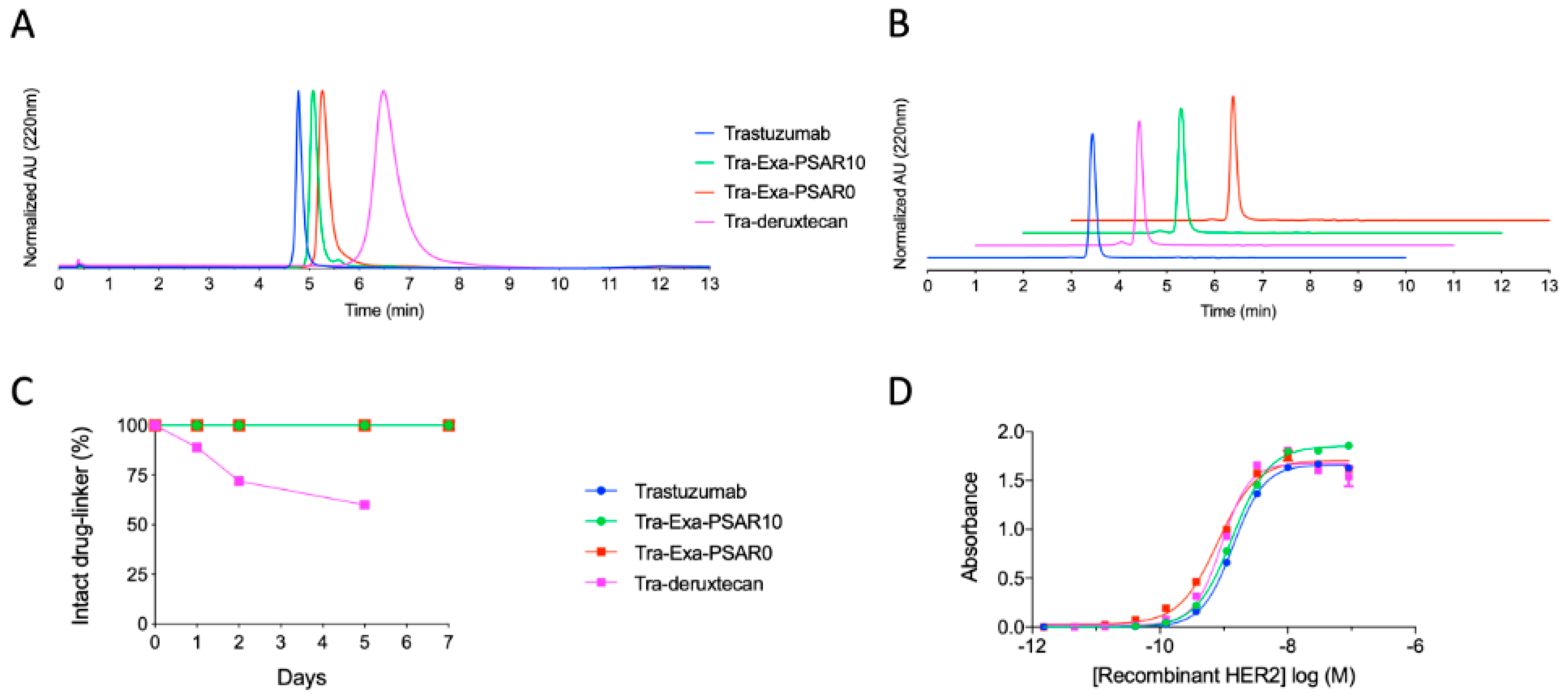
2.2. DAR8 Conjugation and Orthogonal Polysarcosine Inclusion Did Not Negatively Impact Binding to HER2
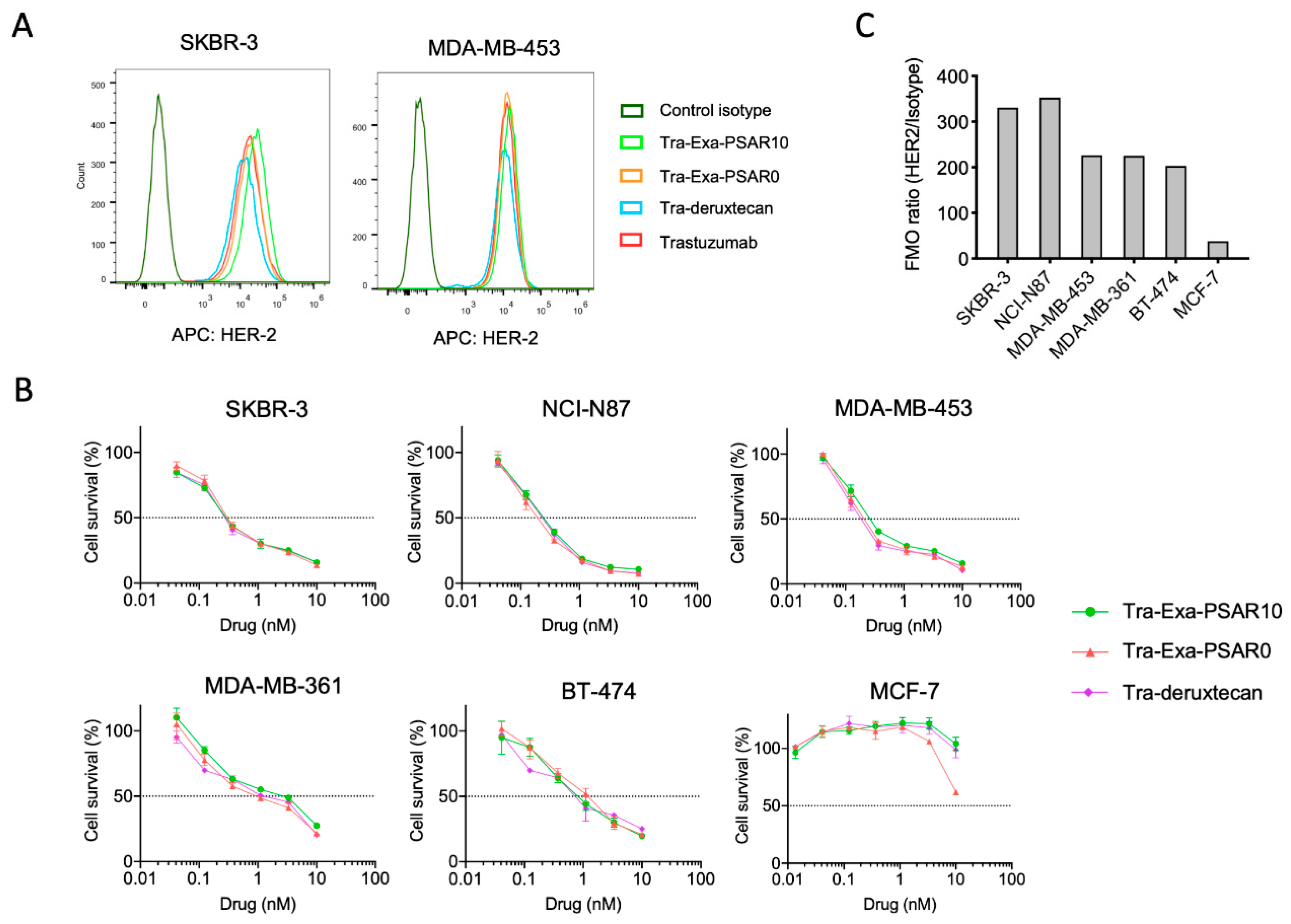
2.3. Tra-Exa-PSAR10 ADC Displayed Strong In Vitro Cytotoxicity in HER2-Positive Cell Lines
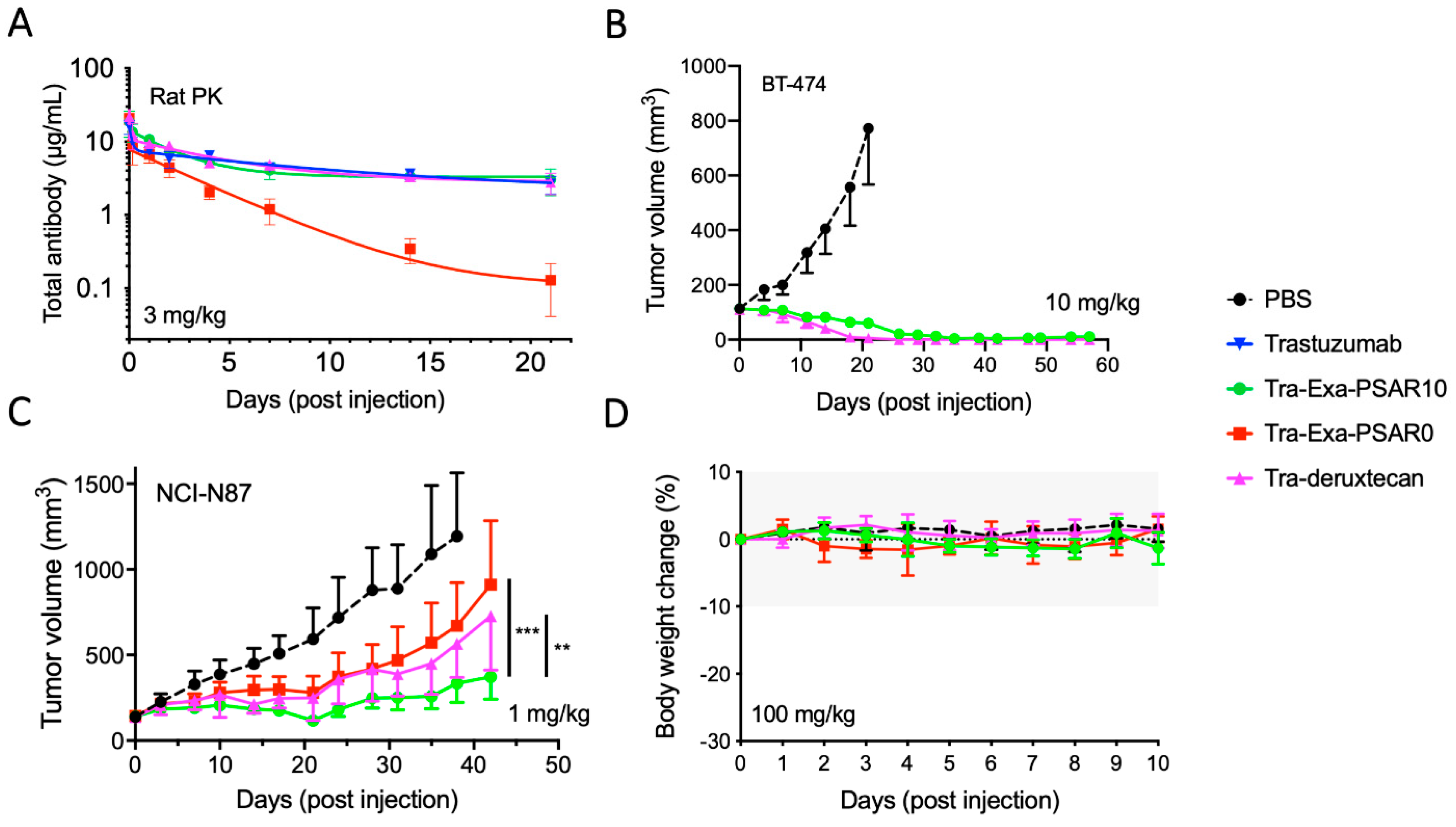
2.4. Tra-Exa-PSAR10 Showed a Favorable PK Profile and Strong In Vivo Activity in HER2+Breast and Gastric Cancer Models
2.5. Tra-Exa-PSAR10 Displayed a Strong Bystander Activity In Vitro
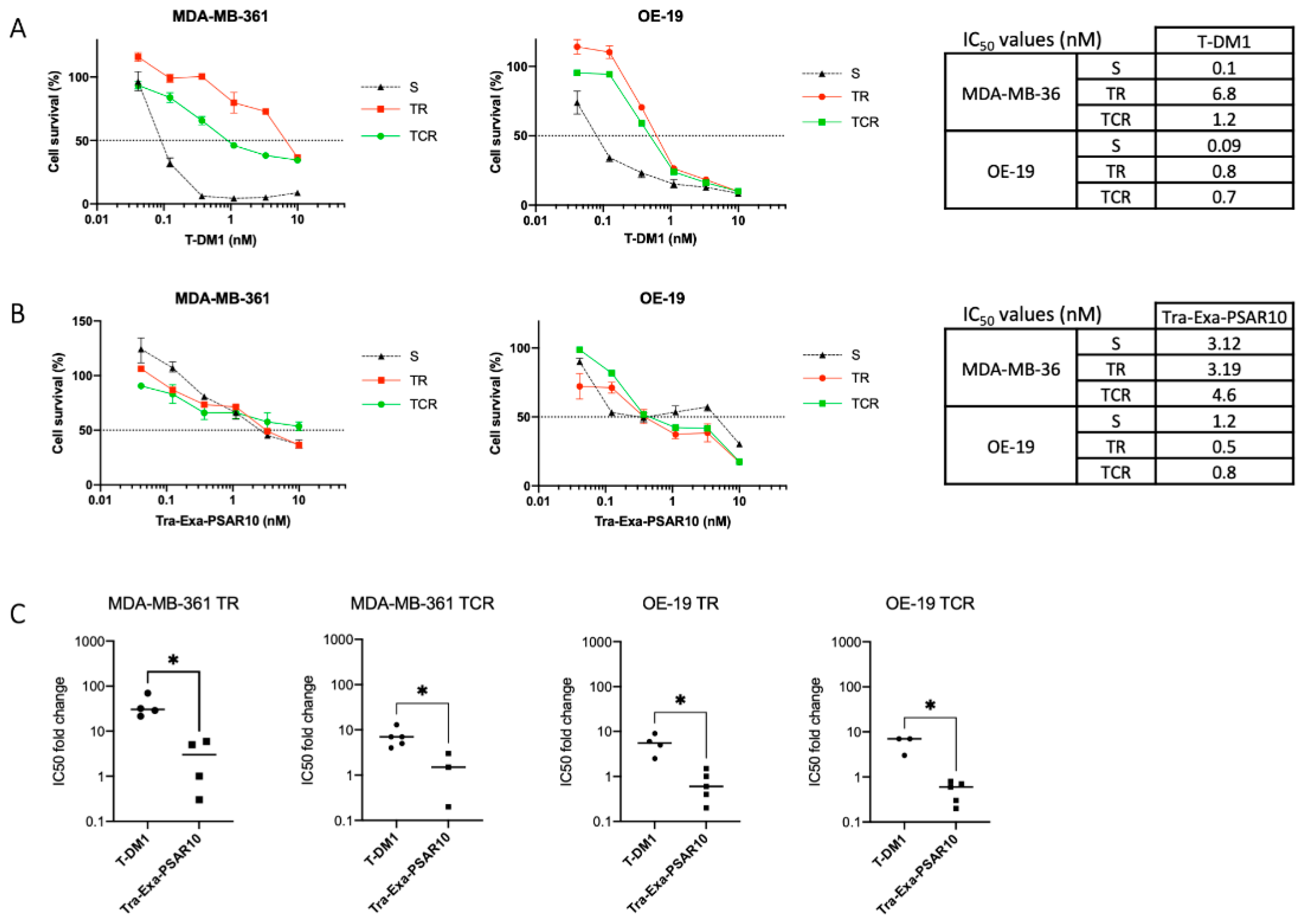
2.6. Tra-Exa-PSAR10 Overcame T-DM1 Resistance in Breast Cancer Models In Vitro
3. Discussion
4. Material and Methods
4.1. Drug-Linker Synthesis
4.2. Preparation of Antibody-Drug-Conjugates
4.3. Characterization of Antibody-Drug Conjugates
4.4. HER2-Binding ELISA Affinity Assay
4.5. Ex Vivo Plasma Stability Assays
4.6. Cell Culture
4.7. Flow Cytometry
4.8. In Vitro Cytotoxicity Assays
4.9. PAMPA Permeability Assay
4.10. In Vitro Bystander Killing Assay
4.11. In Vivo Studies
4.12. Rat PK Study
4.13. In Vivo Efficacy Experiments
4.14. Mice Tolerability Experiments
4.15. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and Challenges for the next Generation of Antibody-Drug Conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef]
- Strebhardt, K.; Ullrich, A. Paul Ehrlich’s Magic Bullet Concept: 100 Years of Progress. Nat. Rev. Cancer 2008, 8, 473–480. [Google Scholar] [CrossRef]
- Keam, S.J. Trastuzumab Deruxtecan: First Approval. Drugs 2020, 80, 501–508. [Google Scholar] [CrossRef]
- Deeks, E.D. Polatuzumab Vedotin: First Global Approval. Drugs 2019, 79, 1467–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, E.; Weinstock, C.; Zhang, L.; Charlab, R.; Dorff, S.E.; Gong, Y.; Hsu, V.; Li, F.; Ricks, T.K.; Song, P.; et al. FDA Approval Summary: Enfortumab Vedotin for Locally Advanced or Metastatic Urothelial Carcinoma. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Syed, Y.Y. Sacituzumab Govitecan: First Approval. Drugs 2020, 80, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Belantamab Mafodotin: First Approval. Drugs 2020, 80, 1607–1613. [Google Scholar] [CrossRef] [PubMed]
- Hafeez, U.; Parakh, S.; Gan, H.K.; Scott, A.M. Antibody-Drug Conjugates for Cancer Therapy. Molecules 2020, 25, 4764. [Google Scholar] [CrossRef] [PubMed]
- Joubert, N.; Beck, A.; Dumontet, C.; Denevault-Sabourin, C. Antibody-Drug Conjugates: The Last Decade. Pharmaceuticals 2020, 13, 245. [Google Scholar] [CrossRef]
- Buecheler, J.W.; Winzer, M.; Tonillo, J.; Weber, C.; Gieseler, H. Impact of Payload Hydrophobicity on the Stability of Antibody-Drug Conjugates. Mol. Pharm. 2018, 15, 2656–2664. [Google Scholar] [CrossRef]
- Ratanji, K.D.; Derrick, J.P.; Dearman, R.J.; Kimber, I. Immunogenicity of Therapeutic Proteins: Influence of Aggregation. J. Immunotoxicol. 2014, 11, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Lyon, R.P.; Bovee, T.D.; Doronina, S.O.; Burke, P.J.; Hunter, J.H.; Neff-LaFord, H.D.; Jonas, M.; Anderson, M.E.; Setter, J.R.; Senter, P.D. Reducing Hydrophobicity of Homogeneous Antibody-Drug Conjugates Improves Pharmacokinetics and Therapeutic Index. Nat. Biotechnol. 2015, 33, 733–735. [Google Scholar] [CrossRef]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.C.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of Drug Loading on the Antitumor Activity of a Monoclonal Antibody Drug Conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef] [Green Version]
- Simmons, J.K.; Burke, P.J.; Cochran, J.H.; Pittman, P.G.; Lyon, R.P. Reducing the Antigen-Independent Toxicity of Antibody-Drug Conjugates by Minimizing Their Non-Specific Clearance through PEGylation. Toxicol. Appl. Pharm. 2020, 392, 114932. [Google Scholar] [CrossRef]
- Teicher, B.A.; Chari, R.V.J. Antibody Conjugate Therapeutics: Challenges and Potential. Clin. Cancer Res. 2011, 17, 6389–6397. [Google Scholar] [CrossRef] [Green Version]
- Diamantis, N.; Banerji, U. Antibody-Drug Conjugates—an Emerging Class of Cancer Treatment. Br. J. Cancer 2016, 114, 362–367. [Google Scholar] [CrossRef]
- Jeffrey, S.C.; Burke, P.J.; Lyon, R.P.; Meyer, D.W.; Sussman, D.; Anderson, M.; Hunter, J.H.; Leiske, C.I.; Miyamoto, J.B.; Nicholas, N.D.; et al. A Potent Anti-CD70 Antibody–Drug Conjugate Combining a Dimeric Pyrrolobenzodiazepine Drug with Site-Specific Conjugation Technology. Bioconjugate Chem. 2013, 24, 1256–1263. [Google Scholar] [CrossRef]
- Mantaj, J.; Jackson, P.J.M.; Rahman, K.M.; Thurston, D.E. From Anthramycin to Pyrrolobenzodiazepine (PBD)-Containing Antibody–Drug Conjugates (ADCs). Angew. Chem. Int. Ed. 2017, 56, 462–488. [Google Scholar] [CrossRef] [Green Version]
- Walsh, S.J.; Bargh, J.D.; Dannheim, F.M.; Hanby, A.R.; Seki, H.; Counsell, A.J.; Ou, X.; Fowler, E.; Ashman, N.; Takada, Y.; et al. Site-Selective Modification Strategies in Antibody–Drug Conjugates. Chem. Soc. Rev. 2021, 50, 1305–1353. [Google Scholar] [CrossRef]
- Harper, J.; Lloyd, C.; Dimasi, N.; Toader, D.; Marwood, R.; Lewis, L.; Bannister, D.; Jovanovic, J.; Fleming, R.; D’Hooge, F.; et al. Preclinical Evaluation of MEDI0641, a Pyrrolobenzodiazepine-Conjugated Antibody-Drug Conjugate Targeting 5T4. Mol. Cancer Ther. 2017, 16, 1576–1587. [Google Scholar] [CrossRef] [Green Version]
- Saber, H.; Simpson, N.; Ricks, T.K.; Leighton, J.K. An FDA Oncology Analysis of Toxicities Associated with PBD-Containing Antibody-Drug Conjugates. Regul. Toxicol. Pharmacol. 2019, 107, 104429. [Google Scholar] [CrossRef] [PubMed]
- Hartley, J.A. Antibody-Drug Conjugates (ADCs) Delivering Pyrrolobenzodiazepine (PBD) Dimers for Cancer Therapy. Expert Opin. Biol. Ther. 2020, 1–13. [Google Scholar] [CrossRef]
- Ogitani, Y.; Aida, T.; Hagihara, K.; Yamaguchi, J.; Ishii, C.; Harada, N.; Soma, M.; Okamoto, H.; Oitate, M.; Arakawa, S.; et al. DS-8201a, A Novel HER2-Targeting ADC with a Novel DNA Topoisomerase I Inhibitor, Demonstrates a Promising Antitumor Efficacy with Differentiation from T-DM1. Clin. Cancer Res. 2016, 22, 5097–5108. [Google Scholar] [CrossRef] [Green Version]
- Goldenberg, D.M.; Cardillo, T.M.; Govindan, S.V.; Rossi, E.A.; Sharkey, R.M. Trop-2 Is a Novel Target for Solid Cancer Therapy with Sacituzumab Govitecan (IMMU-132), an Antibody-Drug Conjugate (ADC). Oncotarget 2015, 6, 22496–22512. [Google Scholar] [CrossRef] [Green Version]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA Topoisomerases and Their Poisoning by Anticancer and Antibacterial Drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef] [Green Version]
- Pommier, Y. Topoisomerase I Inhibitors: Camptothecins and Beyond. Nat. Rev. Cancer 2006, 6, 789–802. [Google Scholar] [CrossRef]
- Zunino, F.; Pratesi, G. Camptothecins in Clinical Development. Expert Opin. Investig. Drugs 2004, 13, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Bailly, C. Irinotecan: 25 Years of Cancer Treatment. Pharm. Res. 2019, 148, 104398. [Google Scholar] [CrossRef]
- Viricel, W.; Fournet, G.; Beaumel, S.; Perrial, E.; Papot, S.; Dumontet, C.; Joseph, B. Monodisperse Polysarcosine-Based Highly-Loaded Antibody-Drug Conjugates. Chem. Sci. 2019, 10, 4048–4053. [Google Scholar] [CrossRef] [Green Version]
- Burke, P.J.; Hamilton, J.Z.; Jeffrey, S.C.; Hunter, J.H.; Doronina, S.O.; Okeley, N.M.; Miyamoto, J.B.; Anderson, M.E.; Stone, I.J.; Ulrich, M.L.; et al. Optimization of a PEGylated Glucuronide-Monomethylauristatin E Linker for Antibody-Drug Conjugates. Mol. Cancer Ther. 2017, 16, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Yurkovetskiy, A.V.; Yin, M.; Bodyak, N.; Stevenson, C.A.; Thomas, J.D.; Hammond, C.E.; Qin, L.; Zhu, B.; Gumerov, D.R.; Ter-Ovanesyan, E.; et al. A Polymer-Based Antibody-Vinca Drug Conjugate Platform: Characterization and Preclinical Efficacy. Cancer Res. 2015, 75, 3365–3372. [Google Scholar] [CrossRef] [Green Version]
- Shao, S.; Tsai, M.-H.; Lu, J.; Yu, T.; Jin, J.; Xiao, D.; Jiang, H.; Han, M.; Wang, M.; Wang, J. Site-Specific and Hydrophilic ADCs through Disulfide-Bridged Linker and Branched PEG. Bioorg. Med. Chem. Lett. 2018, 28, 1363–1370. [Google Scholar] [CrossRef]
- Le Joncour, V.; Martins, A.; Puhka, M.; Isola, J.; Salmikangas, M.; Laakkonen, P.; Joensuu, H.; Barok, M. A Novel Anti-HER2 Antibody-Drug Conjugate XMT-1522 for HER2-Positive Breast and Gastric Cancers Resistant to Trastuzumab Emtansine. Mol. Cancer Ther. 2019, 18, 1721–1730. [Google Scholar] [CrossRef] [Green Version]
- Shao, T.; Chen, T.; Chen, Y.; Liu, X.; Chen, Y.-L.; Wang, Q.; Zhu, T.; Guo, M.; Li, H.; Ju, D.; et al. Construction of Paclitaxel-Based Antibody-Drug Conjugates with a PEGylated Linker to Achieve Superior Therapeutic Index. Signal. Transduct. Target. Ther. 2020, 5, 132. [Google Scholar] [CrossRef]
- Meyer, D.W.; Bou, L.B.; Shum, S.; Jonas, M.; Anderson, M.E.; Hamilton, J.Z.; Hunter, J.H.; Wo, S.W.; Wong, A.O.; Okeley, N.M.; et al. An in Vitro Assay Using Cultured Kupffer Cells Can Predict the Impact of Drug Conjugation on in Vivo Antibody Pharmacokinetics. Mol. Pharm. 2020, 17, 802–809. [Google Scholar] [CrossRef]
- Ogitani, Y.; Hagihara, K.; Oitate, M.; Naito, H.; Agatsuma, T. Bystander Killing Effect of DS-8201a, a Novel Anti-Human Epidermal Growth Factor Receptor 2 Antibody–Drug Conjugate, in Tumors with Human Epidermal Growth Factor Receptor 2 Heterogeneity. Cancer Sci. 2016, 107, 1039–1046. [Google Scholar] [CrossRef]
- Takegawa, N.; Nonagase, Y.; Yonesaka, K.; Sakai, K.; Maenishi, O.; Ogitani, Y.; Tamura, T.; Nishio, K.; Nakagawa, K.; Tsurutani, J. DS-8201a, a New HER2-Targeting Antibody–Drug Conjugate Incorporating a Novel DNA Topoisomerase I Inhibitor, Overcomes HER2-Positive Gastric Cancer T-DM1 Resistance. Int. J. Cancer 2017, 141, 1682–1689. [Google Scholar] [CrossRef]
- Iwata, T.N.; Ishii, C.; Ishida, S.; Ogitani, Y.; Wada, T.; Agatsuma, T. A HER2-Targeting Antibody-Drug Conjugate, Trastuzumab Deruxtecan (DS-8201a), Enhances Antitumor Immunity in a Mouse Model. Mol. Cancer Ther. 2018, 17, 1494–1503. [Google Scholar] [CrossRef] [Green Version]
- Nagai, Y.; Oitate, M.; Shiozawa, H.; Ando, O. Comprehensive Preclinical Pharmacokinetic Evaluations of Trastuzumab Deruxtecan (DS-8201a), a HER2-Targeting Antibody-Drug Conjugate, in Cynomolgus Monkeys. Xenobiotica 2019, 49, 1086–1096. [Google Scholar] [CrossRef]
- Ogitani, Y.; Abe, Y.; Iguchi, T.; Yamaguchi, J.; Terauchi, T.; Kitamura, M.; Goto, K.; Goto, M.; Oitate, M.; Yukinaga, H.; et al. Wide Application of a Novel Topoisomerase I Inhibitor-Based Drug Conjugation Technology. Bioorg. Med. Chem. Lett. 2016, 26, 5069–5072. [Google Scholar] [CrossRef]
- Andrikopoulou, A.; Zografos, E.; Liontos, M.; Koutsoukos, K.; Dimopoulos, M.-A.; Zagouri, F. Trastuzumab Deruxtecan (DS-8201a): The Latest Research and Advances in Breast Cancer. Clin. Breast Cancer 2020. [Google Scholar] [CrossRef]
- Nakada, T.; Masuda, T.; Naito, H.; Yoshida, M.; Ashida, S.; Morita, K.; Miyazaki, H.; Kasuya, Y.; Ogitani, Y.; Yamaguchi, J.; et al. Novel Antibody Drug Conjugates Containing Exatecan Derivative-Based Cytotoxic Payloads. Bioorg. Med. Chem. Lett. 2016, 26, 1542–1545. [Google Scholar] [CrossRef]
- Mitsui, I.; Kumazawa, E.; Hirota, Y.; Aonuma, M.; Sugimori, M.; Ohsuki, S.; Uoto, K.; Ejima, A.; Terasawa, H.; Sato, K. A New Water-Soluble Camptothecin Derivative, DX-8951f, Exhibits Potent Antitumor Activity against Human Tumors in Vitro and in Vivo. Jpn. J. Cancer Res. 1995, 86, 776–782. [Google Scholar] [CrossRef]
- Legigan, T.; Clarhaut, J.; Renoux, B.; Tranoy-Opalinski, I.; Monvoisin, A.; Berjeaud, J.-M.; Guilhot, F.; Papot, S. Synthesis and Antitumor Efficacy of a β-Glucuronidase-Responsive Albumin-Binding Prodrug of Doxorubicin. J. Med. Chem. 2012, 55, 4516–4520. [Google Scholar] [CrossRef]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable Linkers in Antibody-Drug Conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef]
- Tetko, I.V.; Gasteiger, J.; Todeschini, R.; Mauri, A.; Livingstone, D.; Ertl, P.; Palyulin, V.A.; Radchenko, E.V.; Zefirov, N.S.; Makarenko, A.S.; et al. Virtual Computational Chemistry Laboratory – Design and Description. J. Comput. Aided Mol. Des. 2005, 19, 453–463. [Google Scholar] [CrossRef]
- Christie, R.J.; Fleming, R.; Bezabeh, B.; Woods, R.; Mao, S.; Harper, J.; Joseph, A.; Wang, Q.; Xu, Z.-Q.; Wu, H.; et al. Stabilization of Cysteine-Linked Antibody Drug Conjugates with N-Aryl Maleimides. J. Control. Release 2015, 220, 660–670. [Google Scholar] [CrossRef]
- Szijj, P.A.; Bahou, C.; Chudasama, V. Minireview: Addressing the Retro-Michael Instability of Maleimide Bioconjugates. Drug Discov. Today Technol. 2018, 30, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Alley, S.C.; Benjamin, D.R.; Jeffrey, S.C.; Okeley, N.M.; Meyer, D.L.; Sanderson, R.J.; Senter, P.D. Contribution of Linker Stability to the Activities of Anticancer Immunoconjugates. Bioconjugate Chem. 2008, 19, 759–765. [Google Scholar] [CrossRef]
- Ovacik, M.; Lin, K. Tutorial on Monoclonal Antibody Pharmacokinetics and Its Considerations in Early Development. Clin. Transl. Sci. 2018, 11, 540–552. [Google Scholar] [CrossRef]
- Sauveur, J.; Matera, E.-L.; Chettab, K.; Valet, P.; Guitton, J.; Savina, A.; Dumontet, C. Esophageal Cancer Cells Resistant to T-DM1 Display Alterations in Cell Adhesion and the Prostaglandin Pathway. Oncotarget 2018, 9, 21141–21155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauveur, J.; Conilh, L.; Beaumel, S.; Chettab, K.; Jordheim, L.-P.; Matera, E.-L.; Dumontet, C. Characterization of T-DM1-Resistant Breast Cancer Cells. Pharm. Res. Perspect. 2020, 8, e00617. [Google Scholar] [CrossRef] [PubMed]
- Hartimath, S.V.; El-Sayed, A.; Makhlouf, A.; Bernhard, W.; Gonzalez, C.; Hill, W.; Parada, A.C.; Barreto, K.; Geyer, C.R.; Fonge, H. Therapeutic Potential of Nimotuzumab PEGylated-Maytansine Antibody Drug Conjugates against EGFR Positive Xenograft. Oncotarget 2019, 10, 1031–1044. [Google Scholar] [CrossRef] [PubMed]
- Bryden, F.; Martin, C.; Letast, S.; Lles, E.; Viéitez-Villemin, I.; Rousseau, A.; Colas, C.; Brachet-Botineau, M.; Allard-Vannier, E.; Larbouret, C.; et al. Impact of Cathepsin B-Sensitive Triggers and Hydrophilic Linkers on in Vitro Efficacy of Novel Site-Specific Antibody-Drug Conjugates. Org. Biomol. Chem. 2018, 16, 1882–1889. [Google Scholar] [CrossRef]
- Yin, O.; Xiong, Y.; Endo, S.; Yoshihara, K.; Garimella, T.; AbuTarif, M.; Wada, R.; LaCreta, F. Population Pharmacokinetics of Trastuzumab Deruxtecan in Patients With HER2-Positive Breast Cancer and Other Solid Tumors. Clin. Pharm. Ther. 2020. [Google Scholar] [CrossRef]
- Rivas, P.L.; Müller, C.; Breunig, C.; Hechler, T.; Pahl, A.; Arosio, D.; Belvisi, L.; Pignataro, L.; Corso, A.D.; Gennari, C. β-Glucuronidase Triggers Extracellular MMAE Release from an Integrin-Targeted Conjugate. Org. Biomol. Chem. 2019, 17, 4705–4710. [Google Scholar] [CrossRef] [PubMed]
- Renoux, B.; Raes, F.; Legigan, T.; Péraudeau, E.; Eddhif, B.; Poinot, P.; Tranoy-Opalinski, I.; Alsarraf, J.; Koniev, O.; Kolodych, S.; et al. Targeting the Tumour Microenvironment with an Enzyme-Responsive Drug Delivery System for the Efficient Therapy of Breast and Pancreatic Cancers. Chem. Sci. 2017, 8, 3427–3433. [Google Scholar] [CrossRef] [Green Version]
- Kovtun, Y.V.; Audette, C.A.; Ye, Y.; Xie, H.; Ruberti, M.F.; Phinney, S.J.; Leece, B.A.; Chittenden, T.; Blättler, W.A.; Goldmacher, V.S. Antibody-Drug Conjugates Designed to Eradicate Tumors with Homogeneous and Heterogeneous Expression of the Target Antigen. Cancer Res. 2006, 66, 3214–3221. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.M.; Chari, R.V.J. Ado-Trastuzumab Emtansine (T-DM1): An Antibody–Drug Conjugate (ADC) for HER2-Positive Breast Cancer. J. Med. Chem. 2014, 57, 6949–6964. [Google Scholar] [CrossRef]
- Staudacher, A.H.; Brown, M.P. Antibody Drug Conjugates and Bystander Killing: Is Antigen-Dependent Internalisation Required? Br. J. Cancer 2017, 117, 1736–1742. [Google Scholar] [CrossRef]
- Fischer, H.; Kansy, M.; Avdeef, A.; Senner, F. Permeation of Permanently Positive Charged Molecules through Artificial Membranes—Influence of Physico-Chemical Properties. Eur. J. Pharm. Sci. 2007, 31, 32–42. [Google Scholar] [CrossRef]
- Avdeef, A.; Artursson, P.; Neuhoff, S.; Lazorova, L.; Gråsjö, J.; Tavelin, S. Caco-2 Permeability of Weakly Basic Drugs Predicted with the Double-Sink PAMPA PKaflux Method. Eur. J. Pharm. Sci. 2005, 24, 333–349. [Google Scholar] [CrossRef]
- García-Alonso, S.; Ocaña, A.; Pandiella, A. Resistance to Antibody–Drug Conjugates. Cancer Res. 2018. [Google Scholar] [CrossRef] [Green Version]
- Collins, D.M.; Bossenmaier, B.; Kollmorgen, G.; Niederfellner, G. Acquired Resistance to Antibody-Drug Conjugates. Cancers 2019, 11, 394. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conilh, L.; Fournet, G.; Fourmaux, E.; Murcia, A.; Matera, E.-L.; Joseph, B.; Dumontet, C.; Viricel, W. Exatecan Antibody Drug Conjugates Based on a Hydrophilic Polysarcosine Drug-Linker Platform. Pharmaceuticals 2021, 14, 247. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14030247
Conilh L, Fournet G, Fourmaux E, Murcia A, Matera E-L, Joseph B, Dumontet C, Viricel W. Exatecan Antibody Drug Conjugates Based on a Hydrophilic Polysarcosine Drug-Linker Platform. Pharmaceuticals. 2021; 14(3):247. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14030247
Chicago/Turabian StyleConilh, Louise, Guy Fournet, Eric Fourmaux, Angélique Murcia, Eva-Laure Matera, Benoît Joseph, Charles Dumontet, and Warren Viricel. 2021. "Exatecan Antibody Drug Conjugates Based on a Hydrophilic Polysarcosine Drug-Linker Platform" Pharmaceuticals 14, no. 3: 247. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14030247