
Doxepin Exacerbates Renal Damage, Glucose Intolerance, Nonalcoholic Fatty Liver Disease, and Urinary Chromium Loss in Obese Mice

 , ,
, ,
Abstract
:1. Introduction
2. Results
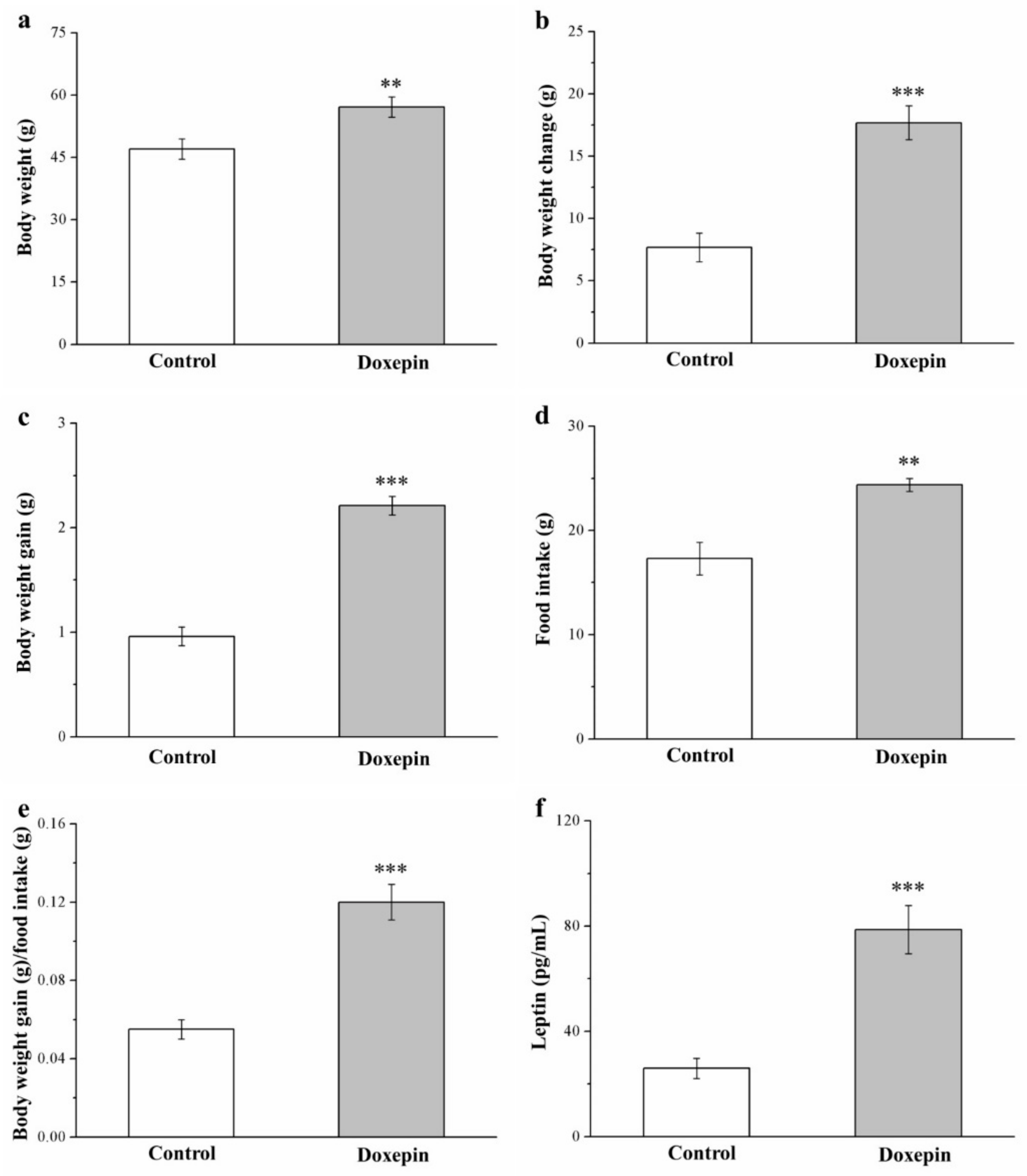
2.1. Effects on Morphometric Parameters and Food Intake and Efficiency
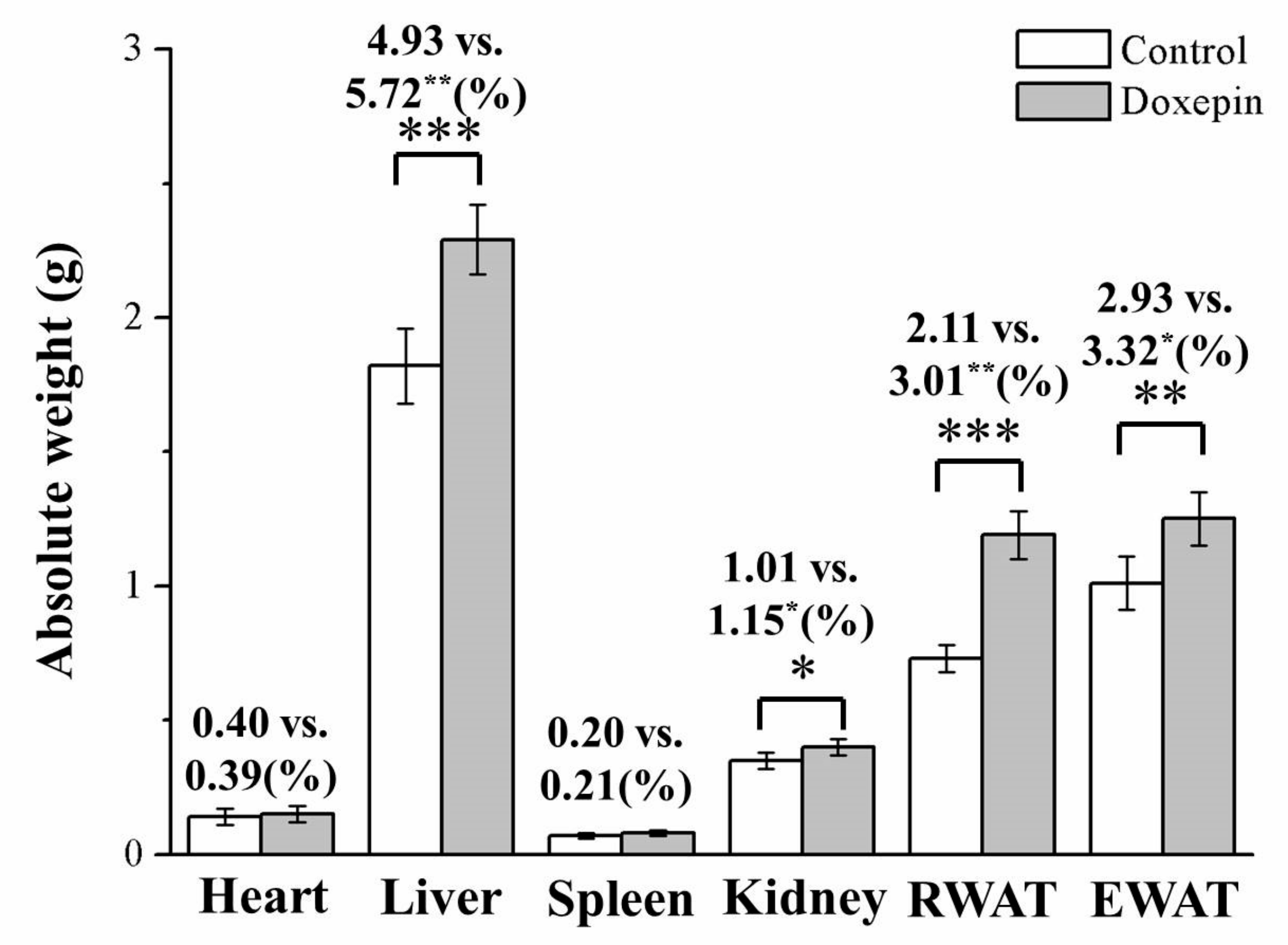
2.2. Effects on Liver, Kidney, and WAT Weights
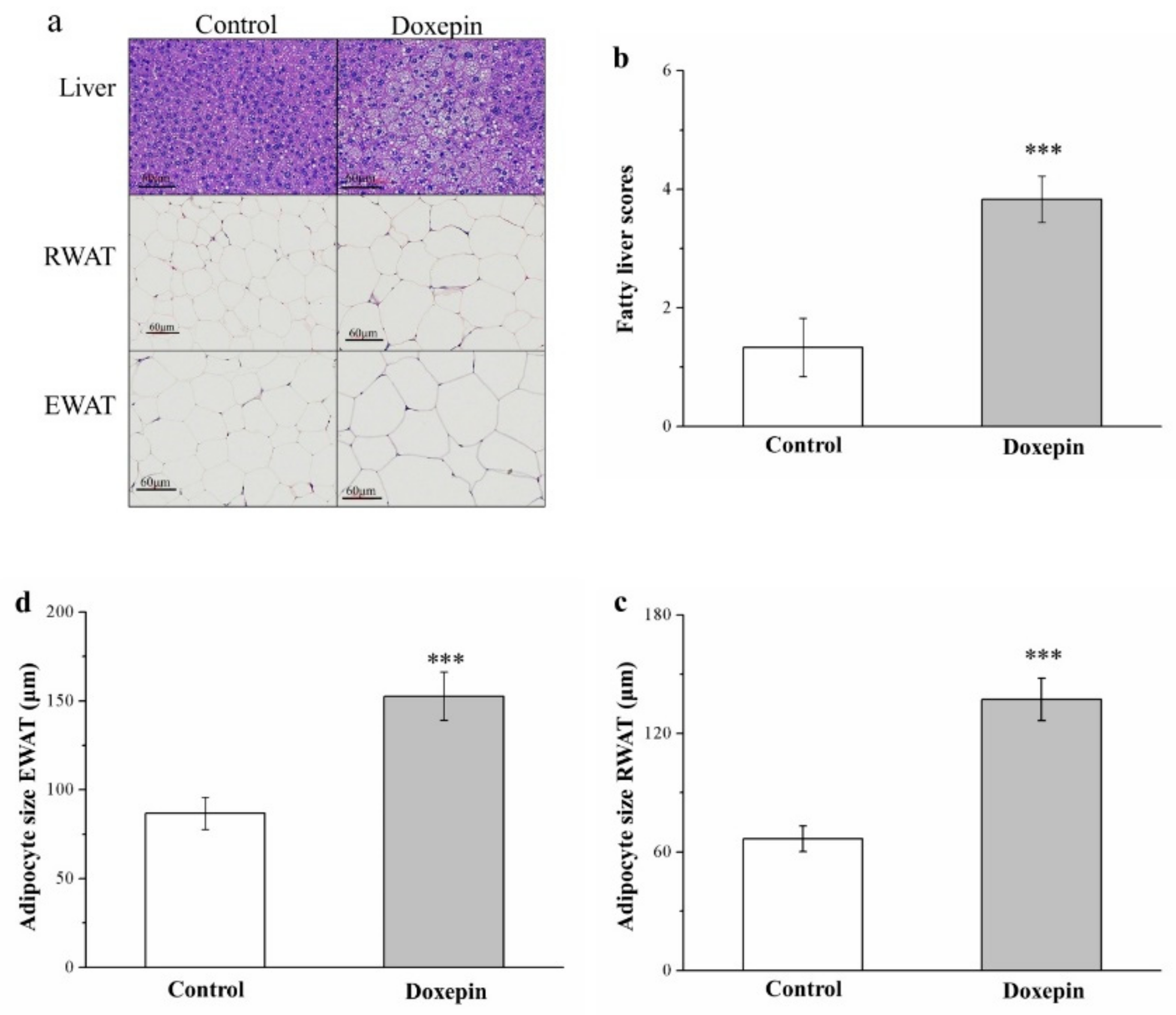
2.3. Effects on Liver Fat Accumulation and Large Adipocyte Proportion
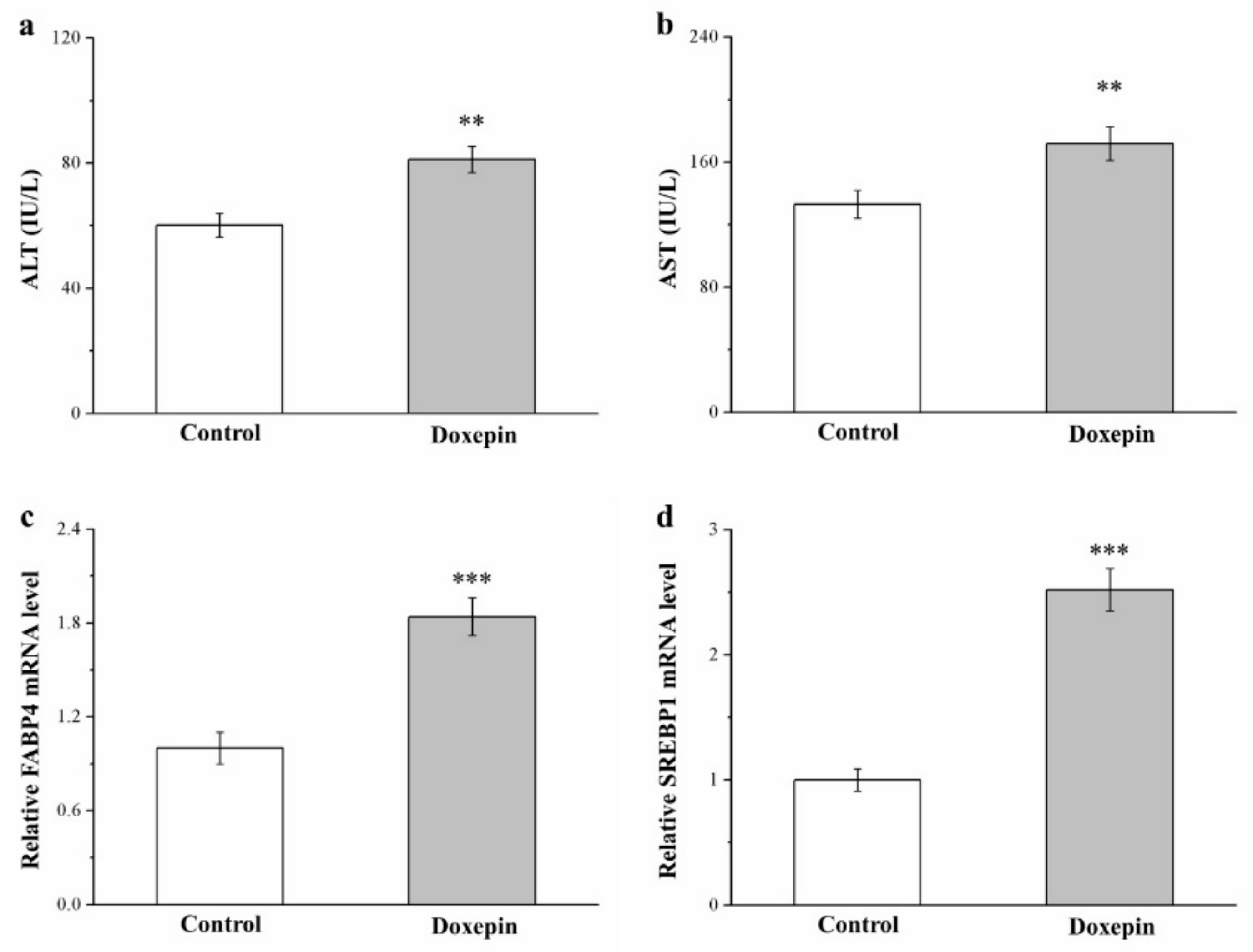
2.4. Effects on Serum ALT and AST Levels and Fatty Acid–Binding Protein 4 and Sterol Regulatory Element–Binding Protein 1 mRNA Levels
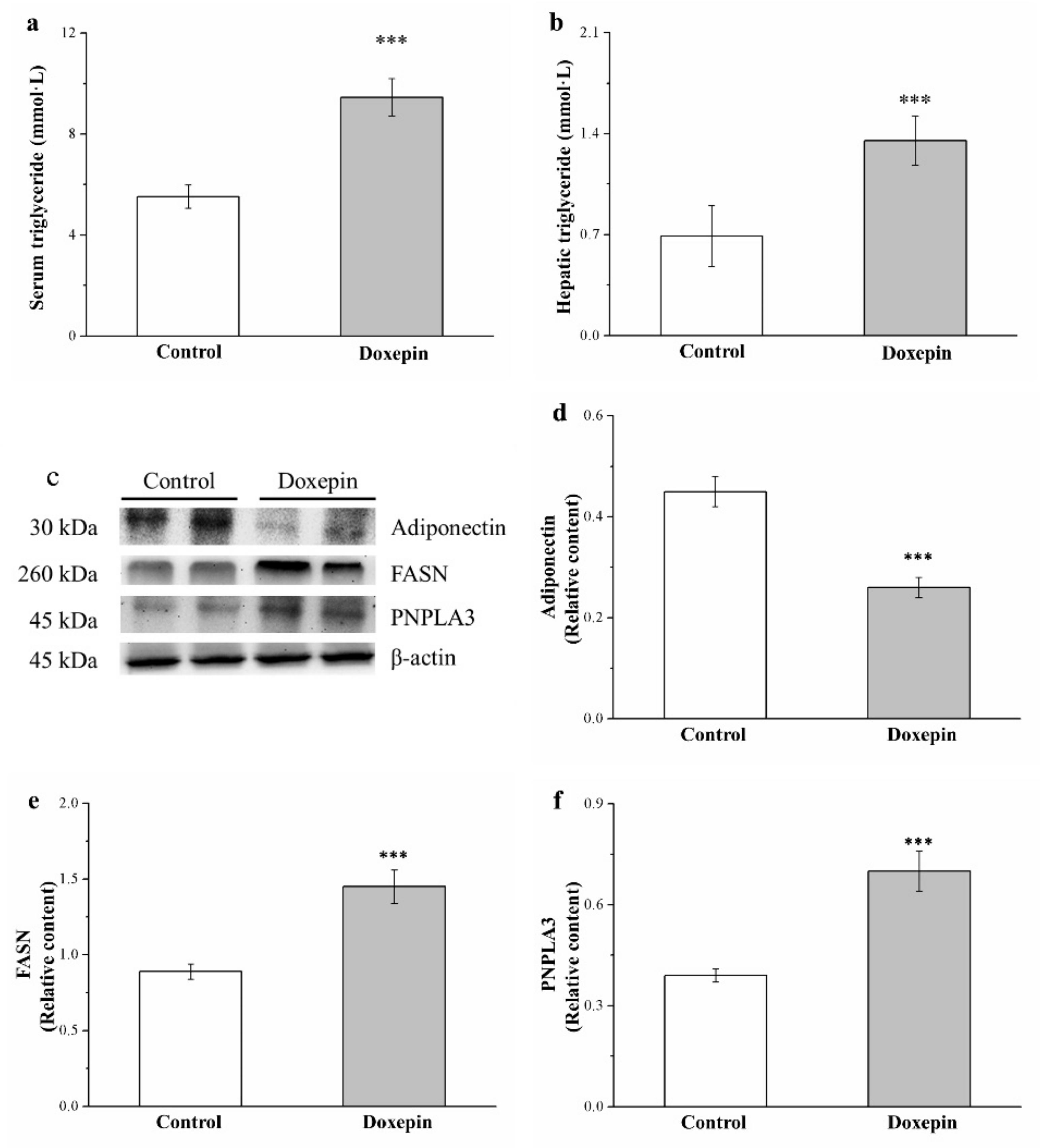
2.5. Effects on Serum and Liver Triglyceride Levels and Fatty Acid Synthase, Adiponectin, and Patatin-Like Phospholipid Domain Containing Protein 3 Expression
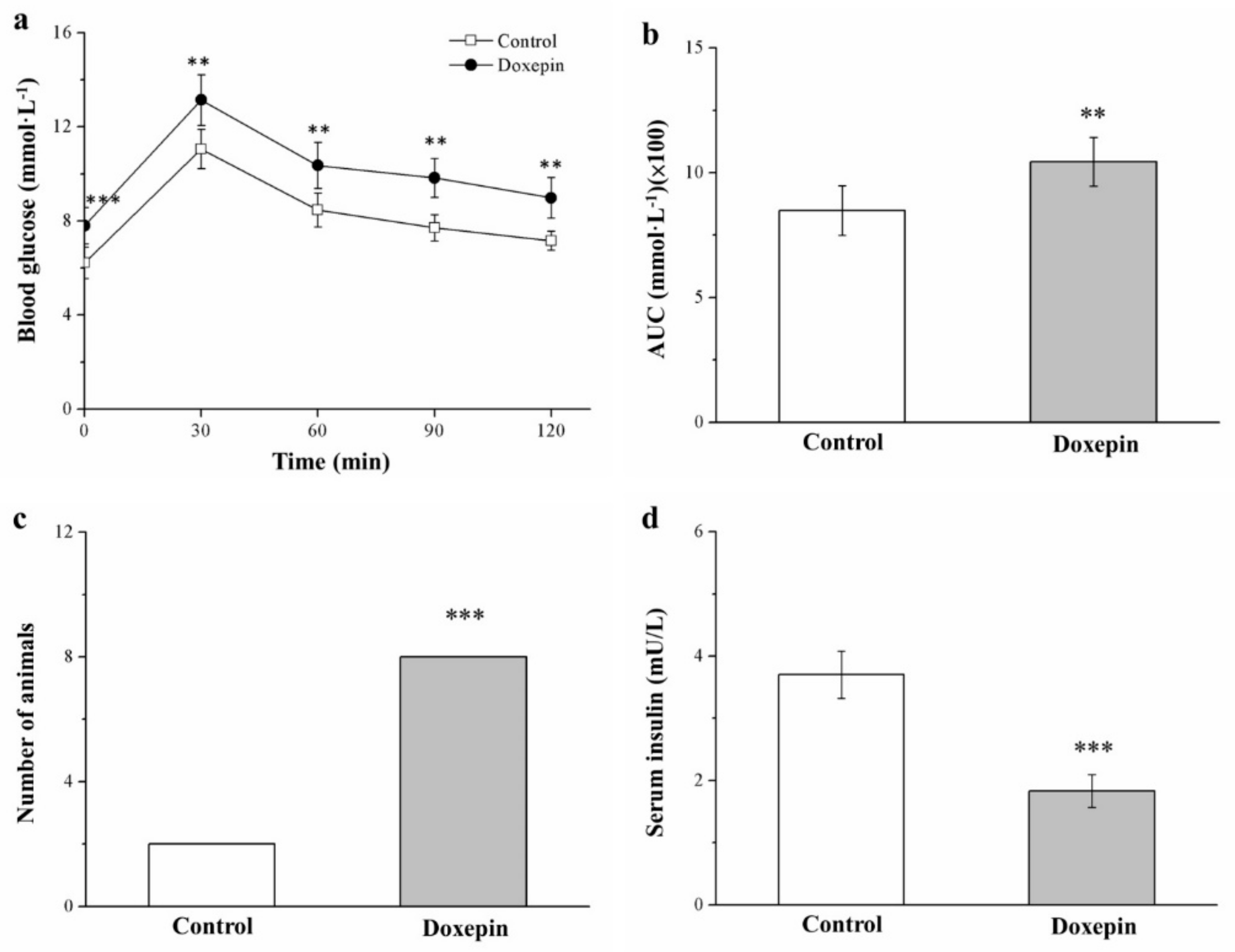
2.6. Influences on Insulin Level and Glucose Tolerance
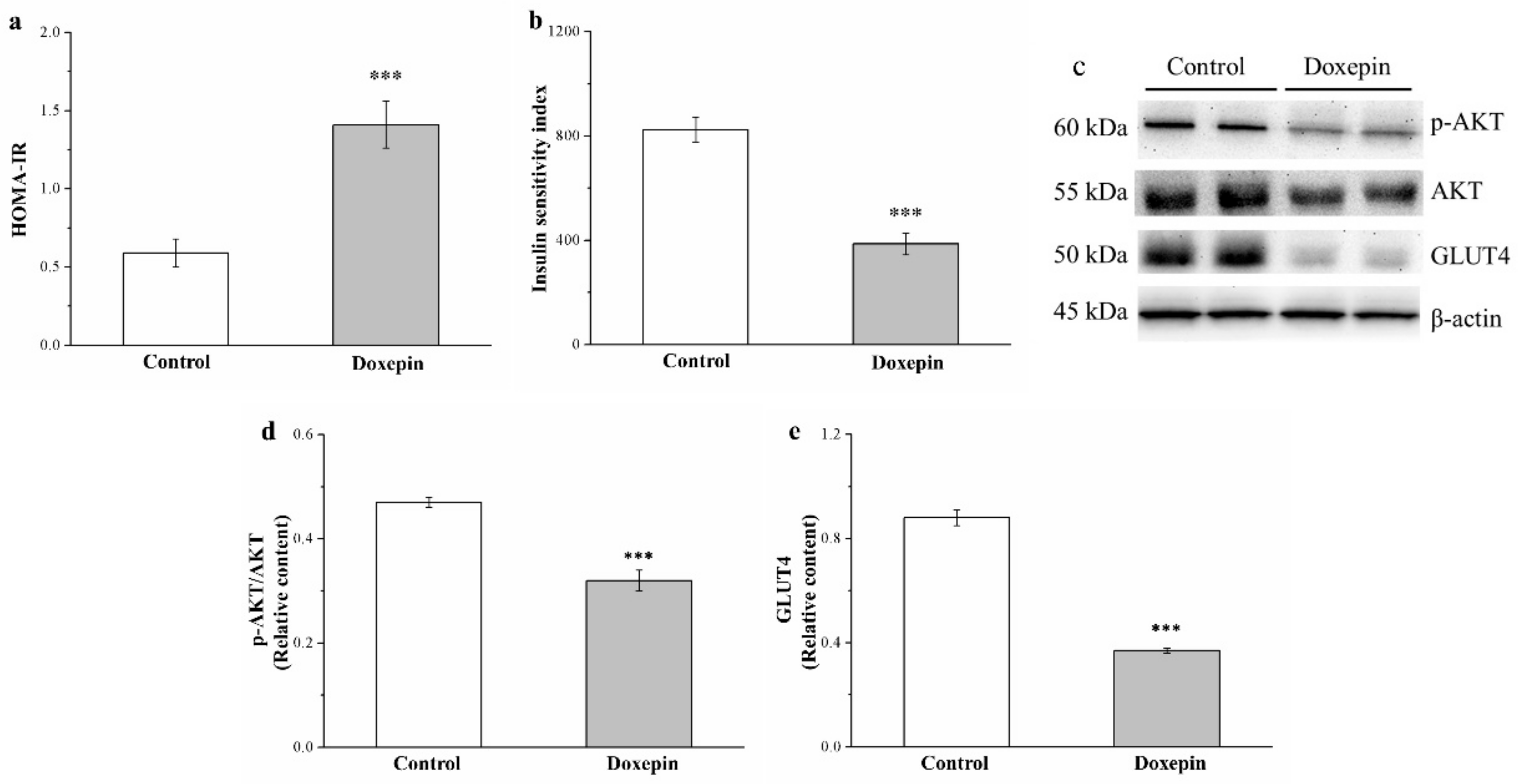
2.7. Effects on IS and IR and Phosphorylated Akt and Glucose Transport 4 Expression
2.8. Effects on Organ and Tissue Cr Levels and Urinary Cr Loss
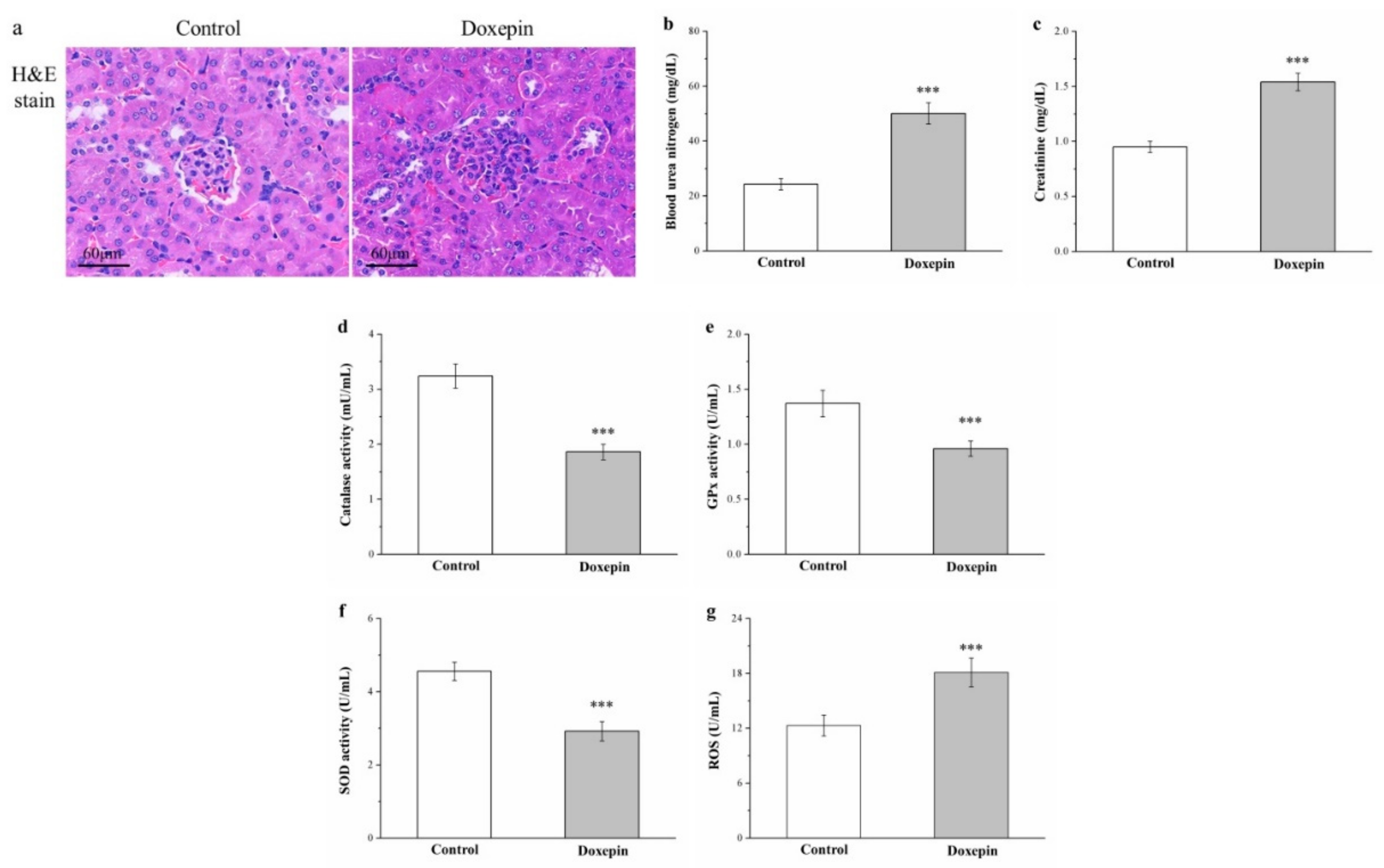
2.9. Effects on Renal Injury, Serum Creatinine and Blood Urea Nitrogen Levels, and Kidney Reactive Oxygen Species, and Antioxidant Enzyme Levels
3. Discussion
4. Materials and Methods
4.1. Animals, Diet, and Doxepin Treatment
4.2. Measurement of Body Weight, Food Intake, and Insulin and Leptin Levels
4.3. Serum Triglyceride, ALT, AST, Creatinine, BUN, and Liver Triglyceride Level Measurement
4.4. IPGTT
4.5. Morphometric and Histological Tissue Analyses
4.6. IR and IS Indexes
4.7. RNA Extraction and Real-Time Quantitative Polymerase Chain Reaction
4.8. Western Blot Analysis
4.9. Analysis of Cr Concentration
4.10. Measurement of Renal GPx, Catalase, SOD, and ROS Levels
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations:
| Cr | Chromium |
| IR | Insulin resistance |
| Type 2 diabetes mellitus | T2DM |
| NAFLD | Nonalcoholic fatty liver disease |
| CKD | Chronic kidney disease |
| WAT | White adipose tissue |
| IS | Insulin sensitivity |
| HFD | High fat diet |
| SD | Standard diet |
| EWAT | Epididymal white adipose tissue |
| RWAT | Retroperitoneal white adipose tissue |
| TBW | Total body weight |
| H&E | Hematoxylin and eosin |
| ALT | Alanine aminotransferase |
| AST | Aspartate aminotransferase |
| FABP4 | Fatty acid-binding protein 4 |
| SREBP1 | Sterol regulatory element–binding protein 1 |
| FASN | Fatty acid synthase |
| PNPLA3 | Patatin-like phospholipid domain containing protein 3 |
| IP | Intraperitoneal |
| GTT | Glucose tolerance test |
| AUC | Area under the curves |
| HOMA | Homeostatic model assessment |
| GLUT4 | Glucose transport 4 |
| BUN | Blood urea nitrogen |
| GPx | Glutathione peroxidase |
| SOD | Superoxide dismutase |
| ROS | Renal reactive oxygen species |
| UCP1 | Uncoupling protein 1 |
| FGF-21 | Fibroblast growth factor-21 |
| TNF-α | Tumor necrosis factor-α |
| CRP | C-reactive protein |
| IL-1β | Interleukin-1β |
| IHC | Immunohistochemical |
| ELISA | Enzyme-linked immunosorbent assay |
| PCR | Polymerase chain reaction |
References
- Cortese, S.; Brown, T.E.; Corkum, P.; Gruber, R.; O’Brien, L.M.; Stein, M.; Weiss, M.; Owens, J. Assessment and Management of Sleep Problems in Youths With Attention-Deficit/Hyperactivity Disorder. J. Am. Acad. Child Adolesc. Psychiatry 2013, 52, 784–796. [Google Scholar] [CrossRef]
- Benasi, G.; Guidi, J.; Offidani, E.; Balon, R.; Rickels, K.; Fava, G.A. Benzodiazepines as a Monotherapy in Depressive Disorders: A Systematic Review. Psychother. Psychosom. 2018, 87, 65–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.R.; Park, B.K.; Kim, Y.H.; Shim, I.; Kang, I.C.; Lee, M.Y. Antidepressant Effect of Fraxinus rhynchophylla Hance Extract in a Mouse Model of Chronic Stress-Induced Depression. Biomed. Res. Int. 2018, 2018, 8249563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azadeh, M.; Meshkati, A.; Hajhashemi, V. A synbiotic mixture augmented the efficacy of doxepin, venlafaxine, and fluvoxamine in a mouse model of depression. Turk. J. Pharm. Sci. 2020, 17, 293–298. [Google Scholar]
- Gahr, M.; Zeiss, R.; Lang, D.; Connemann, B.J.; Hiemke, C.; Muche, R.; Freudenmann, R.W.; Schönfeldt-Lecuona, C. Associ-ation between haemorrhages and treatment with selective and non-selective serotonergic antidepressants: Possible implications of quantitative signal detection. Psychiatry Res. 2015, 229, 257–263. [Google Scholar] [CrossRef]
- Nguyen, H.; Pan, A.; Smollin, C.; Cantrell, L.F.; Kearney, T. An 11-year retrospective review of cyproheptadine use in serotonin syndrome cases reported to the California Poison Control System. J. Clin. Pharm. Ther. 2019, 44, 327–334. [Google Scholar] [CrossRef]
- Abosi, O.; Lopes, S.; Schmitz, S.; Fiedorowicz, J.G. Cardiometabolic effects of psychotropic medications. Horm. Mol. Biol. Clin. Investig. 2018, 36, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, H.-P.; Hou, P.-H.; Mao, F.-C.; Chang, C.-C.; Yang, W.-C.; Wu, C.-F.; Liao, H.-J.; Lin, T.-C.; Chou, L.-S.; Hsiao, L.-W.; et al. Risperidone Exacerbates Glucose Intolerance, Nonalcoholic Fatty Liver Disease, and Renal Impairment in Obese Mice. Int. J. Mol. Sci. 2021, 22, 409. [Google Scholar] [CrossRef]
- Wu, Q.; Xu, Y.; Bao, Y.; Alvarez, J.; Gonzales, M.L. Tricyclic Antidepressant Use and Risk of Fractures: A Meta-Analysis of Cohort Studies through the Use of Both Frequentist and Bayesian Approaches. J. Clin. Med. 2020, 9, 2584. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-F.; Hou, P.-H.; Mao, F.C.; Su, Y.-C.; Wu, C.-Y.; Yang, W.-C.; Lin, C.-S.; Tsai, H.-P.; Liao, H.-J.; Chang, G.-R. Mirtazapine Reduces Adipocyte Hypertrophy and Increases Glucose Transporter Expression in Obese Mice. Animals 2020, 10, 1423. [Google Scholar] [CrossRef]
- Chang, G.-R.; Wu, Y.-Y.; Chiu, Y.-S.; Chen, W.-Y.; Liao, J.-W.; Hsu, H.-M.; Chao, T.-H.; Hung, S.-W.; Mao, F.C. Long-term Administration of Rapamycin Reduces Adiposity, but Impairs Glucose Tolerance in High-Fat Diet-fed KK/HlJ Mice. Basic Clin. Pharmacol. Toxicol. 2009, 105, 188–198. [Google Scholar] [CrossRef]
- Kim, J.W.; Lee, Y.-S.; Park, Y.S.; Kim, B.-H.; Lee, S.Y.; Yeon, J.E.; Lee, C.H. Multiparametric MR Index for the Diagnosis of Non-Alcoholic Steatohepatitis in Patients with Non-Alcoholic Fatty Liver Disease. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Chang, G.-R.; Chiu, Y.-S.; Wu, Y.-Y.; Lin, Y.-C.; Hou, P.-H.; Mao, F.C. Rapamycin impairs HPD-induced beneficial effects on glucose homeostasis. Br. J. Pharmacol. 2015, 172, 3793–3804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudish, L.I.; Reusch, J.E.; Sussel, L. β Cell dysfunction during progression of metabolic syndrome to type 2 diabetes. J. Clin. Investig. 2019, 129, 4001–4008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carney, C.P.; Jones, L.; Woolson, R.F. Medical comorbidity in women and men with schizophrenia: A population-based con-trolled study. J. Gen. Intern. Med. 2006, 21, 1133–1137. [Google Scholar] [CrossRef]
- Xu, H.; Zhuang, X. Atypical antipsychotics-induced metabolic syndrome and nonalcoholic fatty liver disease: A critical review. Neuropsychiatr. Dis. Treat. 2019, 15, 2087–2099. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.-Y.; Huang, C.L.-C.; Feng, I.J.; Tsuang, H.-C. Second-generation antipsychotic medications and risk of chronic kidney disease in schizophrenia: Population-based nested case–control study. BMJ Open 2018, 8, e019868. [Google Scholar] [CrossRef]
- Boyce, P.; Ma, C. Choosing an antidepressant. Aust. Prescr 2021, 44, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Bitan, D.T.; Krieger, I.; Berkovitch, A.; Comaneshter, D.; Cohen, A. Chronic kidney disease in adults with schizophrenia: A nationwide population-based study. Gen. Hosp. Psychiatry 2019, 58, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.-H.; Chien, I.-C.; Lin, C.-H.; Chou, Y.-J.; Chou, P. Increased Risk of Chronic Liver Disease in Patients with Schizophrenia: A Population-Based Cohort Study. J. Psychosom. Res. 2014, 55, 163–171. [Google Scholar] [CrossRef]
- Hazell, P.; Mirzaie, M. Tricyclic drugs for depression in children and adolescents. Cochrane Database Syst. Rev. 2013, 002317. [Google Scholar] [CrossRef] [Green Version]
- Hasnain, M.; Vieweg, W.V.R. Weight Considerations in Psychotropic Drug Prescribing and Switching. Postgrad. Med. 2013, 125, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Krystal, A.D.; Lankford, A.; Durrence, H.H.; Ludington, E.; Jochelson, P.; Rogowski, R.; Roth, T. Efficacy and Safety of Doxepin 3 and 6 mg in a 35-day Sleep Laboratory Trial in Adults with Chronic Primary Insomnia. Sleep 2011, 34, 1433–1442. [Google Scholar] [CrossRef] [Green Version]
- Kaur, K.K.; Gautam Allahbadia, G.; Singh, M. Importance of simultaneous treatment of obesity and diabetes mellitus: A se-quelae to the understanding of diabesity-a review. Obes. Res. Open J. 2018, 6, 1–10. [Google Scholar] [CrossRef]
- Alzgool, S.A. The Effect of Doxepin on Body Mass in Mice: Does It Cause Weight Gain? 2020. Available online: https://0-search-proquest-com.brum.beds.ac.uk/docview/2442579570?pq-origsite=gscholar&fromopenview=true/ (accessed on 5 January 2021).
- True, B.L.; Perry, P.J.; Burns, E.A. Profound hypoglycemia with the addition of a tricyclic antidepressant to maintenance sul-fonylurea therapy. Am. J. Psychiatry 1987, 144, 1220–1221. [Google Scholar]
- Khoza, S.; Barner, J.C. Glucose dysregulation associated with antidepressant agents: An analysis of 17 published case reports. Int. J. Clin. Pharm. 2011, 33, 484–492. [Google Scholar] [CrossRef]
- Gupta, B.; Shakarwal, M.K.; Kumar, A.; Jaju, B.P. Modulation of glucose homeostasis by doxepin. Methods Find. Exp. Clin. Pharmacol. 1992, 14, 61–71. [Google Scholar]
- Chen, Z.; Liu, X.; Luo, Y.; Wang, J.; Meng, Y.; Sun, L.; Chang, Y.; Cui, Q.; Yang, J. Repurposing Doxepin to Ameliorate Steatosis and Hyperglycemia by Activating FAM3A Signaling Pathway. Diabetes 2020, 69, 1126–1139. [Google Scholar] [CrossRef] [PubMed]
- Tran, Y.-H.; Schuiling-Veninga, C.C.M.; Bergman, J.E.H.; Groen, H.; Wilffert, B. Impact of Muscarinic M3 Receptor Antagonism on the Risk of Type 2 Diabetes in Antidepressant-Treated Patients: A Case-Controlled Study. CNS Drugs 2017, 31, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Akinnusi, M.; El Solh, A.A. Drug treatment strategies for insomnia in patients with post-traumatic stress disorder. Expert Opin. Pharmacother. 2019, 20, 691–699. [Google Scholar] [CrossRef]
- Roessner, V.; Demling, J.; Bleich, S. Doxepin increases serum cholesterol levels. Can. J. Psychiatry 2004, 49, 74–75. [Google Scholar] [CrossRef] [Green Version]
- Lechin, F.; van der Dijs, B.; Lechin, A.; Lechin, M.; Coll-García, E.; Jara, H.; Cabrera, A.; Jiménez, V.; Gómez, F.; Tovar, D.; et al. Doxepin therapy for postprandial symptomatic hypoglycaemic patients: Neurochemical, hormonal and metabolic disturbances. Clin. Sci. 1991, 80, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Do, A.; Ilagan-Ying, Y.C.; Mehal, W.Z. Medical Approach for Weight Loss in Nonalcoholic Fatty Liver Disease. Curr. Hepatol. Rep. 2019, 18, 444–454. [Google Scholar] [CrossRef]
- Medici, V.; McClave, S.A.; Miller, K.R. Common Medications Which Lead to Unintended Alterations in Weight Gain or Organ Lipotoxicity. Curr. Gastroenterol. Rep. 2015, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Højgaard, A.D.; Andersen, P.T.; Møller-Petersen, J. Rhabdomyolysis and Acute Renal Failure Following an Overdose of Doxepin and Nitrazepam. Acta Medica Scand. 2009, 223, 79–82. [Google Scholar] [CrossRef]
- Chang, G.-R.; Chen, W.-K.; Hou, P.-H.; Mao, F.C. Isoproterenol exacerbates hyperglycemia and modulates chromium distribution in mice fed with a high fat diet. J. Trace Elements Med. Biol. 2017, 44, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Chang, G.R.; Chiu, Y.S.; Wu, Y.Y.; Chen, W.Y.; Liao, J.W.; Chao, T.H.; Mao, F.C. Rapamycin protects against high fat di-et-induced obesity in C57BL/6J mice. J. Pharmacol. Sci. 2009, 109, 496–503. [Google Scholar] [CrossRef]
- Hou, P.-H.; Chang, G.-R.; Chen, C.-P.; Lin, Y.-L.; Chao, I.-S.; Shen, T.-T.; Mao, F.C. Long-term administration of olanzapine induces adiposity and increases hepatic fatty acid desaturation protein in female C57BL/6J mice. Iran J. Basic Med. Sci. 2018, 21, 495–501. [Google Scholar]
- Chang, G.R.; Hou, P.H.; Wang, C.M.; Wu, C.F.; Su, H.K.; Liao, H.J.; Chen, T.P. Chronic everolimus treatment of high fat diet mice leads to a reduction in obesity but impaired glucose tolerance. Pharmacol. Res. Perspect. 2021, 9, e00732. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesch, G.H.; Lim, A.K.H. Recent insights into diabetic renal injury from the db/db mouse model of type 2 diabetic nephropathy. Am. J. Physiol. Physiol. 2011, 300, F301–F310. [Google Scholar] [CrossRef]
- Nagarajrao, R.; Alharbi, S.A. Relationship between oxidant and antioxidant enzymes status in type 2 diabetic patients with nephropathy in Saudi population. Asian J. Pharm. Clin. Res. 2018, 1, 363–368. [Google Scholar] [CrossRef] [Green Version]
- Cottingham, C.M.; Patrick, T.; Richards, M.A.; Blackburn, K.D. Tricyclic antipsychotics promote adipogenic gene expression to potentiate preadipocyte differentiation in vitro. Hum. Cell 2020, 33, 502–511. [Google Scholar] [CrossRef]
- Li, H.; Li, J.; Qu, Z.; Qian, H.; Zhang, J.; Wang, H.; Xu, X.; Liu, S. Intrauterine exposure to low-dose DBP in the mice induces obesity in offspring via suppression of UCP1 mediated ER stress. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Jensen, G.L. Drug-Induced Hyperphagia: What Can We Learn From Psychiatric Medications? J. Parenter. Enter. Nutr. 2008, 32, 578–581. [Google Scholar] [CrossRef] [PubMed]
- Piao, L.; Park, J.; Li, Y.; Shin, S.; Shin, S.; Kong, G.; Shrestha, R.; Tran, Q.; Hur, G.M.; Kim, J.-L.; et al. SOCS3 and SOCS6 are required for the risperidone-mediated inhibition of insulin and leptin signaling in neuroblastoma cells. Int. J. Mol. Med. 2014, 33, 1364–1370. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.W.; Kim, H.S.; Im, J.H.; Kim, J.W.; Jun, D.W.; Lim, S.C.; Lee, K.; Choi, J.M.; Kim, S.K.; Kang, K.W. GPR119: A promising target for nonalcoholic fatty liver disease. FASEB J. 2015, 30, 324–335. [Google Scholar] [CrossRef] [Green Version]
- Remy, A.J.; Larrey, D.; Pageaux, G.P.; Ribstein, J.; Ramos, J.; Michel, H. Cross hepatotoxicity between tricyclic antidepressants and phenothiazines. Eur. J. Gastroenterol. Hepatol. 1995, 7, 373–376. [Google Scholar] [PubMed]
- Voican, C.S.; Corruble, E.; Naveau, S.; Perlemuter, G. Antidepressant-Induced Liver Injury: A Review for Clinicians. Am. J. Psychiatry 2014, 171, 404–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.; Hanouneh, I.A.; Noureddin, M.; Rolph, T.; Alkhouri, N. Fibroblast growth factor (FGF)-21 based therapies: A magic bullet for nonalcoholic fatty liver disease (NAFLD)? Expert. Opin. Investig. Drugs 2020, 29, 197–204. [Google Scholar] [CrossRef]
- Su, X.; Kong, Y.; Peng, D. Fibroblast growth factor 21 in lipid metabolism and non-alcoholic fatty liver disease. Clin. Chim. Acta 2019, 498, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Asrih, M.; Jornayvaz, F.R. Inflammation as a potential link between nonalcoholic fatty liver disease and insulin resistance. J. Endocrinol. 2013, 218, R25–R36. [Google Scholar] [CrossRef]
- Yuan, S.; Liu, H.; Yuan, D.; Xu, J.; Chen, Y.; Xu, X.; Xu, F.; Liang, H. PNPLA3 I148M mediates the regulatory effect of NF-kB on inflammation in PA-treated HepG2 cells. J. Cell Mol. Med. 2020, 24, 1541–1552. [Google Scholar] [CrossRef] [PubMed]
- Dorn, C.; Riener, M.-O.; Kirovski, G.; Saugspier, M.; Steib, K.; Weiss, T.S.; Gäbele, E.; Kristiansen, G.; Hartmann, A.; Hellerbrand, C. Expression of fatty acid synthase in nonalcoholic fatty liver disease. Int. J. Clin. Exp. Pathol. 2010, 3, 505–514. [Google Scholar] [PubMed]
- Erenmemisoglu, A.; Ozdogan, U.K.; Saraymen, R.; Tutus, A. Effect of Some Antidepressants on Glycaemia and Insulin Levels of Normoglycaemic and Alloxan-induced Hyperglycaemic Mice. J. Pharm. Pharmacol. 1999, 51, 741–743. [Google Scholar] [CrossRef]
- Deuschle, M. Effects of antidepressants on glucose metabolism and diabetes mellitus type 2 in adults. Curr. Opin. Psychiatry 2013, 26, 60–65. [Google Scholar] [CrossRef]
- Joost, H.G.; Poser, W.; Panten, U. Inhibition of insulin release from the rat pancreas by cyproheptadine and tricyclic antide-pressants. Naunyn Schmiedebergs Arch. Pharmacol. 1974, 285, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Ardizzone, T.D.; Bradley, R.J.; Freeman, A.M.; Dwyer, D.S. Inhibition of glucose transport in PC12 cells by the atypical anti-psychotic drugs risperidone and clozapine, and structural analogs of clozapine. Brain Res. 2001, 923, 82–90. [Google Scholar] [CrossRef]
- Derijks, H.J.; Heerdink, E.R.; De Koning, F.H.; Janknegt, R.; Klungel, O.H.; Egberts, A.C. The association between antide-pressant use and hypoglycaemia in diabetic patients: A nested case-control study. Pharmacoepidemiol. Drug Saf. 2008, 17, 336–344. [Google Scholar] [CrossRef]
- Chang-Chen, K.J.; Mullur, R.; Bernal-Mizrachi, E. Beta-cell failure as a complication of diabetes. Rev. Endocr. Metab. Disord. 2008, 9, 329–343. [Google Scholar] [CrossRef] [Green Version]
- Chang, G.-R.; Hou, P.-H.; Chen, W.-K.; Lin, C.-T.; Tsai, H.-P.; Mao, F.C. Exercise Affects Blood Glucose Levels and Tissue Chromium Distribution in High-Fat Diet-Fed C57BL6 Mice. Molecules 2020, 25, 1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajendran, K.; Manikandan, S.; Nair, L.D.; Karuthodiyil, R.; Vijayarajan, N.; Gnanasekar, R.; Kapil, V.V.; Mohamed, A.S. Serum Chromium Levels in Type 2 Diabetic Patients and Its Association with Glycaemic Control. J. Clin. Diagn. Res. 2015, 9, OC05–OC08. [Google Scholar] [CrossRef]
- Vogelzangs, N.; Duivis, H.E.; Beekman, A.; Kluft, C.; Neuteboom, J.; Hoogendijk, W.; Smit, J.H.; De Jonge, P.; Penninx, B.W.J.H. Association of depressive disorders, depression characteristics and antidepressant medication with inflammation. Transl. Psychiatry 2012, 2, e79. [Google Scholar] [CrossRef] [Green Version]
- Tanigaki, K.; Vongpatanasin, W.; Barrera, J.A.; Atochin, D.N.; Huang, P.L.; Bonvini, E.; Shaul, P.W.; Mineo, C. C-reactive protein causes insulin resistance in mice through fcγ receptor iib–mediated inhibition of skeletal muscle glucose delivery. Diabetes 2013, 62, 721–731. [Google Scholar] [CrossRef] [Green Version]
- Molli, A.E.I.; Bergonzi, M.F.; Spalvieri, M.P.; Linari, M.A.; Frechtel, G.D.; Cerrone, G.E. Relationship between the IL-1β serum concentration, mRNA levels and rs16944 genotype in the hyperglycemic normalization of T2D patients. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef]
- Hu, Y.; Yang, C.; Shen, G.; Yang, S.; Cheng, X.; Cheng, F.; Rao, J.; Wang, X. Hyperglycemia-triggered sphingosine-1-phosphate and sphingosine-1-phosphate receptor 3 signaling worsens liver ischemia/reperfusion injury by regulating m1/m2 polarization. Liver Transpl. 2019, 25, 1074–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Wei, S.; Zhou, S.; Qiu, J.; Shi, C.; Liu, R.; Zhou, H.; Lu, L. Hyperglycemia aggravates acute liver injury by promoting liver-resident macrophage NLRP3 inflammasome activation via the inhibition of AMPK/mTOR-mediated autophagy induc-tion. Immunol. Cell Biol. 2020, 98, 54–66. [Google Scholar] [CrossRef] [PubMed]
- García-Ruiz, C.; Colell, A.; Marí, M.; Morales, A.; Calvo, M.; Enrich, C.; Fernández-Checa, J.C. Defective TNF-α–mediated hepatocellular apoptosis and liver damage in acidic sphingomyelinase knockout mice. J. Clin. Investig. 2003, 111, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Abbas, N.; Getachew, A.; You, K.; Shah, Z.; Chen, Y.; Tao, J.; Hussain, M.; Yang, F.; Zhuang, Y.; Xu, Y.; et al. Kupffer cells mediate the recruitment of hepatic stellate cells into the localized liver damage. Biochem. Biophys. Res. Commun. 2020, 529, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.; Dukes, E.; Mercadante, S.; Oster, G. Use of antiepileptics and tricyclic antidepressants in cancer patients with neuropathic pain. Eur. J. Cancer Care 2005, 15, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Achike, F.I.; To, N.-H.P.; Wang, H.; Kwan, C.-Y. Obesity, metabolic syndrome, adipocytes and vascular function: A holistic viewpoint. Clin. Exp. Pharmacol. Physiol. 2010, 38, 1–10. [Google Scholar] [CrossRef]
- Tobita, S.; Sogawa, R.; Murakawa, T.; Kimura, S.; Tasaki, M.; Sakamoto, Y.; Monji, A.; Irie, H. The importance of monitoring renal function and concomitant medication to avoid toxicity in patients taking lithium. Int. Clin. Psychopharmacol. 2020, 36, 34–37. [Google Scholar] [CrossRef]
- Rayego-Mateos, S.; Morgado-Pascual, J.L.; Opazo-Ríos, L.; Guerrero-Hue, M.; García-Caballero, C.; Vázquez-Carballo, C.; Mas, S.; Sanz, A.B.; Herencia, C.; Mezzano, S.; et al. Pathogenic pathways and therapeutic approaches targeting inflammation in diabetic nephropathy. Int. J. Mol. Sci. 2020, 21, 3798. [Google Scholar] [CrossRef]
- Stephens, J.W.; Brown, K.E.; Min, T. Chronic kidney disease in type 2 diabetes: Implications for managing glycaemic control, cardiovascular and renal risk. Diabetes Obes. Metab. 2020, 22, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.-W.; Lee, Y.; Lee, C.-Y.; Lin, P.-Y. Neurotoxicity and nephrotoxicity caused by combined use of lithium and risperidone: A case report and literature review. BMC Pharmacol. Toxicol. 2016, 17, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.-J.; Kong, M.-K.; Kim, Y.-C. Beneficial effects of Phellodendri Cortex extract on hyperglycemia and diabetic nephropathy in streptozotocin-induced diabetic rats. BMB Rep. 2008, 41, 710–715. [Google Scholar] [CrossRef] [Green Version]
- Al Hroob, A.M.; Abukhalil, M.H.; Alghonmeen, R.D.; Mahmoud, A.M. Ginger alleviates hyperglycemia-induced oxidative stress, inflammation and apoptosis and protects rats against diabetic nephropathy. Biomed. Pharmacother. 2018, 106, 381–389. [Google Scholar] [CrossRef]
- Muraro, C.; Tiezza, M.D.; Pavan, C.; Ribaudo, G.; Zagotto, G.; Orian, L. Major Depressive Disorder and Oxidative Stress: In Silico Investigation of Fluoxetine Activity against ROS. Appl. Sci. 2019, 9, 3631. [Google Scholar] [CrossRef] [Green Version]
- Sehonova, P.; Zikova, A.; Blahova, J.; Svobodova, Z.; Chloupek, P.; Kloas, W. mRNA expression of antioxidant and bio-transformation enzymes in zebrafish (Danio rerio) embryos after exposure to the tricyclic antidepressant amitriptyline. Chemosphere 2019, 217, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Azadbakht, A.A.; Radahmadi, M.; Javanmard, S.H.; Reisi, P. The effects of doxepin on stress-induced learning, memory impairments, and TNF-α level in the rat hippocampus. Res. Pharm. Sci. 2016, 10, 460–465. [Google Scholar]
- Chiriță, C.; Ștefănescu, E.; Zbârcea, C.; Mireșan, H.; Negreș, S.; Nuță, D.; Limban, C.; Miulescu, R.; Marineci, C. Experimental pharmacological research regarding the antidepressant effect of associating doxepin and selegiline in normal mice. J. Mind Med Sci. 2019, 6, 261–270. [Google Scholar] [CrossRef]
- Liu, R.; Li, X.; Godbout, R. A novel fatty acid-binding protein (FABP) gene resulting from tandem gene duplication in mammals: Transcription in rat retina and testis. Genomics 2008, 92, 436–445. [Google Scholar] [CrossRef] [Green Version]
- Mkhwanazi, B.N.; Serumula, M.R.; Myburg, R.B.; Van Heerden, F.R.; Musabayane, C.T. Antioxidant effects of maslinic acid in livers, hearts and kidneys of streptozotocin-induced diabetic rats: Effects on kidney function. Ren. Fail. 2013, 36, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Chen, M.-Y.; Liang, W.; Chen, Y.; Guo, M.-Y. Zinc Deficiency Aggravation of ROS and Inflammatory Injury Leading to Renal Fibrosis in Mice. Biol. Trace Elem. Res. 2020, 199, 1–11. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Control | Doxepin |
|---|---|---|
| RWAT | ||
| Adipocyte diameter | ||
| 0–50 μm (%) | 20.00 ± 0.65 | 0 ± 0 *** |
| 50–100 μm (%) | 76.36 ± 1.25 | 10.91 ± 1.07 *** |
| 100–150 μm (%) | 3.64 ± 0.42 | 50.91 ± 1.12 *** |
| >150 μm (%) | 0 ± 0 | 38.18 ± 0.96 *** |
| EWAT | ||
| Adipocyte diameter | ||
| 0–50 μm (%) | 5.45 ± 0.47 | 0 ± 0 *** |
| 50–100 μm (%) | 69.1 ± 0.94 | 12.73 ± 0.57 *** |
| 100–150 μm (%) | 25.45 ± 1.03 | 36.36 ± 1.69 *** |
| >150 μm (%) | 0 ± 0 | 50.91 ± 1.42 *** |
| Variable | Control | Doxepin |
|---|---|---|
| Chromium intake/mouse/week (μg) | 19.37 ± 0.51 | 27.28 ± 0.82 ** |
| Blood (ng/mL) | 162.16 ± 7.67 | 90.43 ± 6.74 *** |
| Bone (ng/g) | 367.19 ± 10.42 | 172.13 ± 8.41 *** |
| Liver (ng/g) | 70.24 ± 6.85 | 51.39 ± 3.82 ** |
| Muscle (ng/g) | 50.69 ± 4.72 | 38.54 ± 3.25 ** |
| Epididymal fat pads (ng/g) | 45.31 ± 2.08 | 34.65 ± 1.94 ** |
| Kidney (ng/g) | 99.78 ± 2.81 | 181.63 ± 3.34 *** |
| Urine (ng/mL) | 56.73± 2.03 | 105.52 ± 2.55 *** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, G.-R.; Hou, P.-H.; Yang, W.-C.; Wang, C.-M.; Fan, P.-S.; Liao, H.-J.; Chen, T.-P. Doxepin Exacerbates Renal Damage, Glucose Intolerance, Nonalcoholic Fatty Liver Disease, and Urinary Chromium Loss in Obese Mice. Pharmaceuticals 2021, 14, 267. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14030267
Chang G-R, Hou P-H, Yang W-C, Wang C-M, Fan P-S, Liao H-J, Chen T-P. Doxepin Exacerbates Renal Damage, Glucose Intolerance, Nonalcoholic Fatty Liver Disease, and Urinary Chromium Loss in Obese Mice. Pharmaceuticals. 2021; 14(3):267. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14030267
Chicago/Turabian StyleChang, Geng-Ruei, Po-Hsun Hou, Wei-Cheng Yang, Chao-Min Wang, Pei-Shan Fan, Huei-Jyuan Liao, and To-Pang Chen. 2021. "Doxepin Exacerbates Renal Damage, Glucose Intolerance, Nonalcoholic Fatty Liver Disease, and Urinary Chromium Loss in Obese Mice" Pharmaceuticals 14, no. 3: 267. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14030267