
Marine-Derived Natural Products as ATP-Competitive mTOR Kinase Inhibitors for Cancer Therapeutics

 ,
,  ,
, 
Abstract
:
1. Introduction
2. Results
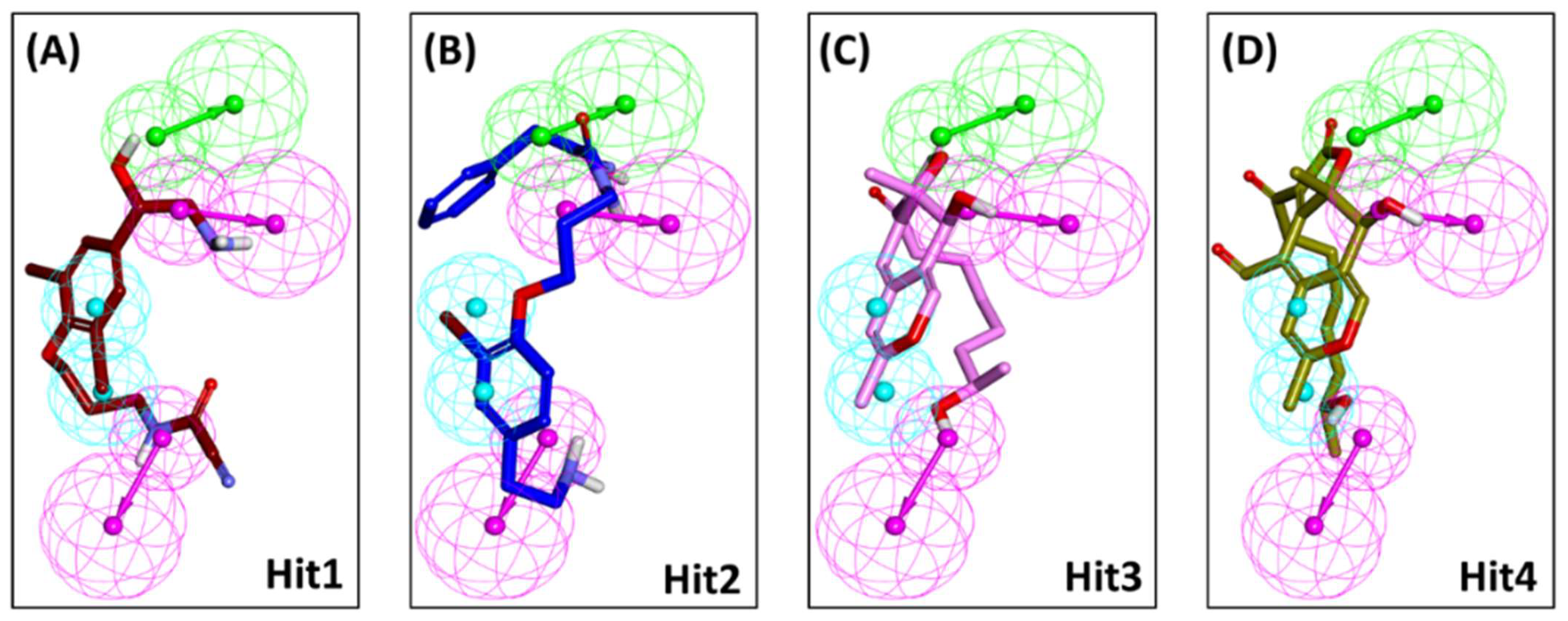
2.1. Structure-Based Pharmacophore Model
2.2. Decoy Set Validation of the Structure-Based Pharmacophore Model
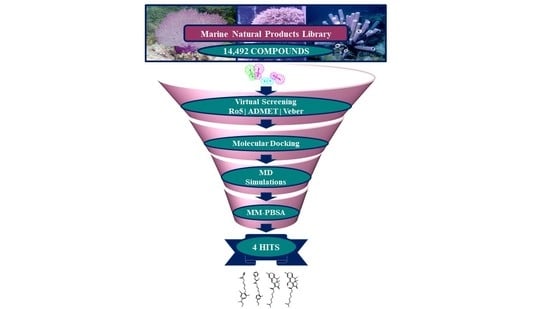
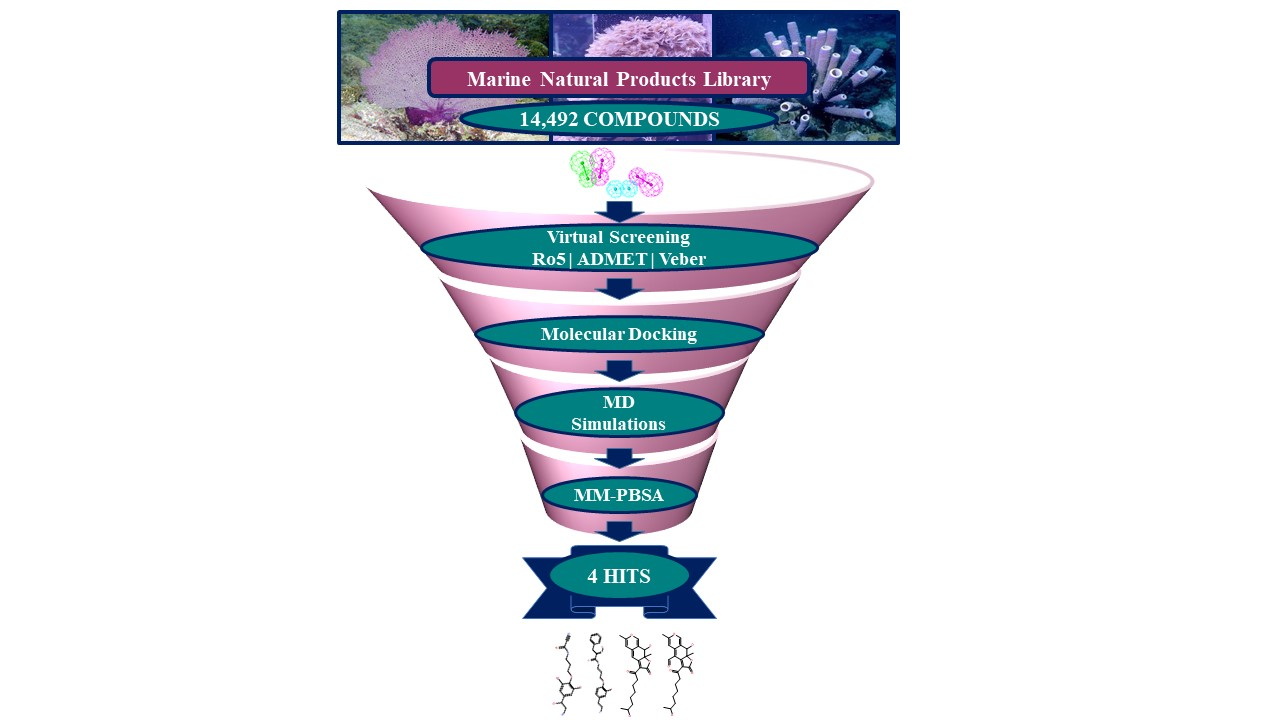
2.3. Drug-Like Marine Compounds Retrieved by Virtual Screening
2.4. Molecular Docking of Retrieved Marine Drug-Like Compounds with mTOR Kinase
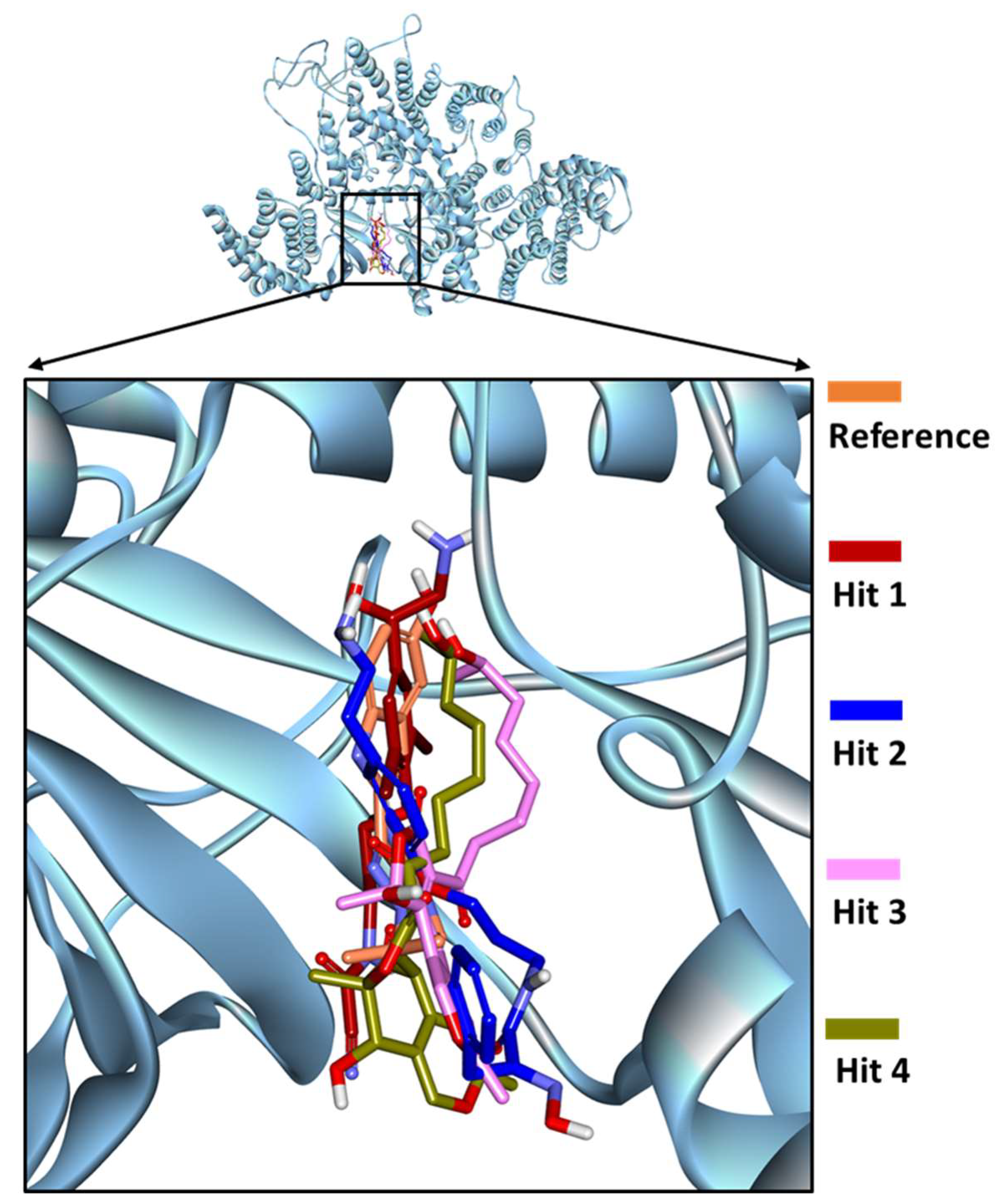
2.5. Binding Mode and Binding Free Energy Analysis of Identified Marine Compounds by Molecular Dynamics Simulations
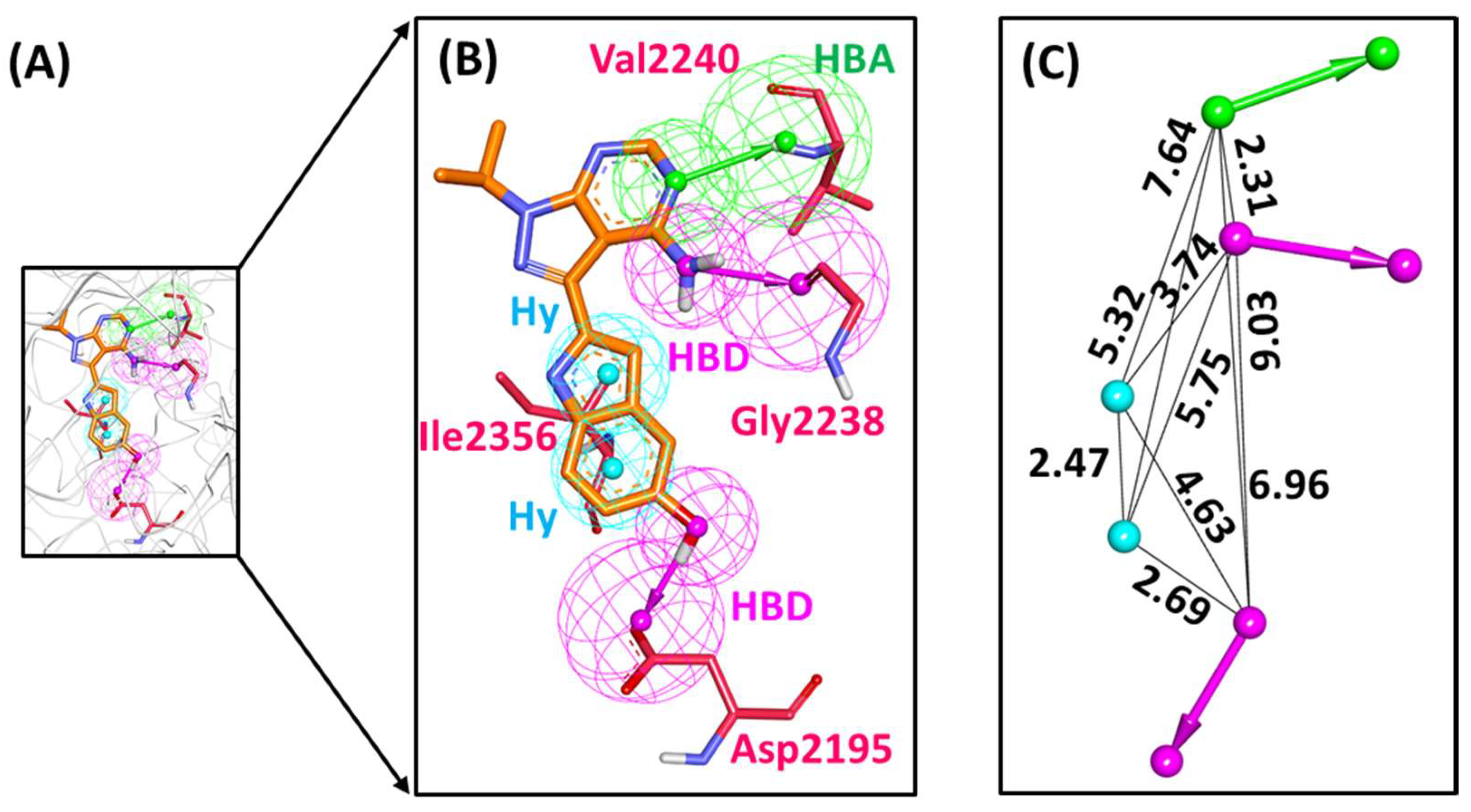
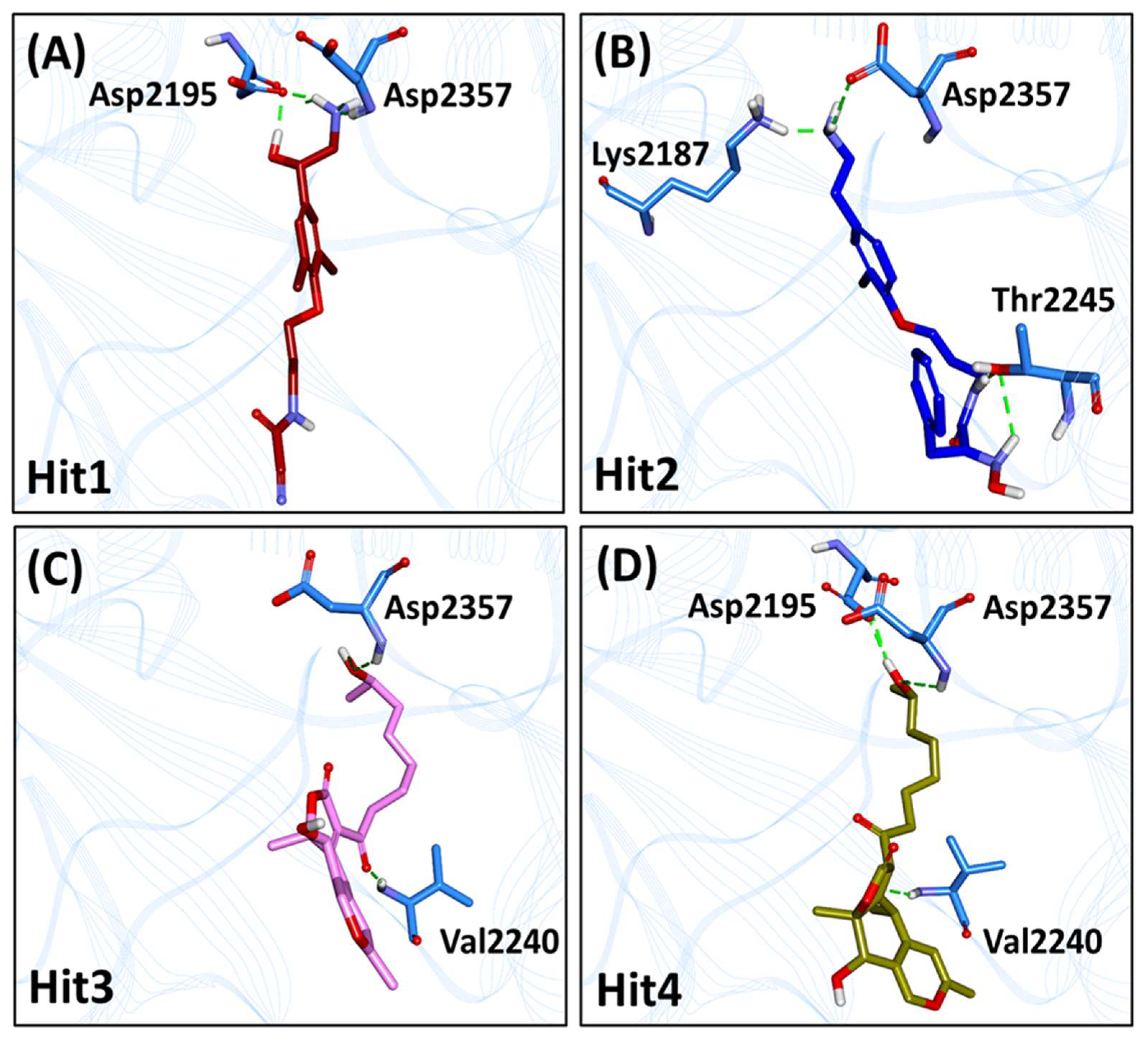
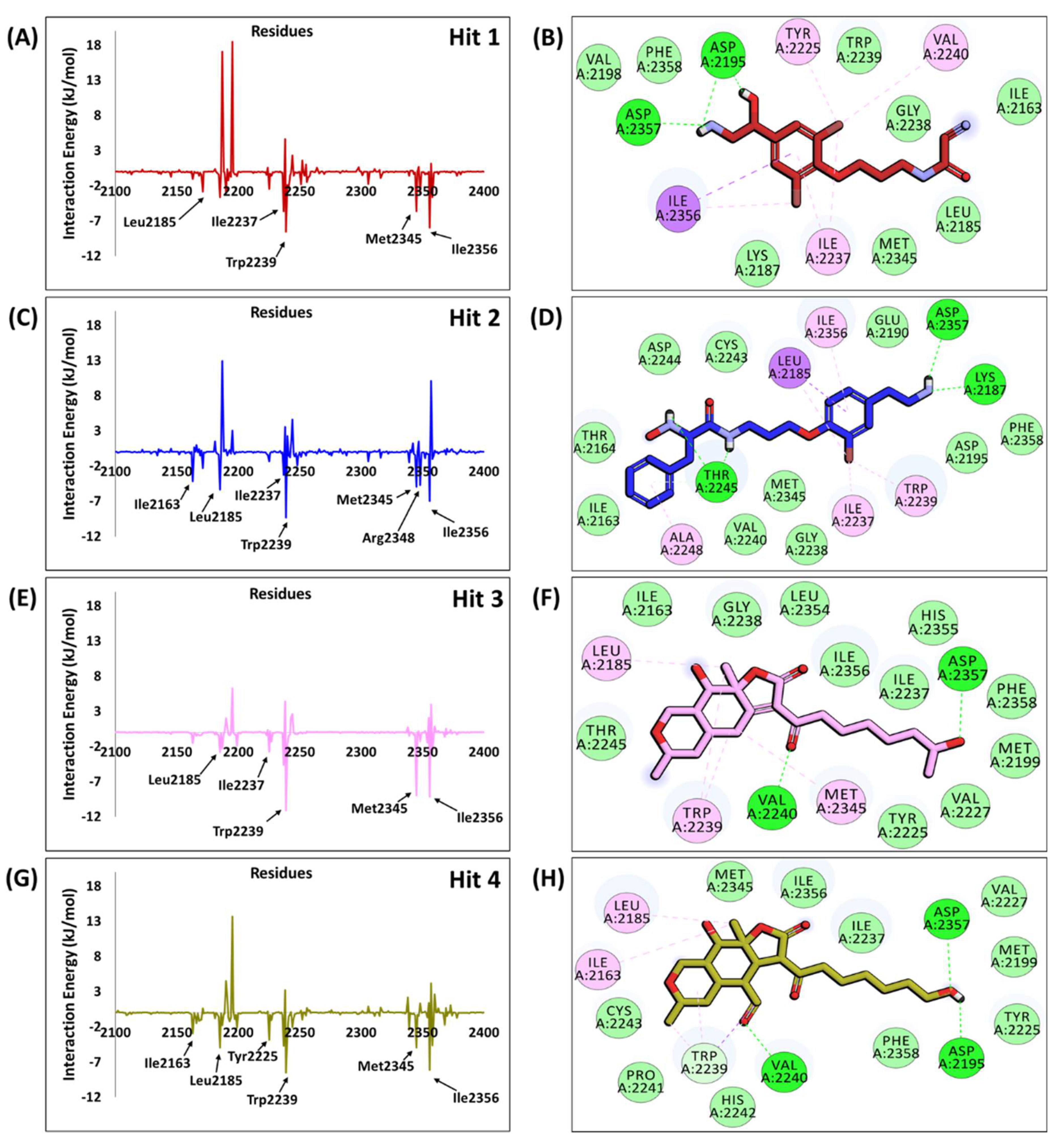
2.6. Characteristic Binding Interaction and Binding Free Energy Analysis of the Confirmed Marine Hits with mTOR ATP-Binding Pocket Residues
2.6.1. mTOR-Hit1 Interaction
2.6.2. mTOR-Hit2 Interaction
2.6.3. mTOR-Hit3 Interaction
2.6.4. mTOR-Hit4 Interaction
2.7. Evaluation of Drug-Likeness, ADME and Toxicity Properties of Identified mTOR Hits
2.8. Novelty and Source Documentation of Identified mTOR Inhibitors
3. Discussion
4. Materials and Methods
4.1. Structure-Based Pharmacophore Model Generation
4.2. Validation of Generated Pharmacophore Model
4.3. Virtual Screening of Marine Natural Product Library
4.4. Molecular Docking of Drug-Like Compounds with mTOR Kinase Domain
4.5. Molecular Dynamics Simulation of Identified Hits
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, Y.C.; Guan, K.L. MTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, A.C. Targeting the PI3K/Akt/mTOR pathway in non-small cell lung cancer (NSCLC). Thorac. Cancer 2020, 11, 511–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fattahi, S.; Amjadi-Moheb, F.; Tabaripour, R.; Ashrafi, G.H.; Akhavan-Niaki, H. PI3K/AKT/mTOR signaling in gastric cancer: Epigenetics and beyond. Life Sci. 2020, 262, 118513. [Google Scholar] [CrossRef]
- Ediriweera, M.K.; Tennekoon, K.H.; Samarakoon, S.R. Role of the PI3K/AKT/mTOR signaling pathway in ovarian cancer: Biological and therapeutic significance. Semin. Cancer Biol. 2019, 59, 147–160. [Google Scholar] [CrossRef]
- Tirrò, E.; Martorana, F.; Romano, C.; Vitale, S.R.; Motta, G.; Di Gregorio, S.; Massimino, M.; Pennisi, M.S.; Stella, S.; Puma, A.; et al. Molecular alterations in thyroid cancer: From bench to clinical practice. Genes 2019, 10, 709. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; Li, X.; Zhang, J. mTOR signaling in cancer and mtor inhibitors in solid tumor targeting therapy. Int. J. Mol. Sci. 2019, 20, 755. [Google Scholar] [CrossRef] [Green Version]
- Guertin, D.A.; Sabatini, D.M. Defining the Role of mTOR in Cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meric-Bernstam, F.; Gonzalez-Angulo, A.M. Targeting the mTOR signaling network for cancer therapy. J. Clin. Oncol. 2009, 27, 2278–2287. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, D.M. mTOR and cancer: Insights into a complex relationship. Nat. Rev. Cancer 2006, 6, 729–734. [Google Scholar] [CrossRef]
- Abraham, R.T.; Gibbons, J.J. The mammalian target of rapamycin signaling pathway: Twists and turns in the road to cancer therapy. Clin. Cancer Res. 2007, 13, 3109–3114. [Google Scholar] [CrossRef] [Green Version]
- Kim, L.C.; Cook, R.S.; Chen, J. mTORC1 and mTORC2 in cancer and the tumor microenvironment. Oncogene 2017, 36, 2191–2201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Jiang, Y. mTOR inhibitors at a glance. Mol. Cell. Pharmacol. 2015, 7, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Faes, S.; Demartines, N.; Dormond, O. Resistance to mTORC1 Inhibitors in Cancer Therapy: From Kinase Mutations to Intratumoral Heterogeneity of Kinase Activity. Oxid. Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.Y.; Huang, S. Le Current development of the second generation of mTOR inhibitors as anticancer agents. Chin. J. Cancer 2012, 31, 8–18. [Google Scholar]
- Rodrik-Outmezguine, V.S.; Okaniwa, M.; Yao, Z.; Novotny, C.J.; McWhirter, C.; Banaji, A.; Won, H.; Wong, W.; Berger, M.; De Stanchina, E.; et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature 2016, 534, 272–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flemming, A. Cancer: Bivalent mTOR inhibitors—The next generation. Nat. Rev. Drug Discov. 2016, 15, 455. [Google Scholar] [CrossRef]
- Xu, J.; Tian, D. Hematologic toxicities associated with mTOR inhibitors temsirolimus and everolimus in cancer patients: A systematic review and meta-analysis. Curr. Med. Res. Opin. 2014, 30, 67–74. [Google Scholar] [CrossRef]
- Calvo, A.S.; Rochefort, J.; Javelot, M.J.; Descroix, V.; Lescaille, G. Management of mTOR inhibitors oral mucositis: Current state of knowledge. J. Oral Med. Oral Surg. 2019, 25, 11. [Google Scholar] [CrossRef] [Green Version]
- Bauer, R.A.; Wurst, J.M.; Tan, D.S. Expanding the range of “druggable” targets with natural product-based libraries: An academic perspective. Curr. Opin. Chem. Biol. 2010, 14, 308–314. [Google Scholar] [CrossRef] [Green Version]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Wang, L.; Gu, Q.; Xu, J. An in silico protocol for identifying mTOR inhibitors from natural products. Mol. Divers. 2014, 18, 841–852. [Google Scholar] [CrossRef]
- Park, H.; Choe, H.; Hong, S. Virtual screening and biochemical evaluation to identify new inhibitors of mammalian target of rapamycin (mTOR). Bioorganic Med. Chem. Lett. 2014, 24, 835–838. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Torres, V.; Losada-Echeberría, M.; Herranz-López, M.; Barrajón-Catalán, E.; Galiano, V.; Micol, V.; Encinar, J.A. New mammalian target of rapamycin (mTOR) modulators derived from natural product databases and marine extracts by using molecular docking techniques. Mar. Drugs 2018, 16, 385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves, C.; Silva, J.; Pinteus, S.; Gaspar, H.; Alpoim, M.C.; Botana, L.M.; Pedrosa, R. From marine origin to therapeutics: The antitumor potential of marine algae-derived compounds. Front. Pharmacol. 2018, 9, 777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez, P.C.; Wilke, D.V.; Costa-Lotufo, L.V. Marine drugs for cancer: Surfacing biotechnological innovations from the oceans. Clinics 2018, 73, e482s. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Honecker, F. Marine compounds and cancer: 2017 updates. Mar. Drugs 2018, 16, 41. [Google Scholar] [CrossRef] [Green Version]
- Sarfaraj, H.M.; Sheeba, F.; Saba, A.; Khan, M.S. Marine natural products: A lead for anti-cancer. Indian J. Geo-Marine Sci. 2012, 41, 27–39. [Google Scholar]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2019, 36, 122–173. [Google Scholar] [CrossRef] [Green Version]
- Blunt, J.W.; Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2018, 35, 8–53. [Google Scholar] [CrossRef] [Green Version]
- Cragg, G.M.; Pezzuto, J.M. Natural Products as a Vital Source for the Discovery of Cancer Chemotherapeutic and Chemopreventive Agents. Med. Princ. Pract. 2016, 25, 41–59. [Google Scholar] [CrossRef]
- Lin, Y.; Qi, X.; Liu, H.; Xue, K.; Xu, S.; Tian, Z. The anti-cancer effects of fucoidan: A review of both in vivo and in vitro investigations. Cancer Cell Int. 2020, 20, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ciftci, H.I.; Can, M.; Ellakwa, D.E.; Suner, S.C.; Ibrahim, M.A.; Oral, A.; Sekeroglu, N.; Özalp, B.; Otsuka, M.; Fujita, M.; et al. Anticancer activity of Turkish marine extracts: A purple sponge extract induces apoptosis with multitarget kinase inhibition activity. Investig. New Drugs 2020, 38, 1326–1333. [Google Scholar] [CrossRef] [PubMed]
- Kauffmann-Guerrero, D.; Huber, R.M. Orphan drugs in development for the treatment of small-cell lung cancer: Emerging data on lurbinectedin. Lung Cancer Targets Ther. 2020, 11, 27–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauritano, C.; Martínez, K.A.; Battaglia, P.; Granata, A.; de la Cruz, M.; Cautain, B.; Martín, J.; Reyes, F.; Ianora, A.; Guglielmo, L. First evidence of anticancer and antimicrobial activity in Mediterranean mesopelagic species. Sci. Rep. 2020, 10, 4929. [Google Scholar] [CrossRef]
- Khanfar, M.A.; Taha, M.O. Elaborate ligand-based modeling coupled with multiple linear regression and k nearest neighbor QSAR analyses unveiled new nanomolar mTOR inhibitors. J. Chem. Inf. Model. 2013, 53, 2587–2612. [Google Scholar] [CrossRef]
- Fu, X.; Schmitz, F.J. 7-Hydroxyceratinamine, a new cyanoformamide-containing metabolite from a sponge, Aplysinella sp. J. Nat. Prod. 1999, 62, 1072–1073. [Google Scholar] [CrossRef]
- Dictionary of Alkaloids with CD-ROM—Google Books. Available online: https://books.google.co.kr/books?id=mynNBQAAQBAJ&pg=PA1601&dq=149636-93-1&hl=en&sa=X&ved=2ahUKEwjdjN36y7vuAhUBc3AKHZyGC8sQ6AEwAHoECAAQAg#v=onepage&q=149636-93-1&f=false (accessed on 27 January 2021).
- Caraballo Rodriguez, A.M. Acesso a Produtos Naturais Mediante a Estrategia de Cultivos Mistos de Endofiticos: O Fungo Colletotrichum boninense FLe 8.1 e a Actinobacteria Streptomyces albospinus Rle 7. Master’s Thesis, Universidade de São Paulo, São Paulo, Brazil, 2013. [Google Scholar] [CrossRef]
- Papadopoli, D.; Boulay, K.; Kazak, L.; Pollak, M.; Mallette, F.A.; Topisirovic, I.; Hulea, L. Mtor as a central regulator of lifespan and aging. F1000Research 2019, 8. [Google Scholar] [CrossRef]
- Tuo, Y.; Xiang, M. mTOR: A double-edged sword for diabetes. J. Leukoc. Biol. 2019, 106, 385–395. [Google Scholar] [CrossRef]
- Costa, R.L.B.; Han, H.S.; Gradishar, W.J. Targeting the PI3K/AKT/mTOR pathway in triple-negative breast cancer: A review. Breast Cancer Res. Treat. 2018, 169, 397–406. [Google Scholar] [CrossRef]
- Marquard, F.E.; Jücker, M. PI3K/AKT/mTOR signaling as a molecular target in head and neck cancer. Biochem. Pharmacol. 2020, 172, 113729. [Google Scholar] [CrossRef]
- Shorning, B.Y.; Dass, M.S.; Smalley, M.J.; Pearson, H.B. The PI3K-AKT-mTOR pathway and prostate cancer: At the crossroads of AR, MAPK, and WNT signaling. Int. J. Mol. Sci. 2020, 21, 4507. [Google Scholar] [CrossRef] [PubMed]
- Arachchige Don, A.S.; Zheng, F.S.X. Recent Clinical Trials of mTOR-Targeted Cancer Therapies. Rev. Recent Clin. Trials 2010, 6, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Gridelli, C.; Maione, P.; Rossi, A. The Potential Role of mTOR Inhibitors in Non-Small Cell Lung Cancer. Oncologist 2008, 13, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Rath, B.; Hochmair, M.; Plangger, A.; Hamilton, G. Anticancer activity of fascaplysin against lung cancer cell and small cell lung cancer circulating tumor cell lines. Mar. Drugs 2018, 16, 383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schenone, S.; Brullo, C.; Musumeci, F.; Radi, M.; Botta, M. ATP-Competitive Inhibitors of mTOR: An Update. Curr. Med. Chem. 2011, 18, 2995–3014. [Google Scholar] [CrossRef]
- Rana, R.M.; Rampogu, S.; Bin Abid, N.; Zeb, A.; Parate, S.; Lee, G.; Yoon, S.; Kim, Y.; Kim, D.; Woo Lee, K. In silico study identified methotrexate analog as potential inhibitor of drug resistant human dihydrofolate reductase for cancer therapeutics. Molecules 2020, 25, 3510. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Wang, L. Discovery and Development of ATP-Competitive mTOR Inhibitors Using Computational Approaches. Curr. Pharm. Des. 2017, 23, 4321–4331. [Google Scholar] [CrossRef]
- Naveed, M. Molecular Docking and Pharmacokinetic of Highly Specific Novel Pan-Mtor Inhibitors against Solid Tumors. MOJ Proteom. Bioinform. 2017, 5, 13–16. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Chen, L.; Yu, M.; Xu, L.H.; Cheng, B.; Lin, Y.S.; Gu, Q.; He, X.H.; Xu, J. Discovering new mTOR inhibitors for cancer treatment through virtual screening methods and in vitro assays. Sci. Rep. 2016, 6, 18987. [Google Scholar] [CrossRef] [Green Version]
- Khanfar, M.A.; Abukhader, M.M.; Alqtaishat, S.; Taha, M.O. Pharmacophore modeling, homology modeling, and in silico screening reveal mammalian target of rapamycin inhibitory activities for sotalol, glyburide, metipranolol, sulfamethizole, glipizide, and pioglitazone. J. Mol. Graph. Model. 2013, 42, 39–49. [Google Scholar] [CrossRef]
- Wu, F.; Hou, X.; Luo, H.; Zhou, M.; Zhang, W.; Ding, Z.; Li, R. Exploring the selectivity of PI3Kα and mTOR inhibitors by 3D-QSAR, molecular dynamics simulations and MM/GBSA binding free energy decomposition. MedChemComm 2013, 4, 1482–1496. [Google Scholar] [CrossRef]
- Tanneeru, K.; Guruprasad, L. Ligand-based 3-D pharmacophore generation and molecular docking of mTOR kinase inhibitors. J. Mol. Model. 2012, 18, 1611–1624. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. MTOR kinase structure, mechanism and regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Wang, J.; Kang, S.A.; Thoreen, C.C.; Hur, W.; Ahmed, T.; Sabatini, D.M.; Gray, N.S. Discovery of 9-(6-aminopyridin-3-yl)-1-(3-(trifluoromethyl)phenyl)benzo[h ][1,6]naphthyridin-2(1 H)-one (torin2) as a potent, selective, and orally available mammalian target of rapamycin (mTOR) inhibitor for treatment of cancer. J. Med. Chem. 2011, 54, 1473–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rampogu, S.; Parate, S.; Parameswaran, S.; Park, C.; Baek, A.; Son, M.; Park, Y.; Park, S.J.; Lee, K.W. Natural compounds as potential Hsp90 inhibitors for breast cancer-Pharmacophore guided molecular modelling studies. Comput. Biol. Chem. 2019, 83, 107113. [Google Scholar] [CrossRef]
- Feldman, M.E.; Apsel, B.; Uotila, A.; Loewith, R.; Knight, Z.A.; Ruggero, D.; Shokat, K.M. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009, 7, e1000038. [Google Scholar] [CrossRef]
- Lin, S.-K. Pharmacophore Perception, Development and Use in Drug Design. Edited by Osman F. Güner. Molecules 2000, 5, 987–989. [Google Scholar] [CrossRef]
- Zeb, A.; Kim, D.; Alam, S.I.; Son, M.; Kumar, R.; Rampogu, S.; Parameswaran, S.; Shelake, R.M.; Rana, R.M.; Parate, S.; et al. Computational Simulations Identify Pyrrolidine-2,3-Dione Derivatives as Novel Inhibitors of Cdk5/p25 Complex to Attenuate Alzheimer’s Pathology. J. Clin. Med. 2019, 8, 746. [Google Scholar] [CrossRef] [Green Version]
- Rampogu, S.; Park, C.; Ravinder, D.; Son, M.; Baek, A.; Zeb, A.; Bavi, R.; Kumar, R.; Lee, G.; Parate, S.; et al. Pharmacotherapeutics and molecular mechanism of phytochemicals in alleviating hormone-responsive breast cancer. Oxid. Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rampogu, S.; Baek, A.; Park, C.; Parate, S.; Parameswaran, S.; Park, Y.; Shaik, B.; Kim, J.H.; Park, S.J.; Lee, K.W.; et al. Discovery of Small Molecules that Target Vascular Endothelial Growth Factor Receptor-2 Signalling Pathway Employing Molecular Modelling Studies. Cells 2019, 8, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins Struct. Funct. Bioinform. 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Lopes, P.E.M.; Mackerell, A.D., Jr. Recent developments and applications of the CHARMM force fields. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 167–185. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A.; Open Source Drug Discovery Consortium. G-mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pharmacophore Models | Number of Features | Feature Set * | Selectivity Score |
|---|---|---|---|
| Pharmacophore_01 | 5 | ADDHH | 9.2973 |
| Pharmacophore_02 | 4 | DDHH | 7.7825 |
| Pharmacophore_03 | 4 | ADDH | 7.7825 |
| Pharmacophore_04 | 4 | ADDH | 7.7825 |
| Pharmacophore_05 | 4 | ADHH | 6.8689 |
| Pharmacophore_06 | 4 | ADHH | 6.8689 |
| S. No. | Parameters | Values |
|---|---|---|
| 1 | Total number of compounds in the database (D) | 300 |
| 2 | Total number of active compounds in the database (A) | 50 |
| 3 | Total number of hits retrieved by pharmacophore model from the database (Ht) | 61 |
| 4 | Total number of active compounds in the hit list (Ha) | 49 |
| 5 | % Yield of active ((Ha/Ht) × 100) | 80 |
| 6 | % Ratio of actives ((Ha/A) × 100) | 98 |
| 7 | False negatives (A-Ha) | 1 |
| 8 | False positives (Ht-Ha) | 12 |
| 9 | Goodness of fit score (GF) | 0.80 |
| Compound No. | MNP ID (CAS No *) | Gold Score | Hydrogen Bond Interactions | Hydrophobic and van der Waals Interactions |
|---|---|---|---|---|
| 1 (MNP1) | 200936-85-2 | 65.48 | Val2240, Asp2357 | Leu2185, Lys2187, Leu2192, Asp2195, Tyr2225, Val2227, Ile2237, Gly2238, Trp2239, His2242, Cys2243, Asp2244, Thr2245, Met2345, Arg2348, Ile2356, Phe2358 |
| 2 (MNP2) | 230295-94-0 | 65.41 | Asp2195, Trp2239, Val2240 | Leu2185, Lys2187, Leu2192, Met2199, Tyr2225, Val2227, Ile2237, Pro2241, His2242, Cys2243, Met2345, Arg2348, Ile2356, Asp2357, Phe2358 |
| 3 (MNP3) | 149636-93-1 | 64.72 | Trp2239, Val2240 | Leu2185, Lys2187, Glu2190, Leu2192, Asp2195, Tyr2225, Ile2237, Trp2239, Val2240, Pro2241, His2242, Cys2243, Met2345, Arg2348, Ile2356, Asp2357, Phe2358 |
| 4 (MNP4) | 200936-84-1 | 63.75 | Asp2195, Val2240, Asp2357 | Ile2163, Leu2185, Leu2192, Met2199, Tyr2225, Val2227, Ile2237, Trp2239, Pro2241, His2242, Cys2243, Asp2244, Thr2245, Met2345, Arg2348, Ile2356 |
| 5 (PP242) | Reference (1092351-67-1) | 63.20 | Asp2195, Gly2238, Val2240 | Ile2163, Leu2185, Lys2187, Met2199, Tyr2225, Ile2237, Trp2239, Cys2243, Thr2245, Met2345, Ile2356, Asp2357, Phe2358 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parate, S.; Kumar, V.; Lee, G.; Rampogu, S.; Hong, J.C.; Lee, K.W. Marine-Derived Natural Products as ATP-Competitive mTOR Kinase Inhibitors for Cancer Therapeutics. Pharmaceuticals 2021, 14, 282. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14030282
Parate S, Kumar V, Lee G, Rampogu S, Hong JC, Lee KW. Marine-Derived Natural Products as ATP-Competitive mTOR Kinase Inhibitors for Cancer Therapeutics. Pharmaceuticals. 2021; 14(3):282. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14030282
Chicago/Turabian StyleParate, Shraddha, Vikas Kumar, Gihwan Lee, Shailima Rampogu, Jong Chan Hong, and Keun Woo Lee. 2021. "Marine-Derived Natural Products as ATP-Competitive mTOR Kinase Inhibitors for Cancer Therapeutics" Pharmaceuticals 14, no. 3: 282. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14030282