Solvents and reagents were obtained from commercial suppliers and were used without further purification. All solvents used were of pure analytical grade. Reactions that needed anhydrous conditions were carried out in flame-dried glassware under a positive pressure of dry N2 using the Schlenk line technique. Column chromatography was carried out using silica gel 40–60 μM. Reaction progress was monitored by TLC using fluorescent pre-coated silica gel plates, and detection of the components was made by short UV light (λ = 254 nm); carbamates were detected using freshly prepared furfural/sulfuric acid detection reagent. Proton nuclear magnetic resonance (1H-NMR spectra and 13C-NMR spectra) were recorded using a Bruker Fourier 300 using Varian Mercury 400 Plus. All spectra were determined in the solvents indicated, and chemical shifts were reported in δ units. Solvent signals were δ 2.50 for Dimethyl sulfoxide (DMSO-d6) and δ7.2 for Chloroform (CDCl3). 13C shifts were referenced to the deuterated solvent signal δ 39.5 for DMSO-d6 and δ 77.0 for CDCl3. Coupling constants (J) are given in hertz (Hz). Multiplicities patterns were as follows: s: singlet; d: doublet; t: triplet; q: quartet; m: multiplet; dd:doublet of doublet; ddd: doublet of doublet of doublet. All NMR spectra were analyzed using MestReNova software version 6.0.2–5475. Mass spectrometric analysis (UPLC-ESI-MS) was performed using Waters ACQUITY Xevo TQD system, which consisted of an ACQUITY UPLC H-Class system and XevoTM TQD triple-quadrupole tandem mass spectrometer with an electrospray ionization (ESI) interface (Waters Corp., Milford, MA, USA). Acquity BEH C18 100 mm × 2.1 mm column (particle size, 1.7 µm) was used to separate analytes (Waters, Ireland). The solvent system consisted of water containing 0.1% TFA (A) and 0.1% TFA in acetonitrile (B). HPLC-method: flow rate 200 μL/min. The percentage of B started at an initial of 5% and maintained for 1 minute, then increased up to 100% during 10 min, kept at 100% for 2 min, and flushed back to 5% in 3 min then kept at 5% for 1 min. The MS scan was carried out at the following conditions: capillary voltage 3.5 kV, cone voltage 20 V, radio frequency (RF) lens voltage 2.5 V, source temperature 150 °C, and desolvation gas temperature 500 °C. Nitrogen was used as the desolvation and cone gas at a flow rate of 1000 and 20 L/h, respectively. System operation and data acquisition were controlled using Mass Lynx 4.1 software (Waters). The purities of the tested compounds were determined by HPLC coupled with mass spectrometry and were higher than 95% purity unless otherwise stated (purity of compound 18 was >90%). Measurements for a melting point were not changed and were done using capillary Büchi B-540 melting point apparatus.
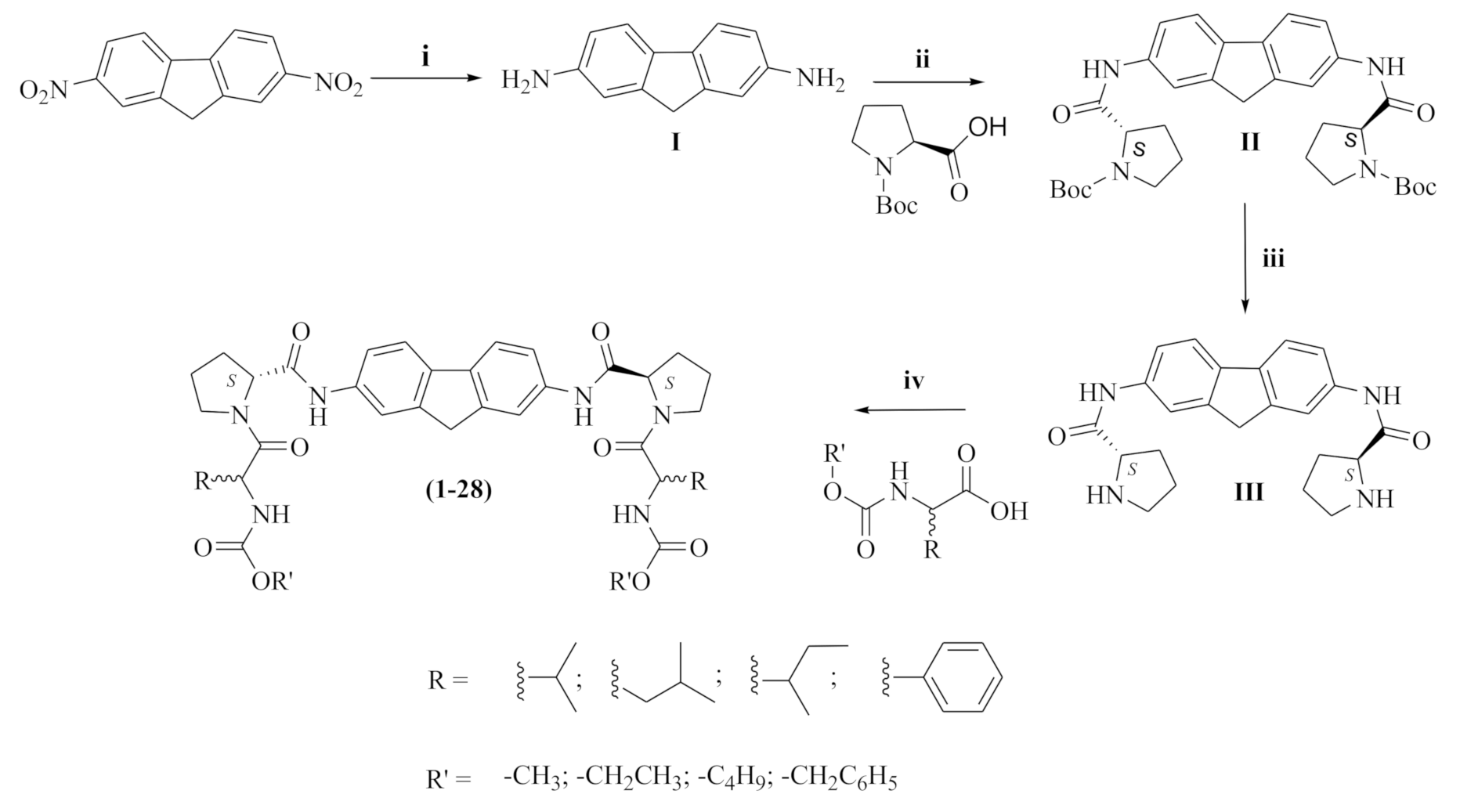
3.1.5. General Procedure for Preparation of Compounds (1–32)
HBTU (1 mmol, 1.554 gm) was added in one portion to the respective amino acid carbamate (3 mmol) and TEA (2 mmol, 0.571 mL), the mixture was stirred for 15 min in DMF under ice-cooling. Compounds III and V (1 mmol) were added portion-wise to the above mixture. The reaction mixture was left to stir at room temperature for 3 h. The crude product was purified by silica gel column chromatography using a mixture of DCM: MeOH.
Di-tert-butyl3,3’-(((9H-fluorene-2,7diyl)bis(azanediyl))bis(carbonyl))bis(piperidine-1-carboxylate) (IV)
Dark red Oil; Yield 41%; 1H NMR (400 MHz, DMSO-d6) δ 9.77 (s, 2H), 7.61 (s, 2H), 7.44 (d, J = 8.2 Hz, 2H), 7.26 (d, J = 8.2 Hz, 2H), 3.69 (d, J = 48.0 Hz, 4H), 3.59 (s, 2H), 2.22 (d, J = 13.7 Hz, 4H), 2.19 (d, J = 10.9 Hz, 2H), 1.72–1.44 (m, 4H), 1.34 (dd, J = 23.9, 12.2 Hz, 4H), 1.14 (s, 18H); 13C NMR (101 MHz, DMSO-d6) δ 172.19, 154.34, 143.95, 138.05, 136.74, 119.94, 118.39, 116.45, 85.18, 60.69, 59.11, 37.04, 28.04, 24.68, 24.54, and 22.89; MS (ESI) m/z: 619 [M + H] +.
N, N’-(9H-fluorene-2,7-diyl)bis(piperidine-3-carboxamide) (V)
Dark red oil; Yield 51%; 1H NMR (400 MHz, DMSO-d6) δ 8.24 (s,2H), 7.91 (s, 2H), 7.69 (d, J = 8.4 Hz, 2H), 7.54–7.52 (m, 2H), 3.48 (s, 2H), 2.79 (d, J = 2.6 Hz, 4H), 2.71–2.69 (m, 4H), 2.58–2.55 (m, 2H), 1.90–1.85 (m, 4H), 1.71–1.67 (m, 2H), 1.48–1.41 (m, 4H); 13C NMR (101 MHz, DMSO-d6) δ 159.46, 125.55, 122.08, 118.91, 115.91, 115.32, 112.95, 63.05, 60.43, 31.17, 24.24, 22.44, and 21.60; MS (ESI) m/z: 419 [M+ H] +.
Dimethyl((2S,2’S)-((2S,2’S)-(((9H-fluorene-2,7-diyl)bis(azanediyl))bis(carbonyl)) bis(pyrrolidine-2,1-diyl))bis(3-methyl-1-oxobutane-1,2-diyl))dicarbamate (1)
Yellow oil; Yield 20.6%; 1H NMR (500 MHz, DMSO-d6) δ 10.07 (s, 2H), 7.89 (s, 2H), 7.72–7.69 (m, 2H), 7.47 (d, J = 8.2 Hz, 2H), 7.33 (d, J = 8.4 Hz, 2H), 4.05 (t, J = 8.5 Hz, 2H), 3.86 (s, 2H), 3.66–3.61 (m, 2H), 3.53 (s, 6H), 2.19–2.13 (m, 6H), 1.97–1.87 (m, 8H), 0.90 (d, J = 6.6 Hz, 12H); 13C NMR (126 MHz, DMSO-d6) δ 170.85, 170.73, 157.26, 144.18, 138.11, 136.64, 119.96, 118.21, 116.28, 60.69, 58.43, 51.89, 47.71, 31.10, 29.98, 25.12, 19.39, and 19.12; MS (ESI) m/z: 705.35 [M+ H]+.
Diethyl((2S,2’S)-((2S,2’S)-(((9H-fluorene-2,7-diyl)bis(azanediyl))bis(carbonyl)) bis(pyrrolidine-2,1-diyl))bis(3-methyl-1-oxobutane-1,2-diyl))dicarbamate (2)
Yellow oil ; Yield 20%; 1H NMR (500 MHz, DMSO-d6) δ 10.06 (s, 2H), 7.89 (s, 2H), 7.70 (d, J = 8.3 Hz, 2H), 7.49–7.46 (m, 2H), 7.23 (d, J = 8.3 Hz, 2H), 4.48 (dd, J = 8.1, 4.8 Hz, 2H), 4.04 (t, J = 8.4 Hz, 2H), 4.01–3.97 (m, 4H), 3.87–3.80 (m, 4H), 3.68–3.60 (m, 2H), 2.18–2.12 (m, 2H), 1.97–1.88 (m, 8H), 1.16 (t, J = 7.1 Hz, 6H), 0.93 (dd, J = 31.3, 6.7 Hz, 12H); 13C NMR (126 MHz, DMSO-d6) δ 170.88, 170.75, 156.98, 144.02, 138.11, 136.64, 119.96, 118.21, 116.28, 60.70, 60.28, 58.33, 47.69, 38.71, 31.16, 25.14, 19.41, 19.11, and 15.11; MS (ESI) m/z: 733.38 [M+ H]+.
Dibutyl((2S,2’S)-((2S,2’S)-(((9H-fluorene-2,7-diyl)bis(azanediyl))bis(carbonyl))bis(pyrrolidine-2,1-diyl))bis(3-methyl-1-oxobutane-1,2-diyl))dicarbamate (3)
Yellow oil; Yield 23.5%; 1H NMR (500 MHz, DMSO-d6) δ 10.05 (s, 2H), 7.89 (s, 2H), 7.70 (d, J = 8.3 Hz, 2H), 7.48 (d, J = 8.2 Hz, 2H), 7.21 (d, J = 8.3 Hz, 2H), 4.48 (dd, J = 8.0, 4.5 Hz, 2H), 4.05 (t, J = 8.4 Hz, 2H), 3.93 (dt, J = 10.7, 5.4 Hz, 4H), 3.86 (s, 2H), 3.82 (d, J = 9.0 Hz, 2H), 3.64 (dd, J = 15.7, 6.3 Hz, 2H), 2.17 (dd, J = 12.8, 8.1 Hz, 2H), 2.00–1.85 (m, 8H), 1.56–1.47 (m, 4H), 1.35–1.28 (m, 4H), 0.96 (d, J = 6.6 Hz, 6H), 0.89 (t, J = 7.4 Hz, 12H); 13C NMR (101 MHz, DMSO-d6) δ 170.87, 170.73, 156.94, 144.01, 138.10, 136.65, 119.94, 118.23, 116.29, 64.09, 58.34, 55.36, 47.70, 38.70, 31.21, 31.13, 29.94, 25.14, 19.40, 19.07, and 14.07; MS (ESI) m/z: 789.45 [M+ H]+.
Dibenzyl ((2S,2’S)-((2S,2’S)-(((9H-fluorene-2,7- diyl)bis(azanediyl))bis(carbonyl)) bis(pyrrolidine-2,1-diyl))bis(3-methyl-1-oxobutane-1,2-diyl))dicarbamate (4)
Yellow oil; Yield 26%; 1H NMR (500 MHz, DMSO-d6) δ 10(s, 2H), 7.89 (s, 2H), 7.70 (d, J = 8.3 Hz, 2H), 7.46 (t, J = 8.4 Hz, 2H), 7.35 (s, 10H), 7.32 (dd, J = 5.6, 3.4 Hz, 2H), 5.03 (d, J = 8.0 Hz, 4H), 4.51–4.44 (m, 2H), 4.08 (t, J = 8.4 Hz, 2H), 3.87–3.82 (m, 4H), 3.63 (dd, J = 15.3, 6 Hz, 2H), 2.21–2.17 (m, 2H), 2.02–1.89 (m, 8H), 0.97 (d, J = 6.7 Hz, 12H); 13C NMR (101 MHz, DMSO-d6) δ 170.74, 170.73, 165.06, 156.70, 144.01, 138.09, 137.56, 136.65, 128.80, 128.11, 119.94, 118.23, 116.30, 65.86, 60.71, 58.46, 47.69, 38.70, 31.14, 25.11, 19.41, and 19.05; MS (ESI) m/z: 857.42 [M+ H]+.
Dimethyl ((2R,2’R)-((2S,2’S)-(((9H-fluorene-2,7-diyl)bis(azanediyl))bis(carbonyl)) bis(pyrrolidine-2,1-diyl))bis(3-methyl-1-oxobutane-1,2-diyl))dicarbamate (5)
Yellow oil; Yield 19%; 1H NMR (500 MHz, DMSO-d6) δ 9.72 (s, 2H), 7.88 (s, 2H), 7.71 (d, J = 8.2 Hz, 2H), 7.54 (d, J = 7.5 Hz, 2H), 7.39 (d, J = 8.1 Hz, 2H), 4.49–4.42 (m, 2H), 4.10 (t, J = 8.1 Hz, 2H), 3.85 (t, J = 10.4 Hz, 4H), 3.57 (s, 6H), 3.52–3.42 (m, 2H), 2.16–2.10 (m, 4H), 1.98–1.92 (m, 4H), 1.45 (s, 2H), 0.93–0.86 (m, 12H); 13C NMR (126 MHz, DMSO-d6) δ 172.26, 170.79, 157.51, 143.95, 136.77, 134.06, 119.97, 118.34, 116.48, 60.82, 58.54, 52.06, 47.54, 30.16, 27.32, 24.82, 19.50, and 18.84; MS (ESI) m/z: 705.35 [M+ H]+.
Diethyl((2R,2’R)-((2S,2’S)-(((9H-fluorene-2,7-diyl)bis(azanediyl))bis(carbonyl)) bis(pyrrolidine-2,1-diyl))bis(3-methyl-1-oxobutane-1,2-diyl))dicarbamate (6)
Yellow oil ; yield 24.3%; 1H NMR (500 MHz, DMSO-d6) δ 9.67 (s, 2H), 7.88 (s, 2H), 7.71 (d, J = 6.2 Hz, 2H), 7.55 (d, J = 8.2 Hz, 2H), 7.31 (d, J = 7.9 Hz, 2H), 4.45 (d, J = 6.5 Hz, 2H), 4.02–3.98 (m, 4H), 3.85 (s, 2H), 3.62 (dd, J = 16.6, 7.5 Hz, 2H), 3.00–2.95 (m, 2H), 2.19–2.08 (m, 4H), 1.99–1.89 (m, 8H), 1.19–1.11 (m, 12H), 0.91–0.85 (m, 6H); 13C NMR (126 MHz, DMSO-d6) δ 170.92, 170.67, 157.08, 143.92, 137.85, 136.80, 119.94, 118.40, 116.49, 60.51, 58.47, 47.56, 46.06, 38.71, 30.11, 24.73, 19.50, 15.08, and 9.48; MS (ESI) m/z: 733.38 [M+ H]+.
Dibutyl((2R,2’R)-((2S,2’S)-(((9H-fluorene-2,7-diyl)bis(azanediyl))bis(carbonyl)) bis(pyrrolidine-2,1-diyl))bis(3-methyl-1-oxobutane-1,2-diyl))dicarbamate (7)
Yellow oily ; Yield 18.5%; 1H NMR (500 MHz, DMSO-d6) δ 9.66 (s, 2H), 7.88 (d, J = 5.5 Hz, 2H), 7.71 (dd, J = 8.3, 2.3 Hz, 2H), 7.54 (d, J = 8.0 Hz, 2H), 7.32 (d, J = 8.0 Hz, 2H), 4.48–4.42 (m, 2H), 4.08 (t, J = 8.1 Hz, 2H), 3.98–3.90 (m, 4H), 3.87–3.80 (m, 4H), 3.62 (dd, J = 16.7, 7.5 Hz, 2H), 2.18–2.10 (m, 2H), 2.00–1.91 (m, 8H), 1.54–1.48 (m, 4H), 1.35–1.27 (m, 4H), 0.91–0.83 (m, 18H); 13C NMR (126 MHz, DMSO-d6) δ 170.95, 170.65, 165.06, 157.20, 143.97, 136.81, 119.91, 118.44, 116.51, 64.31, 60.86, 58.51, 47.56, 38.71, 31.19, 30.09, 24.73, 19.50, 19.02, 18.88, and 14.05; MS (ESI) m/z: 789.45 [M+ H]+.
Dibenzyl((2R,2’R)-((2S,2’S)-(((9H-fluorene-2,7-diyl)bis(azanediyl))bis(carbonyl)) bis(pyrrolidine-2,1-diyl))bis(3-methyl-1-oxobutane-1,2-diyl))dicarbamate (8)
Yellow oil ; Yield 22%; 1H NMR (500 MHz, DMSO-d6) δ 9.73 (s, 2H), 7.88 (d, J = 9.5 Hz, 2H), 7.73–7.68 (m, 2H), 7.53 (dd, J = 8.4, 3.6 Hz, 2H), 7.37–7.27 (m, 12H), 5.11–5.00 (m, 4H), 4.51–4.41 (m, 2H), 4.13 (t, J = 8.2 Hz, 2H), 3.83 (d, J = 11.2 Hz, 4H), 3.52–3.46 (m, 2H), 3.09 (qd, J = 7.3, 4.9 Hz, 2H), 1.99–1.92 (m, 4H), 1.17 (t, J = 7.3 Hz, 4H), 0.94–0.82 (m, 12H); 13C NMR (101 MHz, DMSO-d6) δ 170.75, 170.66, 156.90, 144.03, 143.86, 138.04, 137.43, 137.04, 128.90, 128.08, 119.93, 118.44, 116.50, 70.37, 65.98, 60.85, 58.61, 38.70, 30.13, 24.76, 19.51, and 18.83; MS (ESI) m/z: 857.42 [M+ H]+.
Dimethyl((2S,2’S)-((2S,2’S)-(((9H-fluorene-2,7-diyl)bis(azanediyl))bis(carbonyl))bis(pyrrolidine-2,1-diyl))bis(3-methyl-1-oxopentane-1,2-diyl))dicarbamate (9)
Yellow oil; Yield 19.4%; 1H NMR (400 MHz, CDCl3) δ 9.61 (s, 2H), 7.26 (s, 2H), 7.16 (d, J = 7.9 Hz, 4H), 7.12 (d, J = 8.0 Hz, 2H), 5.22 (d, J = 8.8 Hz, 2H), 4.68 (dd, J = 7.6, 4.4 Hz, 2H), 4.56–4.50 (m, 2H), 3.80–3.68 (m, 6H), 3.59 (s, 4H), 3.17 (s, 2H), 2.53 (dd, J = 11.6, 2.2 Hz, 4H), 2.27–1.99 (m, 4H), 1.76–1.43 (m, 4H), 0.94 (dd, J = 11.9, 6.6 Hz, 6H), 0.89–0.82 (m, 6H);13C NMR (126 MHz, CDCl3) δ 172.10, 170.03, 165.52, 156.91, 143.12, 136.76, 118.78, 117.80, 115.65, 61.02, 52.26, 47.70, 41.53, 38.65, 36.72, 29.05, 25.15, 24.60, 23.39, and 21.77; MS (ESI) m/z: 733.38 [M + H]+.
Diethyl((2S,2’S)-((2S,2’S)-(((9H-fluorene-2,7-diyl)bis(azanediyl))bis(carbonyl))bis(pyrrolidine-2,1-diyl))bis(3-methyl-1-oxopentane-1,2-diyl))dicarbamate (10)
Yellow oil; Yield 29%; 1H NMR (500 MHz, DMSO-d6) δ 9.67 (s, 2H), 7.36 (d, J = 7.7 Hz, 2H), 7.25 (s, 2H), 7.06 (d, J = 8.5 Hz, 2H), 6.90 (s, 2H), 4.40–4.29 (m, 2H), 4.15–4.03 (m, 2H), 3.88–3.80 (m, 2H), 3.57–3.48 (m, 2H), 2.32–2.13 (m, 4H), 2.00–1.96 (m, 4H), 1.89–1.79 (m, 4H), 1.65–1.56 (m, 8H), 1.43–1.35 (m, 6H), 0.88 (dd, J = 12.7, 4.9 Hz, 12H); 13C NMR (126 MHz, DMSO-d6) δ 172.06, 171.54, 156.82, 143.92, 137.87, 136.80, 119.94, 118.41, 116.56, 60.45, 60.19, 53.32, 51.33, 31.13, 24.74, 24.64, 23.67, 23.49, 21.86, 15.07, and 9.09; MS (ESI) m/z: 761.42 [M + H]+.
Dibutyl ((2S,2’S)-((2S,2’S)-(((9H-fluorene-2,7-diyl)bis(azanediyl))bis(carbonyl)) bis(pyrrolidine-2,1-diyl))bis(3-methyl-1-oxopentane-1,2-diyl))dicarbamate (11)
Yellow oil; Yield 28.4%; 1H NMR (500 MHz, DMSO-d6) δ 10.03 (s, 2H), 7.88 (s, 2H), 7.70 (d, J = 8.3 Hz, 2H), 7.47 (d, J = 1.2 Hz, 2H), 7.27 (d, J = 8.0 Hz, 2H), 4.48 (dd, J = 8.0, 4.6 Hz, 2H), 4.32–4.26 (m, 2H), 3.91 (dd, J = 24.2, 17.6 Hz, 8H), 3.77–3.68 (m, 2H), 3.57 (dt, J = 9.2, 5.7 Hz, 2H), 2.69 (s, 8H), 2.18–2.00 (m, 4H), 1.96–1.89 (m, 4H), 1.58–1.44 (m, 4H), 1.37–1.26 (m, 6H), 0.93–0.86 (m, 12H); 13C NMR (126 MHz, DMSO-d6) δ 171.33, 170.75, 156.83, 143.99, 138.06, 136.68, 119.95, 118.27, 116.35, 64.03, 60.65, 51.19, 47.19, 38.71, 37.05, 31.21, 29.80, 25.16, 24.61, 23.68, 21.79, 19.04, and 14.08; MS (ESI) m/z: 817.48 [M + H]+.
Dibenzyl((2S,2’S)-((2S,2’S)-(((9H-fluorene-2,7-diyl)bis(azanediyl))bis(carbonyl))bis(pyrrolidine-2,1-diyl))bis(3-methyl-1-oxopentane-1,2-diyl))dicarbamate (12)
Yellow oil; Yield 23.8%; 1H NMR (500 MHz, DMSO-d6) δ 10.04 (s, 2H), 7.88 (s, 2H), 7.71 (d, J = 8.3 Hz, 2H), 7.48 (d, J = 5.8 Hz, 2H), 7.39–7.29 (m, 12H), 5.02 (s, 4H), 4.48 (dd, J = 8.0, 4.6 Hz, 2H), 4.32 (t, J = 6.6 Hz, 2H), 3.90–3.74 (m, 2H), 3.70–3.49 (m, 4H), 2.21–2.12 (m, 4H), 1.98–1.84 (m, 4H), 1.55–1.34 (m, 6H), 0.91 (d, J = 6.6 Hz, 12H); 13C NMR (126 MHz, DMSO-d6) δ 171.19, 170.74,156.58, 143.99, 138.06, 137.55, 136.68, 128.79, 128.26, 128.15, 119.95, 118.28, 116.35, 65.84, 60.68, 51.32, 47.22, 38.80, 31.15, 29.80, 25.16, 24.62, 23.68, and 21.79; MS (ESI) m/z: 885.45 [M+ H]+.
Dimethyl((2R,2’R)-((2S,2’S)-(((9H-fluorene-2,7-diyl)bis(azanediyl))bis(carbonyl))bis(pyrrolidine-2,1-diyl))bis(4-methyl-1-oxopentane-1,2-diyl))dicarbamate (13)
Yellow oil; Yield 19%; 1H NMR (400 MHz, CDCl3) δ 9.12 (s, 2H), 7.76–7.65 (m, 2H), 7.42 (d, J = 23.6 Hz, 6H), 5.65 (d, J = 5.8 Hz, 2H), 4.67 (d, J = 6.4 Hz, 2H), 4.47 (s, 2H), 3.69–3.57 (m, 6H), 2.00 (dd, J = 11.2, 4.2 Hz, 6H), 1.77–1.64 (m, 4H), 1.60–1.52 (m, 4H), 1.49–1.39 (m, 4H), 1.28 (t, J = 7.3 Hz, 6H), 0.93 (dd, J = 12.1, 6.6 Hz, 6H); 13C NMR (126 MHz, CDCl3) δ 172.79, 172.22, 169.16, 165.71, 157.36, 143.80, 136.84, 119.22, 118.68, 116.87, 52.39, 51.45, 47.33, 46.75, 36.93, 28.66, 24.64, 23.39, 21.78, and 8.68; MS (ESI) m/z: 733.38 [M + H]+.
Diethyl ((2R,2’R)-((2S,2’S)-(((9H-fluorene-2,7-diyl)bis(azanediyl))bis(carbonyl)) bis(pyrrolidine-2,1-diyl))bis(4-methyl-1-oxopentane-1,2-diyl))dicarbamate (14)
Yellow oil; Yield 18.7%; 1H NMR (500 MHz, DMSO-d6) δ 8.85 (s, 2H), 7.39 (dd, J = 26.3, 7.7 Hz, 2H), 7.25 (s, 2H), 7.06 (d, J = 8.4 Hz, 2H), 6.90 (s, 2H), 4.00–3.95 (m, 6H), 3.92–3.78 (m, 4H), 3.48–3.41 (m, 4H), 3.35 (d, J = 2.9 Hz, 6H), 2.04–1.93 (m, 4H), 1.59 (ddd, J = 31.4, 15.6, 9.0 Hz, 4H), 1.49–1.35 (m, 6H), 0.86 (dd, J = 11.9, 6.6 Hz, 12H); 13C NMR (126 MHz, DMSO-d6) δ 175.06, 171.56, 170.66, 156.83, 156.54, 143.94, 119.95, 118.42, 116.49, 70.25, 60.19, 53.33, 46.26, 31.11, 24.73, 23.79, 23.48, 21.85, 15.05, and 9.07; MS (ESI) m/z: 761.42 [M + H]+.
Dibutyl((2R,2’R)-((2S,2’S)-(((9H-fluorene-2,7-diyl)bis(azanediyl))bis(carbonyl)) bis(pyrrolidine-2,1-diyl))bis(4-methyl-1-oxopentane-1,2-diyl))dicarbamate (15)
Yellow oil; Yield 21.4%; 1H NMR (500 MHz, DMSO-d6) δ 10.03 (s, 2H), 7.88 (s, 2H), 7.70 (d, J = 8.3 Hz, 2H), 7.48 (d, J = 8.2 Hz, 2H), 7.26 (d, J = 8.0 Hz, 2H), 4.47 (dd, J = 8.0, 4.6 Hz, 2H), 4.29 (t, J = 7.2 Hz, 2H), 3.93 (dd, J = 13.9, 7.1 Hz, 4H), 3.86 (s, 2H), 3.74–3.53 (m, 6H), 2.25–2.00 (m, 8H), 1.92–1.83 (m, 4H), 1.56–1.45 (m, 8H), 1.36–1.26 (m, 6H), 0.91–0.84 (m, 12H); 13C NMR (126 MHz, DMSO-d6) δ 171.33, 170.75, 165.06, 156.83, 143.99, 138.06, 119.95, 118.27, 116.34, 64.03, 60.65, 52.25, 51.19, 31.21, 29.79, 25.16, 24.61, 23.68, 23.23, 21.79, 19.04, and 14.08; MS (ESI) m/z: 817.48 [M + H]+.
Dibenzyl((2R,2’R)-((2S,2’S)-(((9H-fluorene-2,7-diyl)bis(azanediyl))bis(carbonyl))bis(pyrrolidine-2,1-diyl))bis(4-methyl-1-oxopentane-1,2-diyl))dicarbamate (16)
Yellow oil; Yield 25.5%; 1H NMR (400 MHz, DMSO-d6) δ 10.02 (s, 2H), 7.88 (s, 2H), 7.70 (d, J = 8.3 Hz, 2H), 7.48 (d, J = 8.0 Hz, 2H), 7.40–7.26 (m, 12H), 5.02 (s, 4H), 4.48 (dd, J = 7.8, 4.5 Hz, 2H), 4.39–4.27 (m, 2H), 3.58 (dd, J = 14.3, 7.8 Hz, 2H), 2.18–1.99 (m, 4H), 1.96–1.84 (m, 4H), 1.78–1.62 (m, 4H), 1.55–1.41 (m, 4H), 1.40–1.29 (m, 2H), 0.91 (d, J = 6.5 Hz, 12H); 13C NMR (101 MHz, DMSO-d6) δ 171.19, 170.73, 156.57, 143.98, 138.05, 137.55, 136.68, 128.80, 128.25, 128.14, 119.94, 118.28, 116.36, 65.84, 60.67, 55.37, 51.32, 38.71, 29.79, 25.15, 24.61, 23.67, and 21.79; MS (ESI) m/z: 885.45 [M + H]+.
Dimethyl((2S,2’S)-((2S,2’S)-(((9H-fluorene-2,7-diyl)bis(azanediyl))bis (carbonyl)) bis(pyrrolidine-2,1-diyl))bis(4-methyl-1-oxopentane-1,2-diyl))dicarbamate (17)
Yellow oil; Yield 18.7%; 1H NMR (400 MHz, CDCl3) δ 9.77 (s, 2H), 7.42–7.34 (m, 4H), 7.21 (d, J = 6.9 Hz, 2H), 7.12 (d, J = 8.1 Hz, 2H), 5.33–5.30 (m, 2H), 4.78 (dd, J = 7.7, 4.9 Hz, 2H), 4.47–4.42 (m, 2H), 3.92 (t, J = 6.7 Hz, 4H), 3.69 (d, J = 14.7 Hz, 6H), 2.31–2.19 (m, 4H), 2.13–2.00 (m, 4H), 1.86–1.62 (m, 4H), 1.26–1.19 (m, 2H), 1.17–1.07 (m, 12H); 13C NMR (126 MHz, CDCl3) δ 171.77, 170.03, 157.02, 143.05, 136.77, 128.95, 118.78, 117.78, 115.65, 61.11, 56.93, 52.33, 48.13, 38.64, 26.91, 25.19, 15.53, 11.15, and 10.96; MS (ESI) m/z: 733.38 [M + H]+.
Diethyl((2S,2’S)-((2S,2’S)-(((9H-fluorene-2,7 diyl)bis(azanediyl))bis(carbonyl)) bis(pyrrolidine-2,1-diyl))bis(4-methyl-1-oxopentane-1,2-diyl))dicarbamate (18)
Yellow oil ; Yield 19.5%; 1H NMR (500 MHz, DMSO-d6) δ 10.05 (s, 2H), 7.89 (s, 2H), 7.71 (d, J = 8.3 Hz, 2H), 7.48 (d, J = 8.2 Hz, 2H), 7.27 (d, J = 8.4 Hz, 2H), 4.48 (dd, J = 8.1, 4.6 Hz, 2H), 4.09 (t, J = 8.8 Hz, 2H), 3.97 (tt, J = 10.8, 5.3 Hz, 4H), 3.88–3.78 (m, 4H), 3.68–3.56 (m, 2H), 2.26–2.07 (m, 4H), 1.96–1.67 (m, 6H), 1.57–1.45 (m, 2H), 1.20–1.09 (m, 8H), 0.96–0.80 (m, 12H);13C NMR (101 MHz, DMSO-d6) δ 171.03, 170.72, 156.77, 144.01, 138.10, 136.65, 119.95, 118.23, 116.29, 60.72, 60.28, 56.87, 47.73, 36.31, 29.97, 25.10, 24.86, 15.29, 15.10, and 11.02; MS (ESI) m/z: 761.42 [M + H]+.
Dibutyl((2S,2’S)-((2S,2’S)-(((9H-fluorene-2,7-diyl)bis(azanediyl))bis(carbonyl))bis(pyrrolidine-2,1-diyl))bis(4-methyl-1-oxopentane-1,2-diyl))dicarbamate (19)
Yellow oil ; Yield 24.7%; 1H NMR (500 MHz, DMSO-d6) δ 10.05 (s, 2H), 7.89 (s, 2H), 7.70 (d, J = 4.0 Hz, 2H), 7.47 (d, J = 1.4 Hz, 2H), 7.27 (d, J = 8.4 Hz, 2H), 4.48 (dd, J = 8.0, 4.6 Hz, 2H), 4.09 (t, J = 8.8 Hz, 2H), 2.69 (s, 2H), 2.22–2.02 (m, 4H), 1.99–1.87 (m, 4H), 1.73 (d, J = 8.7 Hz, 4H), 1.55–1.45 (m, 8H), 1.36–1.26 (m, 6H), 1.20–1.07 (m, 4H), 0.93–0.78 (m, 18H); 13C NMR (101 MHz, DMSO-d6) δ 171.03, 170.72, 156.88, 144.01, 138.10, 136.65, 119.95, 118.23, 116.29, 64.09, 60.71, 56.88, 47.70, 36.30, 31.21, 29.96, 25.10, 24.85, 19.03, 15.29, 14.08, and 11.02; MS (ESI) m/z: 817.48 [M + H]+.
Dibenzyl((2S,2’S)-((2S,2’S)-(((9H-fluorene-2,7-diyl)bis(azanediyl))bis(carbonyl))bis(pyrrolidine-2,1-diyl))bis(4-methyl-1-oxopentane-1,2-diyl))dicarbamate (20)
Yellow oil ; Yield 27.4%; 1H NMR (500 MHz, DMSO-d6) δ 10.05 (s, 2H), 7.89 (s, 2H), 7.71 (d, J = 8.3 Hz, 2H), 7.50 (dd, J = 13.0, 8.6 Hz, 4H), 7.40–7.28 (m, 10H), 5.06–4.96 (m, 4H), 4.49 (dd, J = 7.9, 4.6 Hz, 2H), 4.13 (t, J = 8.8 Hz, 2H), 3.86 (s, 4H), 3.70–3.60 (m, 4H), 2.69 (s, 2H), 2.19–1.99 (m, 4H), 1.94–1.86 (m, 4H), 1.59–1.46 (m, 2H), 0.93 (d, J = 6.7 Hz, 12H); 13C NMR (101 MHz, DMSO-d6) δ 170.90, 170.70, 165.06, 164.78, 156.64, 144.00, 138.10, 136.65, 128.80, 128.24, 128.09, 119.94, 118.23, 116.30, 65.87, 60.73, 57.02, 47.75, 36.34, 29.97, 25.09, 24.84, 15.29, and 11.03; MS (ESI) m/z: 885.45 [M + H]+.
Dimethyl((1S,1’S)-((2S,2’S)-(((9H-fluorene-2,7diyl)bis(azanediyl))bis(carbonyl)) bis(pyrrolidine-2,1-diyl))bis(2-oxo-1-phenylethane-2,1-diyl))dicarbamate (21)
Yellow oil; Yield 16%; 1H NMR (500 MHz, CDCl3) δ 9.34 (s, 2H), 8.23 (s, 2H), 7.79 (s, 2H), 7.59 (d, J = 15.7 Hz, 2H), 7.42 (d, J = 10.2 Hz, 2H), 7.28–7.05 (m, 10H), 5.53 (d, J = 7.6 Hz, 2H), 4.50–4.32 (m, 2H), 3.50 (s, 2H), 2.21–2.07 (m, 6H), 1.95–1.80 (m, 4H), 1.59–1.31 (m, 4H), 1.23–1.03 (m, 4H); 13C NMR (126 MHz, CDCl3) δ 172.92, 170.38, 169.49, 168.84, 156.38, 143.77, 137.42, 129.34, 128.88, 127.87, 119.37, 118.34, 116.34, 58.50, 56.90, 52.22, 47.76, 29.43, 24.78, and 22.25; MS (ESI) m/z: 773.32 [M + H]+.
Diethyl((1S,1’S)-((2S,2’S)-(((9H-fluorene-2,7-diyl)bis(azanediyl))bis(carbonyl)) bis(pyrrolidine-2,1-diyl))bis(2-oxo-1-phenylethane-2,1-diyl))dicarbamate (22)
Yellow oil; Yield 24%; 1H NMR (500 MHz, CDCl3) δ 9.46 (s, 2H), 7.78 (s, 2H), 7.50 (d, J = 14.6 Hz, 6H), 7.41 (d, J = 6.8 Hz, 10 H), 5.91 (s, 2H), 5.66–5.51 (m, 2H), 5.00–4.73 (m, 4H), 3.72 (s, 2H), 3.31–3.12 (m, 4H), 2.14–2.00 (m, 8H), 1.43–1.29 (m, 6H); 13C NMR (126 MHz, CDCl3) δ 168.24, 167.20, 163.40, 154.04, 141.67, 135.29, 134.64, 134.39, 127.37, 125.99, 117.32, 116.33, 114.29, 59.32, 54.86, 45.32, 36.85, 26.11, 23.09, 12.63, and 6.79; MS (ESI) m/z: 801.35 [M + H] +.
Dibutyl((1S,1’S)-((2S,2’S)-(((9H-fluorene-2,7-diyl)bis(azanediyl))bis(carbonyl))bis(pyrrolidine-2,1-diyl))bis(2-oxo-1-phenylethane-2,1-diyl))dicarbamate (23)
Yellow oil; Yield 23.5%; 1H NMR (500 MHz, DMSO-d6) δ 10.07 (s, 2H), 7.87 (s, 2H), 7.71 (d, J = 2.6 Hz, 2H), 7.55 (d, J = 8.3 Hz, 2H), 7.44 (d, J = 7.2 Hz, 2H), 7.40–7.29 (m, 10H), 5.47 (d, J = 8.3 Hz, 2H), 4.52 (dd, J = 8.1, 4.6 Hz, 2H), 3.95 (tt, J = 6.5, 3.2 Hz, 4H), 3.88 (s, 2H), 3.72–3.61 (m, 4H), 2.23–2.09 (m, 4H), 1.91–1.85 (m, 4H), 1.55–1.47 (m, 4H), 1.35–1.26 (m, 4H), 0.89–0.85 (m, 6H); 13C NMR (126 MHz, DMSO-d6) δ 170.55, 168.62, 156.46, 144.02, 138.03, 137.43, 136.70, 128.85, 128.54, 128.23, 120.01, 118.30, 116.37, 64.22, 61.03, 57.02, 55.38, 38.72, 31.19, 29.82, 25.12, 19.02, and 14.08; MS (ESI) m/z: 857.42 [M + H]+.
Dibenzyl((1S,1’S)-((2S,2’S)-(((9H-fluorene-2,7-diyl)bis(azanediyl))bis(carbonyl)) bis(pyrrolidine-2,1-diyl))bis(2-oxo-1-phenylethane-2,1-diyl))dicarbamate (24)
Yellow oil; Yield 20.3%; 1H NMR (500 MHz, CDCl3) δ 9.68–9.46 (m, 2H), 7.52 (d, J = 6.9 Hz, 2H), 7.47 (dd, J = 12.8, 6.7 Hz, 2H), 7.40 (dd, J = 14.8, 7.5 Hz, 4H), 7.31 (d, J = 6.7 Hz, 10 H), 6.21–5.90 (m, 2H), 5.65–5.51 (m, 2H), 5.15–5.03 (m, 4H), 4.92–4.63 (m, 2H), 3.81–3.60 (m, 2H), 3.49–3.29 (m, 4H), 3.03–2.87 (m, 8H), 2.23–1.93 (m, 12H); 13C NMR (126 MHz, CDCl3) δ 169.68, 169.24, 165.62, 155.70, 143.42, 137.06, 136.58, 136.18, 129.27, 128.52, 128.19, 128.04, 119.09, 118.08, 116.00, 66.99, 61.22, 56.89, 47.46, 29.70, 28.49, and 25.01; MS (ESI) m/z: 925.38 [M + H]+.
Dimethyl((1R,1’R)-((2S,2’S)-(((9H-fluorene-2,7-diyl)bis(azanediyl))bis(carbonyl)) bis(pyrrolidine-2,1-diyl))bis(2-oxo-1-phenylethane-2,1-diyl))dicarbamate (25)
Yellow oil; Yield 17.75%; 1H NMR (500 MHz, DMSO-d6) δ 8.13–8.00 (m, 6H), 7.86–7.78 (m, 2H), 7.67–7.57 (m, 2H), 7.46–7.19 (m, 10H), 3.75–3.43 (m, 4H), 2.63–2.50 (m, 2H), 2.36 (dt, J = 3.6, 1.8 Hz, 6H), 1.67–1.51 (m, 4H), 1.21–0.99 (m, 4H), 0.96–0.61 (m, 4H); 13C NMR (126 MHz, DMSO-d6) δ 177.29, 171.77, 168.82, 156.86, 142.77, 136.78, 129.95, 129.02, 128.81, 128.24, 125.11, 120.02, 110.60, 66.76, 62.17, 58.47, 52.72, 52.15, 49.83, and 21.68; MS (ESI) m/z: 773.32 [M + H]+.
Diethyl((1R,1’R)-((2S,2’S)-(((9H-fluorene-2,7-diyl)bis(azanediyl))bis(carbonyl))bis(pyrrolidine-2,1-diyl))bis(2-oxo-1-phenylethane-2,1-diyl))dicarbamate (26)
Yellow oil; Yield 18%; 1H NMR (500 MHz, CDCl3-d) δ 9.43 (d, J = 11.9 Hz, 2H), 7.74 (d, J = 16.3 Hz, 2H), 7.65 (s, 2H), 7.47 (d, J = 8.4 Hz, 4H), 7.42–7.35 (m, 10H), 6.05 (dd, J = 13.9, 6.7 Hz, 2H), 5.58–5.52 (m, 2H), 4.89 (s, 2H), 3.69 (dd, J = 16.8, 7.4 Hz, 4H), 3.34 (dd, J = 17.3, 9.2 Hz, 4H), 2.38 (dd, J = 19.5, 11.9 Hz, 4H), 2.13–2.01 (m, 4H), 1.25–1.21 (m, 6H); 13C NMR (126 MHz, CDCl3-d) δ 170.43, 170.19, 169.03, 155.97, 143.71, 136.74, 136.30, 129.22, 128.83, 127.96, 119.34, 116.26, 109.60, 61.60, 61.33, 56.78, 47.47, 36.98, 29.70, 27.47, and 14.66; MS (ESI) m/z: 801.35 [M + H]+.
Dibutyl((1R,1’R)-((2S,2’S)-(((9H-fluorene-2,7-diyl)bis(azanediyl))bis(carbonyl))bis(pyrrolidine-2,1-diyl))bis(2-oxo-1-phenylethane-2,1-diyl))dicarbamate (27)
Yellow oil; Yield 20.6%; 1H NMR (400 MHz, CDCl3-d) δ 9.22 (s, 2H), 7.90 (s, 2H), 7.82 (s, 2H), 7.62 (d, J = 7.9 Hz, 2H), 7.56–7.36 (m, 10H), 7.24–7.13 (m, 2H), 5.90 (s, 2H), 5.50 (s, 2H), 4.93–4.76 (m, 2H), 3.67–3.53 (m, 4H), 3.35–3.17 (m, 4H), 2.44–2.32 (m, 4H), 2.12–1.88 (m, 4H), 1.41–1.31 (m, 8H), 0.98–0.86 (m, 6H),13C NMR (126 MHz, CDCl3-d) δ 170.31, 169.76, 164.89, 162.51, 147.97, 143.78, 130.27, 129.46, 127.88, 125.85, 119.35, 110.75, 94.04, 66.00, 45.68, 32.12, 31.09, 29.77, 25.58, 22.99, 19.08, 14.66, and 13.89; MS (ESI) m/z: 857.42 [M + H]+.
Dibenzyl((1R,1’R)-((2S,2’S)-(((9H-fluorene-2,7-diyl)bis(azanediyl))bis(carbonyl)) Bis(pyrrolidine-2,1-diyl))bis(2-oxo-1-phenylethane-2,1-diyl))dicarbamate (28)
Yellow oil; Yield 22%; 1H NMR (500 MHz, DMSO-d6) δ 10.11 (s, 2H), 7.89 (d, J = 16.4 Hz, 2H), 7.79 (s, 2H), 7.72 (d, J = 3.4 Hz, 2H), 7.55 (d, J = 7.4 Hz, 2H), 7.51–7.41 (m, 20H), 5.51–5.46 (m, 2H), 5.04 (s, 4H), 3.88 (s, 2H), 3.13 (dd, J = 18.0, 11.0 Hz, 2H), 2.25–2.01 (m, 4H), 1.93–1.77 (m, 8H); 13C NMR (126 MHz, DMSO-d6) δ 170.56, 169.18, 168.57, 156.30, 155.98, 149.61, 144.06, 138.43, 137.49, 137.33, 136.70, 129.06, 128.55, 127.88, 120.06, 118.32, 116.36, 66.11, 61.05, 57.15, 47.36, 37.07, 29.82, 25.13; MS (ESI) m/z: 925.38 [M + H]+.
Dimethyl ((((9H-fluorene-2,7-diyl)bis(azanediyl))bis(carbonyl))bis(piperidine-3,1-diyl))bis(3-methyl-1-oxobutane-1,2-diyl))dicarbamate (29)
Yellow oil; Yield 18%;1H NMR (400 MHz, CDCl3-d) δ 7.85 (d, J = 11.0 Hz, 2H), 7.65–7.61 (m, 2H), 7.45 (s, 2H), 7.33 (d, J = 13.4 Hz, 2H), 7.26 (s, 2H), 5.12 (s, 2H), 4.32 (s, 2H), 3.84 (s, 6H), 3.69–3.62 (m, 4H), 3.58–3.44 (m, 4H), 2.58 (t, J = 12 Hz, 2H), 2.37–2.22 (m, 2H), 2.09–1.96 (m, 4H), 1.41–1.24 (m, 4H), 1.23 (s, 12H); 13C NMR (126 MHz, CDCl3-d) δ 171.03, 170.65, 167.73, 132.47, 128.87, 119.69, 118.91, 116.94, 68.17, 49.14, 39.09, 38.74, 37.01, 30.37, 28.93, 23.76, 22.99, 14.05, and 10.96; MS (ESI) m/z: 733.38 [M + H]+.
Dimethyl ((((9H-fluorene-2,7-diyl)bis(azanediyl))bis(carbonyl))bis(piperidine-3,1-diyl))bis(4-methyl-1-oxopentane-1,2-diyl))dicarbamate (30)
Yellow oil; Yield 17.5%; 1H NMR (500 MHz, CDCl3-d) δ 7.75–7.71 (m, 4H), 7.57–7.53 (m, 6H), 4.27–4.19 (m, 4H), 3.79–3.74 (m, 4H), 3.73–3.67 (m, 6H), 1.73–1.71 (m, 2H), 1.70–1.67 (m, 4H), 1.47–1.43 (m, 4H), 1.44–1.41 (m, 2H), 1.40–1.37 (m, 4H), 1.01 (dd, J = 12.1, 5.2 Hz, 4H), 0.92 (s, 12H); 13C NMR (126 MHz, CDCl3-d) δ 171.03, 170.65, 167.73, 132.47, 130.89, 128.87, 119.69, 118.91, 116.94, 68.17, 49.14, 39.09, 38.74, 37.01, 30.37, 28.93, 23.76, 22.99, 14.05, and 10.96; MS (ESI) m/z: 761.42 [M + H]+.
Dimethyl ((((9H-fluorene-2,7-diyl)bis(azanediyl))bis(carbonyl))bis(piperidine-3,1-diyl))bis(3-methyl-1-oxopentane-1,2-diyl))dicarbamate (31)
Yellow oil; Yield 16%; 1H NMR (500 MHz, DMSO-d6) δ 10.05 (s, 2H), 7.88 (s, 2H), 7.70 (d, J = 7.4 Hz, 2H), 7.51 (d, J = 8.3 Hz, 2H), 7.30 (d, J = 8.7 Hz, 2H), 4.12 (s, 2H), 3.86 (s, 2H), 3.17–3.00 (m, 8H), 2.85–2.74 (m, 4H), 2.62 (dd, J = 18.1, 16.4 Hz, 6H), 2.07–1.94 (m, 8H), 1.78–1.65 (m, 4H), 1.46 (d, J = 7.4 Hz, 6H), 0.87–0.79 (m, 6H); 13C NMR (126 MHz, DMSO- d6) δ 173.50, 168.73, 161.01, 152.03, 130.20, 120.21, 115.08, 110.60, 81.39, 66.64, 62.46, 54.79, 47.70, 44.82, 44.17, 37.73, 33.62, 29.42, 24.58, 22.69, and 12.71; MS (ESI) m/z: 761.42 [M + H]+.
Dimethyl((((9H-fluorene-2,7-diyl)bis(azanediyl))bis(carbonyl))bis(piperidine-3,1-diyl))bis(2-oxo-1-phenylethane-2,1-diyl))dicarbamate (32)
Yellow oil; Yield 15%; 1H NMR (400 MHz, CDCl3-d) δ 7.45–7.36 (m, 10H), 7.15–6.99 (m, 5H), 6.80–6.67 (m, 5H), 4.05–3.86 (m, 4H), 3.81–3.57 (m, 8H), 1.39–1.27 (m, 2H), 1.23 (s, 6H), 0.89–0.77 (m, 8H); 13C NMR (126 MHz, CDCl3-d) δ 171.47, 171.25, 165.77, 161.64, 153.89, 145.04, 138.20, 129.97, 129.27, 127.73, 116.82, 115.53, 115.27, 71.13, 63.75, 46.76, 40.13, 31.93, 29.70, 22.70, and 14.12; MS (ESI) m/z: 801.35 [M + H]+.



 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

































