
Neuropeptide S-Mediated Modulation of Prepulse Inhibition Depends on Age, Gender, Stimulus-Timing, and Attention

Abstract
:1. Introduction
2. Results
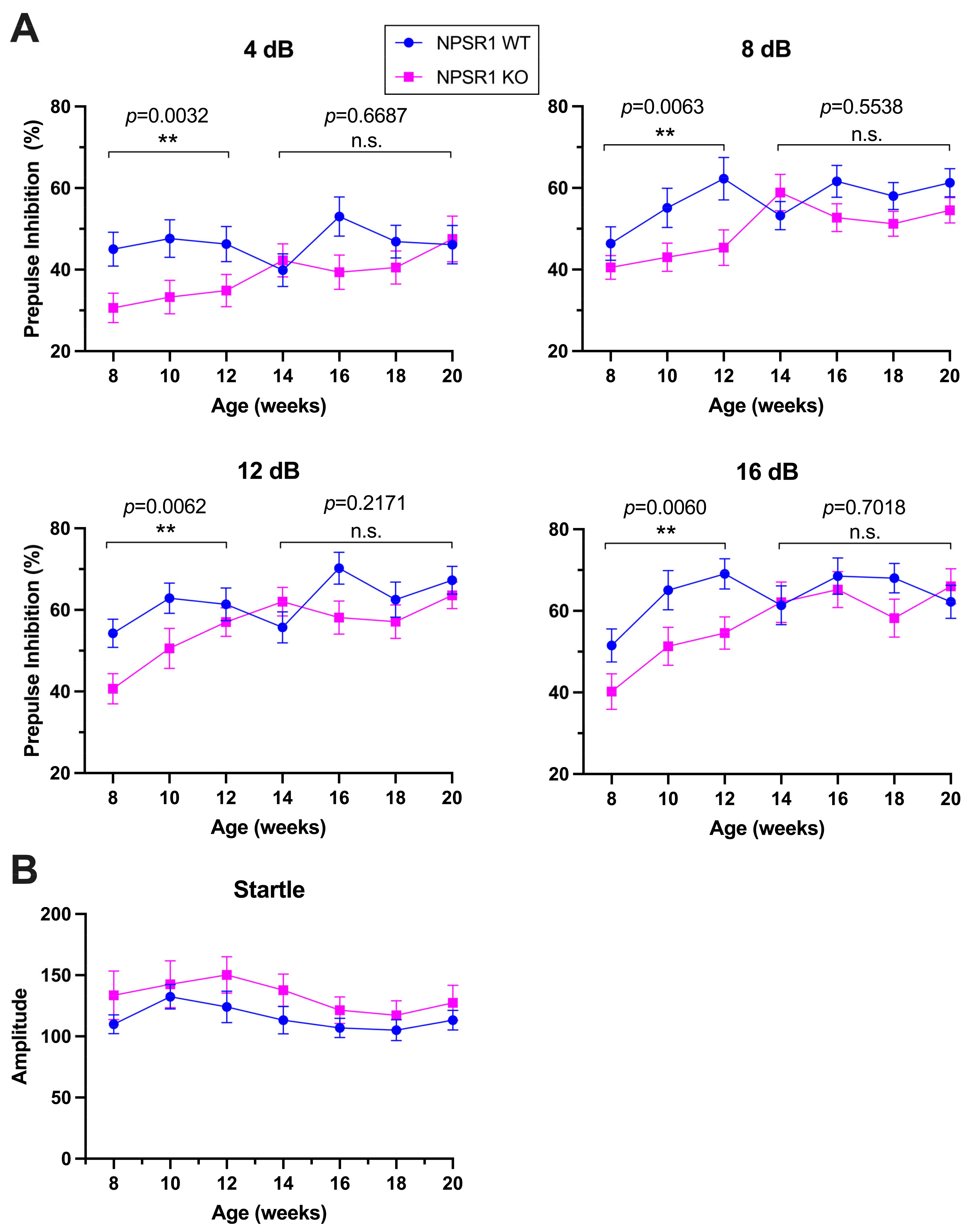
2.1. NPS-Dependent Age Effect on PPI
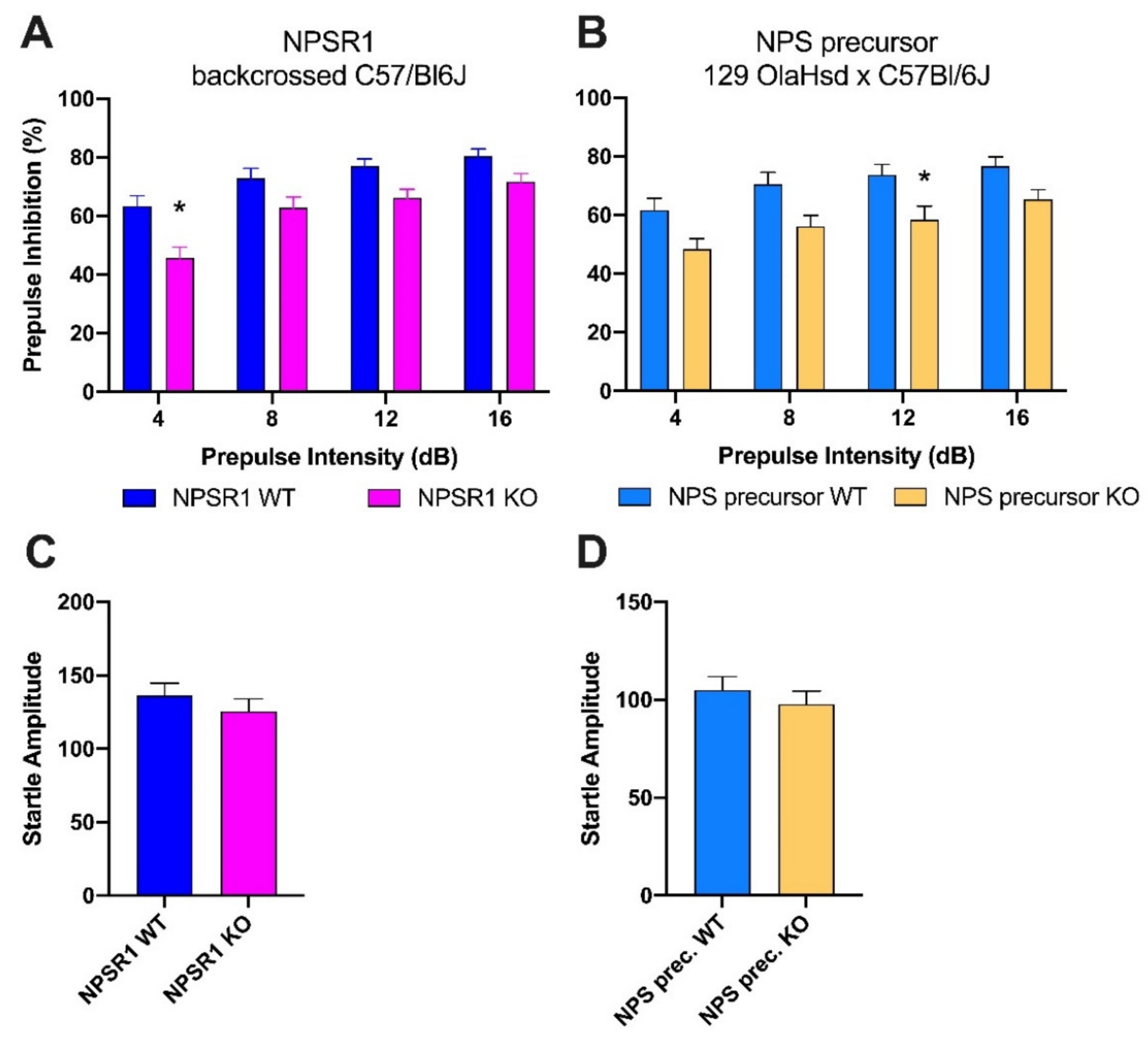
2.2. Comparison of PPI in NPSR1 and NPS KO Mice
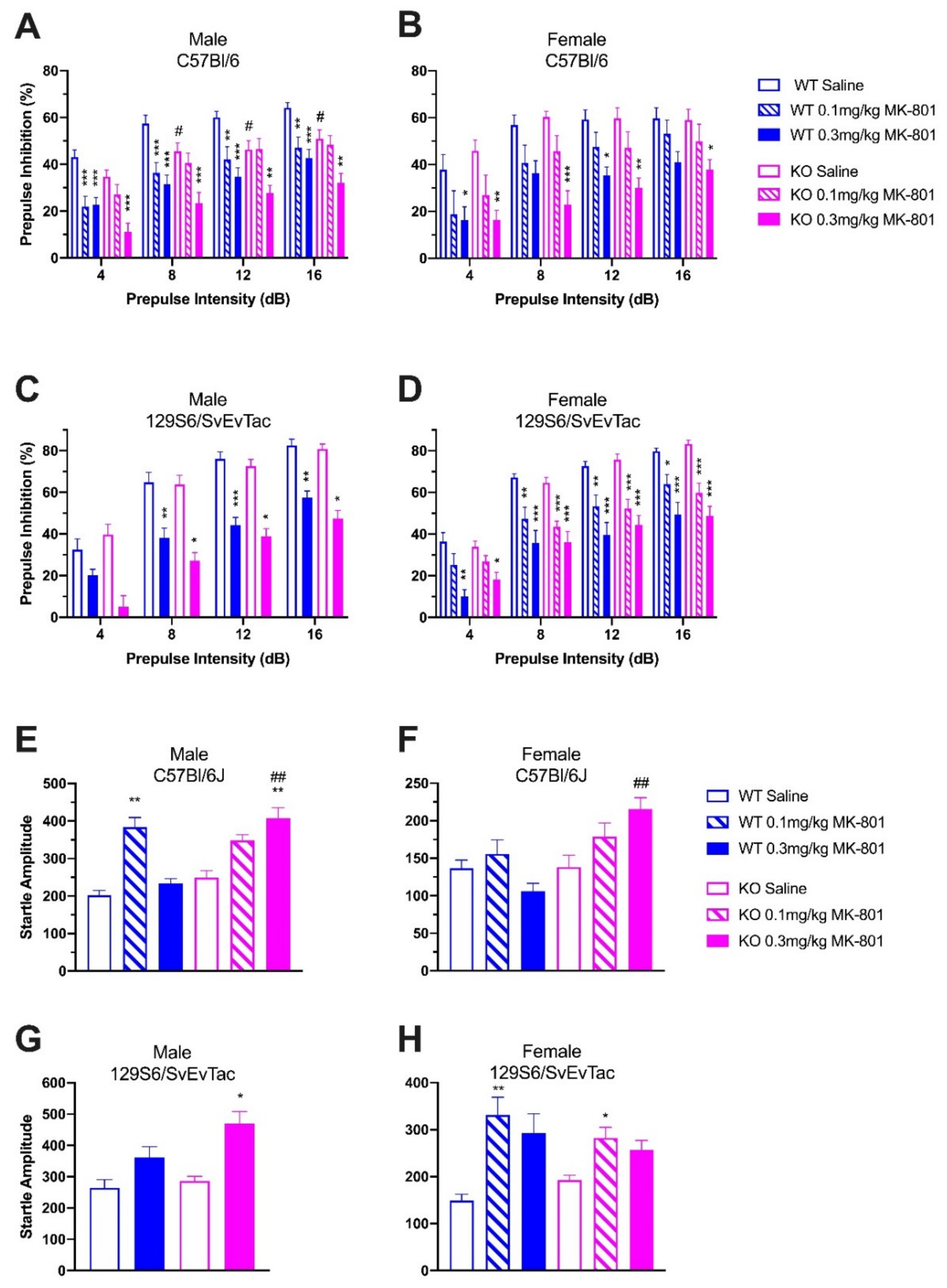
2.3. Gender-Specific Effects and PPI Disruption by MK-801
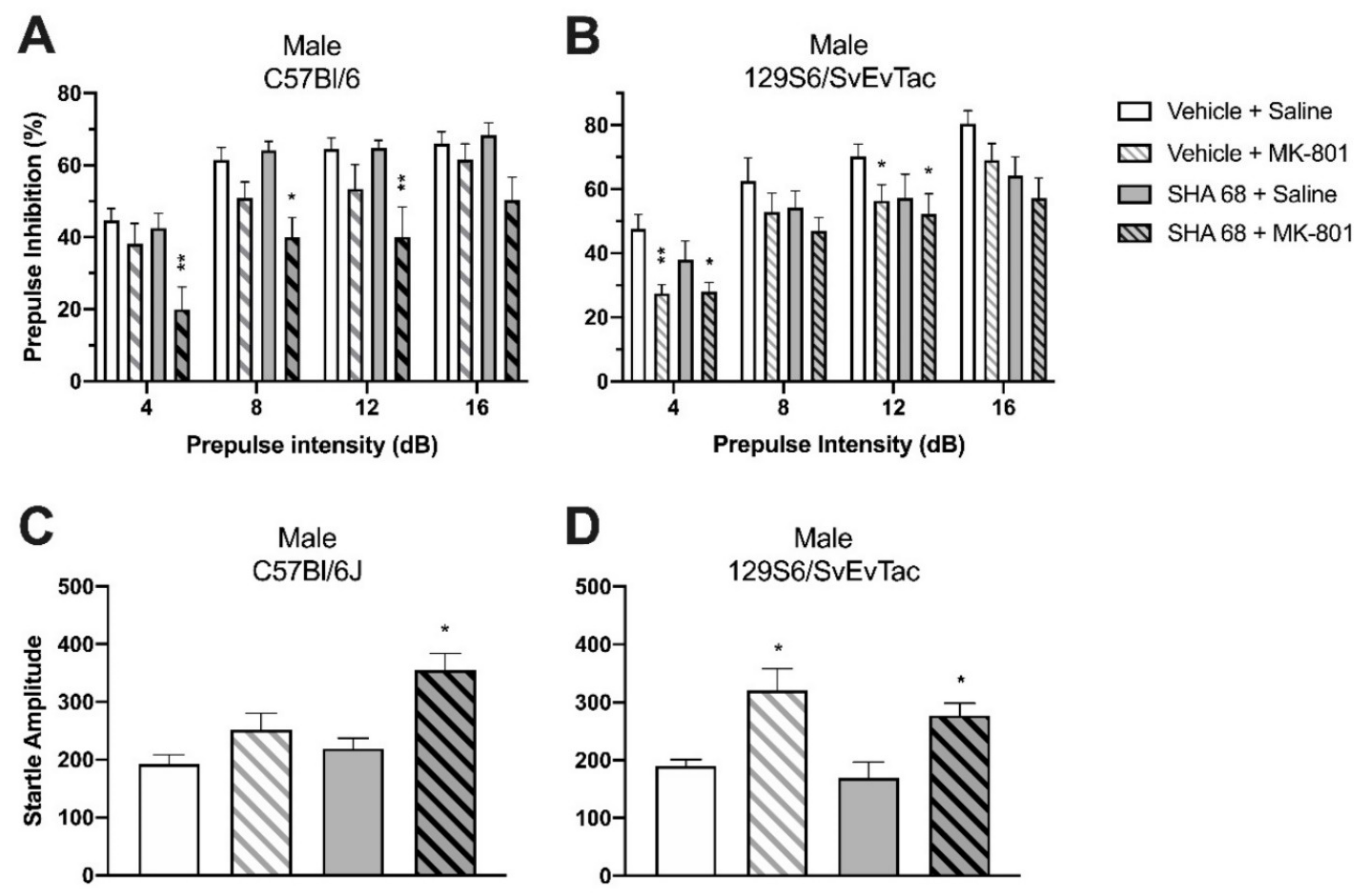
2.4. Effect of SHA 68 on MK-801-Induced PPI Disruption in Wildtype Mice
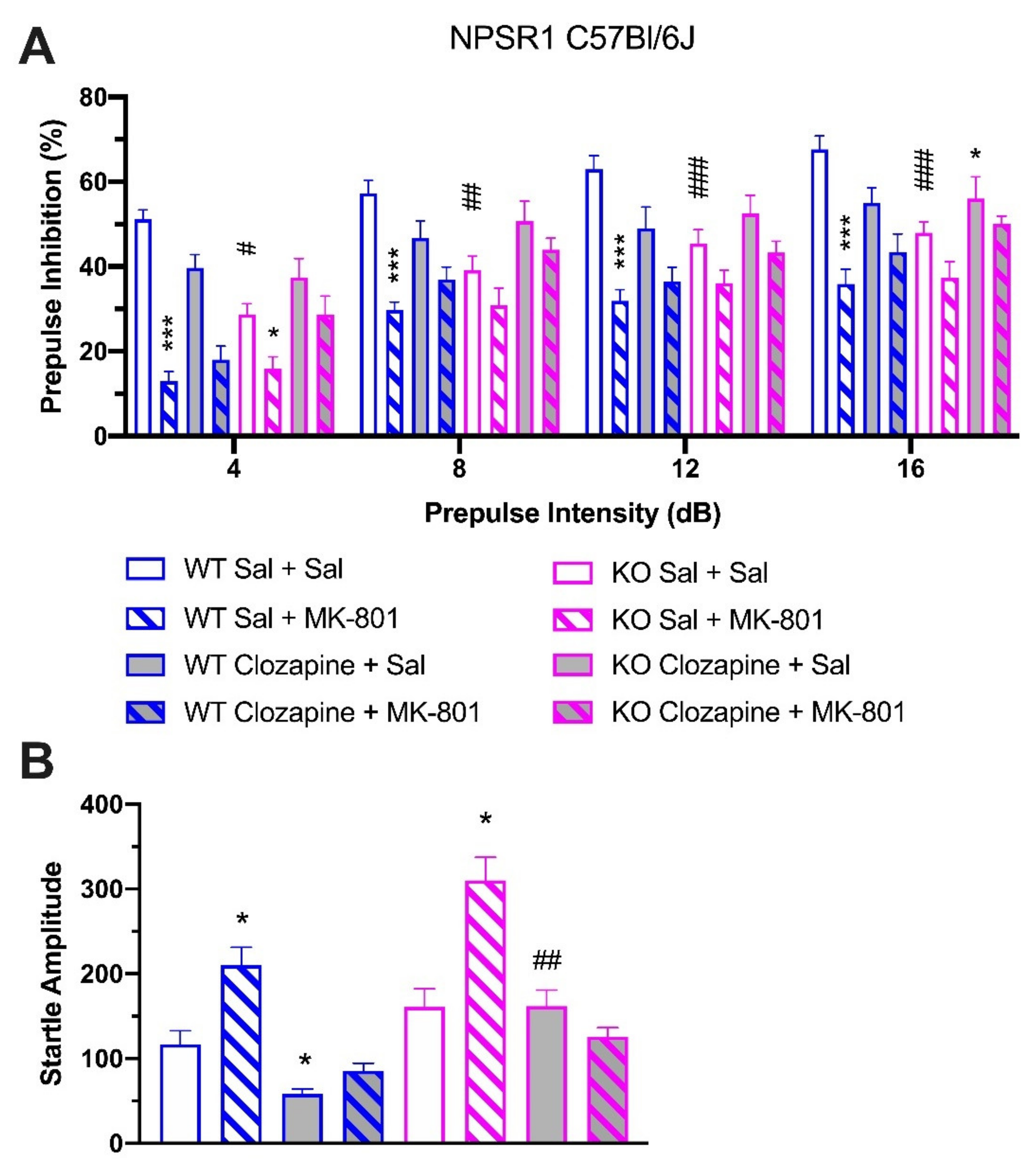
2.5. Effect of Clozapine in NPSR1 WT and KO Mice
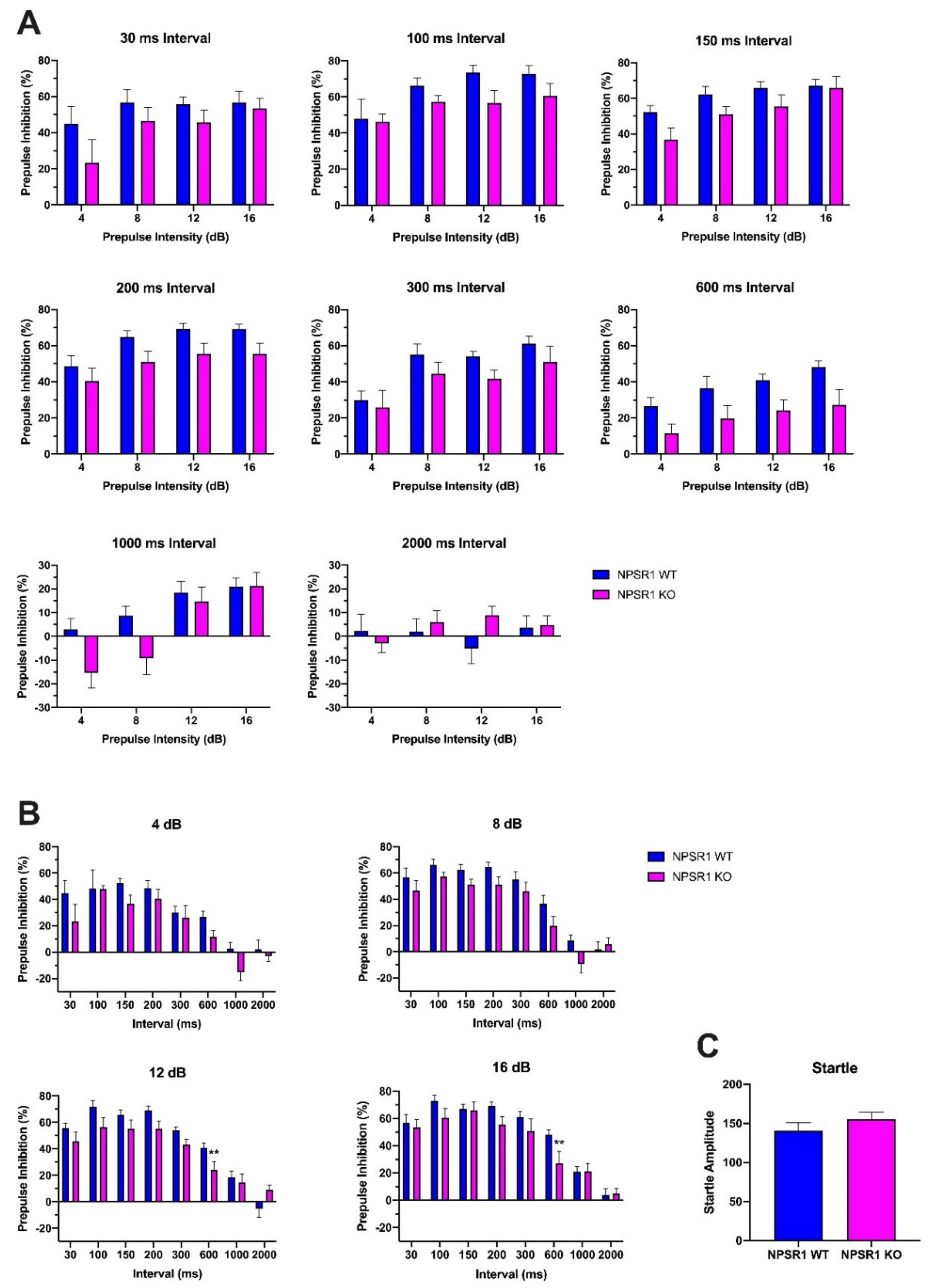
2.6. Role of NPS Signaling in Attentional Control
3. Discussion
3.1. Influences of Age and Strain on PPI
3.2. Gender Differences in PPI
3.3. Sensitivity to Antipsychotic Treatment
3.4. The Role of Attention in PPI Performance
3.5. Effect on Acoustic Startle Responses
3.6. Conclusions
4. Materials and Methods
4.1. Animals
4.2. Drugs
4.3. Drug Administration
4.4. Prepulse Inhibition
4.5. Effect of Interstimulus Interval on PPI
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Okamura, N.; Reinscheid, R.K.; Ohgake, S.; Iyo, M.; Hashimoto, K. Neuropeptide S attenuates neuropathological, neurochemical and behavioral changes induced by the NMDA receptor antagonist MK-801. Neuropharmacology 2010, 58, 166–172. [Google Scholar] [CrossRef] [Green Version]
- Han, R.W.; Yin, X.Q.; Chang, M.; Peng, Y.L.; Li, W.; Wang, R. Neuropeptide S facilitates spatial memory and mitigates spatial memory impairment induced by N-methyl-d-aspartate receptor antagonist in mice. Neurosci. Lett. 2009, 455, 74–77. [Google Scholar] [CrossRef]
- Han, R.W.; Zhang, R.S.; Xu, H.J.; Chang, M.; Peng, Y.L.; Wang, R. Neuropeptide S enhances memory and mitigates memory impairment induced by MK801, scopolamine or Aβ1-42 in mice novel object and object location recognition tasks. Neuropharmacology 2013, 70, 261–267. [Google Scholar] [CrossRef]
- Duangdao, D.M.; Clark, S.D.; Okamura, N.; Reinscheid, R.K. Behavioral phenotyping of Neuropeptide S receptor knockout mice. Behav. Brain Res. 2009, 205, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Mingler, M.K.; McBride, M.L.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Williams, M.T.; Vorhees, C.V.; Rothenberg, M.E. Abnormal response to stress and impaired NPS-induced hyperlocomotion, anxiolytic effect and corticosterone increase in mice lacking NPSR1. Psychoneuroendocrinology 2010, 35, 1119–1132. [Google Scholar] [CrossRef] [Green Version]
- Fendt, M.; Buchi, M.; Bürki, H.; Imobersteg, S.; Ricoux, B.; Suply, T.; Sailer, A.W. Neuropeptide S receptor deficiency modulates spontaneous locomotor activity and the acoustic startle response. Behav. Brain Res. 2011, 217, 1–9. [Google Scholar] [CrossRef]
- Allen, I.C.; Pace, A.J.; Jania, L.A.; Ledford, J.G.; Latour, A.M.; Snouwaert, J.N.; Bernier, V.; Stocco, R.; Therien, A.G.; Koller, B.H. Expression and function of NPSR1/GPRA in the lung before and after induction of asthma-like disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L1005–L1017. [Google Scholar] [CrossRef] [PubMed]
- Okamura, N.; Hashimoto, K.; Iyo, M.; Shimizu, E.; Dempfle, A.; Friedel, S.; Reinscheid, R.K. Gender-specific association of a functional coding polymorphism in the Neuropeptide S receptor gene with panic disorder but not with schizophrenia or attention-deficit/hyperactivity disorder. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2007, 31, 1444–1448. [Google Scholar] [CrossRef]
- Raczka, K.A.; Gartmann, N.; Mechias, M.L.; Reif, A.; Büchel, C.; Deckert, J.; Kalisch, R. A neuropeptide S receptor variant associated with overinterpretation of fear reactions: A potential neurogenetic basis for catastrophizing. Mol. Psychiatry 2010, 15, 1067–1074. [Google Scholar] [CrossRef] [PubMed]
- Donner, J.; Haapakoski, R.; Ezer, S.; Meln, E.; Pirkola, S.; Gratacs, M.; Zucchelli, M.; Anedda, F.; Johansson, L.E.; Sderhll, C.; et al. Assessment of the neuropeptide S system in anxiety disorders. Biol. Psychiatry 2010, 68, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Dannlowski, U.; Kugel, H.; Franke, F.; Stuhrmann, A.; Hohoff, C.; Zwanzger, P.; Lenzen, T.; Grotegerd, D.; Suslow, T.; Arolt, V.; et al. Neuropeptide-S (NPS) receptor genotype modulates basolateral amygdala responsiveness to aversive stimuli. Neuropsychopharmacology 2011, 36, 1879–1885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domschke, K.; Reif, A.; Weber, H.; Richter, J.; Hohoff, C.; Ohrmann, P.; Pedersen, A.; Bauer, J.; Suslow, T.; Kugel, H.; et al. Neuropeptide S receptor gene converging evidence for a role in panic disorder. Mol. Psychiatry 2011, 16, 938–948. [Google Scholar] [CrossRef] [PubMed]
- Laas, K.; Reif, A.; Akkermann, K.; Kiive, E.; Domschke, K.; Lesch, K.P.; Veidebaum, T.; Harro, J. Neuropeptide S receptor gene variant and environment: Contribution to alcohol use disorders and alcohol consumption. Addict. Biol. 2015, 20, 605–616. [Google Scholar] [CrossRef]
- Laas, K.; Reif, A.; Kiive, E.; Domschke, K.; Lesch, K.P.; Veidebaum, T.; Harro, J. A functional NPSR1 gene variant and environment shape personality and impulsive action: A longitudinal study. J. Psychopharmacol. 2014, 28, 227–236. [Google Scholar] [CrossRef]
- Laas, K.; Reif, A.; Akkermann, K.; Kiive, E.; Domschke, K.; Lesch, K.P.; Veidebaum, T.; Harro, J. Interaction of the neuropeptide S receptor gene Asn107Ile variant and environment: Contribution to affective and anxiety disorders, and suicidal behaviour. Int. J. Neuropsychopharmacol. 2014, 17, 541–552. [Google Scholar] [CrossRef] [Green Version]
- Streit, F.; Akdeniz, C.; Haddad, L.; Kumsta, R.; Entringer, S.; Frank, J.; Yim, I.S.; Zänkert, S.; Witt, S.H.; Kirsch, P.; et al. Sex-specific association between functional neuropeptide S receptor gene (NPSR1) variants and cortisol and central stress responses. Psychoneuroendocrinology 2017, 76, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Reinscheid, R.K.; Xu, Y.-L.; Okamura, N.; Zeng, J.; Chung, S.; Pai, R.; Wang, Z.; Civelli, O. Pharmacological characterization of human and murine neuropeptide S receptor variants. J. Pharmacol. Exp. Ther. 2005, 315, 1338–1345. [Google Scholar] [CrossRef]
- Lennertz, L.; Quednow, B.B.; Schuhmacher, A.; Petrovsky, N.; Frommann, I.; Schulze-Rauschenbach, S.; Landsberg, M.W.; Steinbrecher, A.; Höfels, S.; Pukrop, R.; et al. The functional coding variant Asn107Ile of the neuropeptide S receptor gene (NPSR1) is associated with schizophrenia and modulates verbal memory and the acoustic startle response. Int. J. Neuropsychopharmacol. 2012, 15, 1205–1215. [Google Scholar] [CrossRef] [Green Version]
- Okamura, N.; Garau, C.; Duangdao, D.M.; Clark, S.D.; Jüngling, K.; Pape, H.-C.; Reinscheid, R.K. Neuropeptide S enhances memory during the consolidation phase and interacts with noradrenergic systems in the brain. Neuropsychopharmacology 2011, 36, 744–752. [Google Scholar] [CrossRef]
- Swerdlow, N.R.; Braff, D.L.; Geyer, M.A. Sensorimotor gating of the startle reflex: What we said 25 years ago, what has happened since then, and what comes next. J. Psychopharmacol. 2016, 30, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Geyer, M.A.; Swerdlow, N.R.; Mansbach, R.S.; Braff, D.L. Startle response models of sensorimotor gating and habituation deficits in schizophrenia. Brain Res. Bull. 1990, 25, 485–498. [Google Scholar] [CrossRef]
- Grillon, C.; Ameli, R.; Charney, D.S.; Krystal, J.; Braff, D. Startle gating deficits occur across prepulse intensities in schizophrenic patients. Biol. Psychiatry 1992, 32, 939–943. [Google Scholar] [CrossRef]
- Thorne, G.L.; Dawson, M.E.; Schell, A.M. Attention and prepulse inhibition: The effects of task-relevant, irrelevant, and no-task conditions. Int. J. Psychophysiol. 2005, 56, 121–128. [Google Scholar] [CrossRef]
- Schell, A.M.; Wynn, J.K.; Dawson, M.E.; Sinaii, N.; Niebala, C.B. Automatic and controlled attentional processes in startle eyeblink modification: Effects of habituation of the prepulse. Psychophysiology 2000, 37, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Scholes, K.E.; Martin-Iverson, M.T. Relationships between prepulse inhibition and cognition are mediated by attentional processes. Behav. Brain Res. 2009, 205, 456–467. [Google Scholar] [CrossRef]
- Scholes, K.E.; Martin-Iverson, M.T. Disturbed prepulse inhibition in patients with schizophrenia is consequential to dysfunction of selective attention. Psychophysiology 2010, 47, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Du, Y.; Li, N.; Wu, X.; Wu, Y. Top-down modulation of prepulse inhibition of the startle reflex in humans and rats. Neurosci. Biobehav. Rev. 2009, 33, 1157–1167. [Google Scholar] [CrossRef]
- McGhie, A.; Chapman, J. Disorders of attention and perception in early schizophrenia. Br. J. Med. Psychol. 1961, 34, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Venables, P.H. Input dysfunction in schizophrenia. Prog. Exp. Pers. Res. 1964, 72, 1–47. [Google Scholar] [PubMed]
- Swerdlow, N.R.; Weber, M.; Qu, Y.; Light, G.A.; Braff, D.L. Realistic expectations of prepulse inhibition in translational models for schizophrenia research. Psychopharmacology 2008, 199, 331–388. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, N.R.; Braff, D.L.; Taaid, N.; Geyer, M.A. Assessing the validity of an animal model of deficient sensorimotor gating in schizophrenic patients. Arch. Gen. Psychiatry 1994, 51, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Braff, D.L.; Freedman, R.; Schork, N.J.; Gottesman, I.I. Deconstructing schizophrenia: An overview of the use of endophenotypes in order to understand a complex disorder. Schizophr. Bull. 2007, 33, 21–32. [Google Scholar] [CrossRef] [Green Version]
- Pietropaolo, S.; Crusio, W.E. Strain-dependent changes in acoustic startle response and its plasticity across adolescence in mice. Behav. Genet. 2009, 39, 623–631. [Google Scholar] [CrossRef]
- Ouagazzal, A.M.; Reiss, D.; Romand, R. Effects of age-related hearing loss on startle reflex and prepulse inhibition in mice on pure and mixed C57BL and 129 genetic background. Behav. Brain Res. 2006, 172, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Aubert, L.; Reiss, D.; Ouagazzal, A.M. Auditory and visual prepulse inhibition in mice: Parametric analysis and strain comparisons. Genes Brain Behav. 2006, 5, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Young, J.W.; Wallace, C.K.; Geyer, M.A.; Risbrough, V.B. Age-Associated Improvements in Cross-Modal Prepulse Inhibition in Mice. Behav. Neurosci. 2010, 124, 133–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellwanger, J.; Geyer, M.A.; Braff, D.L. The relationship of age to prepulse inhibition and habituation of the acoustic startle response. Biol. Psychol. 2003, 62, 175–195. [Google Scholar] [CrossRef]
- Paylor, R.; Crawley, J.N. Inbred strain differences in prepulse inhibition of the mouse startle response. Psychopharmacology 1997, 132, 169–180. [Google Scholar] [CrossRef]
- Willott, J.F.; Tanner, L.; O’Steen, J.; Johnson, K.R.; Bogue, M.A.; Gagnon, L. Acoustic startle and prepulse inhibition in 40 inbred strains of mice. Behav. Neurosci. 2003, 117, 716–727. [Google Scholar] [CrossRef]
- Kumari, V.; Aasen, I.; Sharma, T. Sex differences in prepulse inhibition deficits in chronic schizophrenia. Schizophr. Res. 2004, 69, 219–235. [Google Scholar] [CrossRef] [PubMed]
- Braff, D.L.; Light, G.A.; Ellwanger, J.; Sprock, J.; Swerdlow, N.R. Female schizophrenia patients have prepulse inhibition deficits. Biol. Psychiatry 2005, 57, 817–820. [Google Scholar] [CrossRef] [PubMed]
- Ison, J.R.; Allen, P.D. Pre- but not post-menopausal female CBA/CaJ mice show less prepulse inhibition than male mice of the same age. Behav. Brain Res. 2007, 185, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Koch, M. Sensorimotor gating changes across the estrous cycle in female rats. Physiol. Behav. 1998, 64, 625–628. [Google Scholar] [CrossRef]
- Lehmann, J.; Pryce, C.R.; Bettschen, D.; Feldon, J. The maternal separation paradigm and adult emotionality and cognition in male and female Wistar rats. Pharmacol. Biochem. Behav. 1999, 64, 705–715. [Google Scholar] [CrossRef]
- Palmer, A.A.; Dulawa, S.C.; Mottiwala, A.A.; Conti, L.H.; Geyer, M.A.; Printz, M.P. Prepulse startle deficit in the brown norway rat: A potential genetic model. Behav. Neurosci. 2000, 114, 374–388. [Google Scholar] [CrossRef]
- Gogos, A.; Kusljic, S.; Thwaites, S.J.; van den Buuse, M. Sex differences in psychotomimetic-induced behaviours in rats. Behav. Brain Res. 2017, 322, 157–166. [Google Scholar] [CrossRef]
- Swerdlow, N.R.; Auerbach, P.; Monroe, S.M.; Hartston, H.; Geyer, M.A.; Braff, D.L. Men are more inhibited than women by weak prepulses. Biol. Psychiatry 1993, 34, 253–260. [Google Scholar] [CrossRef]
- Swerdlow, N.R.; Hartman, P.L.; Auerbach, P.P. Changes in sensorimotor inhibition across the menstrual cycle: Implications for neuropsychiatric disorders. Biol. Psychiatry 1997, 41, 452–460. [Google Scholar] [CrossRef]
- Swerdlow, N.R.; Braff, D.L.; Geyer, M.A. Cross-species studies of sensorimotor gating of the startle reflex. Ann. N. Y. Acad. Sci. 1999, 877, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Kumari, V. Sex differences and hormonal influences in human sensorimotor gating: Implications for schizophrenia. Curr. Top. Behav. Neurosci. 2011, 8, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, T.; Szilagyi, S.; Chakravorty, S.; Fiallos, A.M.; Lewison, B.J.; Parwani, A.; Schwartz, M.P.; Gonzenbach, S.; Rotrosen, J.P.; Duncan, E.J. Menstrual cycle phase effects on prepulse inhibition of acoustic startle. Psychophysiology 2004, 41, 401–406. [Google Scholar] [CrossRef]
- Vaillancourt, C.; Cyr, M.; Rochford, J.; Boksa, P.; Di Paolo, T. Effects of ovariectomy and estradiol on acoustic startle responses in rats. Pharmacol. Biochem. Behav. 2002, 74, 103–109. [Google Scholar] [CrossRef]
- Ornitz, E.M.; Guthrie, D.; Sadeghpour, M.; Sugiyama, T. Maturation of Prestimulation-Induced Startle Modulation in Girls. Psychophysiology 1991, 28, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Kumari, V.; Aasen, I.; Papadopoulos, A.; Bojang, F.; Poon, L.; Halari, R.; Cleare, A.J. A comparison of prepulse inhibition in pre- and postmenopausal women and age-matched men. Neuropsychopharmacology 2008, 33, 2610–2618. [Google Scholar] [CrossRef]
- Si, W.; Aluisio, L.; Okamura, N.; Clark, S.D.; Fraser, I.; Sutton, S.W.; Bonaventure, P.; Reinscheid, R.K. Neuropeptide S stimulates dopaminergic neurotransmission in the medial prefrontal cortex. J. Neurochem. 2010, 115, 475–482. [Google Scholar] [CrossRef] [Green Version]
- Mochizuki, T.; Kim, J.; Sasaki, K. Microinjection of neuropeptide S into the rat ventral tegmental area induces hyperactivity and increases extracellular levels of dopamine metabolites in the nucleus accumbens shell. Peptides 2010, 31, 926–931. [Google Scholar] [CrossRef] [PubMed]
- Adori, C.; Barde, S.; Vas, S.; Ebner, K.; Su, J.; Svensson, C.; Mathé, A.A.; Singewald, N.; Reinscheid, R.R.; Uhlén, M.; et al. Exploring the role of neuropeptide S in the regulation of arousal: A functional anatomical study. Brain Struct. Funct. 2016, 221, 3521–3546. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zeng, J.; Zhou, A.; Theodorsson, E.; Fahrenkrug, J.; Reinscheid, R.K. Molecular fingerprint of neuropeptide s-producing neurons in the mouse brain. J. Comp. Neurol. 2011, 519, 1847–1866. [Google Scholar] [CrossRef]
- Palasz, A.; Rojczyk, E.; Golyszny, M.; Filipczyk, L.; Worthington, J.J.; Wiaderkiewicz, R. Long-term treatment with haloperidol affects neuropeptide S and NPSR mRNA levels in the rat brain. Acta Neuropsychiatr. 2016, 28, 110–116. [Google Scholar] [CrossRef] [Green Version]
- Pałasz, A.; Rojczyk, E. Neuroleptics Affect Neuropeptide S and NPSR mRNA Levels in the Rat Brain. J. Mol. Neurosci. 2015, 57, 352–357. [Google Scholar] [CrossRef]
- Kumari, V.; Sharma, T. Effects of typical and atypical antipsychotics on prepulse inhibition in schizophrenia: A critical evaluation of current evidence and directions for future research. Psychopharmacology 2002, 162, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Geyer, M.A.; Ellenbroek, B. Animal behavior models of the mechanisms underlying antipsychotic atypicality. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2003, 27, 1071–1079. [Google Scholar] [CrossRef]
- Oranje, B.; Van Oel, C.J.; Gispen-de Wied, C.C.; Verbaten, M.N.; Kahn, R.S. Effects of typical and atypical antipsychotics on the prepulse inhibition of the startle reflex in patients with schizophrenia. J. Clin. Psychopharmacol. 2002, 22, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Graham, F.K. The more or less startling effects of weak prestimulation. Psychophysiology 1975, 12, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Delpezzo, E.M.; Hoffman, H.S. Attentional factors in the inhibition of a reflex by a visual stimulus. Science 1980, 210, 673–674. [Google Scholar] [CrossRef]
- Filion, D.L.; Dawson, M.E.; Schell, A.M. Modification of the acoustic startle-reflex eyeblink: A tool for investigating early and late attentional processes. Biol. Psychol. 1993, 35, 185–200. [Google Scholar] [CrossRef]
- Filion, D.L.; Poje, A.B. Selective and nonselective attention effects on prepulse inhibition of startle: A comparison of task and no-task protocols. Biol. Psychol. 2003, 64, 283–296. [Google Scholar] [CrossRef]
- Braff, D.L.; Light, G.A. Preattentional and attentional cognitive deficits as targets for treating schizophrenia. Psychopharmacology 2004, 174, 75–85. [Google Scholar] [CrossRef]
- Heekeren, K.; Meincke, U.; Geyer, M.A.; Gouzoulis-Mayfrank, E. Attentional Modulation of Prepulse Inhibition: A New Startle Paradigm. Neuropsychobiology 2004, 49, 88–93. [Google Scholar] [CrossRef]
- Röskam, S.; Koch, M. Enhanced prepulse inhibition of startle using salient prepulses in rats. Int. J. Psychophysiol. 2006, 60, 10–14. [Google Scholar] [CrossRef]
- Du, Y.; Wu, X.; Li, L. Differentially organized top-down modulation of prepulse inhibition of startle. J. Neurosci. 2011, 31, 13644–13653. [Google Scholar] [CrossRef]
- Xu, Y.-L.; Reinscheid, R.K.; Huitron-Resendiz, S.; Clark, S.D.; Wang, Z.; Lin, S.H.; Brucher, F.A.; Zeng, J.; Ly, N.K.; Henriksen, S.J.; et al. Neuropeptide S: A neuropeptide promoting arousal and anxiolytic-like effects. Neuron 2004, 43, 487–497. [Google Scholar] [CrossRef] [Green Version]
- Reinscheid, R.K.; Xu, Y.-L. Neuropeptide S as a novel arousal promoting peptide transmitter. FEBS J. 2005, 272, 5689–5693. [Google Scholar] [CrossRef]
- Rizzi, A.; Vergura, R.; Marzola, G.; Ruzza, C.; Guerrini, R.; Salvadori, S.; Regoli, D.; Calo, G. Neuropeptide S is a stimulatory anxiolytic agent: A behavioural study in mice. Br. J. Pharmacol. 2008, 154, 471–479. [Google Scholar] [CrossRef] [Green Version]
- Ruzza, C.; Rizzi, A.; Trapella, C.; Pela, M.; Camarda, V.; Ruggieri, V.; Filaferro, M.; Cifani, C.; Reinscheid, R.K.; Vitale, G.; et al. Further studies on the pharmacological profile of the neuropeptide S receptor antagonist SHA 68. Peptides 2010, 31, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Shao, Y.F.; Zhang, M.; Fan, K.; Kong, X.P.; Wang, R.; Hou, Y.P. Neuropeptide S promotes wakefulness through activation of the posterior hypothalamic histaminergic and orexinergic neurons. Neuroscience 2012, 207, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Neufang, S.; Geiger, M.J.; Homola, G.A.; Mahr, M.; Akhrif, A.; Nowak, J.; Reif, A.; Romanos, M.; Deckert, J.; Solymosi, L.; et al. Modulation of prefrontal functioning in attention systems by NPSR1 gene variation. Neuroimage 2015, 114, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.E.; Hazlett, E.A.; Filion, D.L.; Nuechterlein, K.H.; Schell, A.M. Attention and schizophrenia: Impaired modulation of the startle reflex. J. Abnorm. Psychol. 1993, 102, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.E.; Schell, A.M.; Hazlett, E.A.; Nuechterlein, K.H.; Filion, D.L. On the clinical and cognitive meaning of impaired sensorimotor gating in schizophrenia. Psychiatry Res. 2000, 96, 187–197. [Google Scholar] [CrossRef]
- Knight, R.T.; Grabowecky, M.F.; Scabini, D. Role of Human Prefrontal Cortex in Attention Control. In Epilepsy and the functional anatomy of the frontal lobe; Jasper, H.H., Riggio, S., Goldman-Rakic, P.S., Eds.; Raven Press: New York, NY, USA, 1995. [Google Scholar]
- Faw, B. Pre-frontal executive committee for perception, working memory, attention, long-term memory, motor control, and thinking: A tutorial review. Conscious. Cogn. 2003, 12, 83–139. [Google Scholar] [CrossRef]
- Aasen, I.; Kolli, L.; Kumari, V. Sex effects in prepulse inhibition and facilitation of the acoustic startle response: Implications for pharmacological and treatment studies. J. Psychopharmacol. 2005, 19, 39–45. [Google Scholar] [CrossRef]
- Postma, P.; Kumari, V.; Hines, M.; Gray, J.A. The relationship between prepulse detection and prepulse inhibition of the acoustic startle reflex. Psychophysiology 2001, 38, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Wynn, J.K.; Dawson, M.E.; Schell, A.M.; McGee, M.; Salveson, D.; Green, M.F. Prepulse facilitation and prepulse inhibition in schizophrenia patients and their unaffected siblings. Biol. Psychiatry 2004, 55, 518–523. [Google Scholar] [CrossRef]
- Koch, M. The neurobiology of startle. Prog. Neurobiol. 1999, 59, 107–128. [Google Scholar] [CrossRef]
- Liu, X.; Si, W.; Garau, C.; Jüngling, K.; Pape, H.-C.; Schulz, S.; Reinscheid, R.K. Neuropeptide S precursor knockout mice display memory and arousal deficits. Eur. J. Neurosci. 2017, 46, 1689–1700. [Google Scholar] [CrossRef]
- National Research Council. Guidelines for the Care and Use of Mammals in Neuroscience and Behavioral Research; National Academies Press: Washington, DC, USA, 2003. [Google Scholar]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The arrive guidelines for reporting animal research. PLoS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef] [PubMed]
- Okamura, N.; Habay, S.A.; Zeng, J.; Chamberlin, A.R.; Reinscheid, R.K. Synthesis and pharmacological in vitro and in vivo profile of 3-oxo-1,1-diphenyl-tetrahydro-oxazolo [3,4-a] pyrazine-7-carboxylic acid 4-fluoro-benzylamide (SHA 68), a selective antagonist of the neuropeptide S receptor. J. Pharmacol. Exp. Ther. 2008, 325, 893–901. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Si, W.; Liu, X.; Pape, H.-C.; Reinscheid, R.K. Neuropeptide S-Mediated Modulation of Prepulse Inhibition Depends on Age, Gender, Stimulus-Timing, and Attention. Pharmaceuticals 2021, 14, 489. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14050489
Si W, Liu X, Pape H-C, Reinscheid RK. Neuropeptide S-Mediated Modulation of Prepulse Inhibition Depends on Age, Gender, Stimulus-Timing, and Attention. Pharmaceuticals. 2021; 14(5):489. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14050489
Chicago/Turabian StyleSi, Wei, Xiaobin Liu, Hans-Christian Pape, and Rainer K. Reinscheid. 2021. "Neuropeptide S-Mediated Modulation of Prepulse Inhibition Depends on Age, Gender, Stimulus-Timing, and Attention" Pharmaceuticals 14, no. 5: 489. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14050489