
Elucidation of the Molecular Pathways Involved in the Protective Effects of AUY-922 in LPS-Induced Inflammation in Mouse Lungs


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
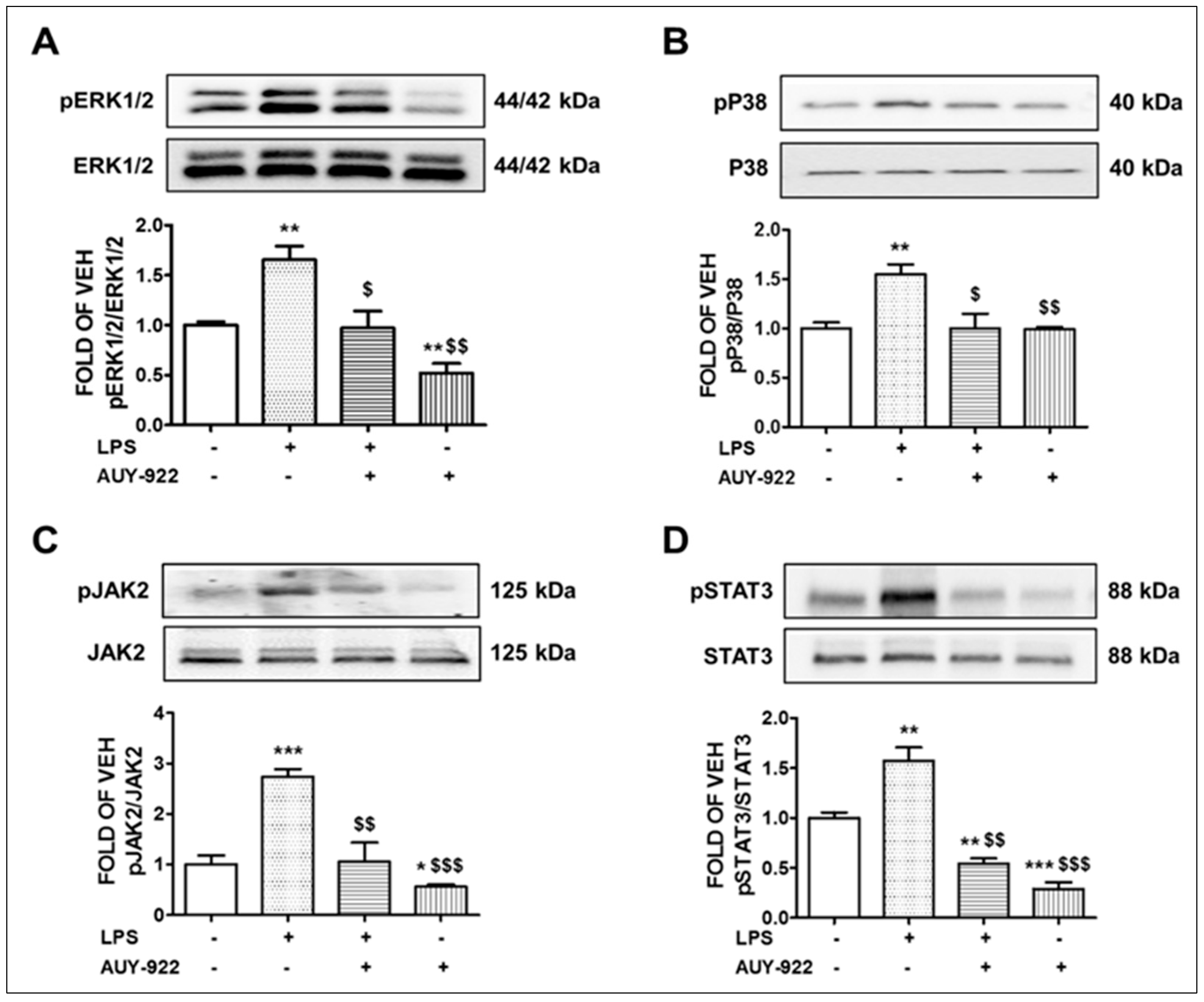
2.1. AUY-922 Inhibits the Activation of ERK1/2 Pathway by LPS
2.2. AUY-922 Counteracts the LPS-Induced Phosphorylation of the P38 MAPK Pathway
2.3. AUY-922 Suppresses the LPS-Induced Activation of JAK2/STAT3 Pathway
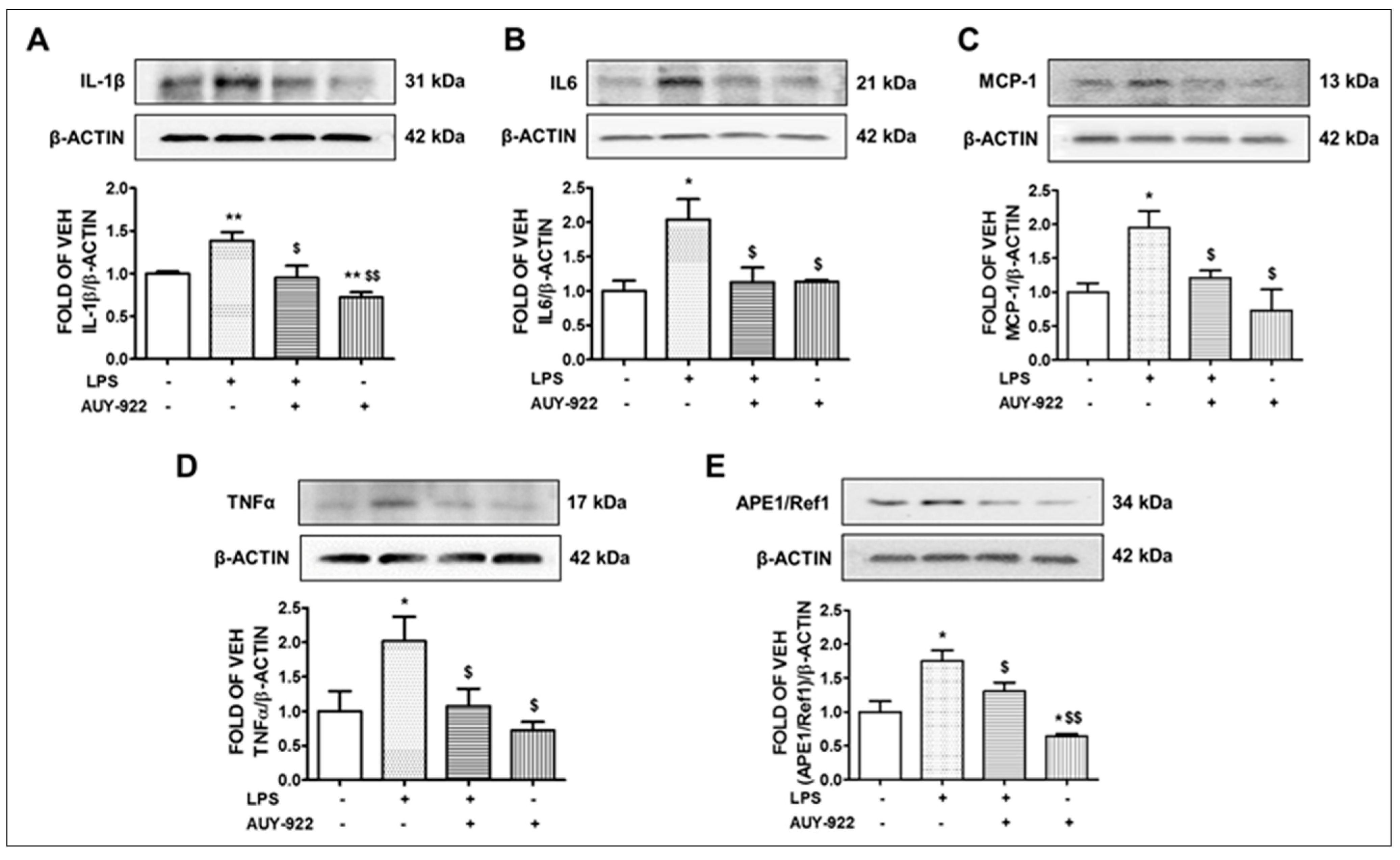
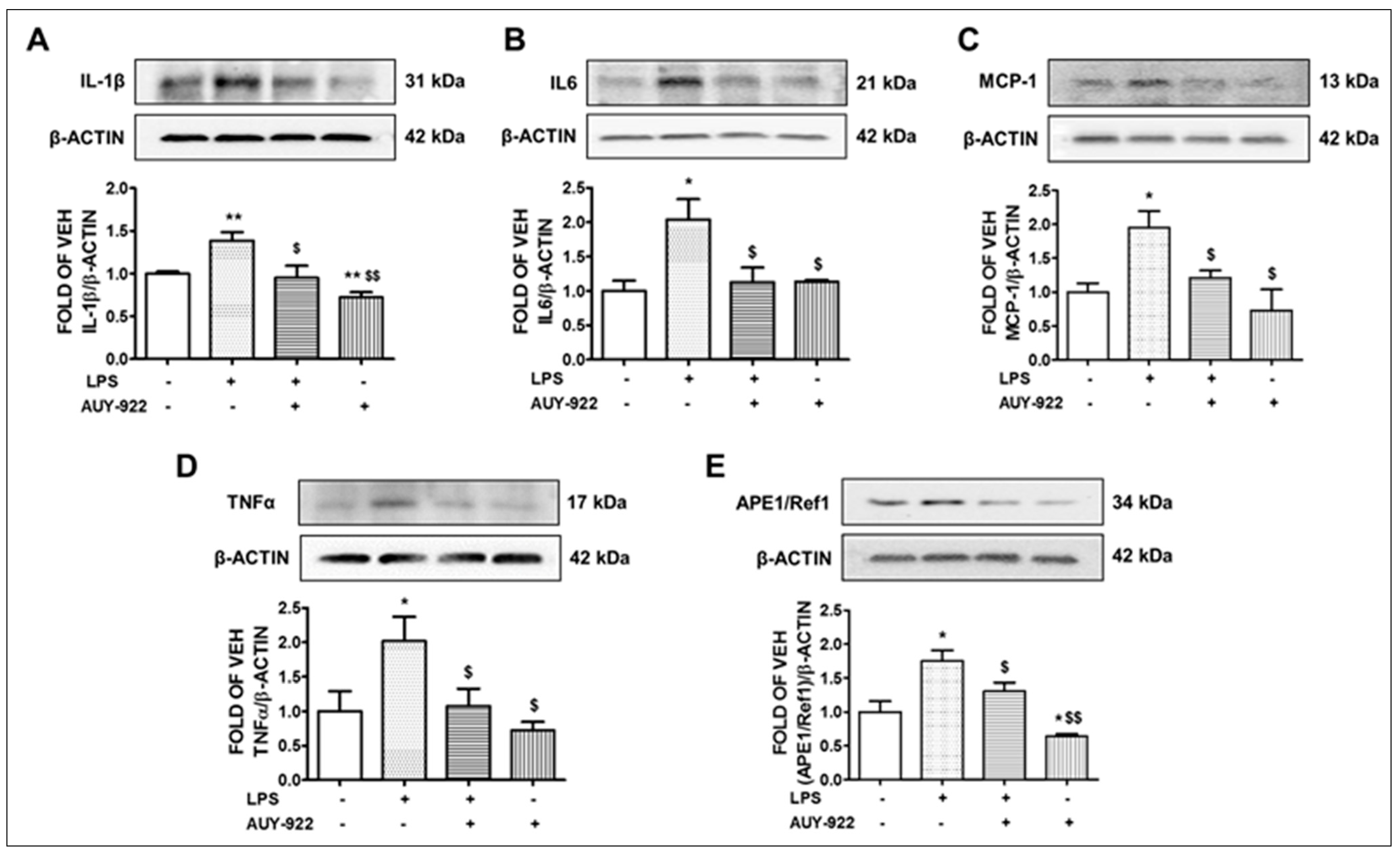
2.4. AUY-922 Counteracts the LPS-Induced IL-1β, IL-6, MCP-1 and TNF-α Expression
2.5. LPS Induces the Expression Levels of APE1/Ref1 and AUY-922 Exerts the Opposite Effects
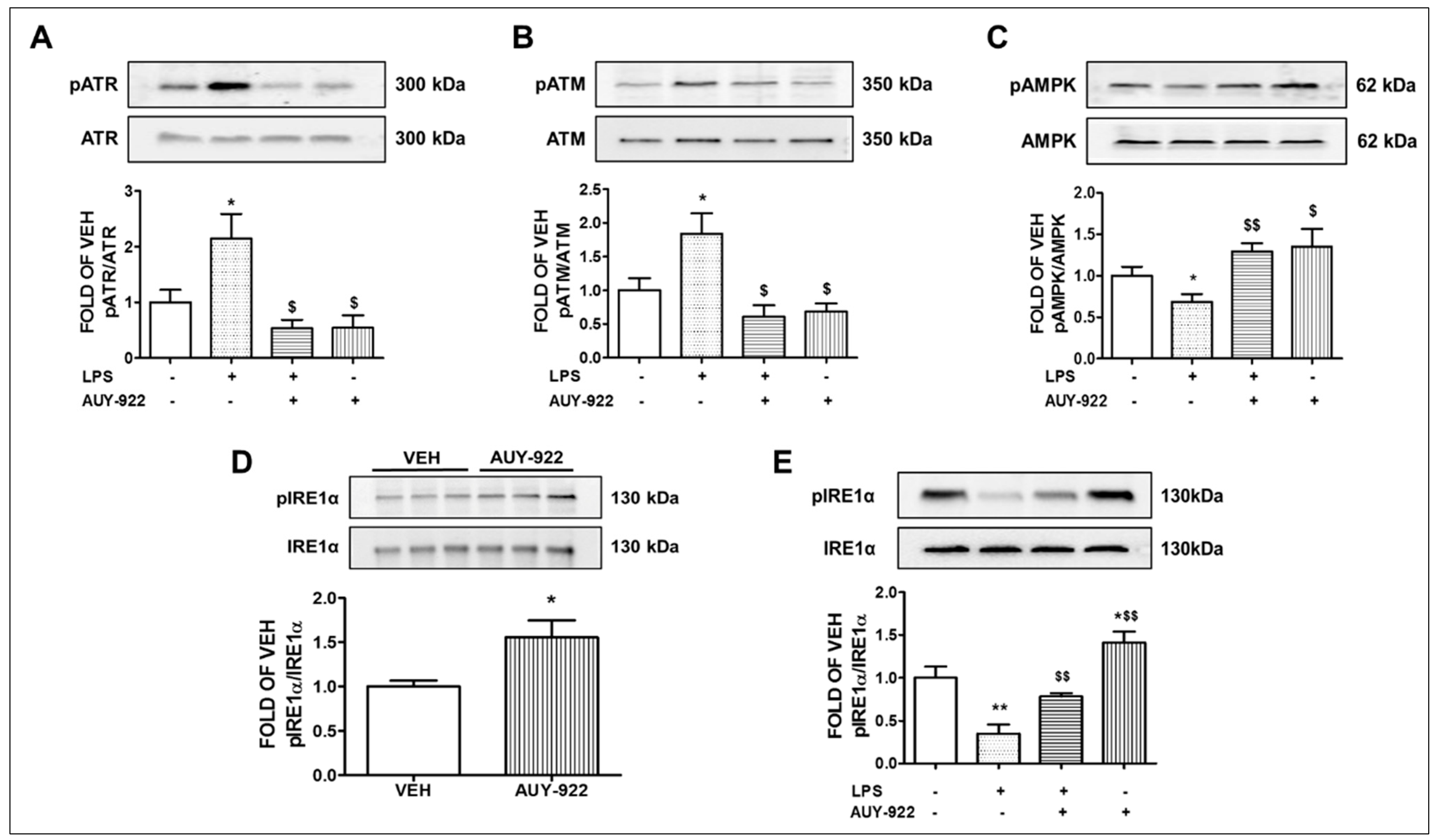
2.6. AUY-922 Suppresses the Levels of the LPS-Induced ATR Phosphorylation
2.7. AUY-922 Reduces the Expression Levels of the Phosphorylated ATM
2.8. AUY-922 Counteracts the LPS-Induced Suppression of pAMPK
2.9. AUY-922 Suppresses the LPS-Induced Deactivation of IRE1α
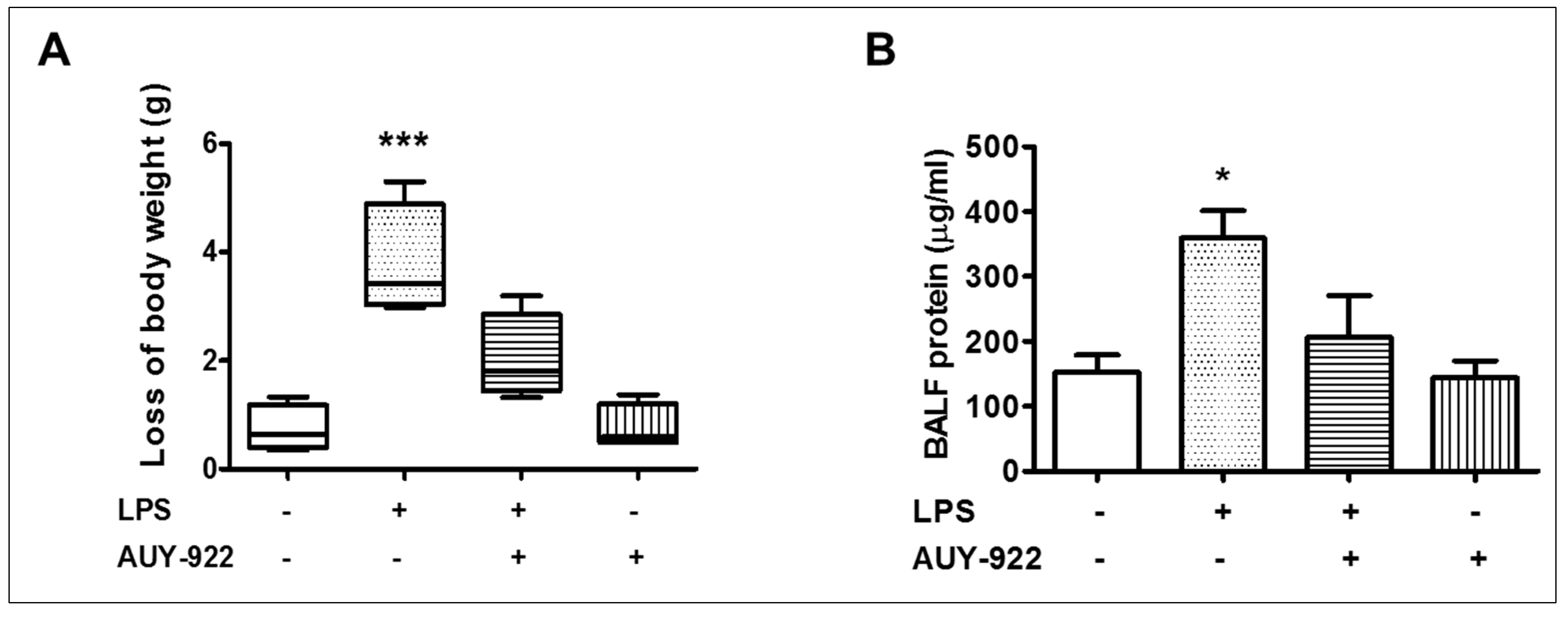
2.10. Effects of AUY-922 on LPS-Induced Body Weight Reduction and BALF Protein Concentration
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Animals
4.3. In Vivo Treatments
4.4. Collection of BALF and Total Protein Measurement
4.5. Western Blot Analysis
4.6. Densitometry and Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Calfee, C.S.; Janz, D.R.; Bernard, G.R.; May, A.K.; Kangelaris, K.N.; Matthay, M.A.; Ware, L.B. Distinct molecular phenotypes of direct vs indirect ARDS in single-center and multicenter studies. Chest 2015, 147, 1539–1548. [Google Scholar] [CrossRef] [Green Version]
- Matthay, M.A.; Zemans, R.L.; Zimmerman, G.A.; Arabi, Y.M.; Beitler, J.R.; Mercat, A.; Herridge, M.; Randolph, A.G.; Calfee, C.S. Acute respiratory distress syndrome. Nat. Rev. Dis Primers 2019, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A. Resolution of pulmonary edema. Thirty years of progress. Am. J. Respir. Crit. Care Med. 2014, 189, 1301–1308. [Google Scholar] [CrossRef] [PubMed]
- Millar, F.R.; Summers, C.; Griffiths, M.J.; Toshner, M.R.; Proudfoot, A.G. The pulmonary endothelium in acute respiratory distress syndrome: Insights and therapeutic opportunities. Thorax 2016, 71, 462–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Paola, R.; Impellizzeri, D.; Fusco, R.; Cordaro, M.; Siracusa, R.; Crupi, R.; Esposito, E.; Cuzzocrea, S. Ultramicronized palmitoylethanolamide (PEA-um((R))) in the treatment of idiopathic pulmonary fibrosis. Pharmacol. Res. 2016, 111, 405–412. [Google Scholar] [CrossRef]
- Akhter, M.S.; Uddin, M.A.; Kubra, K.T.; Barabutis, N. Autophagy, Unfolded Protein Response and Lung Disease. Curr. Res. Cell Biol. 2020, 1. [Google Scholar] [CrossRef]
- Diehl, J.A.; Fuchs, S.Y.; Koumenis, C. The cell biology of the unfolded protein response. Gastroenterology 2011, 141, 38–41. [Google Scholar] [CrossRef] [Green Version]
- Shan, B.; Wang, X.; Wu, Y.; Xu, C.; Xia, Z.; Dai, J.; Shao, M.; Zhao, F.; He, S.; Yang, L.; et al. The metabolic ER stress sensor IRE1alpha suppresses alternative activation of macrophages and impairs energy expenditure in obesity. Nat. Immunol. 2017, 18, 519–529. [Google Scholar] [CrossRef]
- Qiu, Q.; Zheng, Z.; Chang, L.; Zhao, Y.S.; Tan, C.; Dandekar, A.; Zhang, Z.; Lin, Z.; Gui, M.; Li, X.; et al. Toll-like receptor-mediated IRE1alpha activation as a therapeutic target for inflammatory arthritis. EMBO J. 2013, 32, 2477–2490. [Google Scholar] [CrossRef] [Green Version]
- Barabutis, N. Unfolded Protein Response supports endothelial barrier function. Biochimie 2019, 165, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Akhter, M.S.; Kubra, K.T.; Uddin, M.A.; Barabutis, N. Kifunensine compromises lung endothelial barrier function. Microvasc. Res. 2020, 132, 104051. [Google Scholar] [CrossRef] [PubMed]
- Akhter, M.S.; Uddin, M.A.; Schally, A.V.; Kubra, K.T.; Barabutis, N. Involvement of the unfolded protein response in the protective effects of growth hormone releasing hormone antagonists in the lungs. J. Cell Commun. Signal. 2021, 15, 125–129. [Google Scholar] [CrossRef]
- Uddin, M.A.; Akhter, M.S.; Singh, S.S.; Kubra, K.T.; Schally, A.V.; Jois, S.; Barabutis, N. GHRH antagonists support lung endothelial barrier function. Tissue Barriers 2019, 7, 1669989. [Google Scholar] [CrossRef] [Green Version]
- Barabutis, N. Growth Hormone Releasing Hormone in Endothelial Barrier Function. Trends Endocrinol. Metab. 2021, 32, 338–340. [Google Scholar] [CrossRef] [PubMed]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Park, H.K.; Yoon, N.G.; Lee, J.E.; Hu, S.; Yoon, S.; Kim, S.Y.; Hong, J.H.; Nam, D.; Chae, Y.C.; Park, J.B.; et al. Unleashing the full potential of Hsp90 inhibitors as cancer therapeutics through simultaneous inactivation of Hsp90, Grp94, and TRAP1. Exp. Mol. Med. 2020, 52, 79–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tukaj, S.; Wegrzyn, G. Anti-Hsp90 therapy in autoimmune and inflammatory diseases: A review of preclinical studies. Cell Stress Chaperones 2016, 21, 213–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haupt, A.; Joberty, G.; Bantscheff, M.; Frohlich, H.; Stehr, H.; Schweiger, M.R.; Fischer, A.; Kerick, M.; Boerno, S.T.; Dahl, A.; et al. Hsp90 inhibition differentially destabilises MAP kinase and TGF-beta signalling components in cancer cells revealed by kinase-targeted chemoproteomics. BMC Cancer 2012, 12, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, J.; Lin, D.; Xie, X.; Xu, Y.; Xu, L.; Meng, L.; Zhu, Y. NVP-AUY922, a novel HSP90 inhibitor, inhibits the progression of malignant pheochromocytoma in vitro and in vivo. Onco Targets Ther. 2017, 10, 2219–2226. [Google Scholar] [CrossRef] [Green Version]
- Nagaraju, G.P.; Mezina, A.; Shaib, W.L.; Landry, J.; El-Rayes, B.F. Targeting the Janus-activated kinase-2-STAT3 signalling pathway in pancreatic cancer using the HSP90 inhibitor ganetespib. Eur. J. Cancer 2016, 52, 109–119. [Google Scholar] [CrossRef]
- Marubayashi, S.; Koppikar, P.; Taldone, T.; Abdel-Wahab, O.; West, N.; Bhagwat, N.; Caldas-Lopes, E.; Ross, K.N.; Gonen, M.; Gozman, A.; et al. HSP90 is a therapeutic target in JAK2-dependent myeloproliferative neoplasms in mice and humans. J. Clin. Investig. 2010, 120, 3578–3593. [Google Scholar] [CrossRef]
- Uddin, M.A.; Akhter, M.S.; Siejka, A.; Catravas, J.D.; Barabutis, N. P53 supports endothelial barrier function via APE1/Ref1 suppression. Immunobiology 2019, 224, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Kubra, K.T.; Uddin, M.A.; Akhter, M.S.; Barabutis, N. Hsp90 inhibitors induce the unfolded protein response in bovine and mice lung cells. Cell Signal. 2020, 67, 109500. [Google Scholar] [CrossRef]
- Uddin, M.A.; Kubra, K.T.; Sonju, J.J.; Akhter, M.S.; Seetharama, J.; Barabutis, N. Effects of Heat Shock Protein 90 Inhibition In the Lungs. Med. Drug Discov. 2020, 6. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Thangjam, G.S.; Birmpas, C.; Barabutis, N.; Gregory, B.W.; Clemens, M.A.; Newton, J.R.; Fulton, D.; Catravas, J.D. Hsp90 inhibition suppresses NF-kappaB transcriptional activation via Sirt-2 in human lung microvascular endothelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L964–L974. [Google Scholar] [CrossRef] [PubMed]
- Sevin, M.; Girodon, F.; Garrido, C.; de Thonel, A. HSP90 and HSP70: Implication in Inflammation Processes and Therapeutic Approaches for Myeloproliferative Neoplasms. Mediat. Inflamm. 2015, 2015, 970242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitanaka, N.; Nakano, R.; Sugiura, K.; Kitanaka, T.; Namba, S.; Konno, T.; Nakayama, T.; Sugiya, H. Interleukin-1beta promotes interleulin-6 expression via ERK1/2 signaling pathway in canine dermal fibroblasts. PLoS ONE 2019, 14, e0220262. [Google Scholar] [CrossRef]
- Nick, J.A.; Avdi, N.J.; Gerwins, P.; Johnson, G.L.; Worthen, G.S. Activation of a p38 mitogen-activated protein kinase in human neutrophils by lipopolysaccharide. J. Immunol. 1996, 156, 4867–4875. [Google Scholar]
- Stone, M.K.; Kolling, G.L.; Lindner, M.H.; Obrig, T.G. p38 mitogen-activated protein kinase mediates lipopolysaccharide and tumor necrosis factor alpha induction of shiga toxin 2 sensitivity in human umbilical vein endothelial cells. Infect. Immun. 2008, 76, 1115–1121. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Kim, S.C.; Yu, T.; Yi, Y.S.; Rhee, M.H.; Sung, G.H.; Yoo, B.C.; Cho, J.Y. Functional roles of p38 mitogen-activated protein kinase in macrophage-mediated inflammatory responses. Mediat. Inflamm. 2014, 2014, 352371. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.H.; Sung, K.S.; Bartlett, D.L.; Kwon, Y.T.; Lee, Y.J. HSP90 inhibitor NVP-AUY922 enhances TRAIL-induced apoptosis by suppressing the JAK2-STAT3-Mcl-1 signal transduction pathway in colorectal cancer cells. Cell Signal. 2015, 27, 293–305. [Google Scholar] [CrossRef] [Green Version]
- Frossi, B.; Antoniali, G.; Yu, K.; Akhtar, N.; Kaplan, M.H.; Kelley, M.R.; Tell, G.; Pucillo, C.E.M. Endonuclease and redox activities of human apurinic/apyrimidinic endonuclease 1 have distinctive and essential functions in IgA class switch recombination. J. Biol. Chem. 2019, 294, 5198–5207. [Google Scholar] [CrossRef]
- Xia, Y.; Padre, R.C.; De Mendoza, T.H.; Bottero, V.; Tergaonkar, V.B.; Verma, I.M. Phosphorylation of p53 by IkappaB kinase 2 promotes its degradation by beta-TrCP. Proc. Natl. Acad. Sci. USA 2009, 106, 2629–2634. [Google Scholar] [CrossRef] [Green Version]
- Irarrazabal, C.E.; Liu, J.C.; Burg, M.B.; Ferraris, J.D. ATM, a DNA damage-inducible kinase, contributes to activation by high NaCl of the transcription factor TonEBP/OREBP. Proc. Natl. Acad. Sci. USA 2004, 101, 8809–8814. [Google Scholar] [CrossRef] [Green Version]
- Salt, I.P.; Palmer, T.M. Exploiting the anti-inflammatory effects of AMP-activated protein kinase activation. Expert Opin. Investig. Drugs 2012, 21, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Zhang, Y.W.; Lee, J.H.; Zeng, S.X.; Wang, Y.V.; Luo, Z.; Dong, X.C.; Viollet, B.; Wahl, G.M.; Lu, H. AMP-activated protein kinase induces p53 by phosphorylating MDMX and inhibiting its activity. Mol. Cell Biol. 2014, 34, 148–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voruganti, S.; Lacroix, J.C.; Rogers, C.N.; Rogers, J.; Matts, R.L.; Hartson, S.D. The anticancer drug AUY922 generates a proteomics fingerprint that is highly conserved among structurally diverse Hsp90 inhibitors. J. Proteome Res. 2013, 12, 3697–3706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oki, Y.; Younes, A.; Knickerbocker, J.; Samaniego, F.; Nastoupil, L.; Hagemeister, F.; Romaguera, J.; Fowler, N.; Kwak, L.; Westin, J. Experience with HSP90 inhibitor AUY922 in patients with relapsed or refractory non-Hodgkin lymphoma. Haematologica 2015, 100, e272–e274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuno, A.; Lee, M.J.; Lee, S.; Tomita, Y.; Rekhtman, D.; Moore, B.; Trepel, J.B. Clinical Evaluation and Biomarker Profiling of Hsp90 Inhibitors. Methods Mol. Biol. 2018, 1709, 423–441. [Google Scholar] [CrossRef] [PubMed]
- Solopov, P.; Biancatelli, R.; Marinova, M.; Dimitropoulou, C.; Catravas, J.D. The HSP90 Inhibitor, AUY-922, Ameliorates the Development of Nitrogen Mustard-Induced Pulmonary Fibrosis and Lung Dysfunction in Mice. Int. J. Mol. Sci. 2020, 21, 4740. [Google Scholar] [CrossRef]
- Joshi, A.D.; Dimitropoulou, C.; Thangjam, G.; Snead, C.; Feldman, S.; Barabutis, N.; Fulton, D.; Hou, Y.; Kumar, S.; Patel, V.; et al. Heat shock protein 90 inhibitors prevent LPS-induced endothelial barrier dysfunction by disrupting RhoA signaling. Am. J. Respir. Cell Mol. Biol. 2014, 50, 170–179. [Google Scholar] [CrossRef]
- Antonov, A.; Snead, C.; Gorshkov, B.; Antonova, G.N.; Verin, A.D.; Catravas, J.D. Heat shock protein 90 inhibitors protect and restore pulmonary endothelial barrier function. Am. J. Respir. Cell Mol. Biol. 2008, 39, 551–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhter, M.S.; Uddin, M.A.; Barabutis, N. P53 Regulates the Redox Status of Lung Endothelial Cells. Inflammation 2020, 43, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Akhter, M.S.; Uddin, M.A.; Kubra, K.T.; Barabutis, N. P53-induced reduction of lipid peroxidation supports brain microvascular endothelium integrity. J. Pharmacol. Sci. 2019, 141, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Barabutis, N. P53 in RhoA regulation. Cytoskeleton 2020, 77, 197–201. [Google Scholar] [CrossRef]
- Kubra, K.T.; Akhter, M.S.; Uddin, M.A.; Barabutis, N. P53 versus inflammation: An update. Cell Cycle 2020, 19, 160–162. [Google Scholar] [CrossRef]
- Barabutis, N. P53 in lung vascular barrier dysfunction. Vasc. Biol. 2020, 2, E1–E2. [Google Scholar] [CrossRef]
- Barabutis, N.; Uddin, M.A.; Catravas, J.D. Hsp90 inhibitors suppress P53 phosphorylation in LPS-induced endothelial inflammation. Cytokine 2019, 113, 427–432. [Google Scholar] [CrossRef]
- Kubra, K.T.; Uddin, M.A.; Akhter, M.S.; Barabutis, N. P53 is Subjected to Lipoteichoic Acid-Induced Phosphorylation in the Lungs. TH Open 2020, 4, e173–e174. [Google Scholar] [CrossRef]
- Akhter, M.S.; Uddin, M.A.; Barabutis, N. Unfolded protein response regulates P53 expression in the pulmonary endothelium. J. Biochem. Mol. Toxicol. 2019, 33, e22380. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.K.; Lin, Y.H.; Huang, T.C.; Shi, C.S.; Yang, C.T.; Yang, Y.L. VEGF mediates fat embolism-induced acute lung injury via VEGF receptor 2 and the MAPK cascade. Sci. Rep. 2019, 9, 11713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Dai, R.; Zhou, Y.; Fu, H.; Meng, Q. TLR2 Ligand Pam3CSK4 Regulates MMP-2/9 Expression by MAPK/NF-kappaB Signaling Pathways in Primary Brain Microvascular Endothelial Cells. Neurochem. Res. 2018, 43, 1897–1904. [Google Scholar] [CrossRef] [Green Version]
- He, H.; Guo, F.; Li, Y.; Saaoud, F.; Kimmis, B.D.; Sandhu, J.; Fan, M.; Maulik, D.; Lessner, S.; Papasian, C.J.; et al. Adiporedoxin suppresses endothelial activation via inhibiting MAPK and NF-kappaB signaling. Sci. Rep. 2016, 6, 38975. [Google Scholar] [CrossRef]
- Yang, D.; Liu, J.; Tian, C.; Zeng, Y.; Zheng, Y.H.; Fang, Q.; Li, H.H. Epigallocatechin gallate inhibits angiotensin II-induced endothelial barrier dysfunction via inhibition of the p38 MAPK/HSP27 pathway. Acta Pharmacol. Sin. 2010, 31, 1401–1406. [Google Scholar] [CrossRef] [Green Version]
- Birukova, A.A.; Birukov, K.G.; Gorshkov, B.; Liu, F.; Garcia, J.G.; Verin, A.D. MAP kinases in lung endothelial permeability induced by microtubule disassembly. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 289, L75–L84. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Li, Y.; Jin, J.; Zhang, X.; Lopes-Virella, M.F.; Huang, Y. Toll-like receptor 4 activation in microvascular endothelial cells triggers a robust inflammatory response and cross talk with mononuclear cells via interleukin-6. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1696–1706. [Google Scholar] [CrossRef] [Green Version]
- Fang, W.; Bi, D.; Zheng, R.; Cai, N.; Xu, H.; Zhou, R.; Lu, J.; Wan, M.; Xu, X. Identification and activation of TLR4-mediated signalling pathways by alginate-derived guluronate oligosaccharide in RAW264.7 macrophages. Sci. Rep. 2017, 7, 1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandao, S.C.S.; Ramos, J.O.X.; Dompieri, L.T.; Godoi, E.; Figueiredo, J.L.; Sarinho, E.S.C.; Chelvanambi, S.; Aikawa, M. Is Toll-like receptor 4 involved in the severity of COVID-19 pathology in patients with cardiometabolic comorbidities? Cytokine Growth Factor Rev. 2020, 58, 102–110. [Google Scholar] [CrossRef]
- Chatterjee, A.; Dimitropoulou, C.; Drakopanayiotakis, F.; Antonova, G.; Snead, C.; Cannon, J.; Venema, R.C.; Catravas, J.D. Heat shock protein 90 inhibitors prolong survival, attenuate inflammation, and reduce lung injury in murine sepsis. Am. J. Respir. Crit. Care Med. 2007, 176, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Bishop, J.L.; Thaper, D.; Zoubeidi, A. The Multifaceted Roles of STAT3 Signaling in the Progression of Prostate Cancer. Cancers 2014, 6, 829–859. [Google Scholar] [CrossRef] [Green Version]
- Alsaffar, H.; Martino, N.; Garrett, J.P.; Adam, A.P. Interleukin-6 promotes a sustained loss of endothelial barrier function via Janus kinase-mediated STAT3 phosphorylation and de novo protein synthesis. Am. J. Physiol. Cell Physiol. 2018, 314, C589–C602. [Google Scholar] [CrossRef] [Green Version]
- Yun, J.H.; Park, S.W.; Kim, K.J.; Bae, J.S.; Lee, E.H.; Paek, S.H.; Kim, S.U.; Ye, S.; Kim, J.H.; Cho, C.H. Endothelial STAT3 Activation Increases Vascular Leakage Through Downregulating Tight Junction Proteins: Implications for Diabetic Retinopathy. J. Cell Physiol. 2017, 232, 1123–1134. [Google Scholar] [CrossRef]
- Shah, M.; Patel, K.; Fried, V.A.; Sehgal, P.B. Interactions of STAT3 with caveolin-1 and heat shock protein 90 in plasma membrane raft and cytosolic complexes. Preservation of cytokine signaling during fever. J. Biol. Chem. 2002, 277, 45662–45669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleh, A.; Shan, L.; Halayko, A.J.; Kung, S.; Gounni, A.S. Critical role for STAT3 in IL-17A-mediated CCL11 expression in human airway smooth muscle cells. J. Immunol. 2009, 182, 3357–3365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiloh, Y. ATM and related protein kinases: Safeguarding genome integrity. Nat. Rev. Cancer 2003, 3, 155–168. [Google Scholar] [CrossRef]
- Magnussen, H.M.; Ahmed, S.F.; Sibbet, G.J.; Hristova, V.A.; Nomura, K.; Hock, A.K.; Archibald, L.J.; Jamieson, A.G.; Fushman, D.; Vousden, K.H.; et al. Structural basis for DNA damage-induced phosphoregulation of MDM2 RING domain. Nat. Commun. 2020, 11, 2094. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Chen, J. Mechanism of p53 stabilization by ATM after DNA damage. Cell Cycle 2010, 9, 472–478. [Google Scholar] [CrossRef] [Green Version]
- Mei, L.; Zhang, J.; He, K.; Zhang, J. Ataxia telangiectasia and Rad3-related inhibitors and cancer therapy: Where we stand. J. Hematol. Oncol. 2019, 12, 43. [Google Scholar] [CrossRef]
- Kawanishi, S.; Ohnishi, S.; Ma, N.; Hiraku, Y.; Murata, M. Crosstalk between DNA Damage and Inflammation in the Multiple Steps of Carcinogenesis. Int. J. Mol. Sci. 2017, 18, 1808. [Google Scholar] [CrossRef] [Green Version]
- Koll, T.T.; Feis, S.S.; Wright, M.H.; Teniola, M.M.; Richardson, M.M.; Robles, A.I.; Bradsher, J.; Capala, J.; Varticovski, L. HSP90 inhibitor, DMAG, synergizes with radiation of lung cancer cells by interfering with base excision and ATM-mediated DNA repair. Mol. Cancer Ther. 2008, 7, 1985–1992. [Google Scholar] [CrossRef] [Green Version]
- Kaser, A.; Lee, A.H.; Franke, A.; Glickman, J.N.; Zeissig, S.; Tilg, H.; Nieuwenhuis, E.E.; Higgins, D.E.; Schreiber, S.; Glimcher, L.H.; et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 2008, 134, 743–756. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Wang, J.M.; Qiu, Y.; Yang, Z.; Kim, H.; Qian, Q.; Sun, Q.; Zhang, C.; Yin, L.; Fang, D.; Back, S.H.; et al. IRE1alpha prevents hepatic steatosis by processing and promoting the degradation of select microRNAs. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Calay, E.S.; Fan, J.; Arduini, A.; Kunz, R.C.; Gygi, S.P.; Yalcin, A.; Fu, S.; Hotamisligil, G.S. METABOLISM. S-Nitrosylation links obesity-associated inflammation to endoplasmic reticulum dysfunction. Science 2015, 349, 500–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urra, H.; Henriquez, D.R.; Canovas, J.; Villarroel-Campos, D.; Carreras-Sureda, A.; Pulgar, E.; Molina, E.; Hazari, Y.M.; Limia, C.M.; Alvarez-Rojas, S.; et al. IRE1alpha governs cytoskeleton remodelling and cell migration through a direct interaction with filamin A. Nat. Cell Biol. 2018, 20, 942–953. [Google Scholar] [CrossRef]
- Marcu, M.G.; Doyle, M.; Bertolotti, A.; Ron, D.; Hendershot, L.; Neckers, L. Heat shock protein 90 modulates the unfolded protein response by stabilizing IRE1alpha. Mol. Cell Biol. 2002, 22, 8506–8513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ota, A.; Wang, Y. Cdc37/Hsp90 protein-mediated regulation of IRE1alpha protein activity in endoplasmic reticulum stress response and insulin synthesis in INS-1 cells. J. Biol. Chem. 2012, 287, 6266–6274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davenport, E.L.; Moore, H.E.; Dunlop, A.S.; Sharp, S.Y.; Workman, P.; Morgan, G.J.; Davies, F.E. Heat shock protein inhibition is associated with activation of the unfolded protein response pathway in myeloma plasma cells. Blood 2007, 110, 2641–2649. [Google Scholar] [CrossRef] [Green Version]
- Hsueh, Y.S.; Chang, H.H.; Chiang, N.J.; Yen, C.C.; Li, C.F.; Chen, L.T. MTOR inhibition enhances NVP-AUY922-induced autophagy-mediated KIT degradation and cytotoxicity in imatinib-resistant gastrointestinal stromal tumors. Oncotarget 2014, 5, 11723–11736. [Google Scholar] [CrossRef] [Green Version]
- Barabutis, N.; Dimitropoulou, C.; Birmpas, C.; Joshi, A.; Thangjam, G.; Catravas, J.D. p53 protects against LPS-induced lung endothelial barrier dysfunction. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L776–L787. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akhter, M.S.; Uddin, M.A.; Kubra, K.-T.; Barabutis, N. Elucidation of the Molecular Pathways Involved in the Protective Effects of AUY-922 in LPS-Induced Inflammation in Mouse Lungs. Pharmaceuticals 2021, 14, 522. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14060522
Akhter MS, Uddin MA, Kubra K-T, Barabutis N. Elucidation of the Molecular Pathways Involved in the Protective Effects of AUY-922 in LPS-Induced Inflammation in Mouse Lungs. Pharmaceuticals. 2021; 14(6):522. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14060522
Chicago/Turabian StyleAkhter, Mohammad S., Mohammad A. Uddin, Khadeja-Tul Kubra, and Nektarios Barabutis. 2021. "Elucidation of the Molecular Pathways Involved in the Protective Effects of AUY-922 in LPS-Induced Inflammation in Mouse Lungs" Pharmaceuticals 14, no. 6: 522. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14060522