
Design, Synthesis, In Vitro and In Vivo Evaluation of Heterobivalent SiFAlin-Modified Peptidic Radioligands Targeting Both Integrin αvβ3 and the MC1 Receptor—Suitable for the Specific Visualization of Melanomas?

 , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
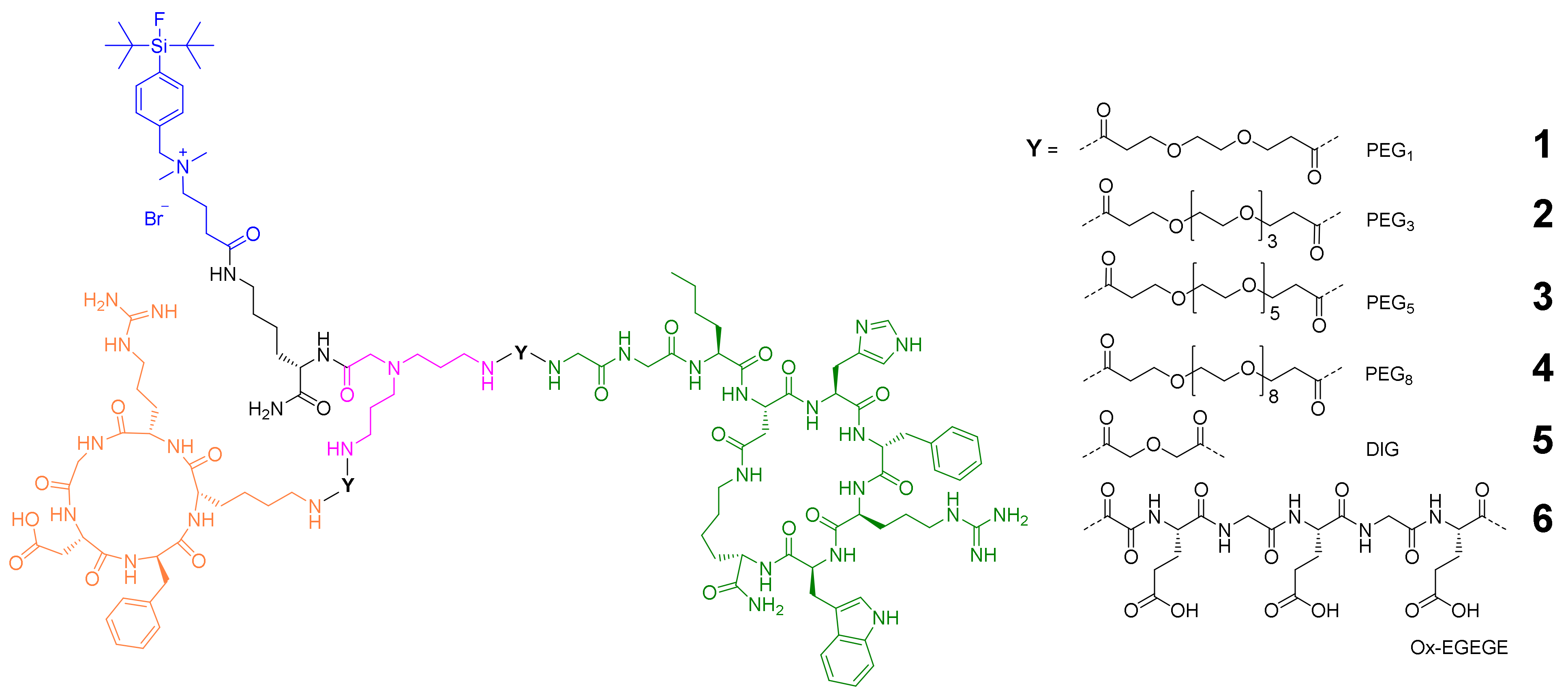
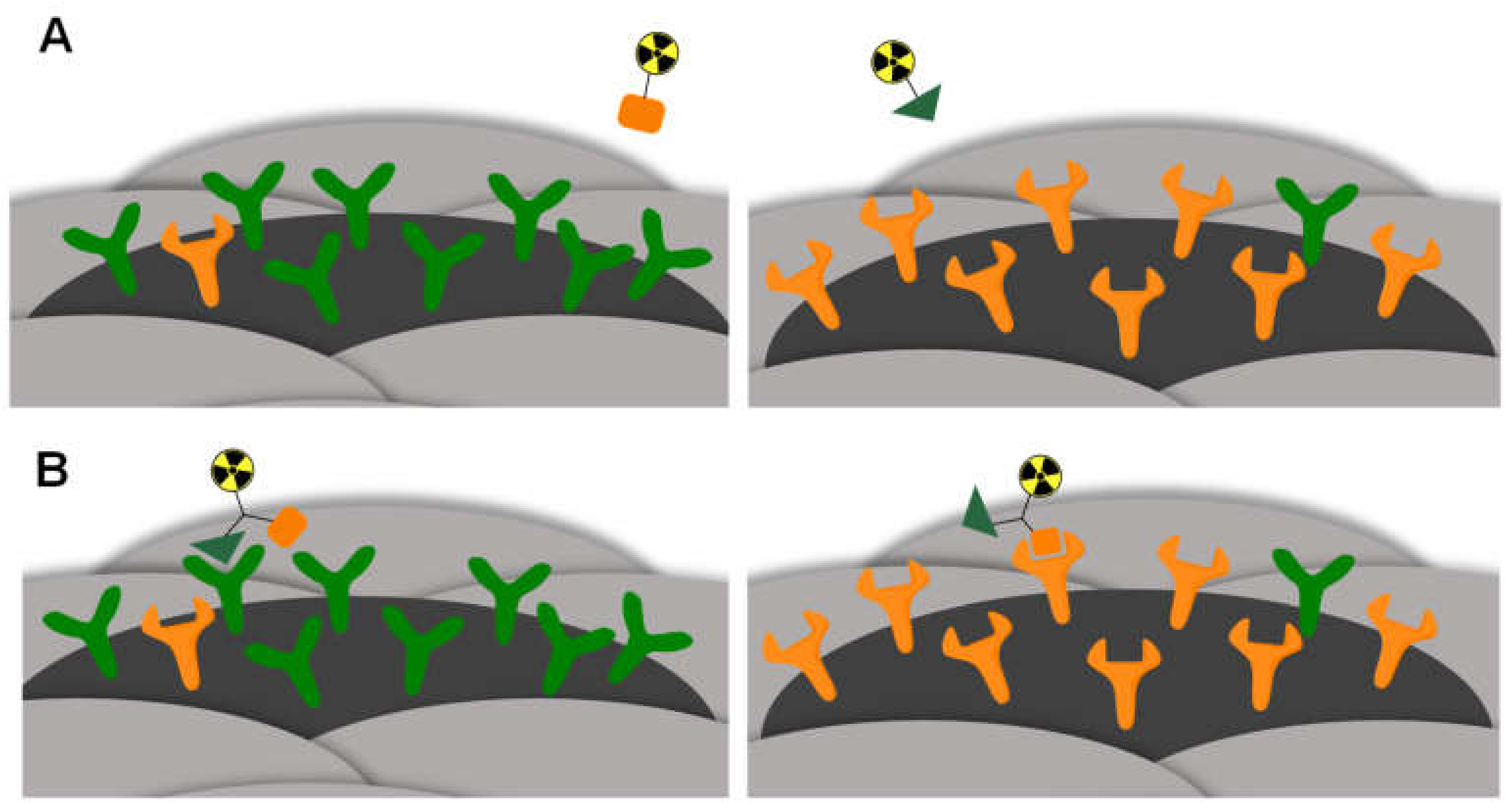
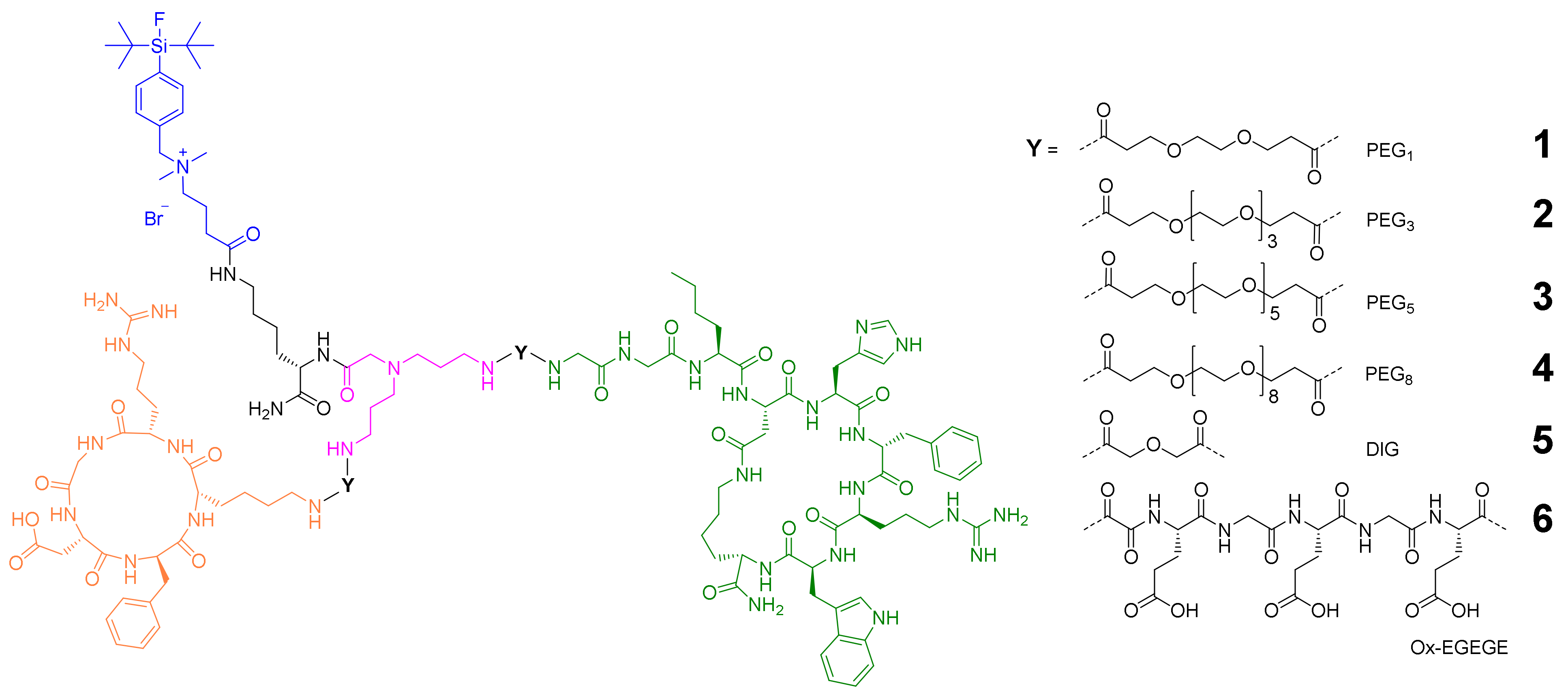
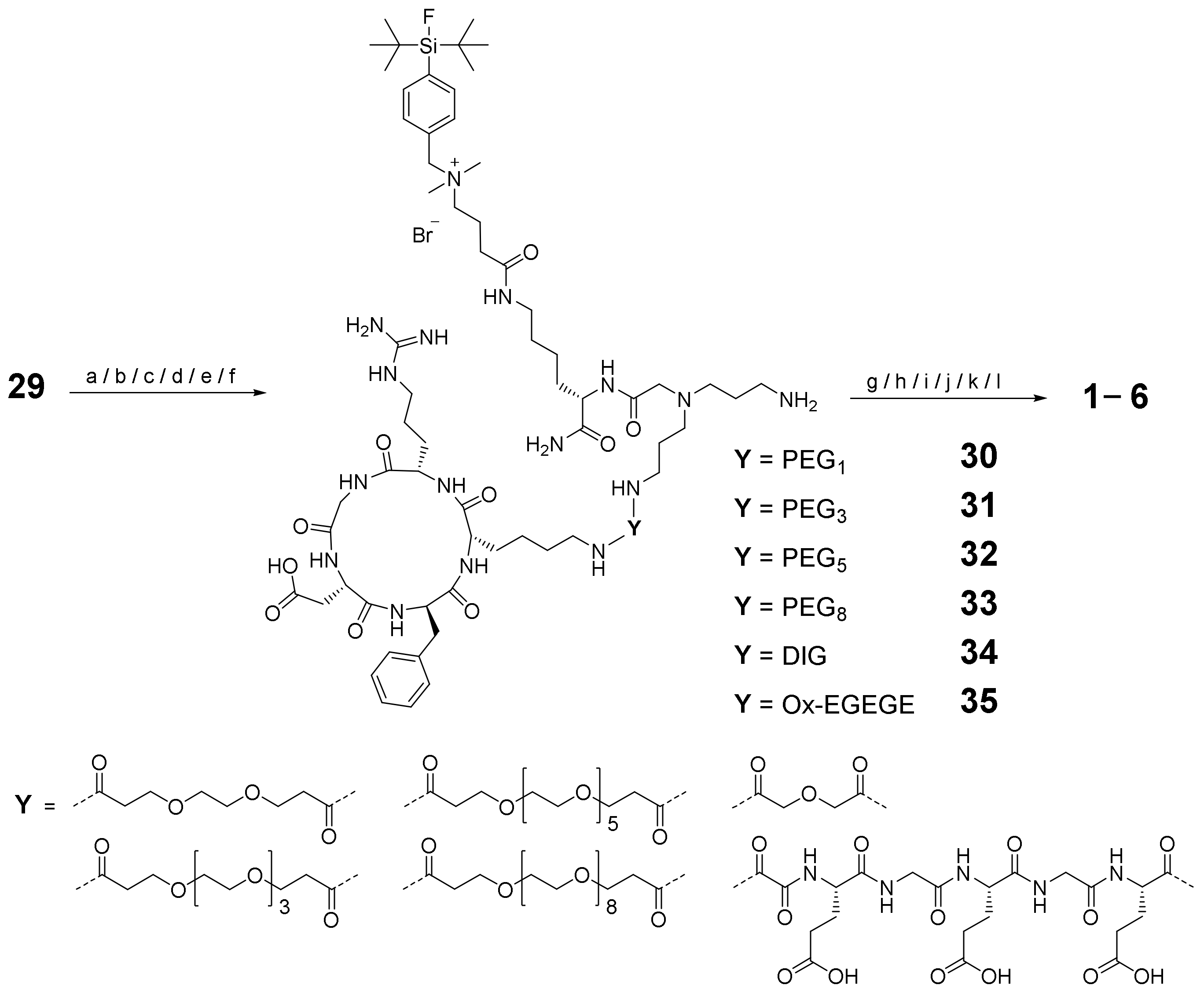
2.1. General Considerations for the Design of the Heterobivalent SiFAlin-Modified Peptidic Ligands
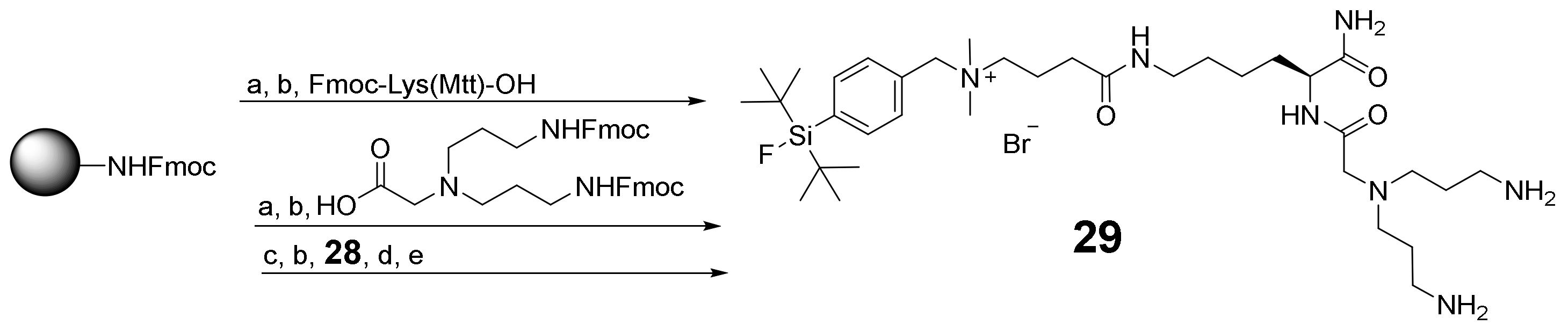
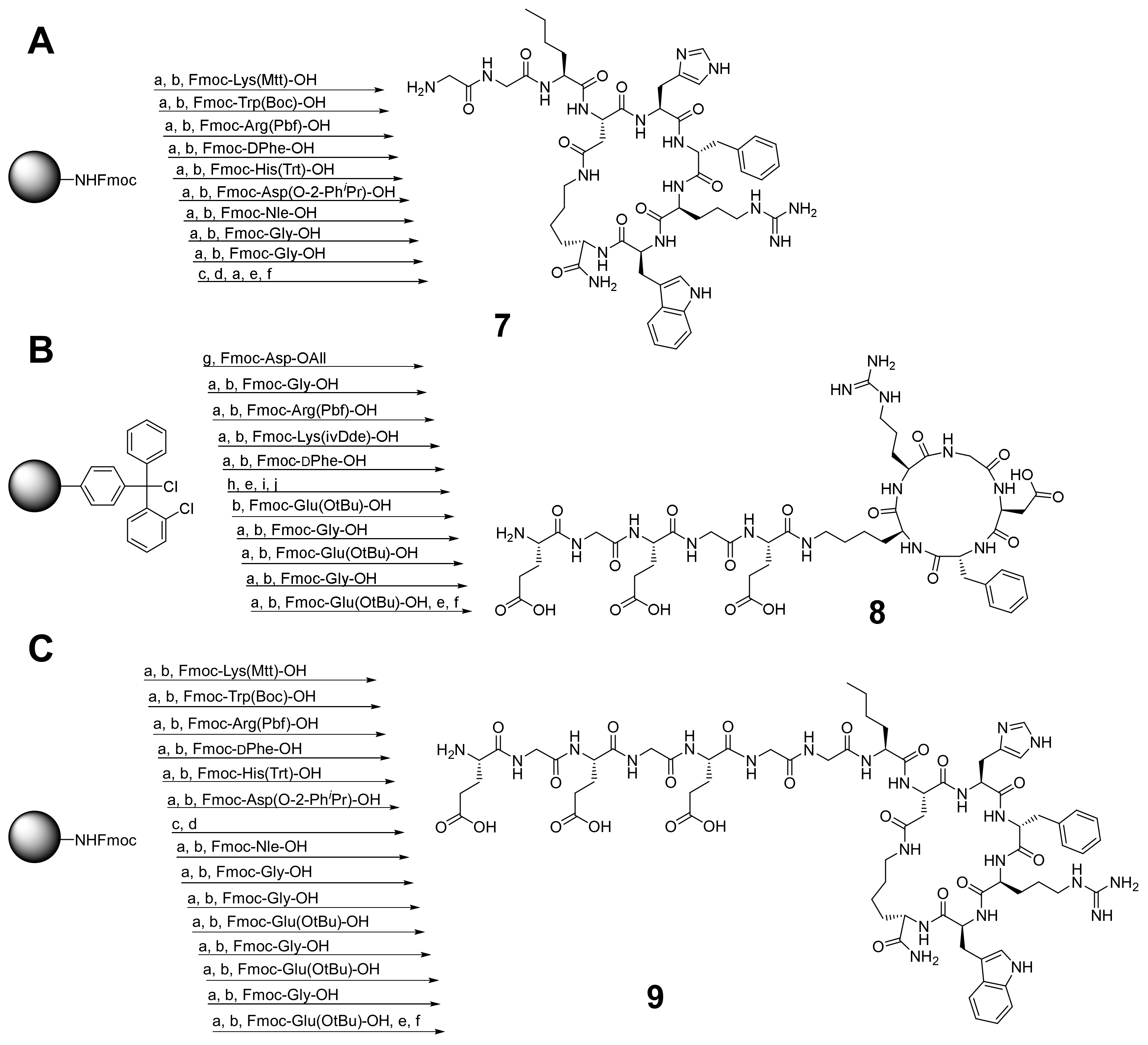
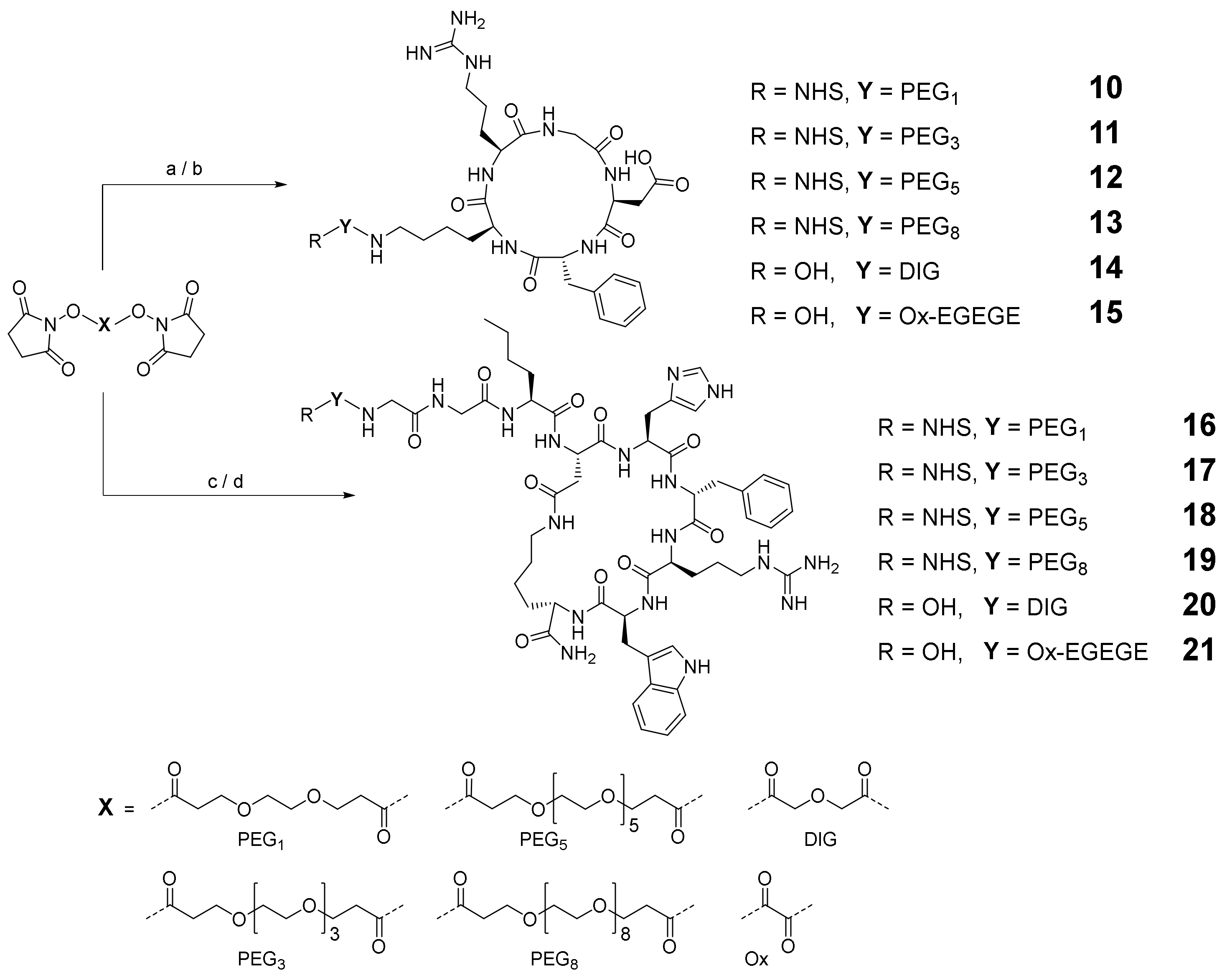
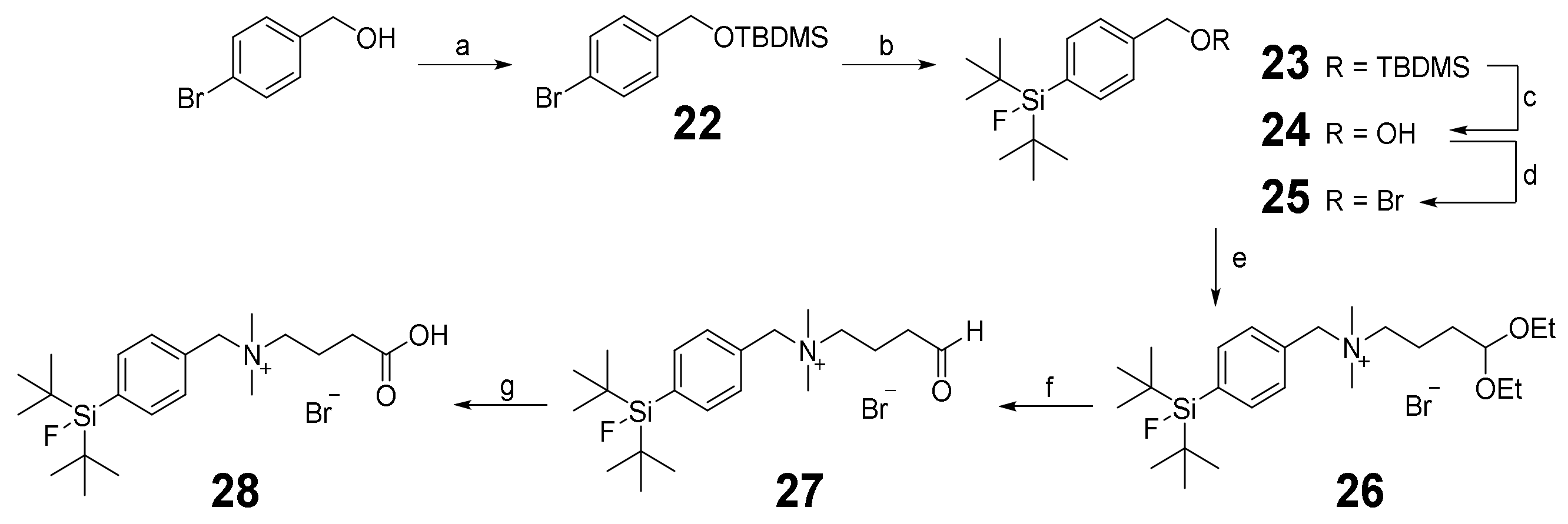
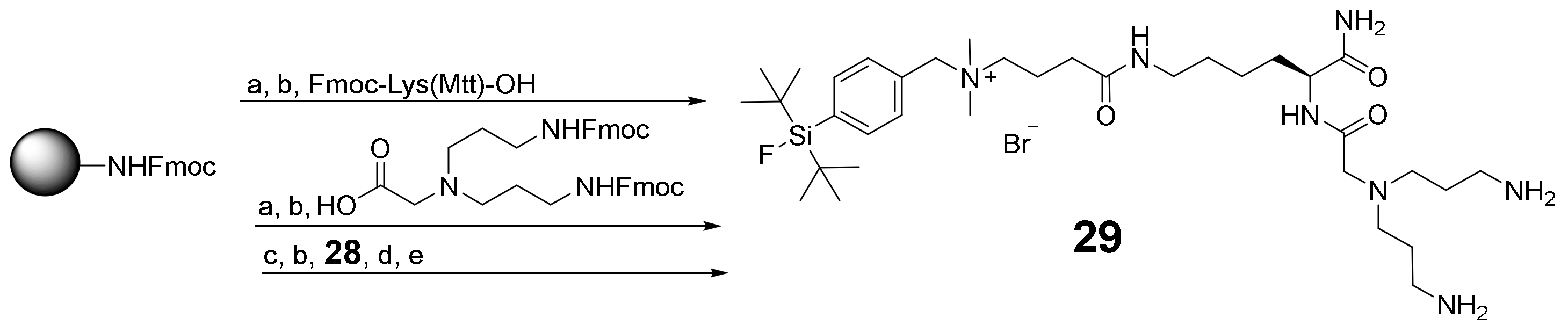
2.2. Synthesis of the Heterobivalent SiFAlin-Modified Peptidic Ligands
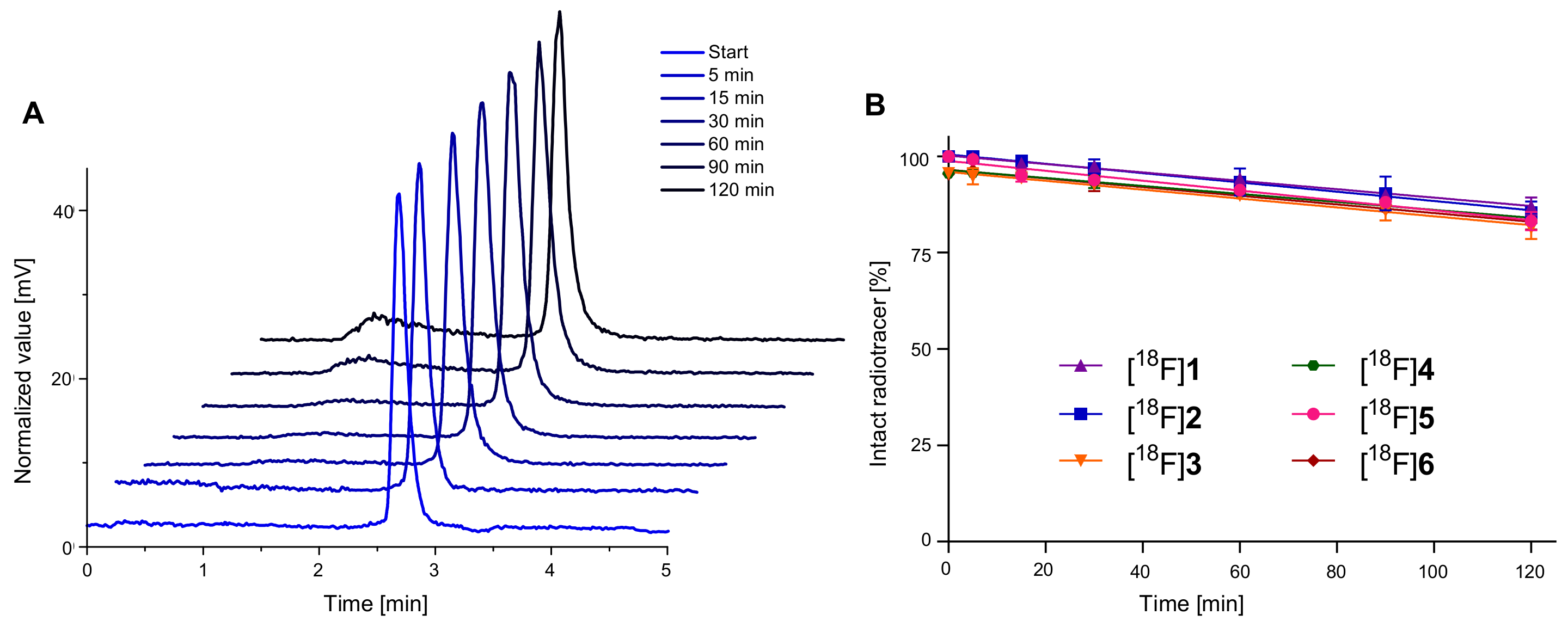
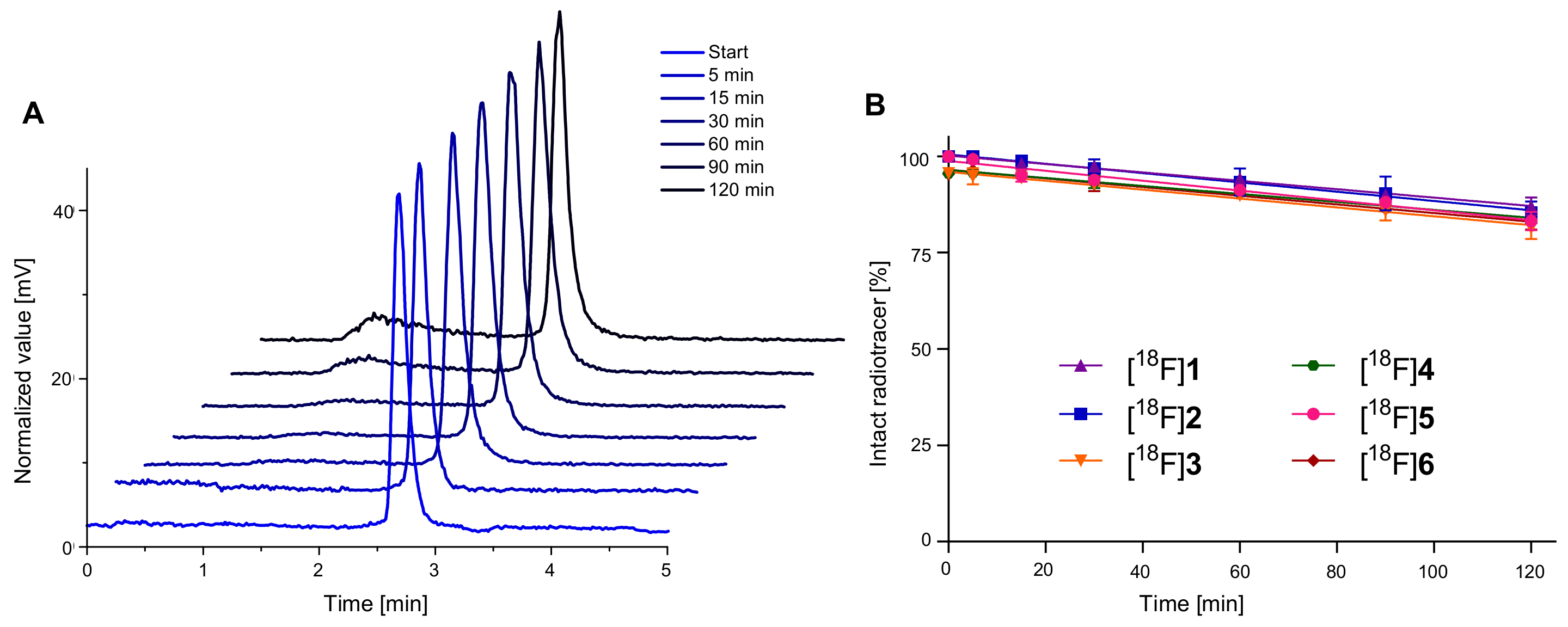
2.3. 18F-Radiolabeling of 1–6 and Determination of Lipophilicity and Stability of [18F]1–[18F]6 in Human Serum
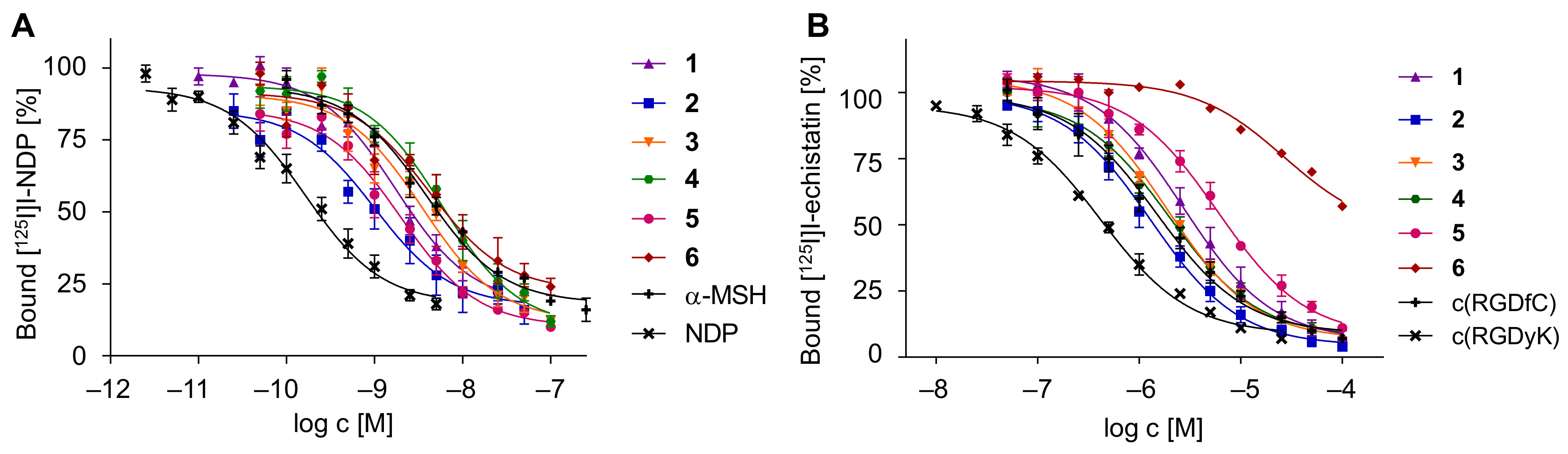
2.4. In Vitro Evaluation of 1–6 Regarding Their Binding Affinities to the Respective Target Receptors
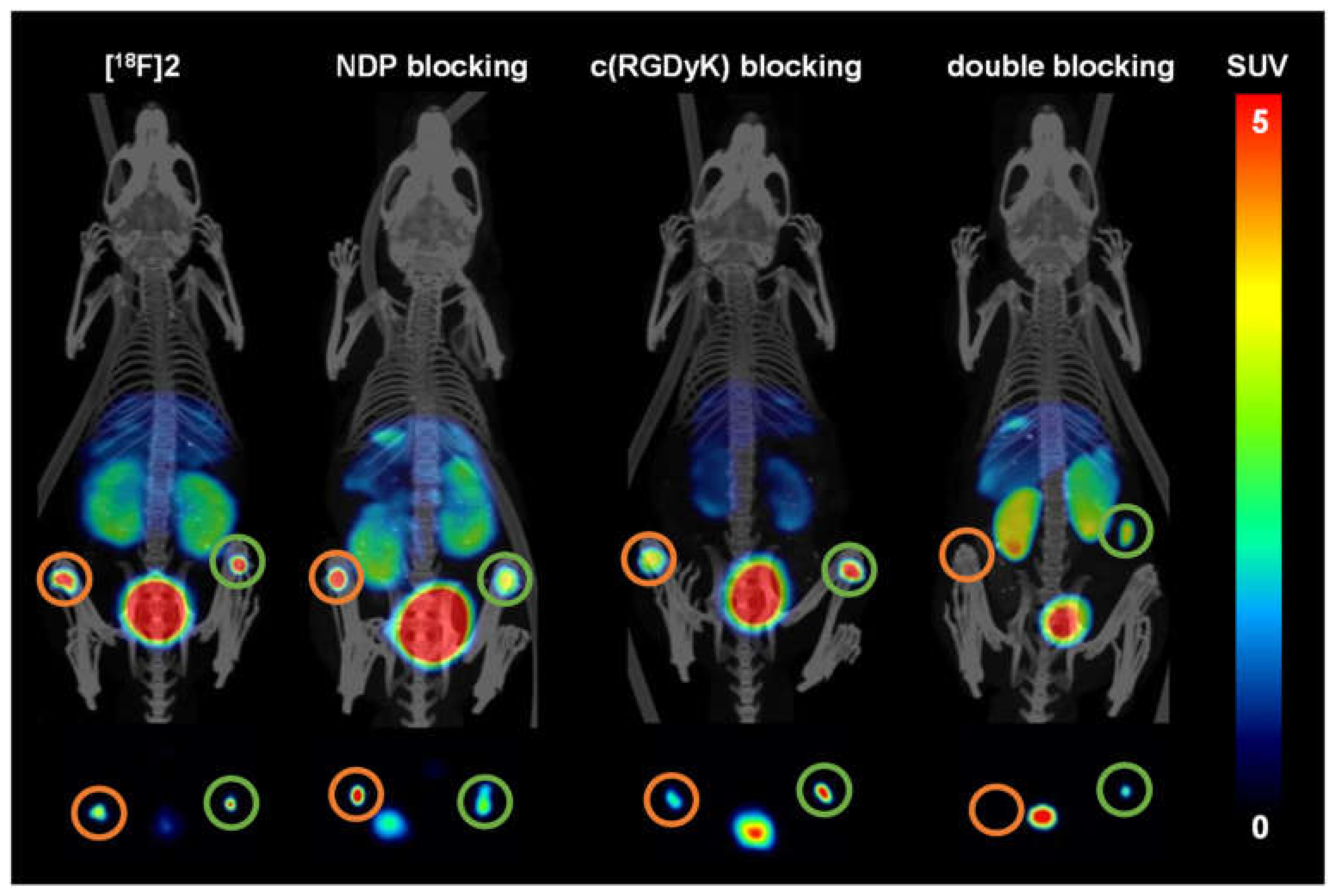
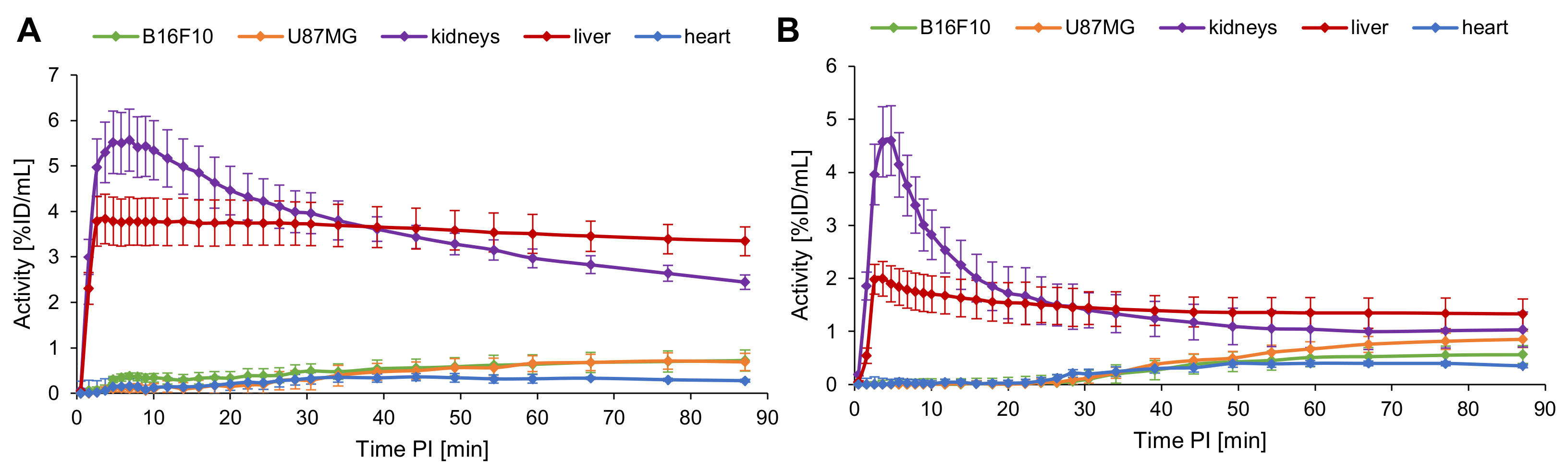
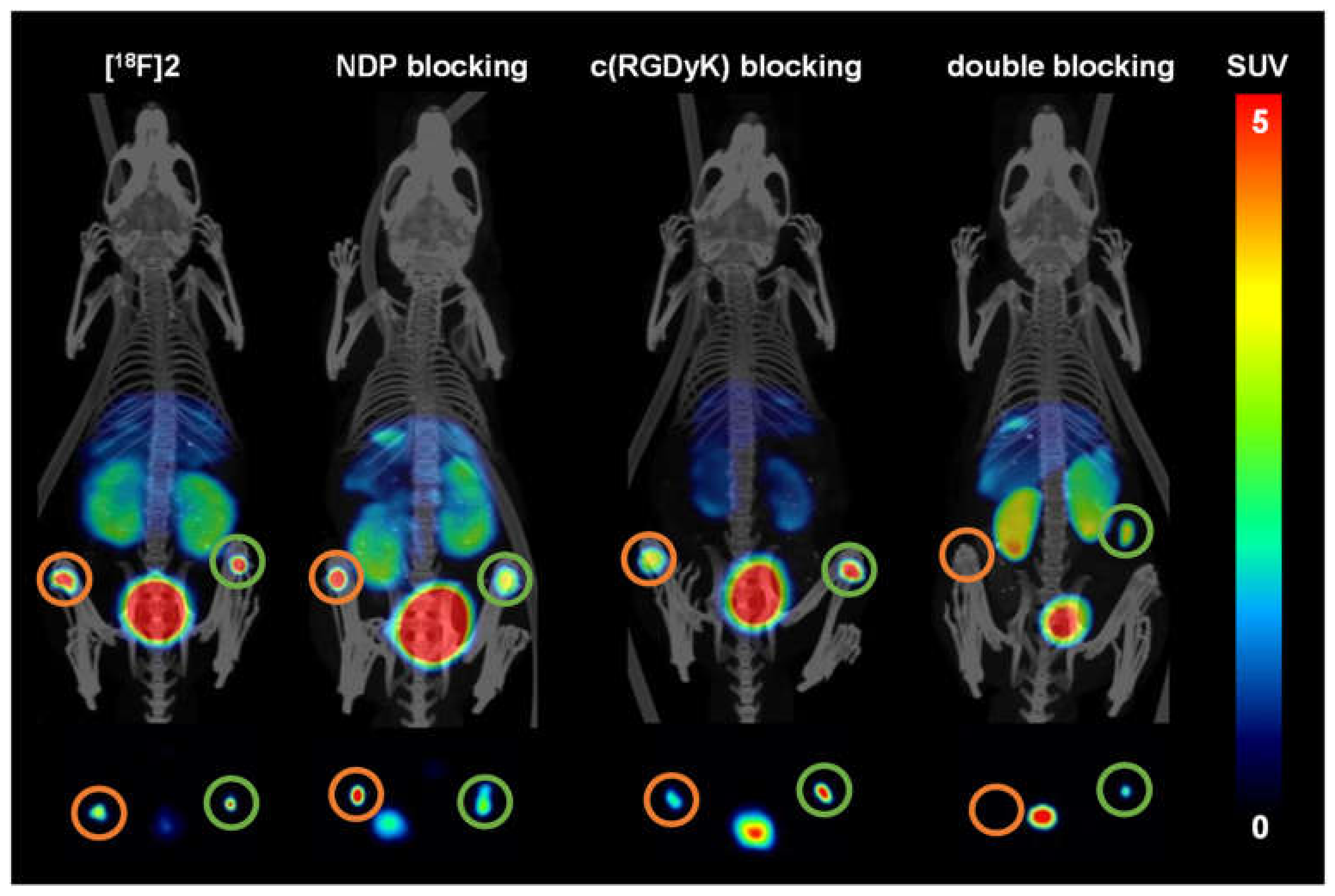
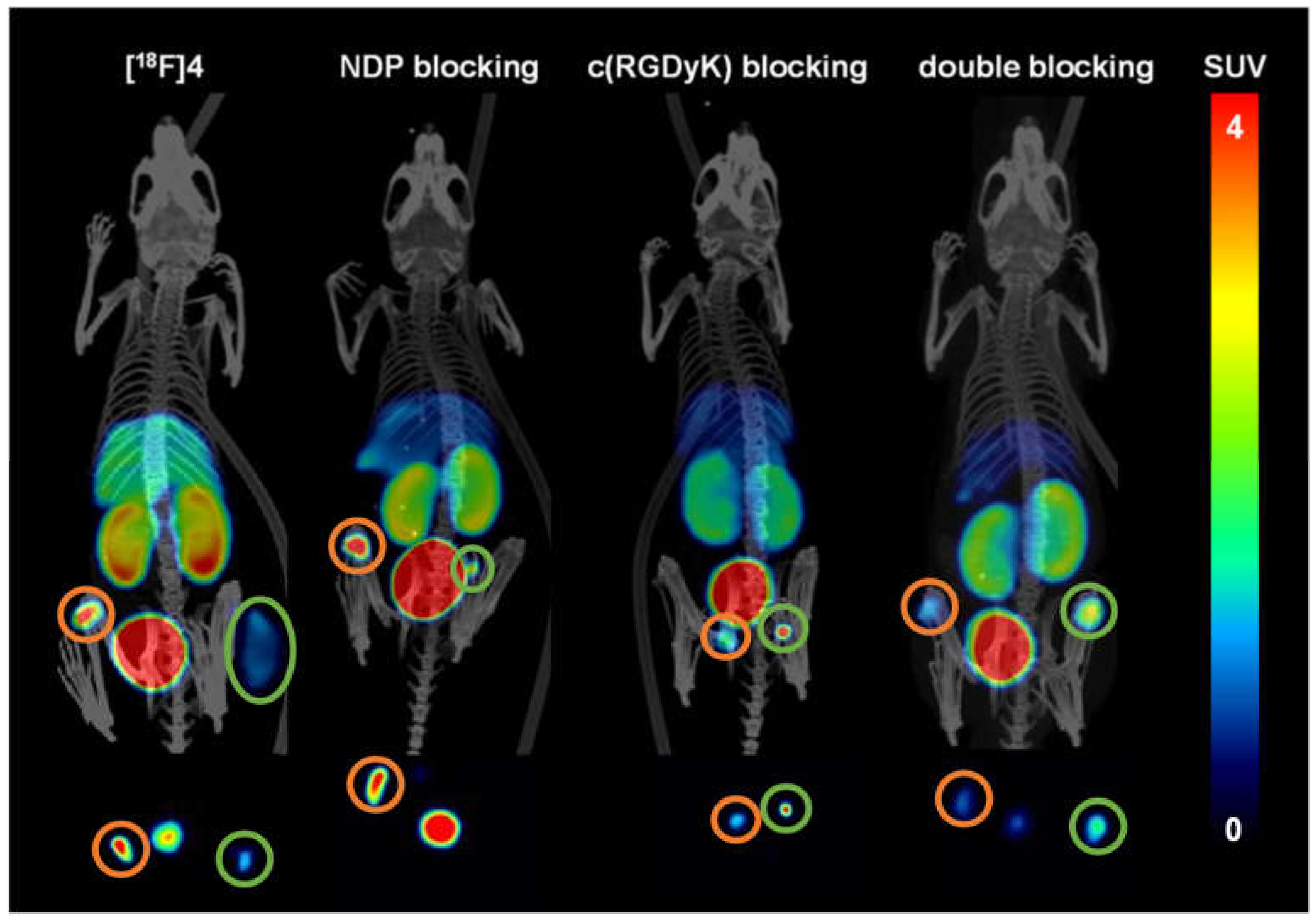
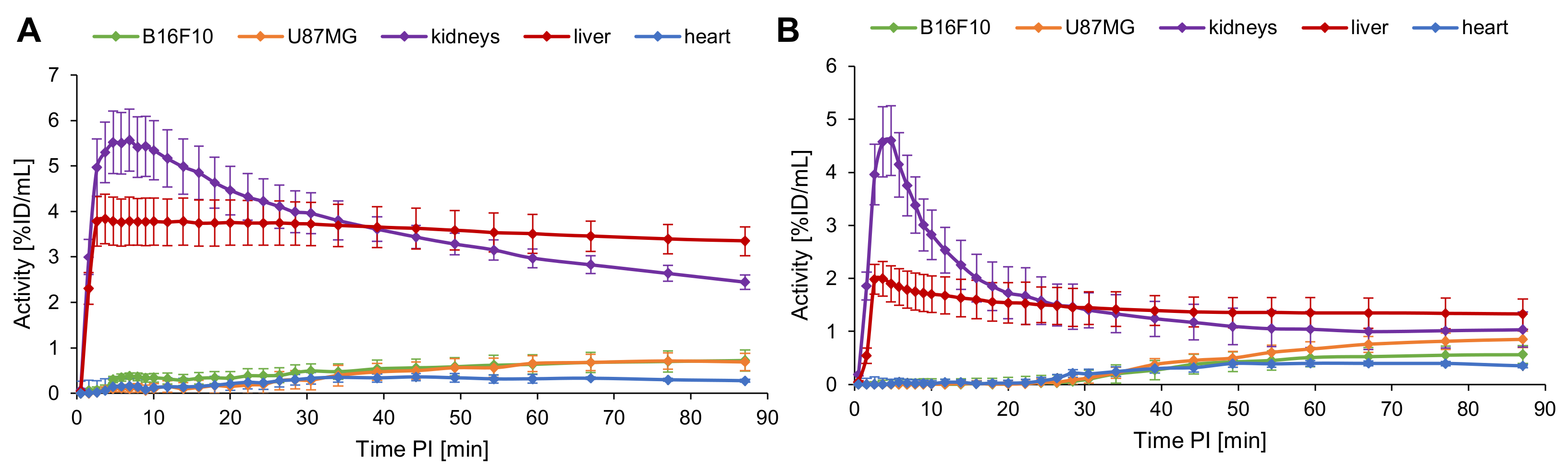
2.5. Evaluation of the In Vivo Pharmacokinetics and Ex Vivo Biodistribution of [18F]2 and [18F]4
3. Materials and Methods
3.1. General
3.1.1. Chemistry
3.1.2. Radiolabeling
3.1.3. Competitive Binding Studies
3.1.4. In Vivo PET Imaging
3.1.5. Statistical Analyses
3.2. Chemical Syntheses
3.2.1. Synthesis of Peptides 7–9
3.2.2. Modification of the Peptides with Linker Structures to Obtain 10–21
3.2.3. Synthesis of SiFAlin-Carboxylic Acid 28 and SiFAlin-Modified Symmetrically Branching Framework 29
3.2.4. Conjugation of 10–21 to the SiFAlin-Modified Framework 29 to Obtain the Target HBPLs 1–6 via the Intermediates 30–35
3.3. 18F-Radiolabeling, Evaluation of logD(7.4), Stability and Binding Affinities for HBPLs 1–6
3.3.1. 18F-Radiolabeling of the HBPLs 1–6
3.3.2. LogD(7.4) Determination of [18F]1–[18F]6
3.3.3. Determination of the Stability of [18F]1–[18F]6 in Human Serum
3.3.4. Cell Culture
3.3.5. Competitive Displacement Studies on B16F10 and U87MG Cells
3.4. In Vivo PET/CT Imaging and Ex Vivo Biodistribution of [18F]2 and [18F]4
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rastrelli, M.; Tropea, S.; Rossi, C.R.; Alaibac, M. Melanoma: Epidemiology, risk factors, pathogenesis, diagnosis and classification. In Vivo 2014, 28, 1005–1012. [Google Scholar]
- Holly, E.A.; Kelly, J.W.; Shpall, S.N.; Chiu, S.-H. Number of melanocytic nevi as a major risk factor for malignant melanoma. J. Am. Acad. Dermatol. 1987, 17, 459–468. [Google Scholar] [CrossRef]
- Grob, J.J.; Gouvernet, J.; Aymar, D.; Mostaque, A.; Romano, M.H.; Collet, A.M.; Noe, M.C.; Diconstanzo, M.P.; Bonerandi, J.J. Count of benign melanocytic nevi as a major indicator of risk for nonfamilial nodular and superficial spreading melanoma. Cancer 1990, 66, 387–395. [Google Scholar] [CrossRef]
- Garbe, C.; Peris, K.; Hauschild, A.; Saiag, P.; Middleton, M.; Spatz, A.; Grob, J.-J.; Malvehy, J.; Newton-Bishop, J.; Stratigos, A.; et al. Diagnosis and treatment of melanoma. European consensus-based interdisciplinary guideline—Update 2012. Eur. J. Cancer 2012, 48, 2375–2390. [Google Scholar] [CrossRef]
- Krieter, M.; Schultz, E.; Debus, D. Das maligne Melanom. MMW Fortschritte Med. 2019, 161, 42–50. [Google Scholar] [CrossRef]
- Gallagher, B.M.; Fowler, J.S.; Gutterson, N.I.; MacGregor, R.R.; Wan, C.-N.; Wolf, A.P. Metabolic trapping as a principle of radiopharmaceutical design: Some factors responsible for the biodistribution of [18F] 2-deoxy-2-fluoro-D-glucose. J. Nucl. Med. 1978, 19, 1154–1161. [Google Scholar]
- Salazar-Onfray, F.; López, M.; Lundqvist, A.; Aguirre, A.; Escobar, A.; Serrano, A.; Korenblit, C.; Petersson, M.; Chhajlani, V.; Larsson, O.; et al. Tissue distribution and differential expression of melanocortin 1 receptor, a malignant melanoma marker. Br. J. Cancer 2002, 87, 414–422. [Google Scholar] [CrossRef] [Green Version]
- Tatro, J.B.; Wen, Z.; Entwistle, M.L.; Atkins, M.B.; Smith, T.J.; Reichlin, S.; Murphy, J.R. Interaction of an α-melanocyte-stimulating hormone-diphtheria toxin fusion protein with melanotropin receptors in human melanoma metastases. Cancer Res. 1992, 52, 2545–2548. [Google Scholar] [PubMed]
- Judmann, B.; Braun, D.; Wängler, B.; Schirrmacher, R.; Fricker, G.; Wängler, C. Current state of radiolabeled heterobivalent peptidic ligands in tumor imaging and therapy. Pharmaceuticals 2020, 13, 173. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C.; Maecke, H.R. Peptide-based probes for cancer imaging. J. Nucl. Med. 2008, 49, 1735–1738. [Google Scholar] [CrossRef] [Green Version]
- Ambrosini, V.; Fani, M.; Fanti, S.; Forrer, F.; Maecke, H.R. Radiopeptide imaging and therapy in Europe. J. Nucl. Med. 2011, 52, 42–55. [Google Scholar] [CrossRef] [Green Version]
- Gharibkandi, N.A.; Conlon, J.M.; Hosseinimehr, S.J. Strategies for improving stability and pharmacokinetic characteristics of radiolabeled peptides for imaging and therapy. Peptides 2020, 133, 170385. [Google Scholar] [CrossRef] [PubMed]
- Liu, S. Radiolabeled Cyclic RGD Peptides as integrin αvβ3-targeted radiotracers: Maximizing binding affinity via bivalency. Bioconjug. Chem. 2009, 20, 2199–2213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.Y.; Liu, S.; Hou, Y.P.; Tohme, M.; Park, R.; Bading, J.R.; Conti, P.S. MicroPET imaging of breast cancer αv-integrin expression with 64Cu-labeled dimeric RGD peptides. Mol. Imaging Biol. 2004, 6, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Fischer, G.; Schirrmacher, R.; Wängler, B.; Wängler, C. Radiolabeled heterobivalent peptidic ligands: An approach with high future potential for in vivo imaging and therapy of malignant diseases. ChemMedChem 2013, 8, 883–890. [Google Scholar] [CrossRef]
- Reubi, J.C.; Gugger, M.; Waser, B. Co-expressed peptide receptors in breast cancer as a molecular basis for in vivo multireceptor tumour targeting. Eur. J. Nucl. Med. Mol. 2002, 29, 855–862. [Google Scholar] [CrossRef]
- Reubi, J.C.; Fleischmann, A.; Waser, B.; Rehmann, R. Concomitant vascular GRP-receptor and VEGF-receptor expression in human tumors: Molecular basis for dual targeting of tumoral vasculature. Peptides 2011, 32, 1457–1462. [Google Scholar] [CrossRef]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef]
- Ananias, H.J.K.; Van den Heuvel, M.C.; Helfrich, W.; De Jong, I.J. Expression of the gastrin-releasing peptide receptor, the prostate stem cell antigen and the prostate-specific membrane antigen in lymph node and bone metastases of prostate cancer. Prostate 2009, 69, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Lindner, S.; Fiedler, L.; Wängler, B.; Bartenstein, P.; Schirrmacher, R.; Wängler, C. Design, synthesis and in vitro evaluation of heterobivalent peptidic radioligands targeting both GRP- and VPAC1-Receptors concomitantly overexpressed on various malignancies—Is the concept feasible? Eur. J. Med. Chem. 2018, 155, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C.; Maecke, H.R. Approaches to multireceptor targeting: Hybrid radioligands, radioligand cocktails, and sequential radioligand applications. J. Nucl. Med. 2017, 58, 10–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, P.C.; Clark, R.A.F.; Cheresh, D.A. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science 1994, 264, 569–571. [Google Scholar] [CrossRef]
- Brooks, P.C.; Montgomery, A.M.P.; Rosenfeld, M.; Reisfeld, R.A.; Hu, T.; Klier, G.; Cheresh, D.A. Integrin alpha v beta 3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell 1994, 79, 1157–1164. [Google Scholar] [CrossRef]
- Niu, G.; Chen, X. Why integrin as a primary target for imaging and therapy. Theranostics 2011, 1, 30–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seftor, R.E.; Seftor, E.A.; Hendrix, M.J. Molecular role(s) for integrins in human melanoma invasion. Cancer Metast. Rev. 1999, 18, 359–375. [Google Scholar] [CrossRef]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.P. Cell adhesion molecules in the development and progression of malignant melanoma. Cancer Metast. Rev. 1999, 18, 345–357. [Google Scholar] [CrossRef]
- Danen, E.H.J.; Jansen, K.F.J.; Van Kraats, A.A.; Cornelissen, I.M.H.A.; Ruiter, D.J.; Van Muijen, G.N.P. αv-Integrins in human melanoma: Gain of αvβ3 and loss OF αvβ5 are related to tumor progression in situ but not to metastatic capacity of cell lines in nude mice. Int. J. Cancer 1995, 61, 491–496. [Google Scholar] [CrossRef]
- Felding-Habermann, B.; Fransvea, E.; O’Toole, T.E.; Manzuk, L.; Faha, B.; Hensler, M. Involvement of tumor cell integrin alpha v beta 3 in hematogenous metastasis of human melanoma cells. Clin. Exp. Metastas. 2002, 19, 427–436. [Google Scholar] [CrossRef]
- Albelda, S.M.; Mette, S.A.; Elder, D.E.; Stewart, R.; Damjanovich, L.; Herlyn, M.; Buck, C.A. Integrin distribution in malignant melanoma: Association of the β3 subunit with tumor progression. Cancer Res. 1990, 50, 6757–6764. [Google Scholar] [PubMed]
- Yang, J.Q.; Guo, H.X.; Gallazzi, F.; Berwick, M.; Padilla, R.S.; Miao, Y.B. Evaluation of a novel Arg-Gly-Asp-conjugated α-melanocyte stimulating hormone hybrid peptide for potential melanoma therapy. Bioconjug. Chem. 2009, 20, 1634–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haubner, R.; Gratias, R.; Diefenbach, B.; Goodman, S.L.; Jonczyk, A.; Kessler, H. Structural and functional aspects of RGD-containing cyclic pentapeptides as highly potent and selective integrin αVβ3 antagonists. J. Am. Chem. Soc. 1996, 118, 7461–7472. [Google Scholar] [CrossRef]
- Al-Obeidi, F.; Castucci, A.M.; Hadley, M.E.; Hruby, V.J. Potent and prolonged acting cyclic lactam analogues of α-melanotropin: Design based on molecular dynamics. J. Med. Chem. 1989, 32, 2555–2561. [Google Scholar] [CrossRef] [PubMed]
- Von Kiedrowski, V.; Hübner, R.; Kail, D.; Cheng, X.; Schirrmacher, R.; Wängler, C.; Wängler, B. Synthesis, characterization and optimization of in vitro properties of NIR-fluorescent cyclic a-MSH peptides for melanoma imaging. J. Mater. Chem. B 2020, 8, 10602–10608. [Google Scholar] [CrossRef] [PubMed]
- Wängler, C.; Niedermoser, S.; Chin, J.; Orchovski, K.; Schirrmacher, E.; Jurkschat, K.; Iovkova-Berends, L.; Kostikov, A.P.; Schirrmacher, R.; Wängler, B. One-step (18)F-labeling of peptides for positron emission tomography imaging using the SiFA methodology. Nat. Protoc. 2012, 7, 1946–1955. [Google Scholar] [CrossRef]
- Vall-Sagarra, A.; Litau, S.; Decristoforo, C.; Wängler, B.; Schirrmacher, R.; Fricker, G.; Wängler, C. Design, synthesis, in vitro, and initial in vivo evaluation of heterobivalent peptidic ligands targeting both NPY(Y₁)- and GRP-receptors—An improvement for breast cancer imaging? Pharmaceuticals 2018, 11, 65. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.F.; Yan, Y.J.; Chin, F.T.; Wang, F.; Chen, X.Y. Dual integrin and gastrin-releasing peptide receptor targeted tumor imaging using 18F-labeled PEGylated RGD-bombesin heterodimer 18F-FB-PEG3-Glu-RGD-BBN. J. Med. Chem. 2009, 52, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-B.; Wu, Z.; Chen, K.; Ryu, E.K.; Chen, X. 18F-labeled BBN-RGD heterodimer for prostate cancer imaging. J. Nucl. Med. 2008, 49, 453–461. [Google Scholar] [CrossRef] [Green Version]
- Lindner, S.; Michler, C.; Wängler, B.; Bartenstein, P.; Fischer, G.; Schirrmacher, R.; Wängler, C. PESIN multimerization improves receptor avidities and in vivo tumor targeting properties to GRPR-overexpressing tumors. Bioconjug. Chem. 2014, 25, 489–500. [Google Scholar] [CrossRef]
- Fischer, G.; Lindner, S.; Litau, S.; Schirrmacher, R.; Wängler, B.; Wängler, C. Next step toward optimization of GRP receptor avidities: Determination of the minimal distance between BBN (7–14) units in peptide homodimers. Bioconjug. Chem. 2015, 26, 1479–1483. [Google Scholar] [CrossRef]
- Josan, J.S.; Handl, H.L.; Sankaranarayanan, R.; Xu, L.P.; Lynch, R.M.; Vagner, J.; Mash, E.A.; Hruby, V.J.; Gillies, R.J. Cell-specific targeting by heterobivalent ligands. Bioconjug. Chem. 2011, 22, 1270–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vagner, J.; Xu, L.P.; Handl, H.L.; Josan, J.S.; Morse, D.L.; Mash, E.A.; Gillies, R.J.; Hruby, V.J. Heterobivalent ligands crosslink multiple cell-surface receptors: The human melanocortin-4 and δ-opioid receptors. Angew. Chem. Int. Ed. 2008, 47, 1685–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacher, L.; Fischer, G.; Litau, S.; Schirrmacher, R.; Wängler, B.; Baller, M.; Wängler, C. Improving the stability of peptidic radiotracers by the introduction of artificial scaffolds: Which structure element is most useful? J. Label. Compd. Radiopharm. 2015, 58, 395–402. [Google Scholar] [CrossRef]
- Wängler, C.; Maschauer, S.; Prante, O.; Schafer, M.; Schirrmacher, R.; Bartenstein, P.; Eisenhut, M.; Wängler, B. Multimerization of cRGD peptides by click chemistry: Synthetic strategies, chemical limitations, and influence on biological properties. ChemBioChem 2010, 11, 2168–2181. [Google Scholar] [CrossRef] [PubMed]
- Del Gatto, A.; De Simone, M.; De Paola, I.; Saviano, M.; Zaccaro, L. Investigation of the best conditions to obtain c(RGDfK) peptide on solid phase. Int. J. Pept. Res. Ther. 2011, 17, 39–45. [Google Scholar] [CrossRef]
- Niedermoser, S.; Chin, J.; Wängler, C.; Kostikov, A.; Bernard-Gauthier, V.; Vogler, N.; Soucy, J.-P.; McEwan, A.J.; Schirrmacher, R.; Wängler, B. In vivo evaluation of 18F-SiFAlin–modified TATE: A potential challenge for 68Ga-DOTATATE, the clinical gold standard for somatostatin receptor imaging with PET. J. Nucl. Med. 2015, 56, 1100–1105. [Google Scholar] [CrossRef] [Green Version]
- Iovkova, L.; Wängler, B.; Schirrmacher, E.; Schirrmacher, R.; Quandt, G.; Böning, G.; Schurmann, M.; Jurkschat, K. Para-functionalized Aryl-di-tert-butylfluorosilanes as potential labeling synthons for 18F radiopharmaceuticals. Chem. Eur. J. 2009, 15, 2140–2147. [Google Scholar] [CrossRef]
- Kostikov, A.P.; Iovkova, L.; Chin, J.; Schirrmacher, E.; Wängler, B.; Wängler, C.; Jurkschat, K.; Cosa, G.; Schirrmacher, R. N-(4-(di-tert-butyl[18F]fluorosilyl)benzyl)-2-hydroxy-N,N-dimethylethylammonium bromide ([18F]SiFAN+Br−): A novel lead compound for the development of hydrophilic SiFA-based prosthetic groups for 18F-labeling. J. Fluor. Chem. 2011, 132, 27–34. [Google Scholar] [CrossRef]
- Damrauer, R.; Simon, R.A. Synthesis of fluorosilanes from chlorosilanes: The use of hexafluorosilicates. Organometallics 1988, 7, 1161–1164. [Google Scholar] [CrossRef]
- Lerner, H.-W.; Scholz, S.; Bolte, M. Synthese, Struktur und Eigenschaften des Natriumsilanids tBu2PhSiNa-ein Zugang zu Di-tert-butyl-phenylsilyl-substituierten Verbindungen. Anorg. Allg. Chem. 2001, 627, 1638–1642. [Google Scholar] [CrossRef]
- Lindner, S.; Wängler, C.; Bailey, J.J.; Jurkschat, K.; Bartenstein, P.; Wängler, B.; Schirrmacher, R. Radiosynthesis of [18F]SiFAlin-TATE for clinical neuroendocrine tumor positron emission tomography. Nature Protoc. 2020, 15, 3827–3843. [Google Scholar] [CrossRef]
- Wessmann, S.H.; Henriksen, G.; Wester, H.-J. Cryptate mediated nucleophilic 18F-fluorination without azeotropic drying. Nuklearmedizin 2012, 51, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Glaser, M.; Morrison, M.; Solbakken, M.; Arukwe, J.; Karlsen, H.; Wiggen, U.; Champion, S.; Kindberg, G.M.; Cuthbertson, A. Radiosynthesis and biodistribution of cyclic RGD peptides conjugated with novel [18F]fluorinated aldehyde-containing prosthetic groups. Bioconjug. Chem. 2008, 19, 951–957. [Google Scholar] [CrossRef]
- Varasteh, Z.; Rosenström, U.; Velikyan, I.; Mitran, B.; Altei, M.; Honarvar, H.; Rosestedt, M.; Lindeberg, G.; Sörensen, J.; Larhed, M.; et al. The effect of mini-PEG-based spacer length on binding and pharmacokinetic properties of a 68Ga-labeled NOTA-conjugated antagonistic analog of bombesin. Molecules 2014, 19, 10455–10472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flori, E.; Rosati, E.; Cardinali, G.; Kovacs, D.; Bellei, B.; Picardo, M.; Maresca, V. The α-melanocyte stimulating hormone/peroxisome proliferator activated receptor-γ pathway down-regulates proliferation in melanoma cell lines. J. Exp. Clin. Cancer Res. 2017, 36, 142–154. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Jia, B.; Zhao, H.; Chen, X.; Wang, F. Specific targeting of human integrin αvβ3 with 111in-labeled Abegrin™ in nude mouse models. Mol. Imaging Biol. 2011, 13, 112–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S. Radiolabeled cyclic RGD peptide bioconjugates as radiotracers targeting multiple integrins. Bioconjug. Chem. 2015, 26, 1413–1438. [Google Scholar] [CrossRef] [Green Version]
- Hübner, R.; Cheng, X.; Wängler, B.; Wängler, C. Functional hybrid molecules for the visualization of cancer: PESIN-homodimers combined with multimodal molecular imaging probes for positron emission tomography and optical imaging: Suited for tracking of GRPR-positive malignant tissue. Chem. Eur. J. 2020, 26, 16349–16356. [Google Scholar] [CrossRef]
- Cemazar, M.; Golzio, M.; Escoffre, J.-M.; Couderc, B.; Sersa, G.; Teissié, J. In vivo imaging of tumor growth after electrochemotherapy with cisplatin. Biochem. Biophys. Res. Commun. 2006, 348, 997–1002. [Google Scholar] [CrossRef]
- Dandekar, M.; Tseng, J.T.; Gambhir, S.S. Reproducibility of 18F-FDG microPET studies in mouse tumor xenografts. J. Nucl. Med. 2007, 48, 602–607. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HBPL | Linker | RCP [%] | RCY [%] | Am [GBq/µmol] | A0 [GBq] | logD(7.4) | Stability * [%] |
|---|---|---|---|---|---|---|---|
| [18F]1 | PEG1 | ≥98 | 27.2 ± 1.4 | 17.5–28.6 | 1.0–1.6 | −1.08 ± 0.07 | 87.0 ± 2.3 |
| [18F]2 | PEG3 | ≥98 | 40.3 ± 2.6 | 20.0–31.2 | 0.8–1.2 | −1.19 ± 0.05 | 85.4 ± 2.8 |
| [18F]3 | PEG5 | ≥97 | 50.4 ± 3.1 | 25.1–49.8 | 0.9–2.0 | −1.15 ± 0.01 | 81.4 ± 2.9 |
| [18F]4 | PEG8 | ≥97 | 38.4 ± 0.6 | 25.7–42.7 | 1.1–1.9 | −1.39 ± 0.03 | 83.6 ± 0.8 |
| [18F]5 | DIG | ≥95 | 27.3 ± 9.9 | 17.0–47.6 | 1.8–2.2 | −1.21 ± 0.01 | 83.2 ± 2.4 |
| [18F]6 | Ox-EGEGE | ≥97 | 43.2 ± 1.6 | 39.3–51.4 | 1.2–1.9 | −1.52 ± 0.01 | 82.7 ± 1.8 |
| Compound | IC50 (B16F10) [nM] | Compound | IC50 (U87MG) [nM] |
|---|---|---|---|
| 1 | 1.74 ± 0.25 | 1 | 2881 ± 757 |
| 2 | 0.99 ± 0.11 | 2 | 1300 ± 288 |
| 3 | 3.44 ± 0.09 | 3 | 1911 ± 70 |
| 4 | 6.00 ± 0.47 | 4 | 2034 ± 323 |
| 5 | 2.05 ± 0.35 | 5 | 5895 ± 722 |
| 6 | 4.18 ± 0.32 | 6 | >100,000 |
| α-MSH | 3.75 ± 0.61 | c(RGDfC) | 1493 ± 210 |
| NDP | 0.17 ± 0.04 | c(RGDfK) | 427 ± 37 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, X.; Hübner, R.; von Kiedrowski, V.; Fricker, G.; Schirrmacher, R.; Wängler, C.; Wängler, B. Design, Synthesis, In Vitro and In Vivo Evaluation of Heterobivalent SiFAlin-Modified Peptidic Radioligands Targeting Both Integrin αvβ3 and the MC1 Receptor—Suitable for the Specific Visualization of Melanomas? Pharmaceuticals 2021, 14, 547. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14060547
Cheng X, Hübner R, von Kiedrowski V, Fricker G, Schirrmacher R, Wängler C, Wängler B. Design, Synthesis, In Vitro and In Vivo Evaluation of Heterobivalent SiFAlin-Modified Peptidic Radioligands Targeting Both Integrin αvβ3 and the MC1 Receptor—Suitable for the Specific Visualization of Melanomas? Pharmaceuticals. 2021; 14(6):547. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14060547
Chicago/Turabian StyleCheng, Xia, Ralph Hübner, Valeska von Kiedrowski, Gert Fricker, Ralf Schirrmacher, Carmen Wängler, and Björn Wängler. 2021. "Design, Synthesis, In Vitro and In Vivo Evaluation of Heterobivalent SiFAlin-Modified Peptidic Radioligands Targeting Both Integrin αvβ3 and the MC1 Receptor—Suitable for the Specific Visualization of Melanomas?" Pharmaceuticals 14, no. 6: 547. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14060547