
Targeting the Copper Transport System to Improve Treatment Efficacies of Platinum-Containing Drugs in Cancer Chemotherapy


{kind=link}
{kind=link}
Abstract
:1. Introduction
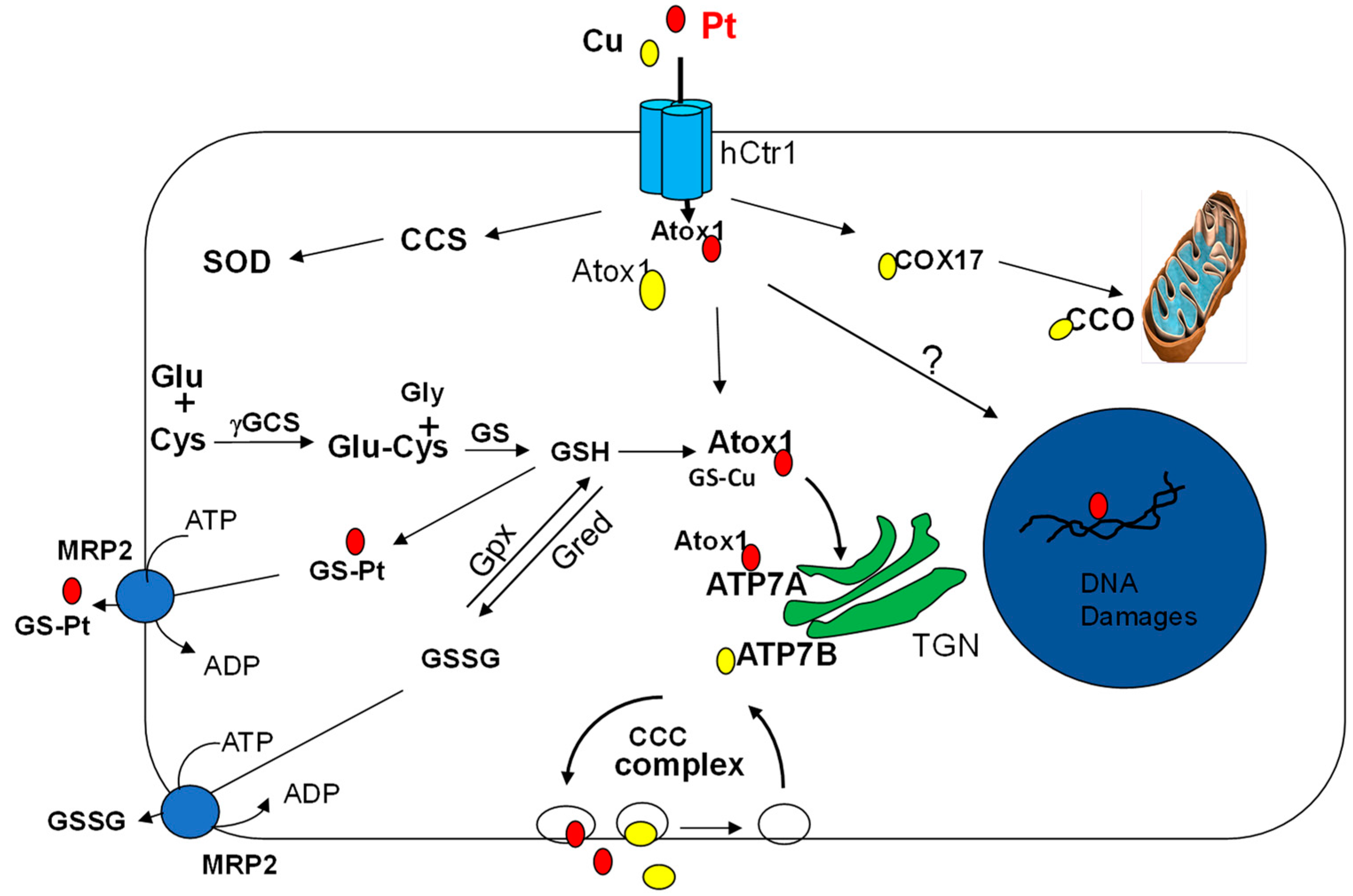
2. The Transport Mechanisms of Pt Drugs
3. Connecting the Essential Trace Element Cu to Pt-Based Antitumor Drugs
3.1. Identification of the High-Affinity Copper Transporter (Ctr1) as cDDP Importer
3.2. Cu Chaperones in Intracellular cDDP Trafficking
3.3. Cu-ATPases in Pt Drugs Efflux
3.4. Redox Regulation of the Cu Transport System in Pt Drug Pharmacology
4. Modulations of Copper Homeostasis and Pt Drug Cancer Chemotherapy
4.1. Roles of the Copper Transport System in Pt Drug Cancer Chemotherapy in Clinical Settings
4.1.1. hCtr1
4.1.2. Atox1, ATP7A and ATP7B
4.1.3. COMMD1
4.2. Enhanced cDDP Cell-Killing Activity through Upregulation of hCtr1 Expression
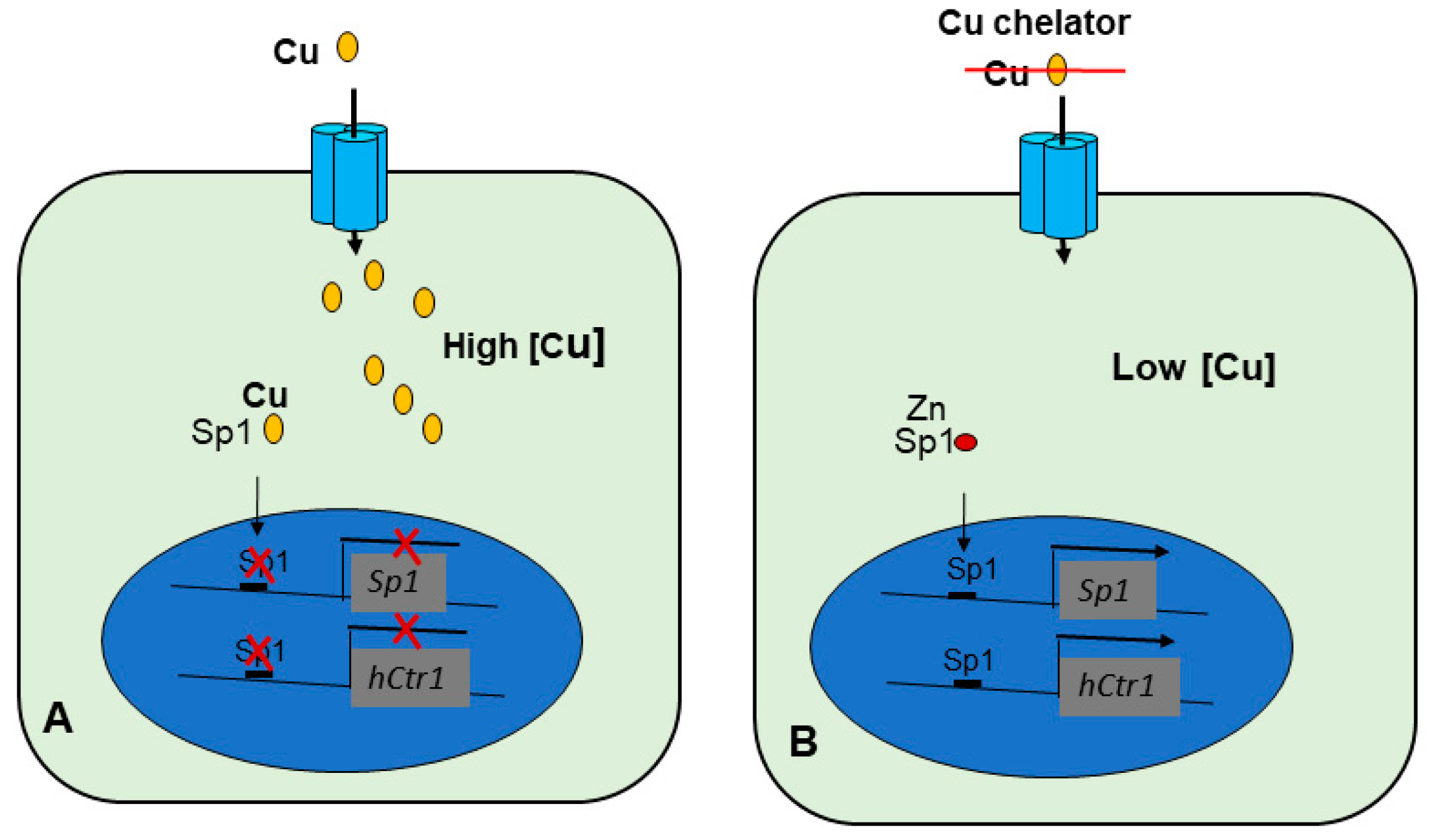
4.2.1. Transcriptional Regulation of hCtr1 Expression and cDDP Cancer Chemotherapy
4.2.2. The Cu-Sp1-hCtr1 Inter-Regulatory Loop in Humans
5. Overcoming cDDP Resistance by Copper Chelators
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Atox1 | antioxidant protein 1 |
| ATP7A, ATP7B | two P-type ATPases involved in Cu and cDDP export |
| CCS | superoxide dismutase 1 Cu carrier |
| Ctr1 | high-affinity copper transporter 1 |
| cDDP | cisplatin (cis-diamminedichloroplatinum II) |
| COMMD1 | the copper metabolism MURR1 domain 1 |
| Cox17 | cytochrome C oxidase copper chaperone |
| D-Pen. | D-penicillamine |
| γGCS | γ-glutamylcysteine synthesis |
| GSH | glutathione, |
| GSSG | oxidized form of GSH |
| Gpx | glutathione peroxidase |
| Gred | glutathione reductase |
| LRRC | leucine-rich repeat-containing protein |
| SOD1 | superoxide dismutase 1 |
| Sp1 | specificity protein 1 |
| OCT | organic cation transporter system |
| ROS | reactive oxygen species |
| TCG | The Cancer Genome Atlas |
| TGN. | Trans-golgi network |
| TM | tetrathiomolybdate |
| VRAC | volume-regulated anion channel |
| ZFs | zinc fingers |
References
- Rottenberg, S.; Disler, C.; Perego, B. The rediscovery of platinum-based cancer therapy. Nat. Rev. Cancer 2021, 21, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Muggia, F.M.; Bonetti, A.; Hoeschele, J.D.; Rozencweig, M.; Howell, S.B. Platinum antitumor complexes: 50 years since Barnett Rosenberg′s discovery. J. Clin. Oncol. 2015, 33, 4219–4226. [Google Scholar] [CrossRef] [PubMed]
- Chiruvella, V.; Annamaraju, P.; Guddati, P.A.K. Management of nephrotoxicity of chemotherapy and targeted agents: 2020. Am. J. Cancer. Res. 2020, 10, 4151–4164. [Google Scholar] [PubMed]
- Wertman, J.N.; Melong, N.; Stoyek, M.R.; Piccolo, O.; Langley, S.; Orr, B.; Steele, S.L.; Razaghi, B.; Berman, J.N. The identification of dual protective agents against cisplatin-induced oto- and nephrotoxicity using the zebrafish model. eLife 9 2020, 9. [Google Scholar] [CrossRef]
- Rabik, C.A.; Dolan, M.E. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat. Rev. 2007, 33, 9–23. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef]
- Kim, E.S.; Lee, J.J.; He, G.; Chow, C.W.; Fujimoto, J.; Kalhor, N.; Swisher, S.G.; Wistuba, I.I.; Stewart, D.J.; Siddik, Z.H. Tissue platinum concentration and tumor response in non-small-cell lung cancer. J. Clin. Oncol. 2012, 30, 3345–3352. [Google Scholar] [CrossRef] [Green Version]
- Gately, D.P.; Sharma, A.; Christen, R.D. Howell, S.B. Cisplatin and taxol activate different signal pathways regulating cellular injury-induced expression of GADD153. Br. J. Cancer 1996, 73, 18–23. [Google Scholar] [CrossRef] [Green Version]
- Planells-Cases, R.; Lutter, D.; Guyader, C.; Gerhards, N.M.; Ullrich, F.; Elger, D.A.; Kucukosmanoglu, A.; Xu, G.; Voss, F.K.; Reincke, S.M.; et al. Subunit composition of VRAC channels determines substrate specificity and cellular resistance to Pt-based anti-cancer drugs. EMBO J. 2015, 34, 2993–3008. [Google Scholar] [CrossRef]
- Konig, B.; Stauber, T. Biophysics and structure-function relationships of LRRC8-formed volume-regulated anion channels. Biophys. J. 2019, 116, 1185–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorensen, B.H.; Dam, C.S.; Sturup, S.; Lambert, I.H. Dual role of LRRC8A-containing transporters on cisplatin resistance in human ovarian cancer cells. J. Inorg. Biochem. 2016, 160, 287–295. [Google Scholar] [CrossRef]
- Ruprecht, N.; Hofmann, L.; Hungerbuhler, M.N.; Kempf, C.; Heverhagen, J.T.; von Tengg-Kobligk, H. Generation of stable cisPt resistant lung adenocarcinoma cells. Pharmaceuticals 2020, 13, 109. [Google Scholar] [CrossRef] [PubMed]
- Sprowl, J.A.; Ciarimboli, G.; Lancaster, C.S.; Giovinazzo, H.; Gibson, A.A.; Du, G.; Janke, L.J.; Cavaletti, G.; Shields, A.F.; Sparreboom, A. Oxaliplatin-induced neurotoxicity is dependent on the organic cation transporter OCT2. Proc. Natl. Acad. Sci. USA 2013, 110, 11199–11204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, J.; Wang, L.; Li, T.; Tang, S.; Wang, Y.; Zhang, W.; Jiang, X. Role and mechanism of organic cation transporter 3 in oxaliplatin treatment of colon cancer in vitro and in vivo. Oncol. Rep. 2019, 42, 1355–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jong, N.N.; Nakanishi, T.; Liu, J.J.; Tamai, I.; McKeage, M.J. Oxaliplatin transport mediated by organic cation/carnitine transporters OCTN1 and OCTN2 in overexpressing human embryonic kidney 293 cells and rat dorsal root ganglion neurons. J. Pharmacol. Exp. Ther. 2011, 338, 537–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, M.; Tsurudome, Y.; Kanemitsu, T.; Yasukochi, S.; Kanado, Y.; Ogino, T.; Matsunaga, N.; Koyanagi, S.; Ohdo, S. Diurnal expression of MRP4 in bone marrow cells underlies the dosing-time dependent changes in the oxaliplatin-induced myelotoxicity. Sci. Rep. 2020, 10, 13484. [Google Scholar] [CrossRef] [PubMed]
- Samodelov, S.L.; Kullak-Ublick, G.A.; Gai, Z.; Visentin, M. Organic cation transporters in human physiology, pharmacology, and toxicology. Int. J. Mol. Sci. 2020, 21, 7890. [Google Scholar] [CrossRef]
- Tatsumi, S.; Matsuoka, H.; Hashimoto, Y.; Hatta, K.; Maeda, K.; Kamoshida, S. Organic cation transporter 2 and tumor budding as independent prognostic factors in metastatic colorectal cancer patients treated with oxaliplatin-based chemotherapy. Int. J. Clin. Exp. Pathol. 2014, 7, 204–212. [Google Scholar]
- Ishida, S.; Lee, J.; Thiele, D.J.; Herskowitz, I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc. Natl. Acad. Sci. USA 2002, 99, 14298–14302. [Google Scholar] [CrossRef] [Green Version]
- Georgatsou, E.; Mavrogiannis, L.A.; Fragiadakis, G.S.; Alexandraki, V. The yeast Fre1p/Fre2p cupric reductases facilitate copper uptake and are regulated by the copper-modulated Mac1p activator. J. Biol. Chem. 1997, 272, 13786–13792. [Google Scholar] [CrossRef] [Green Version]
- Schwab, S.; Shearer, J.; Conklin, S.E.; Alies, B.; Haas, K.L. Sequence proximity between Cu(II) and Cu(I) binding sites of human copper transporter 1 model peptides defines reactivity with ascorbate and O2. J. Inorg. Biochem. 2016, 158, 70–76. [Google Scholar] [CrossRef] [Green Version]
- Puig, S.; Lee, J.; Lau, M.; Thiele, D.J. Biochemical and genetic analyses of yeast and human high affinity copper transporters suggest a conserved mechanism for copper uptake. J. Biol. Chem. 2002, 227, 26021–26030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Z.D.; Stockton, D.; Savaraj, N.; Tien Kuo, M. Mechanistic comparison of human high-affinity copper transporter 1-mediated transport between copper ion and cisplatin. Mol. Pharmacol. 2009, 76, 843–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvaraj, A.; Balamurugan, K.; Yepiskoposyan, H.; Zhou, H.; Egli, D.; Georgiev, O.; Thiele, D.J.; Schaffner, W. Metal-responsive transcription factor (MTF-1) handles both extremes, copper load and copper starvation, by activating different genes. Genes Dev. 2005, 19, 891–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, F.; Logeman, B.L.; Zhang, X.; Liu, Y.; Thiele, D.J.; Yuan, P. X-ray structures of the high-affinity copper transporter Ctr1. Nat. Commun. 2019, 10, 1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delmar, J.A.; Su, C.C.; Yu, E.W. Structural mechanisms of heavy-metal extrusion by the Cus efflux system. Biometals 2013, 26, 593–607. [Google Scholar] [CrossRef] [Green Version]
- Sinani, D.; Adle, D.J.; Kim, H.; Lee, J. Distinct mechanisms for Ctr1-mediated copper and cisplatin transport. J. Biol. Chem. 2007, 282, 26775–26785. [Google Scholar] [CrossRef] [Green Version]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The next generation of platinum drugs: Targeted Pt(II) agents, nanoparticle delivery, and Pt(IV) prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef] [Green Version]
- Barresi, V.; Trovato-Salinaro, A.; Spampinato, G.; Musso, N.; Castorina, S.; Rizzarelli, E.; Condorelli, D.F. Transcriptome analysis of copper homeostasis genes reveals coordinated upregulation of SLC31A1, SCO1, and COX11 in colorectal cancer. FEBS Open Bio 2016, 6, 794–806. [Google Scholar] [CrossRef] [Green Version]
- Perkal, O.; Qasem, Z.; Turgeman, M.; Schwartz, R.; Gevorkyan-Airapetov, L.; Pavlin, M.; Magistrato, A.; Major, D.T.; Ruthstein, S. Cu(I) Controls Conformational States in Human ATOX1 Metallochaperone: An EPR and Multiscale Simulation Study. J. Phys. Chem. B 2020, 124, 4399–4411. [Google Scholar] [CrossRef]
- Wu, X.; Yuan, S.; Wang, E.; Tong, Y.; Ma, G.; Wei, K.; Liu, Y. Platinum transfer from hCTR1 to Atox1 is dependent on the type of platinum complex. Metallomics 2017, 9, 546–555. [Google Scholar] [CrossRef]
- Kahra, D.; Kovermann, M.; Wittung-Stafshede, P. The C-terminus of human copper importer Ctr1 Acts as a binding site and transfers copper to Atox1. Biophys. J. 2016, 110, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Guo, W.; Wu, K.; Zhao, Y.; Luo, Q.; Zhang, Q.; Liu, J.; Xiong, S.; Wang, F. Identification of binding sites of cisplatin to human copper chaperone protein Cox17 by high-resolution FT-ICR-MS. Rapid Commun. Mass Spectrom. 2016, 30, 168–172. [Google Scholar] [CrossRef] [Green Version]
- Theotoki, E.I.; Velentzas, A.D.; Katarachia, S.A.; Papandreou, N.C.; Kalavros, N.I.; Pasadaki, S.N.; Giannopoulou, A.F.; Giannios, P.; Iconomidou, V.A.; Konstantakou, E.G.; et al. Targeting of copper-trafficking chaperones causes gene-specific systemic pathology in Drosophila melanogaster: Prospective expansion of mutational landscapes that regulate tumor resistance to cisplatin. Biol. Open 2019, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boal, A.K.; Rosenzweig, A.C. Crystal structures of cisplatin bound to a human copper chaperone. J. Am. Chem. Soc. 2009, 131, 14196–14197. [Google Scholar] [CrossRef] [Green Version]
- Itoh, S.; Kim, H.W.; Nakagawa, O.; Ozumi, K.; Lessner, S.M.; Aoki, H.; Akram, K.; McKinney, R.D.; Ushio-Fukai, M.; Fukai, T. Novel role of antioxidant-1 (Atox1) as a copper-dependent transcription factor involved in cell proliferation. J. Biol. Chem. 2008, 283, 9157–9167. [Google Scholar] [CrossRef] [Green Version]
- Kamiya, T.; Takeuchi, K.; Fukudome, S.; Hara, H.; Adachi, T. Copper chaperone antioxidant-1, Atox-1, is involved in the induction of SOD3 in THP-1 cells. Biometals 2018, 31, 61–68. [Google Scholar] [CrossRef]
- Celauro, E.; Mukaj, A.; Fierro-Gonzalez, J.C.; Wittung-Stafshede, P. Copper chaperone ATOX1 regulates pluripotency factor OCT4 in preimplantation mouse embryos. Biochem. Biophys. Res. Commun. 2017, 491, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.F.; Sudhahar, V.; Youn, S.W.; Das, A.; Cho, J.; Kamiya, T.; Urao, N.; McKinney, R.D.; Surenkhuu, B.; Hamakubo, T.; et al. Transport protein antioxidant-1 promotes inflammatory neovascularization via chaperone and transcription factor function. Sci. Rep. 2015, 5, 14780. [Google Scholar] [CrossRef] [Green Version]
- Hua, H.; Gunther, V.; Georgiev, O.; Schaffner, W. Distorted copper homeostasis with decreased sensitivity to cisplatin upon chaperone Atox1 deletion in Drosophila. Biometals 2011, 24, 445–453. [Google Scholar] [CrossRef]
- Safaei, R.; Maktabi, M.H.; Blair, B.G.; Larson, C.A.; Howell, S.B. Effects of the loss of Atox1 on the cellular pharmacology of cisplatin. J. Inorg. Biochem. 2009, 103, 333–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inkol, J.M.; Poon, A.C.; Mutsaers, A.J. Inhibition of copper chaperones sensitizes human and canine osteosarcoma cells to carboplatin chemotherapy. Vet. Comp. Oncol. 2020, 18, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.W.; Moore, S.D. Copper transporting P-type ATPases and human disease. J. Bioenerg. Biomembr. 2002, 34, 333–338. [Google Scholar] [CrossRef]
- Gudekar, N.; Shanbhag, V.; Wang, Y.; Ralle, M.; Weisman, G.A.; Petris, M.J. Metallothioneins regulate ATP7A trafficking and control cell viability during copper deficiency and excess. Sci. Rep. 2020, 10, 7856. [Google Scholar] [CrossRef]
- Nardella, M.; Rosato, A.; Belviso, B.; Caliardro, R.; Natile, G.; Arnesano, F. Oxidation of human copper chaperone Atoxi and disusulfide bond cleavage by cisplatin and glutathione. Int. J. Mol. Sci. 2019, 20, 4390. [Google Scholar] [CrossRef] [Green Version]
- Lutsenko, S.; Barnes, N.L.; Bartee, M.Y.; Dmitriev, O.Y. Function and regulation of human copper-transporting ATPases. Physiol. Rev. 2007, 87, 1011–1046. [Google Scholar] [CrossRef]
- Inesi, G.; Pilankatta, R.; Tadini-Buoninsegni, F. Biochemical characterization of P-type copper ATPases. Biochem. J. 2014, 463, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.H.; Yang, N.; Bothe, J.; Tonelli, M.; Nokhrin, S.; Dolgova, N.V.; Braiterman, L.; Lutsenko, S.; Dmitriev, O.Y. The metal chaperone Atox1 regulates the activity of the human copper transporter ATP7B by modulating domain dynamics. J. Biol. Chem. 2017, 292, 18169–18177. [Google Scholar] [CrossRef] [Green Version]
- Mattle, D.; Zhang, L.; Sitsel, O.; Pedersen, L.T.; Moncelli, M.R.; Tadini-Buoninsegni, F.; Gourdon, P.; Rees, D.C.; Nissen, P.; Meloni, G. A sulfur-based transport pathway in Cu+-ATPases. EMBO Rep. 2015, 16, 728–740. [Google Scholar] [CrossRef] [Green Version]
- Singla, A.; Chen, Q.; Suzuki, K.; Song, J.; Fedoseienko, A.; Wijers, M.; Lopez, A.; Billadeau, D.D.; van de Sluis, B.; Burstein, E. Regulation of copper homeostasis by members of the COMMD protein family. Dis. Model Mech. 2020, 14. [Google Scholar] [CrossRef]
- Phillips-Krawczak, C.A.; Singla, A.; Starokadomskyy, P.; Deng, Z.; Osborne, D.G.; Li, H.; Dick, C.J.; Gomez, T.S.; Koenecke, M.; Zhang, J.S.; et al. COMMD1 is linked to the WASH complex and regulates endosomal trafficking of the copper transporter ATP7A. Mol. Biol. Cell 2015, 26, 91–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, D.J.; Short, K.K.; Maniaci, B.N.; Burkhead, J.L. COMMD1 and PtdIns(4,5)P2 interaction maintain ATP7B copper transporter trafficking fidelity in HepG2 cells. J. Cell. Sci. 2019, 132. [Google Scholar] [CrossRef]
- Materia, S.; Cater, M.A.; Klomp, L.W.; Mercer, J.F.; La Fontaine, S. Clusterin and COMMD1 independently regulate degradation of the mammalian copper ATPases ATP7A and ATP7B. J. Biol. Chem. 2012, 287, 2485–2499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tadini-Buoninsegni, F.; Bartolommei, G.; Moncelli, M.R.; Inesi, G.; Galliani, A.; Sinisi, M.; Losacco, M.; Natile, G.; Arnesano, F. Translocation of platinum anticancer drugs by human copper ATPases ATP7A and ATP7B. Angew. Chem. 2014, 126, 1321–1325. [Google Scholar] [CrossRef] [Green Version]
- Tadini-Buoninsegni, F.; Palchetti, I. Label-free bioelectrochemical methods for evaluation of anticancer drug effects at a molecular level. Sensors 2020, 20, 1812. [Google Scholar] [CrossRef] [Green Version]
- Tapia, L.; Gonzalez-Aguero, M.; Cisternas, M.F.; Suazo, M.; Cambiazo, V.; Uauy, R.; Gonzalez, M. Metallothionein is crucial for safe intracellular copper storage and cell survival at normal and supra-physiological exposure levels. Biochem. J. 2004, 378, 617–624. [Google Scholar] [CrossRef] [Green Version]
- Kozlowski, H.; Kolkowska, P.; Watly, J.; Krzywoszynska, K.; Potocki, S. General aspects of metal toxicity. Curr. Med. Chem. 2014, 21, 3721–3740. [Google Scholar] [CrossRef]
- Margis, R.; Dunand, C.; Teixeira, F.K.; Margis-Pinheiro, M. Glutathione peroxidase family—an evolutionary overview. FEBS J. 2008, 275, 3959–3970. [Google Scholar] [CrossRef]
- Chen, H.H.; Kuo, M.T. Role of glutathione in the regulation of Cisplatin resistance in cancer chemotherapy. Met. Based Drugs 2010, 2010. [Google Scholar] [CrossRef]
- Lai, Y.H.; Kuo, C.; Kuo, M.T.; Chen, H.H.W. Modulating chemosensitivity of tumors to platinum-based antitumor drugs by transcriptional regulation of copper homeostasis. Int. J. Mol. Sci. 2018, 19, 1486. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Wang, Z.; Wu, H.; Xi, Z.; Liu, Y. Glutathione selectively modulates the binding of platinum drugs to human copper chaperone Cox17. Biochem. J. 2015, 472, 217–223. [Google Scholar] [CrossRef]
- Brozovic, A.; Ambriovic-Ristov, A.; Osmak, M. The relationship between cisplatin-induced reactive oxygen species, glutathione, and BCL-2 and resistance to cisplatin. Crit. Rev. Toxicol. 2010, 40, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.J. Mechanisms of resistance to cisplatin and carboplatin. Crit. Rev. Oncol. Hematol. 2007, 63, 12–31. [Google Scholar] [CrossRef]
- Chen, H.H.; Song, I.S.; Hossain, A.; Choi, M.K.; Yamane, Y.; Liang, Z.D.; Lu, J.; Wu, L.Y.; Siddik, Z.H.; Klomp, L.W.; et al. Elevated glutathione levels confer cellular sensitization to cisplatin toxicity by up-regulation of copper transporter hCtr1. Mol. Pharmacol. 2008, 74, 697–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attar, N.; Campos, O.A.; Vogelauer, M.; Cheng, C.; Xue, Y.; Schmollinger, S.; Salwinski, L.; Mallipeddi, N.V.; Boone, B.A.; Yen, L.; et al. The histone H3-H4 tetramer is a copper reductase enzyme. Science 2020, 369, 59–64. [Google Scholar] [CrossRef]
- Silva, M.M.; Rocha, C.R.R.; Kinker, G.S.; Pelegrini, A.L.; Menck, C.F.M. The balance between NRF2/GSH antioxidant mediated pathway and DNA repair modulates cisplatin resistance in lung cancer cells. Sci. Rep. 2019, 9, 17639. [Google Scholar] [CrossRef]
- Ishida, S.; McCormick, F.; Smith-McCune, K.; Hanahan, D. Enhancing tumor-specific uptake of the anticancer drug cisplatin with a copper chelator. Cancer Cell 2010, 17, 574–583. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Cai, J.; Yang, Q.; Zhao, S.; Wang, Z. The association between copper transporters and the prognosis of cancer patients undergoing chemotherapy: A meta-analysis of literatures and datasets. Oncotarget 2017, 8, 16036–16051. [Google Scholar] [CrossRef] [Green Version]
- Hanna, N.H.; Einhorn, L.H. Testicular cancer--discoveries and updates. N. Engl. J. Med. 2014, 371, 2005–2016. [Google Scholar] [CrossRef] [Green Version]
- Ghaffari, R.; Di Bona, K.R.; Riley, C.L.; Richburg, J.H. Copper transporter 1 (CTR1) expression by mouse testicular germ cells, but not Sertoli cells, is essential for functional spermatogenesis. PLoS ONE 2019, 14, e0215522. [Google Scholar] [CrossRef] [Green Version]
- Ghaffari, R.; Richburg, J.H. Mice with a Sertoli cell-specific knockout of the Ctr1 gene exhibit a reduced sensitivity to cisplatin-induced testicular germ cell apoptosis. Toxicol. Res. 2019, 8, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Petruzzelli, R.; Polishchuk, R.S. Activity and trafficking of copper-transporting ATPases in tumor development and defense against platinum-based drugs. Cells 2019, 8, 1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, M.; Sumizawa, T.; Mutoh, M.; Chen, Z.S.; Terada, K.; Furukawa, T.; Yang, X.L.; Gao, H.; Miura, N.; Sugiyama, T.; et al. Copper-transporting P-type adenosine triphosphatase (ATP7B) is associated with cisplatin resistance. Cancer Res. 2000, 60, 1312–1316. [Google Scholar] [PubMed]
- Leonhardt, K.; Gebhardt, R.; Mossner, J.; Lutsenko, S.; Huster, D. Functional interactions of Cu-ATPase ATP7B with cisplatin and the role of ATP7B in the resistance of cells to the drug. J. Biol. Chem. 2009, 284, 7793–7802. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, T.; Abe, M.; Yamazaki, T.; Miyashita, H.; Niwa, H.; Kokubun, S.; Sato, Y. HEX acts as a negative regulator of angiogenesis by modulating the expression of angiogenesis-related gene in endothelial cells in vitro. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 231–237. [Google Scholar] [CrossRef] [Green Version]
- Samimi, G.; Safaei, R.; Katano, K.; Holzer, A.K.; Rochdi, M.; Tomioka, M.; Goodman, M.; Howell, S.B. Increased expression of the copper efflux transporter ATP7A mediates resistance to cisplatin, carboplatin, and oxaliplatin in ovarian cancer cells. Clin. Cancer Res. 2004, 10, 4661–4669. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Balibrea, E.; Martinez-Cardus, A.; Musulen, E.; Gines, A.; Manzano, J.L.; Aranda, E.; Plasencia, C.; Neamati, N.; Abad, A. Increased levels of copper efflux transporter ATP7B are associated with poor outcome in colorectal cancer patients receiving oxaliplatin-based chemotherapy. Int. J. Cancer 2009, 124, 2905–2910. [Google Scholar] [CrossRef]
- Nakayama, K.; Kanzaki, A.; Terada, K.; Mutoh, M.; Ogawa, K.; Sugiyama, T.; Takenoshita, S.; Itoh, K.; Yaegashi, N.; Miyazaki, K.; et al. Prognostic value of the Cu-transporting ATPase in ovarian carcinoma patients receiving cisplatin-based chemotherapy. Clin. Cancer Res. 2004, 10, 2804–2811. [Google Scholar] [CrossRef] [Green Version]
- Weiskirchen, R.; Penning, L.C. COMMD1, a multi-potent intracellular protein involved in copper homeostasis, protein trafficking, inflammation, and cancer. J. Trace Elem. Med. Biol. 2021, 65, 126712. [Google Scholar] [CrossRef]
- Fedoseienko, A.; Wieringa, H.W.; Wisman, G.B.; Duiker, E.; Reyners, A.K.; Hofker, M.H.; van der Zee, A.G.; van de Sluis, B.; van Vugt, M.A. Nuclear COMMD1 is associated with cisplatin sensitivity in ovarian cancer. PLoS ONE 2016, 11, e0165385. [Google Scholar]
- Eid, C.; Hemadi, M.; Ha-Duong, N.T.; El Hage Chahine, J.M. Iron uptake and transfer from ceruloplasmin to transferrin. Biochim. Biophys. Acta 2014, 1840, 1771–1781. [Google Scholar] [CrossRef] [PubMed]
- Herman, S.; Lipinski, P.; Ogorek, M.; Starzynski, R.; Grzmil, P.; Bednarz, A.; Lenartowicz, M. Molecular regulation of copper homeostasis in the male gonad during the process of spermatogenesis. Int. J. Mol. Sci. 2020, 21, 9053. [Google Scholar] [CrossRef]
- Dodani, S.C.; Firl, A.; Chan, J.; Nam, C.I.; Aron, A.T.; Onak, C.S.; Ramos-Torres, K.M.; Paek, J.; Webster, C.M.; Feller, M.B.; et al. Copper is an endogenous modulator of neural circuit spontaneous activity. Proc. Natl. Acad. Sci. USA 2014, 111, 16280–16285. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Jiang, Y.; Yang, Y.; Peng, Y.; Li, C. Copper metabolism in Saccharomyces cerevisiae: An update. Biometals 2020, 34, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Ooi, C.E.; Rabinovich, E.; Dancis, A.; Bonifacino, J.S.; Klausner, R.D. Copper-dependent degradation of the Saccharomyces cerevisiae plasma membrane copper transporter Ctr1p in the apparent absence of endocytosis. EMBO J. 1996, 15, 3515–3523. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, J.C.; Bird, A.J. Metal-responsive transcription factors that regulate iron, zinc, and copper homeostasis in eukaryotic cells. Eukaryot. Cell 2004, 3, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosman, D.J. For Cryptococcus neoformans, responding to the copper status in a colonization niche is not just about copper. Mol. Microbiol. 2018, 1108, 463–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Santamarina, S.; Festa, R.A.; Smith, A.D.; Yu, C.H.; Probst, C.; Ding, C.; Homer, C.M.; Yin, J.; Noonan, J.P.; Madhani, H.; et al. Genome-wide analysis of the regulation of Cu metabolism in Cryptococcus neoformans. Mol. Microbiol. 2018, 108, 473–494. [Google Scholar] [CrossRef] [Green Version]
- Merchant, S.S.; Schmollinger, S.; Strenkert, D.; Moseley, J.L.; Blaby-Haas, C.E. From economy to luxury: Copper homeostasis in Chlamydomonas and other algae. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118822. [Google Scholar] [CrossRef]
- Araki, R.; Mermod, M.; Yamasaki, H.; Kamiya, T.; Fujiwara, T.; Shikanai, T. SPL7 locally regulates copper-homeostasis-related genes in Arabidopsis. J. Plant Physiol. 2018, 224, 137–143. [Google Scholar] [CrossRef]
- Bird, A.J. Cellular sensing and transport of metal ions: Implications in micronutrient homeostasis. J. Nutr. Biochem. 2015, 26, 1103–1115. [Google Scholar] [CrossRef]
- Liang, Z.D.; Tsai, W.B.; Lee, M.Y.; Savaraj, N.; Kuo, M.T. Specificity protein 1 (sp1) oscillation is involved in copper homeostasis maintenance by regulating human high-affinity copper transporter 1 expression. Mol. Pharmacol. 2012, 81, 455–464. [Google Scholar] [CrossRef] [Green Version]
- Song, I.S.; Chen, H.H.; Aiba, I.; Hossain, A.; Liang, Z.D.; Klomp, L.W.; Kuo, M.T. Transcription factor Sp1 plays an important role in the regulation of copper homeostasis in mammalian cells. Mol. Pharmacol. 2008, 74, 705–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, D.; Aiba, I.; Chen, H.H.; Kuo, M.T. Effects of Cu(II) and cisplatin on the stability of Specific protein 1 (Sp1)-DNA binding: Insights into the regulation of copper homeostasis and platinum drug transport. J. Inorg. Biochem. 2016, 161, 37–39. [Google Scholar] [CrossRef] [Green Version]
- Shimberg, G.D.; Ok, K.; Neu, H.M.; Splan, K.E.; Michel, S.L.J. Cu(I) disrupts the structure and function of the nonclassical zinc finger protein tristetraprolin (TTP). Inorg. Chem. 2017, 56, 6838–6848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, S.; Chen, S.; Xi, Z.; Liu, Y. Copper-finger protein of Sp1: The molecular basis of copper sensing. Metallomics 2017, 9, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Collins, J.F. Transcription factors Sp1 and Hif2alpha mediate induction of the copper-transporting ATPase (Atp7a) gene in intestinal epithelial cells during hypoxia. J. Biol. Chem. 2013, 288, 23943–23952. [Google Scholar] [CrossRef] [Green Version]
- Kudo, E.; Taura, M.; Suico, M.A.; Goto, H.; Kai, H.; Okada, S. Transcriptional regulation of HIV-1 host factor COMMD1 by the Sp family. Int. J. Mol. Med. 2018, 41, 2366–2374. [Google Scholar] [CrossRef]
- Liang, Z.D.; Long, Y.; Chen, H.H.; Savaraj, N.; Kuo, M.T. Regulation of the high-affinity copper transporter (hCtr1) expression by cisplatin and heavy metals. J. Biol. Inorg. Chem. 2014, 19, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Safe, S.; Abbruzzese, J.; Abdelrahim, M.; Hedrick, E. Specificity protein transcription factors and cancer: Opportunities for drug development. Cancer Prev. Res. 2018, 11, 371–382. [Google Scholar] [CrossRef]
- Helsel, M.E.; Franz, K.J. Pharmacological activity of metal binding agents that alter copper bioavailability. Dalton Trans. 2015, 44, 8760–8770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldari, S.; Di Rocco, G.; Toietta, G. Current biomedical use of copper chelation therapy. Int. J. Mol. Sci. 2020, 21, 1069. [Google Scholar] [CrossRef] [Green Version]
- Cen, D.; Gonzalez, R.I.; Buckmeier, J.A.; Kahlon, R.S.; Tohidian, N.B.; Meyskens, F.L., Jr. Disulfiram induces apoptosis in human melanoma cells: A redox-related process. Mol. Cancer Ther. 2002, 1, 197–204. [Google Scholar] [PubMed]
- Chen, D.; Cui, Q.C.; Yang, H.; Dou, Q.P. Disulfiram, a clinically used anti-alcoholism drug and copper-binding agent, induces apoptotic cell death in breast cancer cultures and xenografts via inhibition of the proteasome activity. Cancer Res. 2006, 66, 10425–10433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasinoff, B.B.; Wu, X.; Yadav, A.A.; Patel, D.; Zhang, H.; Wang, D.S.; Chen, Z.S.; Yalowich, J.C. Cellular mechanisms of the cytotoxicity of the anticancer drug elesclomol and its complex with Cu(II). Biochem. Pharmacol. 2015, 93, 266–276. [Google Scholar] [CrossRef]
- Guthrie, L.M.; Soma, S.; Yuan, S.; Silva, A.; Zulkifli, M.; Snavely, T.C.; Greene, H.F.; Nunez, E.; Lynch, B.; De Ville, C.; et al. Elesclomol alleviates Menkes pathology and mortality by escorting Cu to cuproenzymes in mice. Science 2020, 368, 620–625. [Google Scholar] [CrossRef]
- Long, Y.; Tsai, W.B.; Chang, J.T.; Estecio, M.; Wangpaichitr, M.; Savaraj, N.; Feun, L.G.; Chen, H.H.; Kuo, M.T. Cisplatin-induced synthetic lethality to arginine-starvation therapy by transcriptional suppression of ASS1 is regulated by DEC1, HIF-1alpha, and c-Myc transcription network and is independent of ASS1 promoter DNA methylation. Oncotarget 2016, 7, 82658–82670. [Google Scholar] [CrossRef] [Green Version]
- Liang, Z.D.; Long, Y.; Tsai, W.B.; Fu, S.; Kurzrock, R.; Gagea-Iurascu, M.; Zhang, F.; Chen, H.H.; Hennessy, B.T.; Mills, G.B.; et al. Mechanistic basis for overcoming platinum resistance using copper chelating agents. Mol. Cancer Ther. 2012, 11, 2483–2494. [Google Scholar] [CrossRef] [Green Version]
- Kita, Y.; Hamada, A.; Saito, R.; Teramoto, Y.; Tanaka, R.; Takano, K.; Nakayama, K.; Murakami, K.; Matsumoto, K.; Akamatsu, S.; et al. Systematic chemical screening identifies disulfiram as a repurposed drug that enhances sensitivity to cisplatin in bladder cancer: A summary of preclinical studies. Br. J. Cancer 2019, 121, 1027–1038. [Google Scholar] [CrossRef]
- Barca, A.; Ippati, S.; Urso, E.; Vetrugno, C.; Storelli, C.; Maffia, M.; Romano, A.; Verri, T. Carnosine modulates the Sp1-Slc31a1/Ctr1 copper-sensing system and influences copper homeostasis in murine CNS-derived cells. Am. J. Physiol. Cell Physiol. 2019, 316, C235–C245. [Google Scholar] [CrossRef]
- Zhang, W.; Shi, H.; Chen, C.; Ren, K.; Xu, Y.; Liu, X.; He, L. Curcumin enhances cisplatin sensitivity of human NSCLC cell lines through influencing Cu-Sp1-CTR1 regulatory loop. Phytomedicine 2018, 48, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Hou, M.M.; Wheler, J.; Hong, D.; Naing, A.; Tsimberidou, A.; Janku, F.; Zinner, R.; Piha-Paul, S.; Falchook, G.; et al. Exploratory study of carboplatin plus the copper-lowering agent trientine in patients with advanced malignancies. Investig. New Drugs 2014, 32, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Naing, A.; Fu, C.; Kuo, M.T.; Kurzrock, R. Overcoming platinum resistance through the use of a copper-lowering agent. Mol. Cancer Ther. 2012, 11, 1221–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.F.; Kuo, M.T.; Liu, Y.S.; Cheng, Y.M.; Wu, P.Y.; Chou, C.Y. A dose escalation study of trientine plus carboplatin and pegylated liposomal doxorubicin in women with a first relapse of epithelial ovarian, tubal, and peritoneal cancer within 12 months after platinum-based chemotherapy. Front. Oncol. 2019, 9, 437. [Google Scholar] [CrossRef] [PubMed]
- Leitao, M.M., Jr.; Hummer, A.; Dizon, D.S.; Aghajanian, C.; Hensley, M.; Sabbatini, P.; Venkatraman, E.; Spriggs, D.R. Platinum retreatment of platinum-resistant ovarian cancer after nonplatinum therapy. Gynecol. Oncol. 2003, 91, 123–129. [Google Scholar] [CrossRef]
- Kavanagh, J.; Tresukosol, D.; Edwards, C.; Freedman, R.; Gonzalez de Leon, C.; Fishman, A.; Mante, R.; Hord, M.; Kudelka, A. Carboplatin reinduction after taxane in patients with platinum-refractory epithelial ovarian cancer. J. Clin. Oncol. 1995, 13, 1584–1588. [Google Scholar] [CrossRef]
- Mariniello, M.; Petruzzelli, R.; Wanderlingh, L.G.; La Montagna, R.; Carissimo, A.; Pane, F.; Amoresano, A.; Ilyechova, E.Y.; Galagudza, M.M.; Catalano, F.; et al. Synthetic lethality screening identifies FDA-approved drugs that overcome ATP7B-mediated tolerance of tumor cells to cisplatin. Cancers 2020, 12, 608. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuo, M.T.; Huang, Y.-F.; Chou, C.-Y.; Chen, H.H.W. Targeting the Copper Transport System to Improve Treatment Efficacies of Platinum-Containing Drugs in Cancer Chemotherapy. Pharmaceuticals 2021, 14, 549. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14060549
Kuo MT, Huang Y-F, Chou C-Y, Chen HHW. Targeting the Copper Transport System to Improve Treatment Efficacies of Platinum-Containing Drugs in Cancer Chemotherapy. Pharmaceuticals. 2021; 14(6):549. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14060549
Chicago/Turabian StyleKuo, Macus Tien, Yu-Fang Huang, Cheng-Yang Chou, and Helen H. W. Chen. 2021. "Targeting the Copper Transport System to Improve Treatment Efficacies of Platinum-Containing Drugs in Cancer Chemotherapy" Pharmaceuticals 14, no. 6: 549. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14060549