
Unexpected Salt/Cocrystal Polymorphism of the Ketoprofen–Lysine System: Discovery of a New Ketoprofen–l-Lysine Salt Polymorph with Different Physicochemical and Pharmacokinetic Properties

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results and Discussion
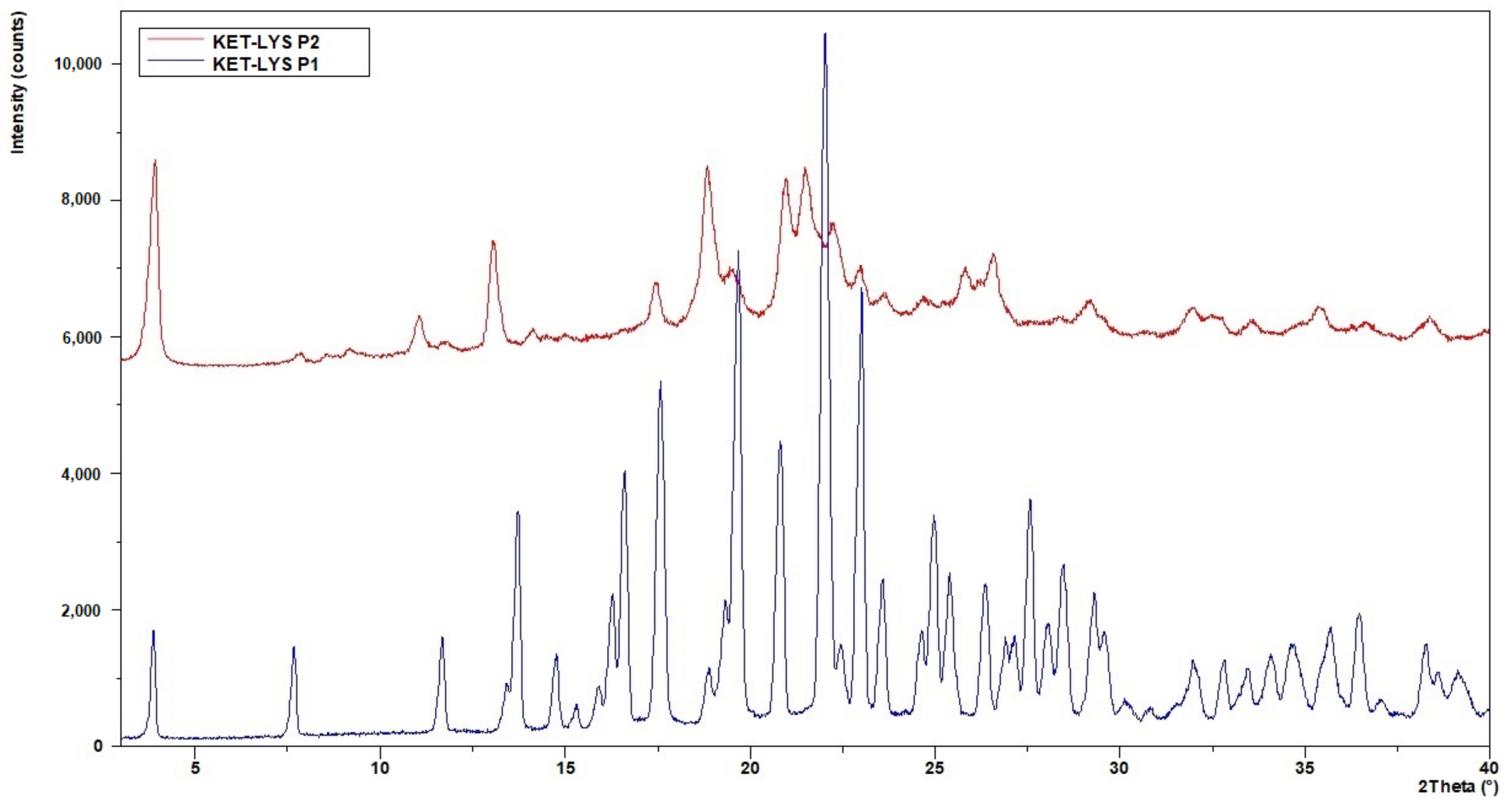
2.1. Crystallization Conditions and Identification of KET–LYS Polymorphs
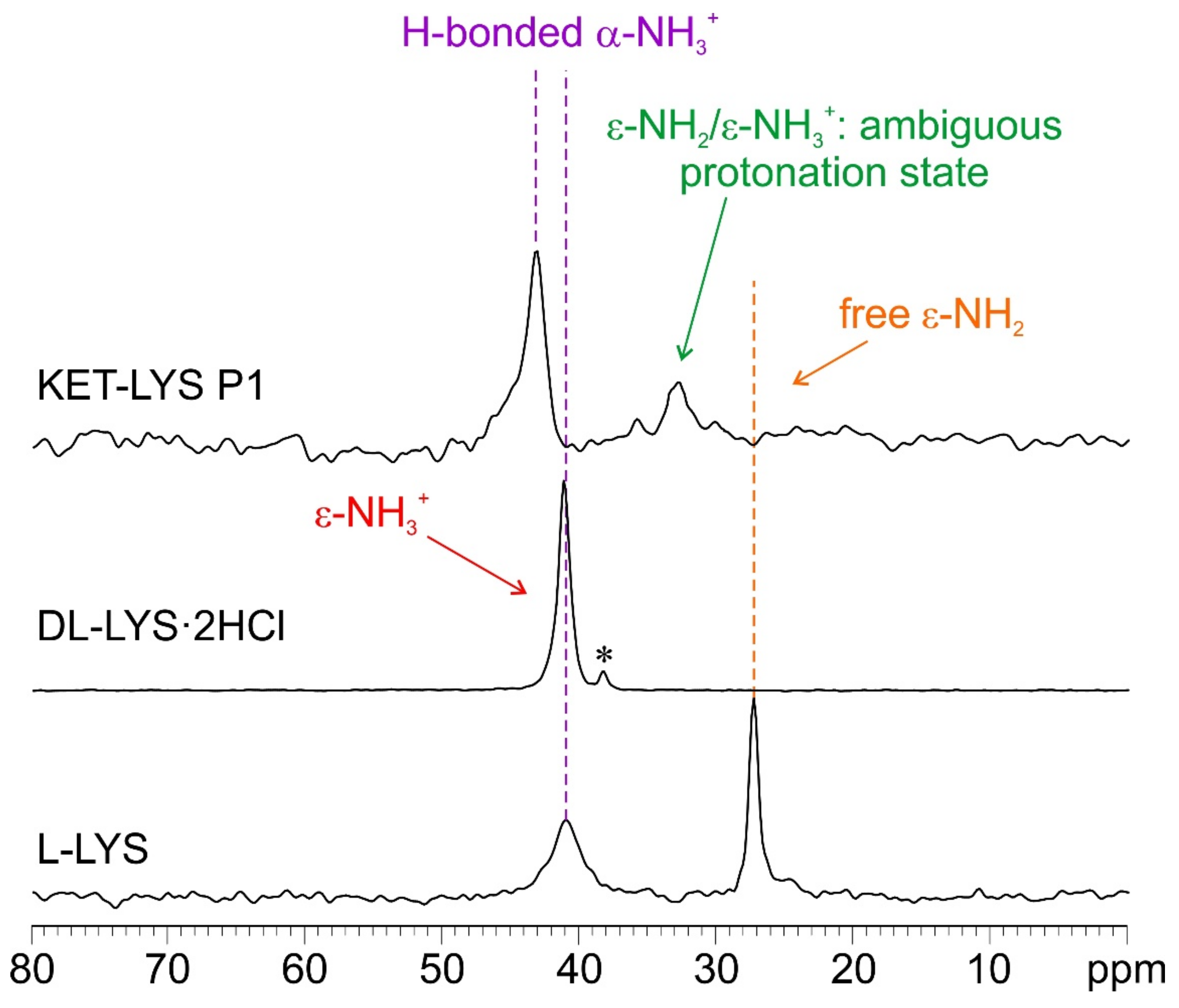
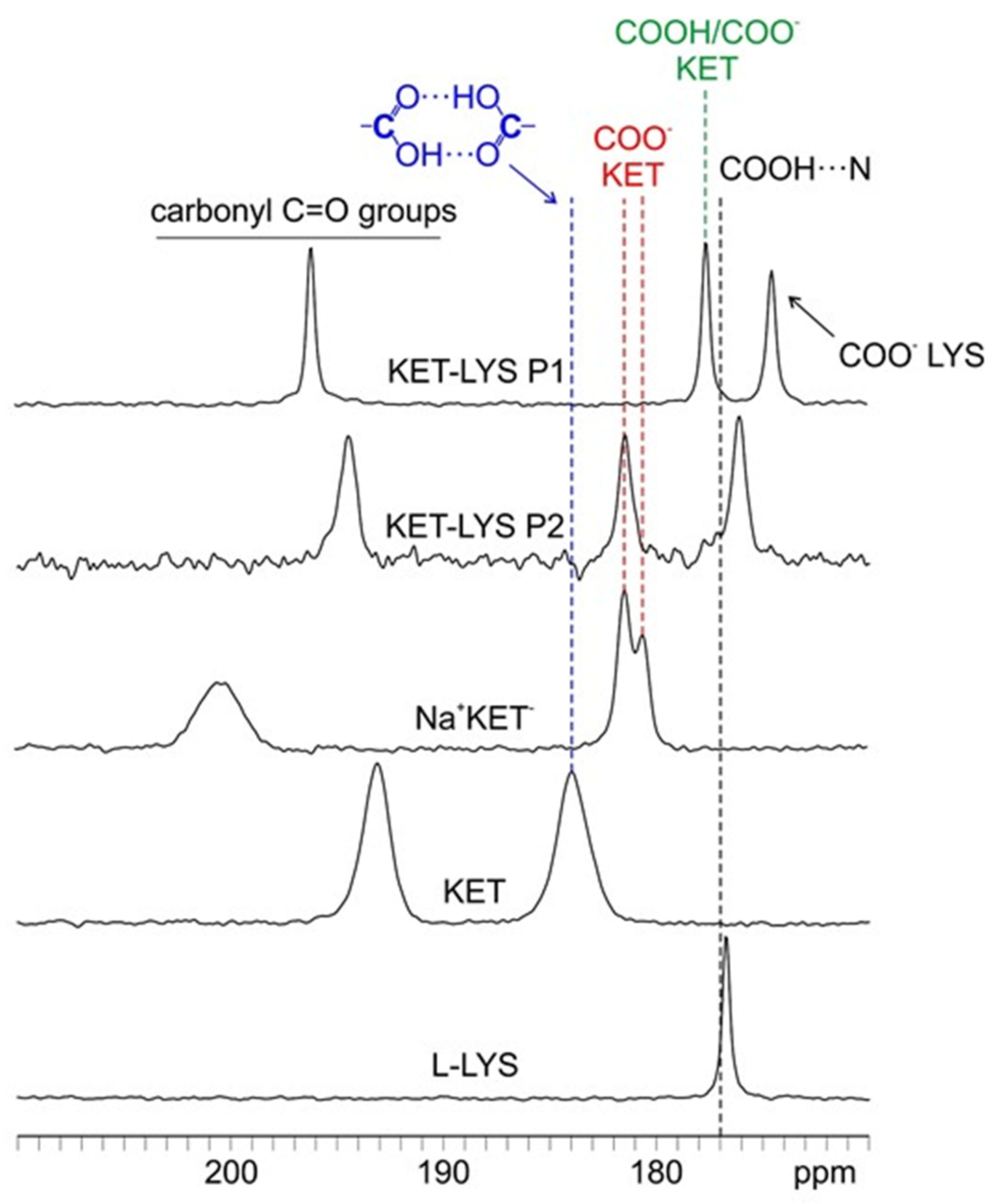
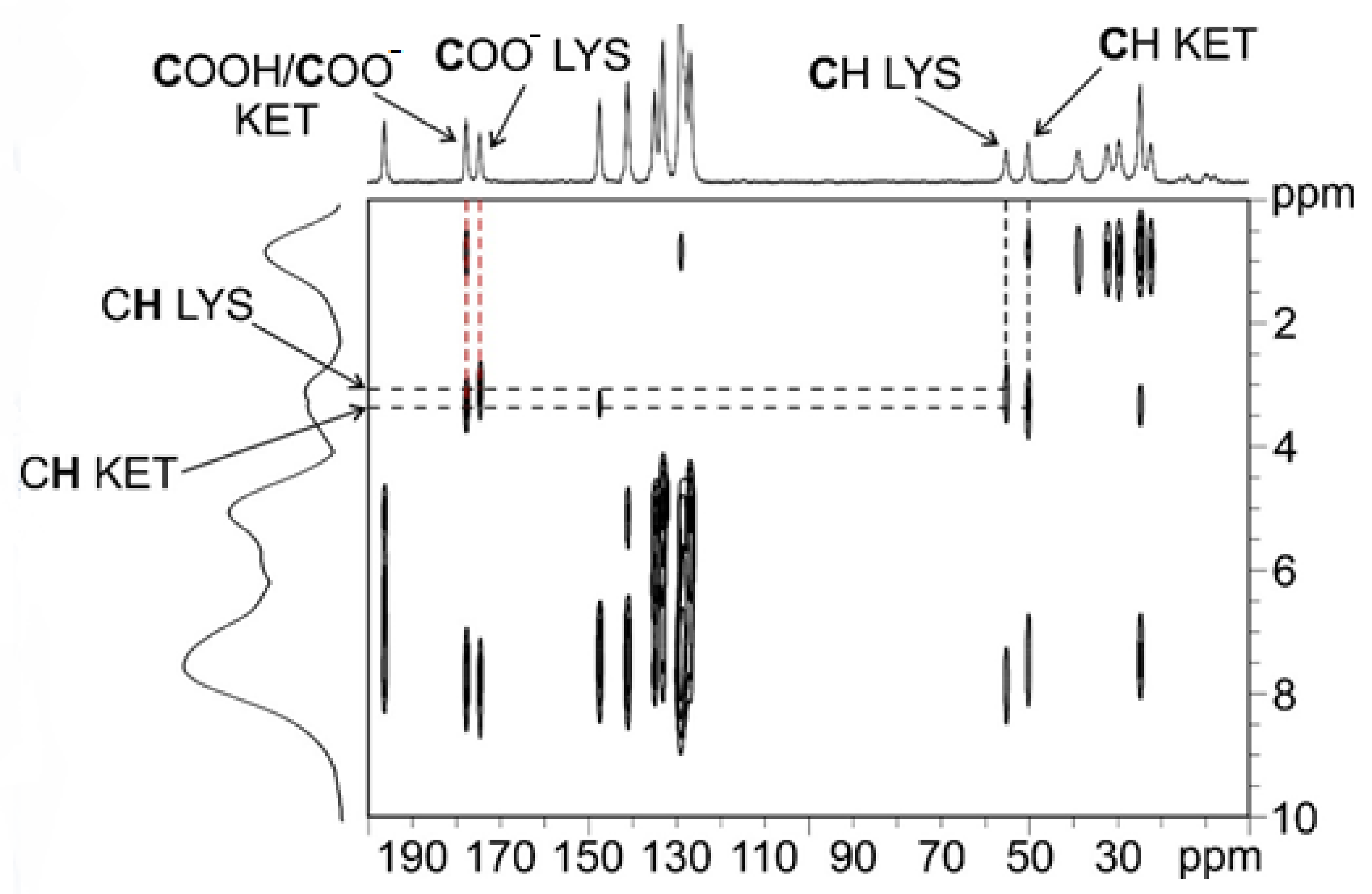
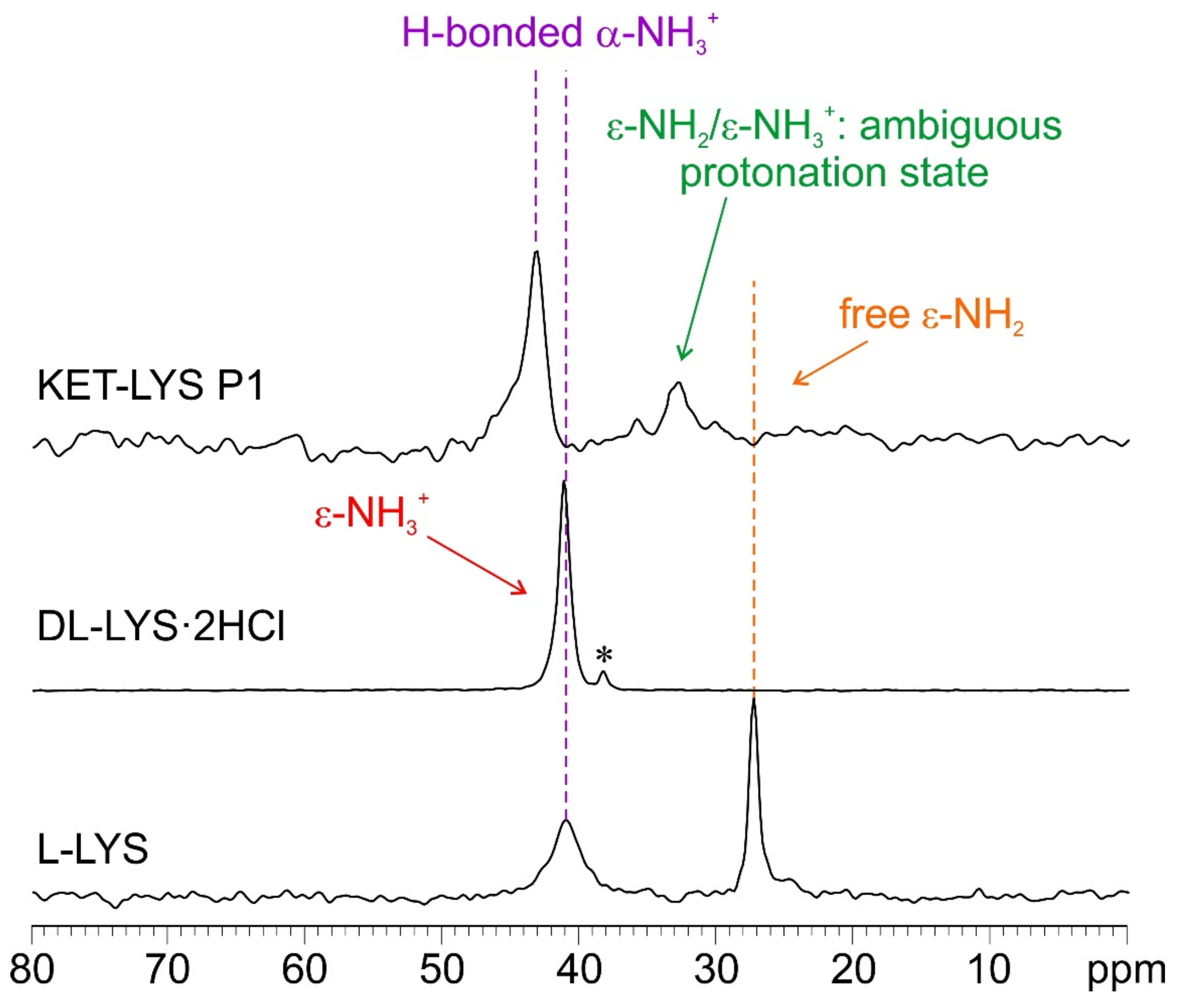
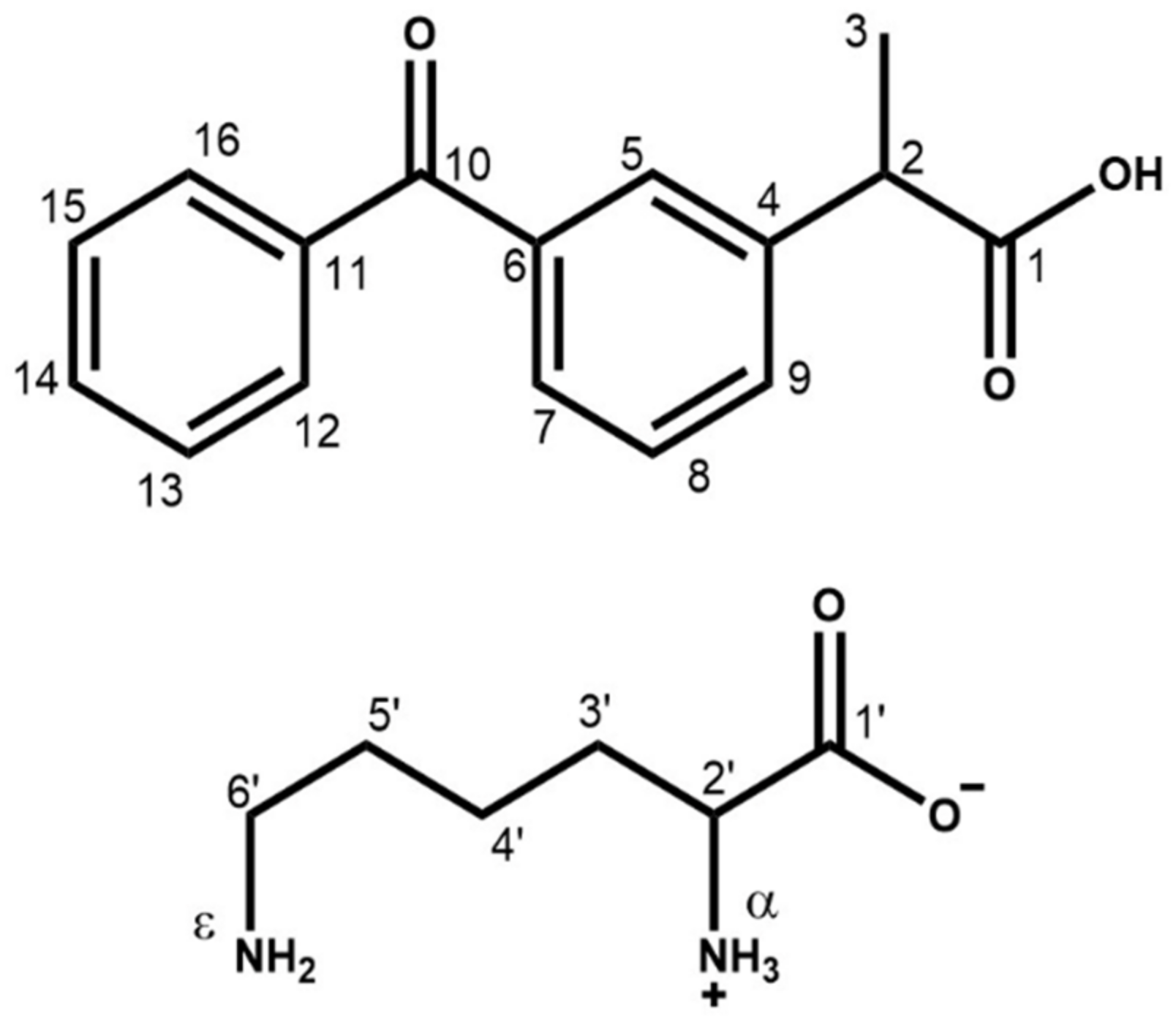
2.2. Characterization of Synthesized KET–LYS P1 and P2
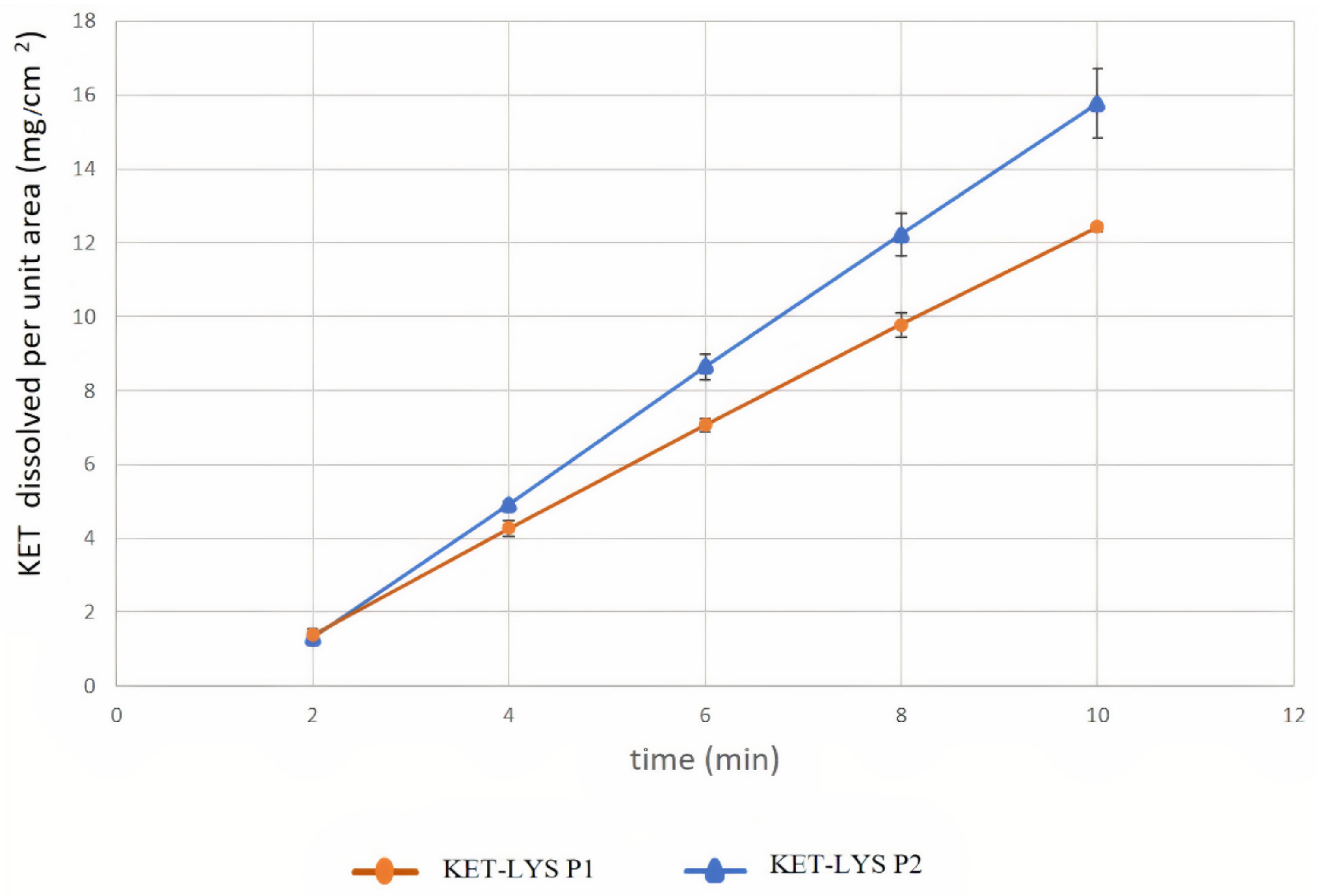
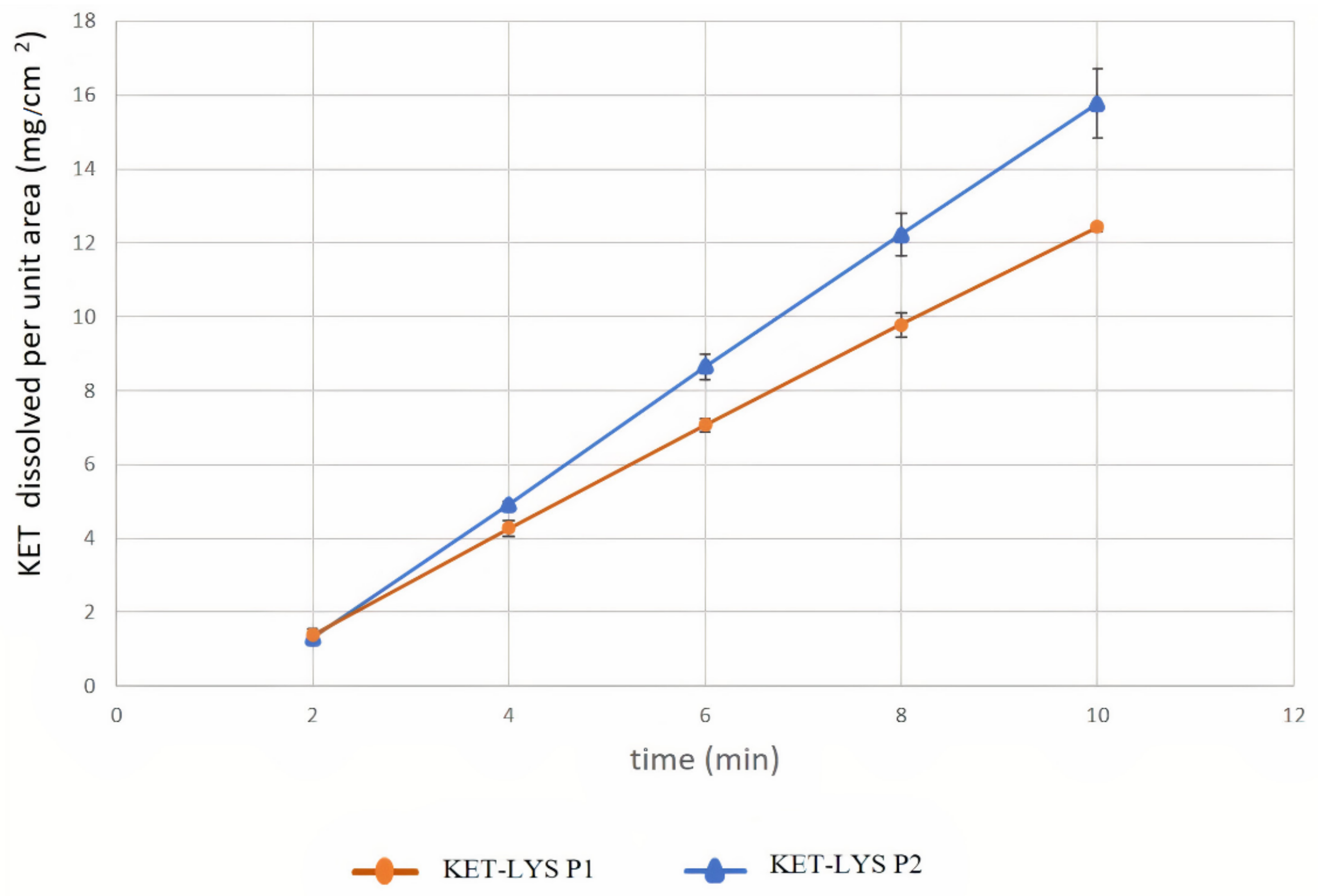
2.3. Intrinsic Dissolution Rates of Cocrystal KET–LYS P1 and Salt KET–LYS P2
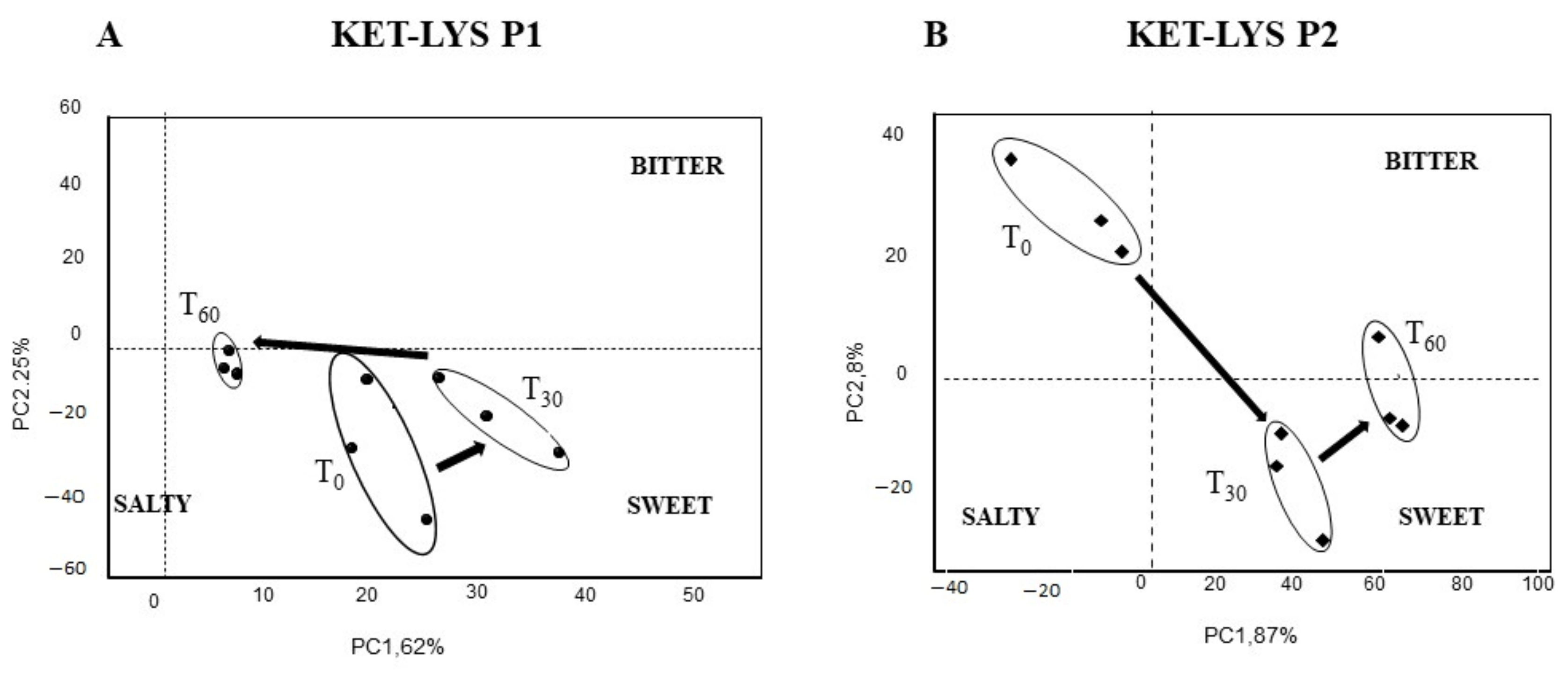
2.4. Taste and Sensorial Kinetic Analysis of KET–LYS P1 and KET–LYS P2
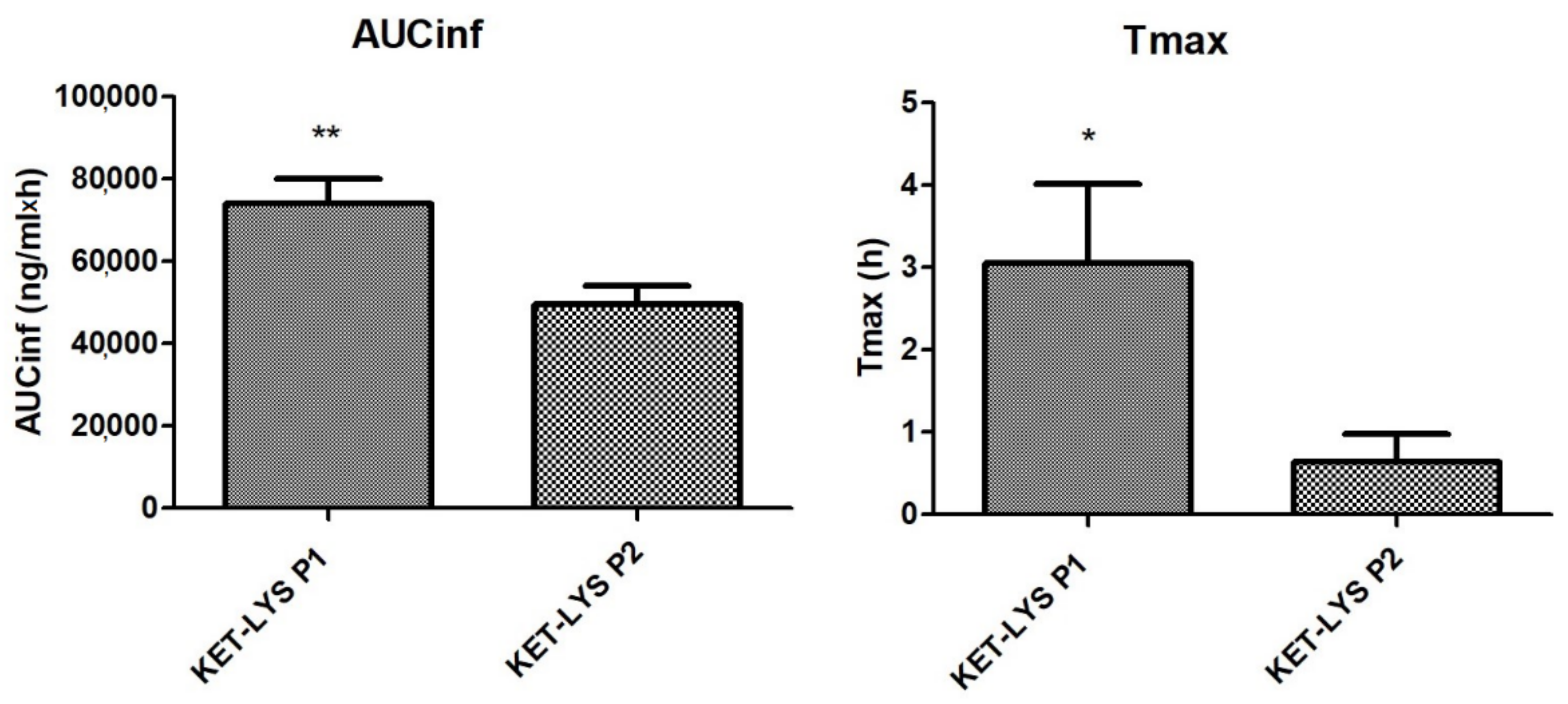
2.5. Pharmacokinetics In Vivo of KET–LYS P1 and KET–LYS P2
3. Materials and Methods
3.1. Crystallization Conditions Screened to Identify Potential Polymorphs
3.2. General Procedure for the Preparation of KET-LYS Polymorph 1 (P1)
3.3. General Procedure for the Preparation of KET–LYS Polymorph 2 (P2)
3.4. X-ray Powder Diffraction (XRPD)
3.5. Thermal Analyses
3.6. Fourier-Transform Infrared Spectroscopy (FT-IR)
3.7. Solid-State NMR Characterization
3.8. Intrinsic Dissolution Rate
3.9. Multisensory Analysis
3.10. Pharmacokinetics
3.11. Statistics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Patents
References
- Duggirala, N.K.; Perry, M.L.; Almarsson, Ö.; Zaworotko, M.J. Pharmaceutical cocrystals: Along the path to improved medi-cines. Chem. Commun. 2015, 52, 640–655. [Google Scholar] [CrossRef]
- Karimi-Jafari, M.; Padrela, L.; Walker, G.; Croker, D.M. Creating Cocrystals: A Review of Pharmaceutical Cocrystal Preparation Routes and Applications. Cryst. Growth Des. 2018, 18, 6370–6387. [Google Scholar] [CrossRef]
- Dai, X.-L.; Chen, J.-M.; Lu, T.-B. Pharmaceutical cocrystallization: An effective approach to modulate the physicochemical properties of solid-state drugs. CrystEngComm 2018, 20, 5292–5316. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Fasulo, M.E.; Desper, J. Cocrystal or Salt: Does It Really Matter? Mol. Pharm. 2007, 4, 317–322. [Google Scholar] [CrossRef]
- Chadha, R.; Bhandari, S.; Haneef, J.; Khullar, S.; Mandal, S. Cocrystals of telmisartan: Characterization, structure elucidation, in vivo and toxicity studies. CrystEngComm 2014, 16, 8375–8389. [Google Scholar] [CrossRef]
- Zaworotko, M. Crystal engineering of the composition of pharmaceutical phases. Do pharmaceutical co-crystals represent a new path to improved medicines? Chem. Commun. 2004, 35, 1889–1896. [Google Scholar] [CrossRef]
- Tothadi, S.; Shaikh, T.R.; Gupta, S.; Dandela, R.; Chathakudath, P.V.; Nangia, A.K. Can We Identify the Salt–Cocrystal Continuum State Using XPS? Cryst. Growth Des. 2021, 21, 735–747. [Google Scholar] [CrossRef]
- Bernasconi, D.; Bordignon, S.; Rossi, F.; Priola, E.; Nervi, C.; Gobetto, R.; Voinovich, D.; Hasa, D.; Duong, N.T.; Nishiyama, Y.; et al. Selective Synthesis of a Salt and a Cocrystal of the Ethionamide–Salicylic Acid System. Cryst. Growth Des. 2019, 20, 906–915. [Google Scholar] [CrossRef]
- Bhogala, B.R.; Basavoju, S.; Nangia, A. Tape and layer structures in cocrystals of some di- and tricarboxylic acids with 4,4′-bipyridines and isonicotinamide. From binary to ternary cocrystals. CrystEngComm 2005, 7, 551–562. [Google Scholar] [CrossRef]
- Cruz-Cabeza, A.J. Acid–base crystalline complexes and the pKa rule. CrystEngComm 2012, 14, 6362–6365. [Google Scholar] [CrossRef]
- Childs, S.L.; Stahly, A.G.P.; Park, A. The Salt−Cocrystal Continuum: The Influence of Crystal Structure on Ionization State. Mol. Pharm. 2007, 4, 323–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stainton, P.; Grecu, T.; McCabe, J.; Munshi, T.; Nauha, E.; Scowen, I.J.; Blagden, N. First Comparative Study of the Three Polymorphs of Bis(isonicotinamide) Citric Acid Cocrystals and the Concomitant Salt 4-Carbamoylpyridinium Citrate Isonicotinamide. Cryst. Growth Des. 2018, 18, 4150–4159. [Google Scholar] [CrossRef] [Green Version]
- Losev, E.; Boldyreva, E. The effect of amino acid backbone length on molecular packing: Crystalline tartrates of glycine, β-alanine, γ-aminobutyric acid (GABA) and DL-α-aminobutyric acid (AABA). Acta Crystallogr. C Struct. Chem. 2018, 74, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Losev, E.A.; Boldyreva, E.V. A salt or a co-crystal—When crystallization protocol matters. CrystEngComm 2018, 20, 2299–2305. [Google Scholar] [CrossRef]
- Perumalla, S.R.; Wang, C.; Guo, Y.; Shi, L.; Sun, C.C. Robust bulk preparation and characterization of sulfamethazine and saccharine salt and cocrystal polymorphs. CrystEngComm 2019, 21, 2089–2096. [Google Scholar] [CrossRef]
- Jones, C.L.; Skelton, J.; Parker, S.C.; Raithby, P.R.; Walsh, A.; Wilson, C.C.; Thomas, L.H. Living in the salt-cocrystal continuum: Indecisive organic complexes with thermochromic behaviour. CrystEngComm 2019, 21, 1626–1634. [Google Scholar] [CrossRef] [Green Version]
- Trask, A.V. An Overview of Pharmaceutical Cocrystals as Intellectual Property. Mol. Pharm. 2007, 4, 301–309. [Google Scholar] [CrossRef] [Green Version]
- Raza, K.; Kumar, P.; Ratan, S.; Malik, R.; Arora, S. Polymorphism: The Phenomenon Affecting the Performance of Drugs. SOJ Pharm. Pharm. Sci. 2014, 1, 10. [Google Scholar]
- Seo, K.-S.; Bajracharya, R.; Lee, S.H.; Han, H.-K. Pharmaceutical Application of Tablet Film Coating. Pharmaceutics 2020, 12, 853. [Google Scholar] [CrossRef]
- Miles, L.; Hall, J.; Jenner, B.; Addis, R.; Hutchings, S. Predicting rapid analgesic onset of ibuprofen salts compared with ibu-profen acid: Tlag, Tlow, Tmed, and a novel parameter, TCmaxRef. Curr. Med. Res. Opin. 2018, 34, 1483–1490. [Google Scholar] [CrossRef]
- Altman, R.; Bosch, B.; Brune, K.; Patrignani, P.; Young, C. Advances in NSAID Development: Evolution of Diclofenac Products Using Pharmaceutical Technology. Drugs 2015, 75, 859–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varrassi, G.; Alon, E.; Bagnasco, M.; Lanata, L.; Mayoral-Rojals, V.; Paladini, A.; Pergolizzi, J.V.; Perrot, S.; Scarpignato, C.; Tölle, T. Towards an Effective and Safe Treatment of Inflammatory Pain: A Delphi-Guided Expert Consensus. Adv. Ther. 2019, 36, 2618–2637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverstein, R.M.; Webster, F.X.; Kiemle, D.J.; Bryce, D.L. Spectrometric Identification of Organic Compounds; Wiley: Hoboken, NJ, USA, 2015. [Google Scholar]
- Cerreia Vioglio, P.; Chierotti, M.R.; Gobetto, R. Pharmaceutical aspects of salt and cocrystal forms of APIs and characteri-zation challenges. Adv. Drug Deliv. Rev. 2017, 117, 86–110. [Google Scholar] [CrossRef]
- Chierotti, M.R.; Gobetto, R. Solid-state NMR studies of weak interactions in supramolecular systems. Chem. Commun. 2008, 14, 1621–1634. [Google Scholar] [CrossRef]
- Briard, P.; Rossi, J.C. Kétoprofène. Acta Crystallogr. C 1990, 46, 1036–1038. [Google Scholar] [CrossRef]
- Chen, S.; Xi, H.; Henry, R.F.; Marsden, I.; Zhang, G.G.Z. Chiral co-crystal solid solution: Structures, melting point phase diagram, and chiral enrichment of (ibuprofen)2(4,4-dipyridyl). CrystEngComm 2010, 12, 1485–1493. [Google Scholar] [CrossRef]
- Pa, W.; Ce, H.; Kd, H. L-Lysine: Exploiting powder X-ray diffraction to complete the set of crystal structures of the 20 directly encoded proteinogenic amino acids. Angew. Chem. Int. Ed. Engl. 2015, 54, 3973–3977. [Google Scholar]
- Pudjiastuti, P.; Wafiroh, S.; Hendradi, E.; Darmokoesoemo, H.; Harsini, M.; Fauzi, M.A.R.D.; Nahar, L.; Sarker, S.D. Disintegration, In vitro Dissolution, and Drug Release Kinetics Profiles of k-Carrageenan-based Nutraceutical Hard-shell Capsules Containing Salicylamide. Open Chem. 2020, 18, 226–231. [Google Scholar] [CrossRef]
- Chauhan, R. Taste Masking: A Unique Approach for Bitter Drugs. J. Stem Cell Biol. Transplant. 2017, 1, 12. [Google Scholar] [CrossRef] [Green Version]
- Sohi, H.; Sultana, Y.; Khar, R.K. Taste Masking Technologies in Oral Pharmaceuticals: Recent Developments and Approaches. Drug Dev. Ind. Pharm. 2004, 30, 429–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baharuddin, A.R.; Shaarani, S. The impact of geographical location on taste sensitivity and preference. Int. Food Res. J. 2015, 22, 731–738. [Google Scholar]
- Maniruzzaman, M.; Boateng, J.; Chowdhry, B.Z.; Snowden, M.J.; Douroumis, D. A review on the taste masking of bitter APIs: Hot-melt extrusion (HME) evaluation. Drug Dev. Ind. Pharm. 2013, 40, 145–156. [Google Scholar] [CrossRef]
- Arafa, M.F.; El-Gizawy, S.A.; Osman, M.A.; El Maghraby, G. Sucralose as co-crystal co-former for hydrochlorothiazide: Development of oral disintegrating tablets. Drug Dev. Ind. Pharm. 2015, 42, 1225–1233. [Google Scholar] [CrossRef]
- Maeno, Y.; Fukami, T.; Kawahata, M.; Yamaguchi, K.; Tagami, T.; Ozeki, T.; Suzuki, T.; Tomono, K. Novel pharmaceutical cocrystal consisting of paracetamol and trimethylglycine, a new promising cocrystal former. Int. J. Pharm. 2014, 473, 179–186. [Google Scholar] [CrossRef]
- Aitipamula, S.; Wong, A.B.; Kanaujia, P. Evaluating Suspension Formulations of Theophylline Cocrystals with Artificial Sweeteners. J. Pharm. Sci. 2018, 107, 604–611. [Google Scholar] [CrossRef]
- Lvova, L.; Denis, S.; Barra, A.; Mielle, P.; Salles, C.; Vergogian, C.; Di Natalle, C.; Paolesse, R.; Temple-Boyer, P.; Feron, G. Salt release monitoring with specific sensors in ‘in vitro’ oral and digestive environments from soft cheeses. Talanta 2012, 97, 171–180. [Google Scholar] [CrossRef]
- Lvova, L.; Gonçalves, C.G.; Petropoulos, K.; Micheli, L.; Volpe, G.; Kirsanov, D.; Legin, A.; Viaggiu, E.; Congestri, R.; Guzzella, L.; et al. Electronic tongue for microcystin screening in waters. Biosens. Bioelectron. 2016, 80, 154–160. [Google Scholar] [CrossRef]
- Lvova, L.; Jahatspanian, I.; Mattoso, L.H.; Correa, D.S.; Oleneva, E.; Legin, A.; Di Natale, C.; Paolesse, R. Potentiometric E-Tongue System for Geosmin/Isoborneol Presence Monitoring in Drinkable Water. Sensors 2020, 20, 821. [Google Scholar] [CrossRef] [Green Version]
- Hack, J.; Grills, D.C.; Miller, J.R.; Mani, T. Identification of Ion-Pair Structures in Solution by Vibrational Stark Effects. J. Phys. Chem. B 2016, 120, 1149–1157. [Google Scholar] [CrossRef]
- Falk, J.E. Porphyrins and Metalloporphyrins: A New Edition Based on the Original Volume by J. E. Falk; Elsevier: Amsterdam, The Netherlands, 1975. [Google Scholar]
- Levitchev, S.; Smirnova, A.; Khitrova, V.; Lvova, L.; Bratov, A.; Vlasov, Y. Photocurable carbonate-selective membranes for chemical sensors containing lipophilic additives. Sens. Actuators B Chem. 1997, 44, 397–401. [Google Scholar] [CrossRef]
- Lvova, L.; Di Natale, C.; D’Amico, A.; Paolesse, R. Corrole-based ion-selective electrodes. J. Porphyr. Phthalocyanines 2009, 13, 1168–1178. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KET–LYS P1 | KET–LYS P2 | ||
|---|---|---|---|
| 13C | |||
| 13C δ (ppm) | C atom | 13C δ (ppm) | C atom |
| 196.1 | 10 | 194.5 | 10 |
| 177.6 | 1 | 181.5 | 1 |
| 174.5 | 1′ | 176.1 | 1′ |
| 147.4 | 6 | 145.1 | 6 |
| 141.0 | 11 | 139.7 | 11 |
| 134.8 | 4 | 138.1 | 4 |
| 133.0 | Aromatic CH | 133.4 | Aromatic CH |
| 128.8 | Aromatic CH | 131.8 | Aromatic CH |
| 128.3 | Aromatic CH | 129.8 | Aromatic CH |
| 128.0 | Aromatic CH | 128.8 | Aromatic CH |
| 126.8 | Aromatic CH | 127.7 | Aromatic CH |
| 55.1 | 2′ | 53.2 | 2′ |
| 50.2 | 2 | 47.4 | 2 |
| 38.8 | 6′ | 34.5 | 6′ |
| 32.2 | 5′ | 25.9 | 5′ + 3′ |
| 29.6 | 3′ | 21.2 | 3 |
| 24.7 | 3 | 16.6 | 4′ |
| 22.3 | 4′ | / | / |
| 15N | |||
| 15N δ (ppm) | N atom | ||
| 43.0 | α | ||
| 32.8 | ε | ||
| Compound | IDR (Slope) mg/(cm2 × min) | Confidence Interval (95%) | |
|---|---|---|---|
| Cocrystal KET–LYS P1 | 1.453 | 1.411 | 1.495 |
| Salt KET–LYS P2 | 1.907 | 1.813 | 2.002 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aramini, A.; Bianchini, G.; Lillini, S.; Bordignon, S.; Tomassetti, M.; Novelli, R.; Mattioli, S.; Lvova, L.; Paolesse, R.; Chierotti, M.R.; et al. Unexpected Salt/Cocrystal Polymorphism of the Ketoprofen–Lysine System: Discovery of a New Ketoprofen–l-Lysine Salt Polymorph with Different Physicochemical and Pharmacokinetic Properties. Pharmaceuticals 2021, 14, 555. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14060555
Aramini A, Bianchini G, Lillini S, Bordignon S, Tomassetti M, Novelli R, Mattioli S, Lvova L, Paolesse R, Chierotti MR, et al. Unexpected Salt/Cocrystal Polymorphism of the Ketoprofen–Lysine System: Discovery of a New Ketoprofen–l-Lysine Salt Polymorph with Different Physicochemical and Pharmacokinetic Properties. Pharmaceuticals. 2021; 14(6):555. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14060555
Chicago/Turabian StyleAramini, Andrea, Gianluca Bianchini, Samuele Lillini, Simone Bordignon, Mara Tomassetti, Rubina Novelli, Simone Mattioli, Larisa Lvova, Roberto Paolesse, Michele Remo Chierotti, and et al. 2021. "Unexpected Salt/Cocrystal Polymorphism of the Ketoprofen–Lysine System: Discovery of a New Ketoprofen–l-Lysine Salt Polymorph with Different Physicochemical and Pharmacokinetic Properties" Pharmaceuticals 14, no. 6: 555. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14060555