
Anti-Tumoral Effects of a (1H-Pyrrol-1-yl)Methyl-1H-Benzoimidazole Carbamate Ester Derivative on Head and Neck Squamous Carcinoma Cell Lines


 ,
,  , , , , , ,
, , , , , , 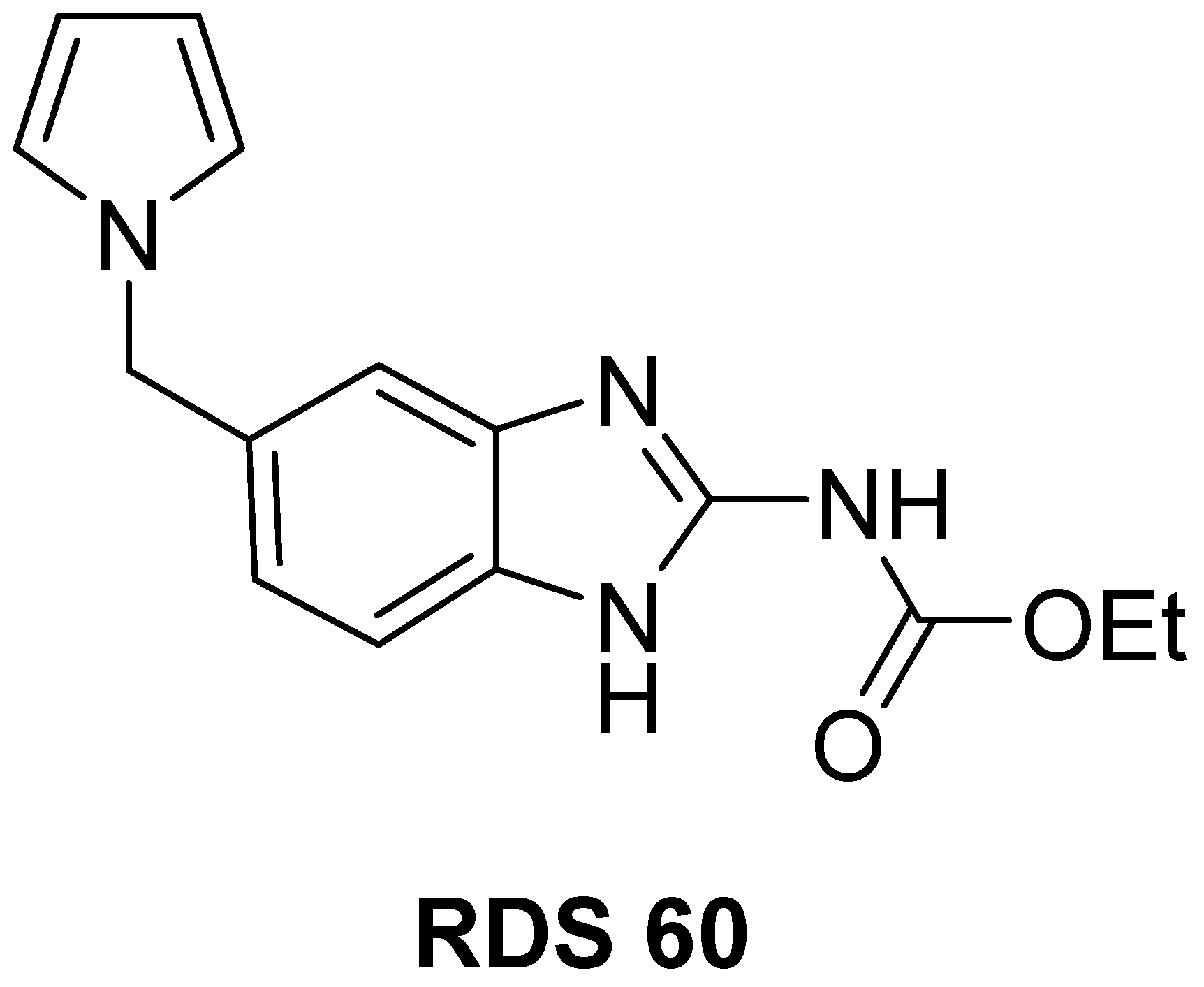{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
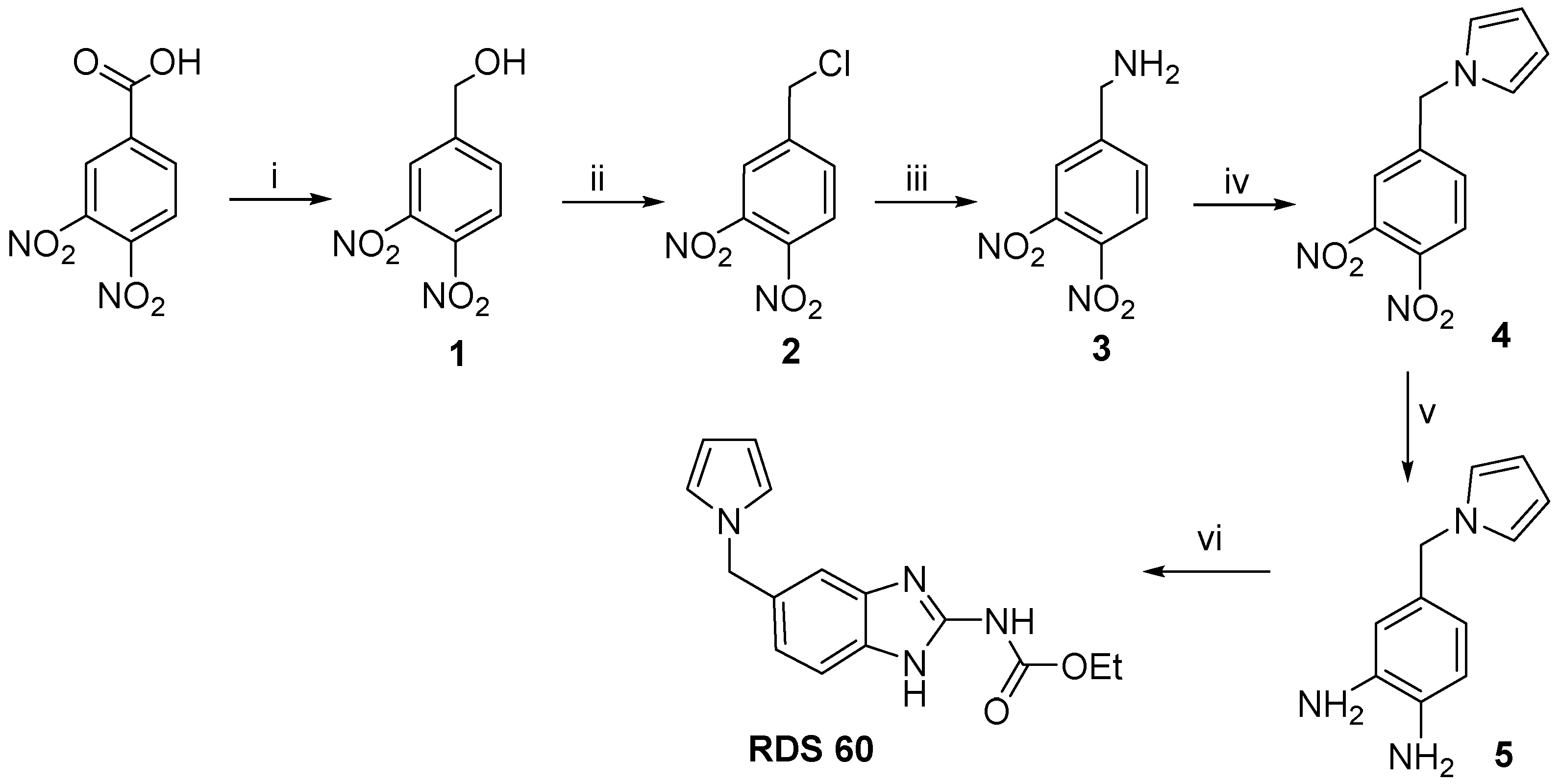
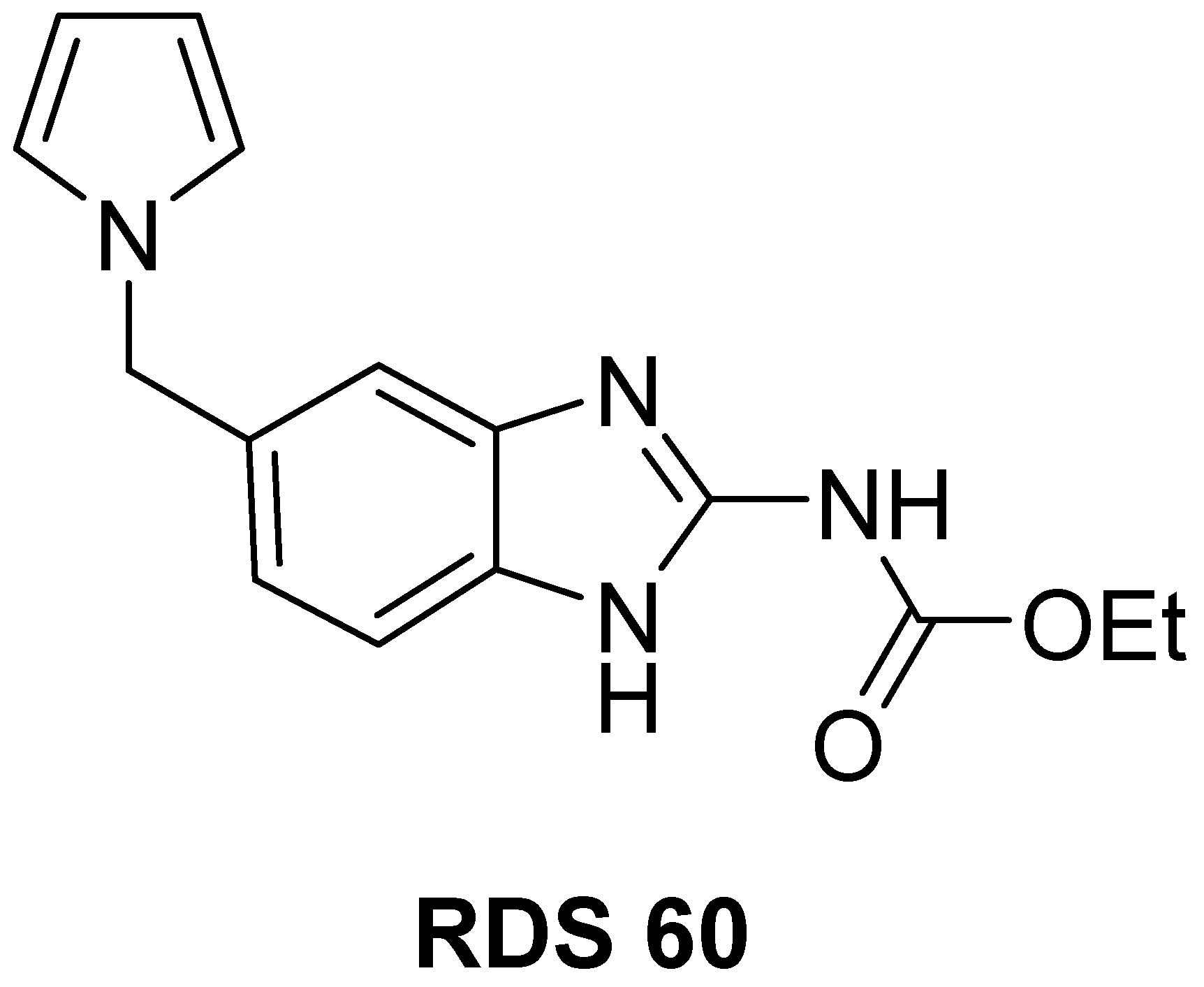
2.1. Chemistry
2.2. RDS 60 Inhibits Proliferation and Impairs Mitotic Spindle Formation in HNSCC Cell Lines
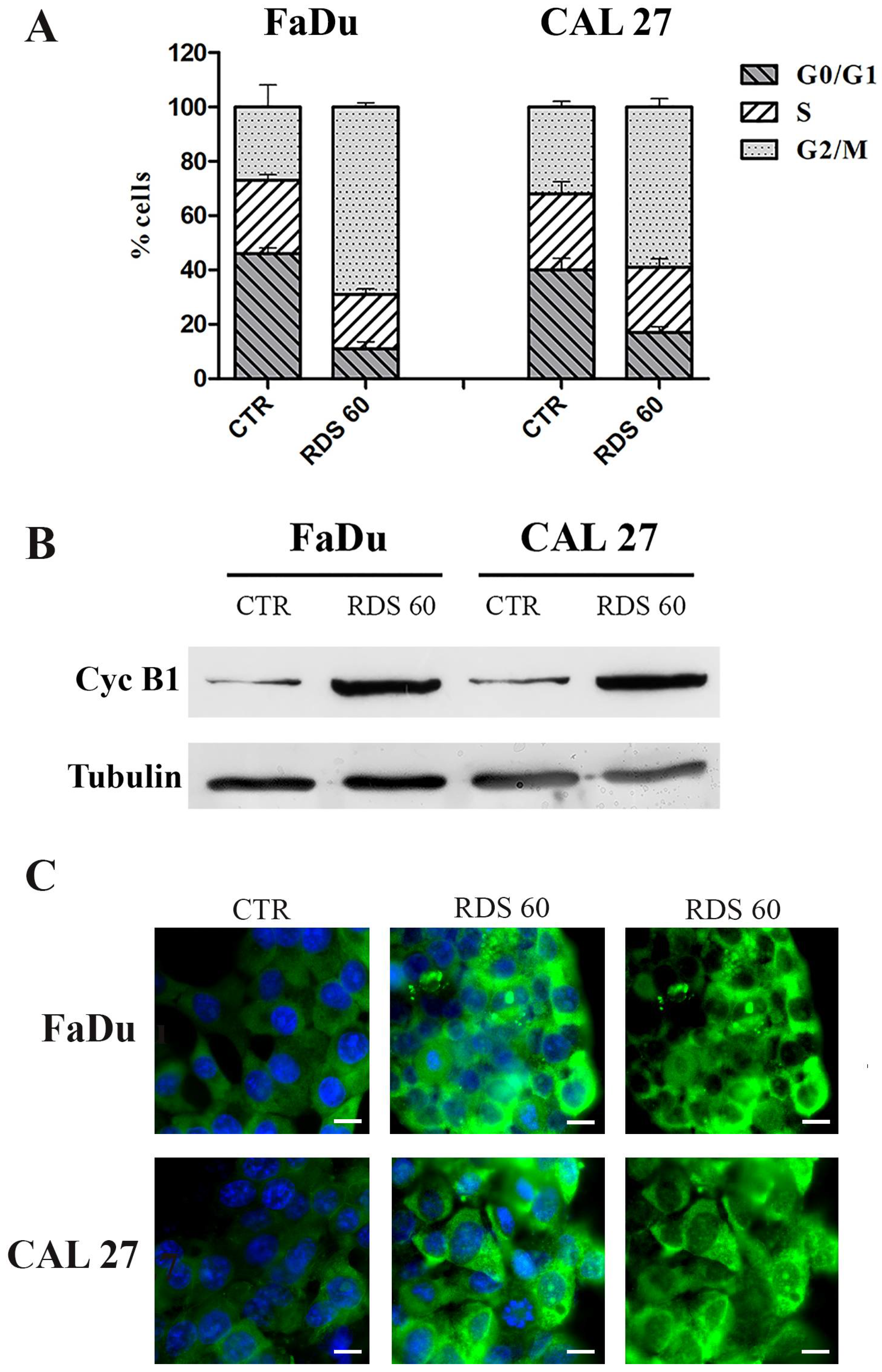
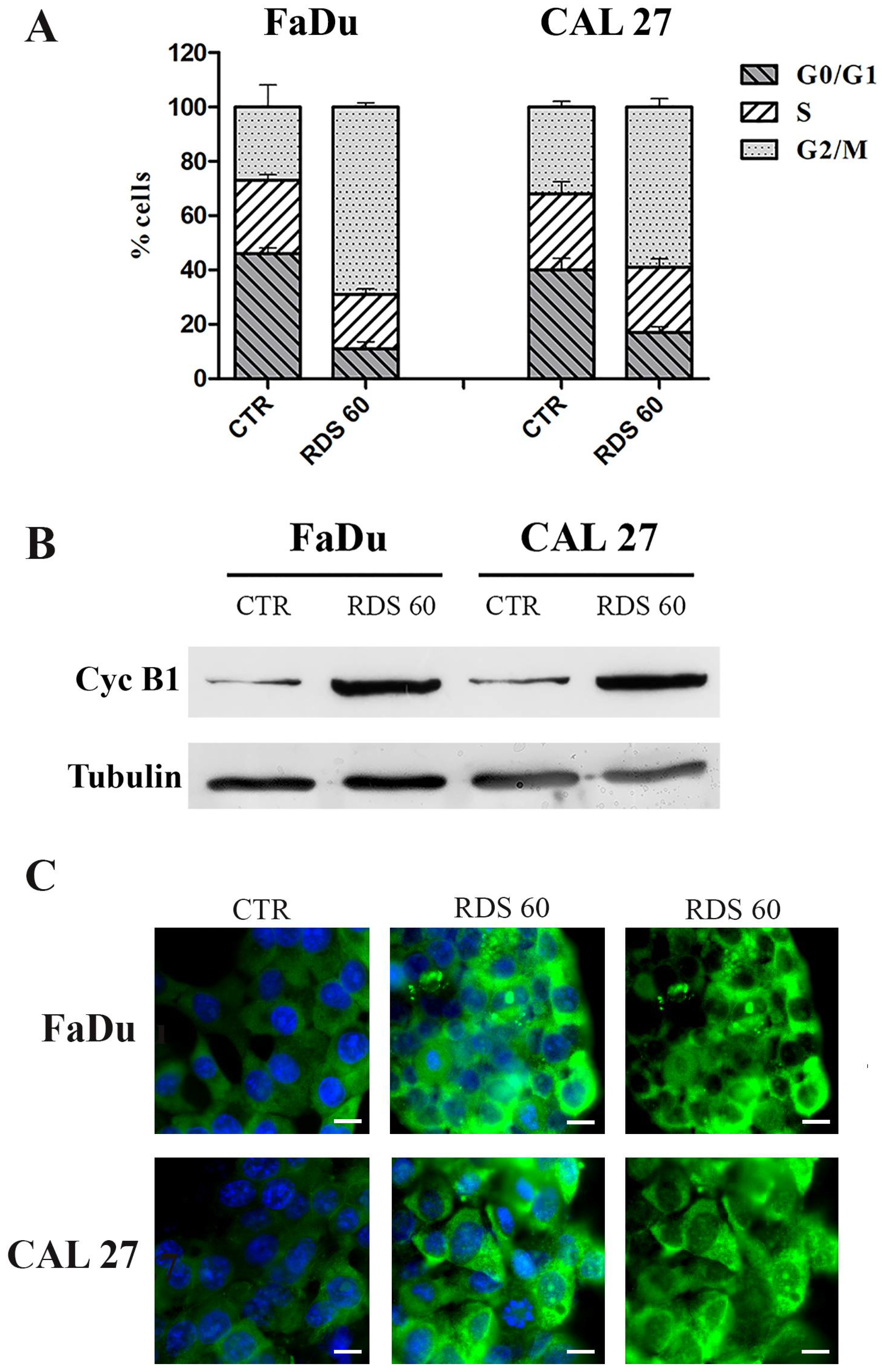
2.3. RDS 60 Determines a Block in G2/M and Up-Regulates Cytoplasmic Cyclin B1 in HNSCC Cell Lines
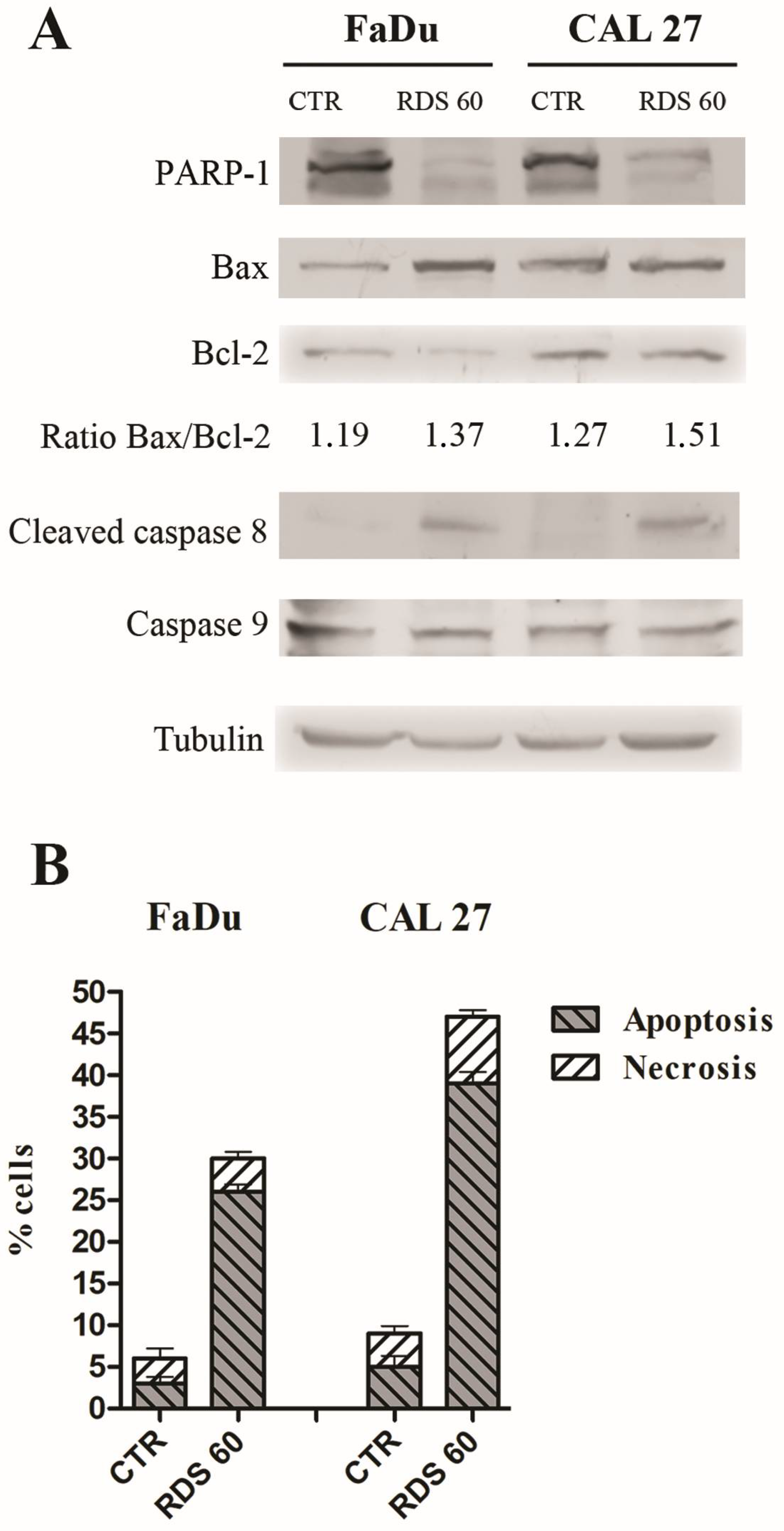
2.4. RDS 60 Induces Apoptosis in HNSCC Cell Lines
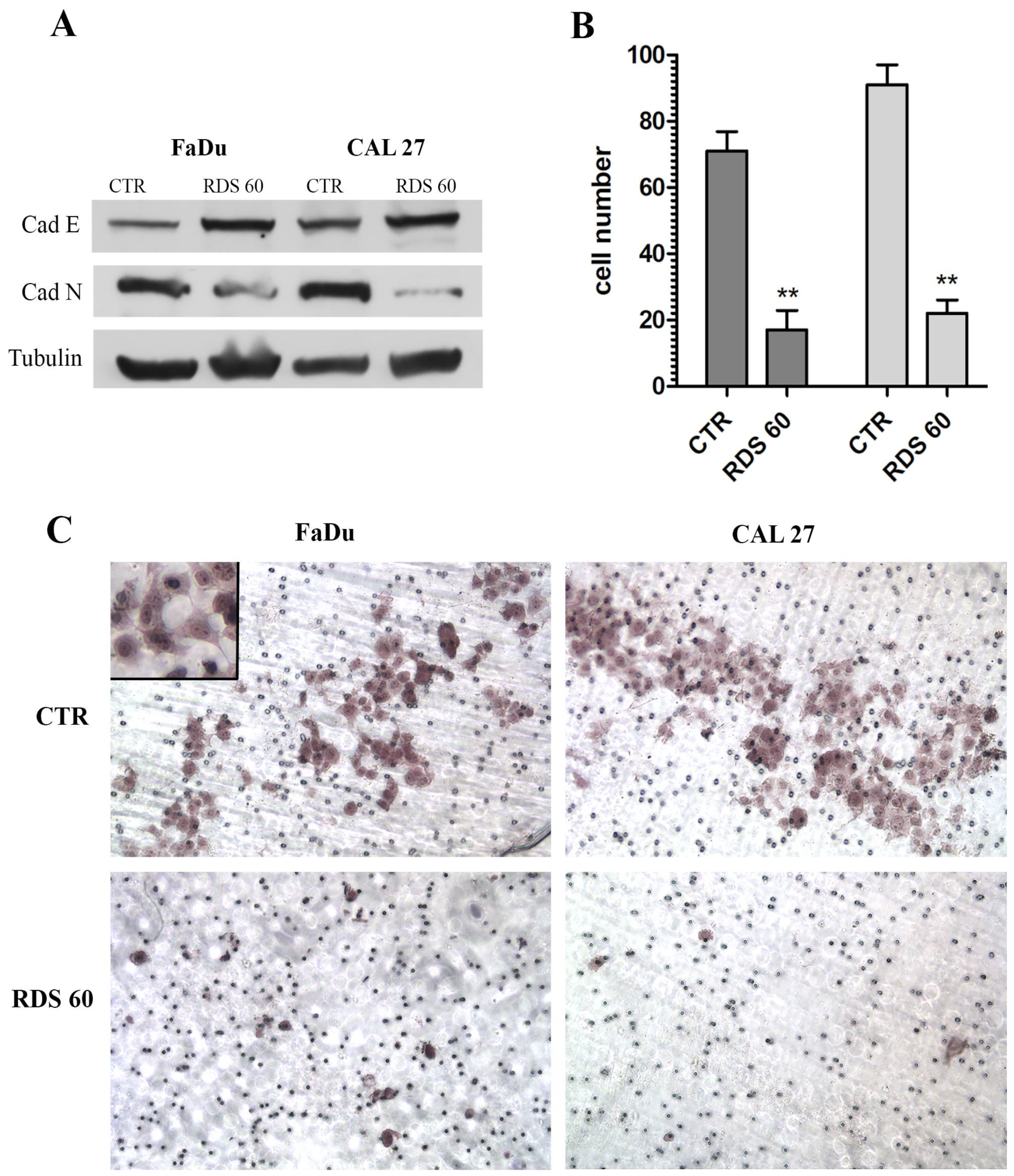
2.5. RDS 60 Reverses EMT and Inhibits Migration of HNSCC Cell Lines
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. General Instrumentation
4.1.2. Specific Procedures and Characterization
4.2. Cell Cultures and Treatment
4.3. Cytotoxicity Assay
4.4. Flow Cytometry Analysis
4.5. Immunofluorescence
4.6. Western Blot Analysis
4.7. Invasion Assay
4.8. Statistical Analysis and Graphic Programs
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Florian, S.; Mitchison, T.J. Anti-microtubule drugs. Methods Mol. Biol. 2016, 1413, 403–421. [Google Scholar] [CrossRef]
- Vasquez, R.J.; Howell, B.; Yvon, A.M.; Wadsworth, P.; Cassimeris, L. Nanomolar concentrations of nocodazole alter microtubule dynamic instability in vivo and in vitro. Mol. Biol. Cell 1997, 8, 973–985. [Google Scholar] [CrossRef] [Green Version]
- Jordan, M.A.; Thrower, D.; Wilson, L. Effects of vinblastine, podophyllotoxin and nocodazole on mitotic spindles. Implications for the role of microtubule dynamics in mitosis. J. Cell Sci. 1992, 102, 401–416. [Google Scholar] [CrossRef] [PubMed]
- Fraschini, R. Factors that control mitotic spindle dynamics. Adv. Exp. Med. Biol. 2017, 925, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Giannakakou, P. Targeting microtubules for cancer chemotherapy. Curr. Med. Chem. Anticancer Agents 2005, 5, 65–71. [Google Scholar] [CrossRef] [Green Version]
- Frezzato, F.; Trimarco, V.; Martini, V.; Gattazzo, C.; Ave, E.; Visentin, A.; Cabrelle, A.; Olivieri, V.; Zambello, R.; Facco, M.; et al. Leukemic cells from chonic lymphocytic leaukemia patients undergo apoptosis following microtubule depolymerization and Lyn inhibition by nocodazole. Br. J. Haematol. 2014, 165, 659–672. [Google Scholar] [CrossRef]
- Wang, Y.; Jeng, J.; Chen, R.; Tseng, H.; Chen, L.; Liang, Y.; Lin, C.; Chen, C.; Chu, J.; Ho, W.; et al. Ketoconazole potentiates the antitumor effects of nocodazole: In vivo therapy for human tumor xenografts in nude mice. Mol. Carcinog. 2002, 34, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, H.; Gigant, B.; Yu, Y.; Wu, Y.; Chen, X.; Lai, Q.; Yang, Z.; Chen, Q.; Yang, J. Structure of a diverse set of colchicine binding site inhibitors in complex with tubulin provide a rationale for drug discovery. FEBS J. 2016, 283, 102–111. [Google Scholar] [CrossRef]
- Signoretto, E.; Honisch, S.; Briglia, M.; Faggio, C.; Castagna, M.; Lang, F. Nocodazole induced suicidal death of human erythocytes. Cell. Physiol. Biochem. 2016, 38, 379–392. [Google Scholar] [CrossRef]
- Shrivastava, N.; Naim, M.J.; Alam, M.J.; Nawaz, F.; Ahmed, S.; Alam, O. Benzimidazole scaffold as anticancer agent: Synthetic approaches and structure-activity relationship. Arch. Pharm. 2017, 350. [Google Scholar] [CrossRef]
- Madia, V.N.; Messore, A.; Pescatori, L.; Saccoliti, F.; Tudino, V.; De Leo, A.; Bortolami, M.; Scipione, L.; Costi, R.; Rivara, S.; et al. Novel benzazole derivatives endowed with potent antiheparanase activity. J. Med. Chem. 2018, 61, 6918–6936. [Google Scholar] [CrossRef] [PubMed]
- Messore, A.; Madia, V.N.; Pescatori, L.; Saccoliti, F.; Tudino, V.; De Leo, A.; Bortolami, M.; De Vita, D.; Scipione, L.; Pepi, F.; et al. Novel symmetrical benzazolyl derivatives endowed with potent anti-heparanase activity. J. Med. Chem. 2018, 61, 10834–10859. [Google Scholar] [CrossRef] [Green Version]
- Khattab, M.; Al-Karmalawy, A.A. Revisiting activity of some nocodazole analogues as potential anticancer drugs using molecular docking and DFT calculations. Front. Chem. 2021. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, J.; Xiao, M.; Li, W.; Miller, D.D. An overview of tubulin inhibitors that interact with the colchicine binging site. Pharm. Res. 2012, 29, 2943–2971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massa, S.; Di Santo, R.; Artico, M. Potential antitumor agents. IV. Pyrrole analogues of oncodazole. J. Heterocycl. Chem. 1990, 27, 1131–1133. [Google Scholar] [CrossRef]
- Yu, A.J.; Choi, J.S.; Swanson, M.S.; Kokot, N.C.; Brown, T.N.; Yan, G.; Sinha, U.K. Association of race/ethnicity, stage, and survival in oral cavity squamous cell carcinoma: A SEER study. OTO Open 2019, 3. [Google Scholar] [CrossRef]
- Gaździcka, J.; Golabek, K.; Strzelczyk, J.K.; Ostrowska, Z. Epigenetic modifications in head and neck cancer. Biochem. Genet. 2019, 58, 213–244. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.L.; Zhu, Z.L.; Huang, M.C.; Tang, Y.J.; Tang, Y.L.; Liang, X.H. Susceptibility of multiple primary cancers in patients with head and neck cancer: Nature or nurture? Front. Oncol. 2019, 9, 1275. [Google Scholar] [CrossRef] [PubMed]
- Tabor, M.P.; Brakenhoff, R.H.; Ruijter-Schippers, H.J.; van der Wal, J.E.; Snow, G.B.; Leemans, C.R.; Braakhuis, B.J.M. Multiple head and neck tumors frequently originate from a single preneoplastic lesion. Am. J. Pathol. 2002, 161, 1051–1060. [Google Scholar] [CrossRef] [Green Version]
- Leemans, C.R.; Braakhuis, B.J.; Brakenhoff, R.H. The molecular biology of head and neck cancer. Nat. Rev. Canc. 2011, 11, 9–22. [Google Scholar] [CrossRef]
- Di Villeneuve, L.; Souza, I.L.; Tolentino, F.D.S.; Ferrarotto, R.; Schvartsman, G. Salivary gland carcinoma: Novel targets to overcome treatment resistance in advanced disease. Front. Oncol. 2020, 10, 580141. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Gholipour, M.; Taheri, M.; Shirvani Farsani, Z. MicroRNA profile in the squamous cell carcinoma: Prognostic and diagnostic roles. Heliyon 2020, 6, e05436. [Google Scholar] [CrossRef]
- Moore, G.F.; Mambourger, M.; Gervaldo, M.; Poluektov, O.G.; Rajh, T.; Gust, D.; Moore, T.A.; Moore, A.L. A bioinspired construct that mimics the proton coupled electron transfer between P680 and the Tyrz-His190 pair of photosystems II. JACS Comm. 2008, 130, 10466–10467. [Google Scholar] [CrossRef] [PubMed]
- Hagting, A.; Jackman, M.; Simpson, K.; Pines, J. Translocation of cyclin B1 to the nucleus at phophase requires a phosphorylation-dependent nuclear import signal. Curr. Biol. 1999, 9, 680–689. [Google Scholar] [CrossRef] [Green Version]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M. The microtubule depolymerizing drugs nocodazole and colchicine inhibit the uptake of Listeria monocytogenes by P388D1 macrophages. FEMS Microbiol. Lett. 1998, 160, 87–90. [Google Scholar] [CrossRef] [Green Version]
- Liarte, S.; Bernabé-García, A.; Nicolás, F.J. Human skin keratinocytes on sustained TGF-β stimulation reveal partial EMT features and weaken growth arrest responses. Cells 2020, 9, 306. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, M.; Christofori, G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef] [Green Version]
- Boeve, K.; Melchers, L.J.; Schuuring, E.; Roodenburg, J.L.; Halmos, G.B.; van Dijk, B.A.; van der Vegt, B.; Witjes, M.J. Addition of tumour infiltration depth and extranodal extension improves the prognostic value of the pathological TNM classification for early-stage oral squamous cell carcinoma. Histopathology 2019, 75, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HADAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, A.; Ackerman, B.; Gundersen, G.G. Tubulin acetylation and cell motility. Nature 2003, 421, 230. [Google Scholar] [CrossRef]
- Mukhtar, E.; Adhami, V.A.; Mukhtar, H. Targeting microtubules by natural agents for cancer therapy. Mol. Cancer Ther. 2014, 13, 275–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taglieri, L.; Saccoliti, F.; Nicolai, A.; Peruzzi, G.; Madia, V.N.; Tudino, V.; Messore, A.; Di Santo, R.; Artico, M.; Taurone, S.; et al. Discovery of a pyrimidine compound endowed with antitumor activity. Investig. New Drugs 2020, 38, 39–49. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nicolai, A.; Madia, V.N.; Messore, A.; De Vita, D.; De Leo, A.; Ialongo, D.; Tudino, V.; Tortorella, E.; Scipione, L.; Taurone, S.; et al. Anti-Tumoral Effects of a (1H-Pyrrol-1-yl)Methyl-1H-Benzoimidazole Carbamate Ester Derivative on Head and Neck Squamous Carcinoma Cell Lines. Pharmaceuticals 2021, 14, 564. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14060564
Nicolai A, Madia VN, Messore A, De Vita D, De Leo A, Ialongo D, Tudino V, Tortorella E, Scipione L, Taurone S, et al. Anti-Tumoral Effects of a (1H-Pyrrol-1-yl)Methyl-1H-Benzoimidazole Carbamate Ester Derivative on Head and Neck Squamous Carcinoma Cell Lines. Pharmaceuticals. 2021; 14(6):564. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14060564
Chicago/Turabian StyleNicolai, Alice, Valentina Noemi Madia, Antonella Messore, Daniela De Vita, Alessandro De Leo, Davide Ialongo, Valeria Tudino, Elisabetta Tortorella, Luigi Scipione, Samanta Taurone, and et al. 2021. "Anti-Tumoral Effects of a (1H-Pyrrol-1-yl)Methyl-1H-Benzoimidazole Carbamate Ester Derivative on Head and Neck Squamous Carcinoma Cell Lines" Pharmaceuticals 14, no. 6: 564. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14060564